

Abstract
BACKGROUND
Congenital analbuminemia (CAA) is a very rare disorder. Our data describes the clinical features and laboratory results of a new case established by mutation analysis of the albumin gene in a 39-year-old woman presenting with hypercholesterolemia. Our findings contribute to shed light on the molecular genetics of the disorder and confirm that safe and well tolerated hypocholesterolemic treatment with atorvastatin may be administered in dislipidemic patient with CAA in order to reduce their cardiovascular risk.
CASE SUMMARY
Our patient presented with a history of hypercholesterolemia and referred asthenia and heaviness in both legs. She was born from healthy and non-consanguineous parents and her development was normal. She had not familiarity for early cardiovascular disease, and did not report personal history of hypertension, chronic kidney or liver diseases. Clinical laboratories results showed critically reduced value of albumin whereas other serum proteins were elevated. Main causes of hypoalbuminemia (proteinuria, inflammatory state and insufficient hepatic synthesis) were ruled out by normal procedures and laboratory tests. So the hypothesis of a CAA was tested through mutation analysis of the albumin gene that revealed a homozygous CA deletion in exon 12, at nucleotide positions c1614-1615. This finding brought to the diagnosis of CAA. Currently the patient receives Atorvastatin 20 mg od and undergoes clinical and laboratory follow-up every six months. She never needed albumin infusions.
CONCLUSION
Our experience shows how treatment with atorvastatin may be safely administered and well tolerated in patients affected by CAA.
Keywords: Congenital analbuminemia, Hypercholesterolemia, Hypoalbuminemia, Rare disease, Case report
Core tip: The role of albumin in lipids transport and metabolism makes hypercholesterolemia the most detectable biochemical sign in analbuminemic patients. Despite patients affected by congenital analbuminemia have no severe clinical symptoms in adulthood; they might have high cardiovascular risk, morbility and perinatal mortality as suggested in our patients’ familiar and personal medical history.
INTRODUCTION
Congenital analbuminemia (CAA; Online Mendelian Inheritance in Man database, OMIM # 616000) is a rare autosomal recessive disorder characterized by the complete absence, or severe reduction of circulating serum albumin. Only about ninety cases have been reported worldwide without gender or ethnic predilection, most of which are listed in the Register of analbuminemia cases[1]. Although diagnosis is usually made by serum protein electrophoresis, mutation analysis of ALB gene is needed for confirmation. No severe clinical phenotype is detected in adult analbuminemic individuals because of the compensatory increase of other serum proteins[2]. The most common clinical signs are edema, hypotension and fatigue, while typical biochemical sign is hypercholesterolemia.
In this report we describe the clinical findings of a dislipidemic young woman and the molecular characterisation of the ALB gene, which confirmed the diagnosis of CAA.
CASE PRESENTATION
Chief complaints
A 39-year-old woman with history of hypercholesterolemia referred asthenia and heaviness in both legs. She had sedentary lifestyle, balanced diet and did not complain intestinal issues.
Physical examination upon admission
Her weight was 56 kg, height 170 cm, body mass index 19 kg/m2, waist circumference 73 cm and her blood pressure was tested at a value of 13/10 KPa.
Personal and family history
She was born from healthy and non-consanguineous parents and her development was normal. She had not familiarity for early cardiovascular disease, and did not report personal history of hypertension, chronic kidney or liver diseases.
During her life she needed hospitalizations for a miscarriage (28 years old) and a delivery of a healthy male newborn by caesarean section (30 years old). The pedigree of her family is reported in Figure 1 and shows the premature death of one of her brothers for unknown reasons at the age of 1 wk.
Figure 1.
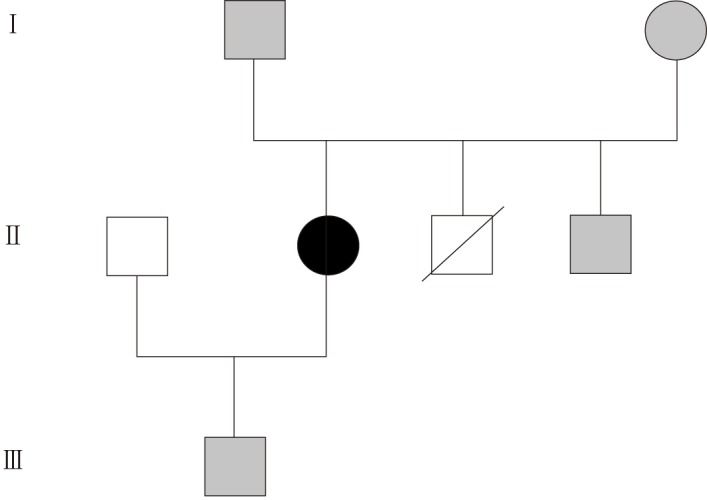
Family pedigree. The analbuminemic patient is represented by a black symbol. Grey symbols denote heterozygous subjects. Void symbols indicate individuals not examined in the present study. One of the three siblings of generation II died for unknown reasons in his first week of life.
Laboratory examinations
The patient’s lipid profile showed a significant elevation of the total (321 mg/dL), low-density lipoprotein (LDL) (161 mg/dL) and high-density lipoprotein (HDL) (118 mg/dL) cholesterol, whereas triglyceride levels were normal. Serum apolipoprotein-A and -B were 291 mg/dL (n.v. 115 mg/dL-210 mg/dL) and 145 mg/dL (n.v. 55 mg/dL-135 mg/dL) respectively.
Since the patient and her relatives had neither clinical history nor physical signs (xanthoma and xanthelasma) of a genetic hypercholesterolemia, we studied other possible causes of her dyslipidemia through further laboratory tests. Blood count, liver and kidney function, coagulation tests, autoimmunity assays, PCR and urine dipstick were normal. Thyroid function tests displayed a subclinical hypothyroidism. Albumin level, detected by nephelometric method, was critically reduced (0.2 g/dL, n.v. 3.4 g/dL-5 g/dL) and total bilirubin was undetectable. Calcium level was 7.5 mg/dL (n.v. 8.5 mg/dL-10.1 mg/dL), total protein 5.8 g/dL (n.v. 6.4 g/dL-8.2 g/dL), pre-albumin 0.46 g/L (n.v. 0.2 g/L-0.4 g/L). Finally, serum protein electrophoresis (Figure 2) showed the presence of a minimal amount of serum albumin (1.6%, n.v.55.8%-66.1%), with a simultaneous increase of the other serum protein fractions (alpha-1, alpha-2, beta-1, beta-2 and gamma), including transferrin (607 mg/dL, n.v. 200 mg/dL-360 mg/dL), alpha 1-antitrypsin (2.56 g/L, n.v. 0.9 g/L-2.0 g/L) and complement C4 (0.46 g/L, n.v. 0.10 g/L-0.40 g/L). Those seemed to compensate the lack of albumin[1] .
Figure 2.
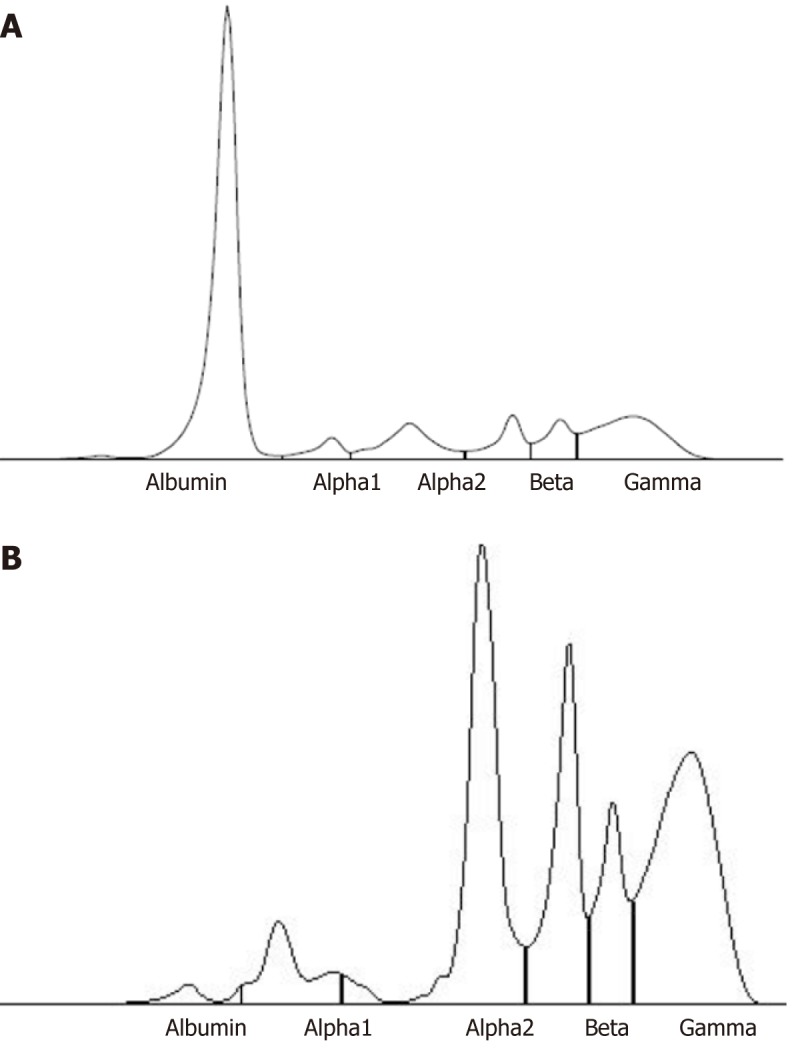
Capillary electrophoresis profile of serum proteins in a control (A) and in our patient (B). The latter shows the presence of a minimal amount of serum albumin (1.6%, n.v. 55.8%-66.1%), with a simultaneous increase of the other serum protein fractions: alpha 1 (7%, n.v. 2.9%-4.9%), alpha 2 (29.7%, n.v. 7.1%-11.8%), beta 1 (18.4%, n.v. 4.7%-7.2%), beta 2 (10.7%, n.v. 3.2%-6.5%), gamma (32.6%, n.v. 11.1%-18.8%).
These findings led us to investigate on the patient’s hypoalbuminemia. Proteinuria was excluded by normal spot urine albumin/creatinine ratio and total protein/creatinine ratio. Other possible secondary causes of hypoalbuminemia, such as inflammatory state and insufficient hepatic synthesis, were ruled out by normal laboratory tests and abdominal ultrasonography. The latter was negative except for mild hepatic steatosis. All the other examined members of the family (her parents, her brother, and her son) showed albumin level close to the upper limit of the normal range, suggesting that they may be heterozygous for a variation in the ALB[3] . For the above reasons, we tested the hypothesis of CAA by mutation analysis of the ALB[4]. After we obtained informed consent, we collected blood samples from all the available members of the family and two unrelated healthy volunteers as a control, and extracted genomic DNA from whole blood. For a rapid identification of variations in the ALB, we used a gel-based mutation detection strategy, which we developed and applied to the identification of many other cases of CAA[2]. Shortly, we PCR amplified the fourteen genomic fragments of the ALB encompassing the fourteen coding exons and their intron-exon junctions, using the specific primer pairs described by Watkins et al[5] . The fragments were then examined by heteroduplex and SSCP analysis: The combination of these two techniques usually allowed us to identify the region of ALB containing the molecular defect, which was then submitted to direct DNA sequencing[2]. In the present case heteroduplex analysis clearly indicated that the only detectable change in both homozygous and heterozygous samples occurred in the 386 bp long region amplified by using PCR primers A23A and A24A encompassing exon 12 and the intron 11-exon 12 and exon 12-intron 13 junctions (Figure 3A). All the other members of the family are heterozygous, since they show the presence of four bands corresponding to homoduplex and heteroduplex PCR products (Figure 3A, lanes 2, 2′, 3, 3′, 4, 4’ 5, 5’). The homozygous sample (Figure 3A, lanes 1 and 1′) revealed only one band but with a different mobility when compared with controls (Figure 3A, lanes 6, 7, 6′, and 7′). No variation due to conformation polymorphism could be seen under these electrophoretic conditions (data not shown). The results of the DNA sequence analysis performed on the abnormal fragment showed that our patient is homozygous for a CA deletion near the 3′ end of exon 12, at nucleotide positions c. 1614-1615, according to the Human Genome Variation Society rules, i.e,. starting from the initiator codon (Figure 3B). The subsequent frame-shift should give rise to a predicted translation product of 516 amino acid residues instead of the 585 found in the mature protein (p.Leu540Phefs*2), in which the sequence Cys(538)-Thr-Leu-Ser has been changed to Cys(538)-Thr-Phe-Stop. Unfortunately, we could not perform a search for this truncated variant in the serum of our patient, but no evidence was found so far for the presence of the putative albumin molecule produced in all the cases of CAA studied at the molecular level[1,6]. The electropherograms from the parents confirmed that they are both heterozygous for the same mutation (data not shown). This result brought to the molecular diagnosis of CAA in our patient.
Figure 3.

HA and DNA sequence analysis of exon 12 of ALB in the affected family. A: HA analysis. The DNA encompassing exon 12 and the exon-intron junction from the patient, her mother and father, and two controls were amplified with primers A23A and A24A and the fragments were electrophoresed onto a non-denaturing polyacrylamide gel: lane 1, proband; lane 2, mother; lane 3, father; lane 4, brother, lane 5, son, and lanes 6-7, controls. The same samples were denatured and cooled before loading: lane 1′, proband; lane 2′, mother; lane 3′, father; lane 4’, brother, lane 5’, son, and lanes 6′-7′, controls. Heteroduplexes are evident in the parents, in the brother, and in the son (lanes 2, 3, 4, 5, and 2′ 3′, 4’ and 5’), indicating a heterozygous condition, while an abnormal homoduplex is present in the homozygous proband. No variation due to conformation polymorphism could be seen under these electrophoretic conditions and therefore the result of SSCP analysis are not reported in the figure; B: DNA sequence of the mutated region of exon 12 in the patient. The arrows indicate the two bases that are deleted in the patient: CA at nucleotide positions c. 1614_1615. The patient is homozygous for this deletion.
FINAL DIAGNOSIS
Based on the biochemical findings and on the mutation analysis of the ALB the final diagnosis was CAA and hypercholesterolemia, the latter not of genetic origin.
TREATMENT
CAA did not require albumin infusions, while hypercholesterolemia was successfully treated with atorvastatin. A low-cholesterol, low-saturated fat diet was prescribed without any significant effect on serum lipid (total cholesterol increased from 321 mg/dL to 334 mg/dL, LDL from 161 mg/dL to 192 mg/dL). Successful and well tolerated treatment with atorvastatin was started at the initial daily dose of 10 mg od, increased to 20 mg od: From baseline total and LDL cholesterol dropped by 23% and 47% respectively, HDL cholesterol increased by 22% while apolipoprotein-B decreased by 25%. Apolipoprotein-A showed no relevant modifications. This observation together with the low blood pressure might explain why, despite the high lipid levels, evident clinical signs of early atherosclerosis have not been observed in the patient[3]: No plaques on carotid artery walls where found at ultrasound imaging study.
OUTCOME AND FOLLOW-UP
The patient undergoes clinical and laboratory follow-up twice a year. Atorvastatin treatment appears to be successful and well tolerated and so far she never needed albumin infusions.
DISCUSSION
CAA is a rare autosomal recessive disorder which has an estimated prevalence of less than one in one million[3]. The so far reported data on the about 50 cases of CAA studied at the molecular level allowed to identify twenty-five different variations within the ALB[1]. Among them, the most common are splicing defects, nonsense variants, and frame shift/deletions, as was in the here reported case. Most of the albumin variants are unique and identified only in single individuals or within the same family[1]. The variation we identified in our patient was previously found in a young Turkish woman (analbuminemia Safranbolu)[2]. The presence of the same defect in unrelated analbuminemic individuals might indicate hypermutable regions in the gene, as it probably is for the relatively more commons variants, as analbuminemia Guimarães (c.1289+1G>A), identified in three unrelated families, and especially analbuminemia Kayseri, which accounts for about one-third of the cases characterized at the molecular level[1].
The absence or severe reduction of circulating serum albumin is usually shown by serum protein electrophoresis[7], but the final diagnosis of CAA is based on the mutational analysis of the ALB.
The benignancy of clinical manifestations and laboratory tests of analbuminemic patients often lead to a misdiagnosis or to a delayed diagnosis of this rare condition. Despite data of analbuminemic patients’ follow-up are not available, early diagnosis of this rare condition may prevent hypercholesterolemia related cardiovascular events[8]. Thus, treatment with statins seems to be recommended to reduce long term cardiovascular risk, according to experiences reported in literature[9,10] .
In this case the diagnosis of CAA was not suspected by clinical examination: No edema or lipodystrophy were found. The typical pattern of serum protein electrophoresis and the presence of markedly low albumin level brought to the assumption of the analbuminemia as possible cause of her hypercholesterolemia. Furthermore, the mechanism by which this possible interaction may occur remains equivocal. In vitro data suggest that a key role is played by the increased liver production of other serum proteins such as Apo A1, Apo B, and Apo E and the contextual reduction of the triglycerides-rich lipoprotein clearance. The latter mechanism have been explained through the inhibition of the lipoprotein lipase by free fatty acids not bounded to albumin[11].
In Nagase rats with hereditary analbuminemia increased LDL and HDL cholesterol levels result from HMG-CoA reductase and Hepatic ACAT-2 protein abundance, while LDL-HDL cholesterol liver receptors are normally expressed. Hence, ACAT-2 hepatic protein catalyzes packaging and secretion of apolipoprotein B-containing lipoproteins by the esterification of cholesterol in the liver[12] .
On the other hand, normal plasma HDL/total cholesterol ratio in analbuminemia is due to normal activity of converting free cholesterol to cholesterol ester in HDL played by LCAT which seems to be more expressed in female analbuminemic rats.
According to the evidence, CAA should be associated with premature atherosclerosis and cardiovascular events. Even if there is no proved strategy for decreasing cardiovascular risk in analbuminemic patients with hypecholesterolemia, other statin treatment in human analbuminemics are described in literature[13,14].
Treatment with simvastatin 40 mg od in dislipidemic patients with CAA has been administered in two African adults, a 21-year-old caucasian male and a 61-year-old Afro-American male. Serum lipid profile showed a decrease of LDL-cholestrol of 38% and 48% respectively after 20 wk of treatment discontinued for three- to five-fold increase in creatine kinase[9].
The latter consideration explains why albumin-bound drugs should be carefully administrered and monitored during treatment in these patients[13]. Long term treatment with atorvastatin was tested in one other Italian patient with CAA: a 38-year-old man received atorvastatin 40 mg od with a decrease of total and LDL cholesterol from baseline by 37.7% and 50.6% respectively. HDL cholesterol increased by 13.4%[10]. Treatment was safe and no elevated values of creatine kinase, liver enzymes were detected as in our case.
CONCLUSION
According to our experience, safe and well tolerated hypocholesterolemic treatment with atorvastatin may be administered in dislipidemic patient with CAA in order to reduce their cardiovascular risk.
Despite analbuminemic patients could be asymptomatic, parents’ screening and clinical follow-up is warrant for conditions such as hypercholesterolemia, atherosclerosis, hypercoagulability, osteoporosis, respiratory tract infections, obstetrical complications and pharmacodynamics consequences[13,14]. In contrast with the relatively benign presentation of CAA in adult individuals, findings like preterm birth, low birth weight, presence of miscarriages, respiratory distress with frequent hospital admissions, and mild developmental delay have been often described in analbuminemic families[15]. Indeed, our patients’ familiar and personal medical history seems to confirm perinatal and intrauterine complications, and the crucial role of albumin in the first periods of life.
Footnotes
Informed consent statement: The study participants provided informed written consent prior to their treatments and study enrollment.
Conflict-of-interest statement: All authors declare no conflict of interest related to this study or its publication.
CARE Checklist (2016): The manuscript was prepared and revised according to the CARE Checklist (2016).
Manuscript source: Unsolicited manuscript
Peer-review started: October 18, 2018
First decision: November 15, 2018
Article in press: December 12, 2018
Specialty type: Medicine, research and experimental
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Caceres-Loriga FM, Pan SL S- Editor: Wang JL L- Editor: A E- Editor: Tan WW
Contributor Information
Patrizia Suppressa, Department of Internal Medicine and Rare Disease Centre, University Hospital of Bari, Bari 70124, Italy. patrizia.suppressa@gmail.com.
Concetta Carbonara, Department of Medicine, University of Bari, School of Medicine, Bari 70124, Italy.
Francesca Lugani, Laboratory of Molecular Nephrology, Istituto Giannini Gaslini, IRCCS, Genova 16148, Italy.
Monica Campagnoli, Department of Molecular Medicine, University of Pavia, Pavia 27100, Italy.
Teresa Troiano, Department of Clinical Pathology, University Hospital of Bari, Bari 70124, Italy.
Lorenzo Minchiotti, Department of Molecular Medicine, University of Pavia, Pavia 27100, Italy.
Carlo Sabbà, Department of Interdisciplinary Medicine, Geriatric Unit and Rare Disease Center, "Aldo Moro" University, Bari 70125, Italy.
References
- 1.Kragh-Hansen U, Minchiotti L, Campagnoli M. The albumin website. Available from: https://www.albumin.org.
- 2.Dagnino M, Caridi G, Aydin Z, Ozturk S, Karaali Z, Kazancioglu R, Cefle K, Gursu M, Campagnoli M, Galliano M, Minchiotti L. A novel frameshift deletion in the albumin gene causes analbuminemia in a young Turkish woman. Clin Chim Acta. 2010;411:1711–1715. doi: 10.1016/j.cca.2010.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Peters T., Jr . All About Albumin: Biochemistry, Genetics, and Medical Applications, 1st ed. San Diego: Academic Press; 1995. [Google Scholar]
- 4.Minghetti PP, Ruffner DE, Kuang WJ, Dennison OE, Hawkins JW, Beattie WG, Dugaiczyk A. Molecular structure of the human albumin gene is revealed by nucleotide sequence within q11-22 of chromosome 4. J Biol Chem. 1986;261:6747–6757. [PubMed] [Google Scholar]
- 5.Watkins S, Madison J, Galliano M, Minchiotti L, Putnam FW. A nucleotide insertion and frameshift cause analbuminemia in an Italian family. Proc Natl Acad Sci U S A. 1994;91:2275–2279. doi: 10.1073/pnas.91.6.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minchiotti L, Galliano M, Caridi G, Kragh-Hansen U, Peters T., Jr Congenital analbuminaemia: molecular defects and biochemical and clinical aspects. Biochim Biophys Acta. 2013;1830:5494–5502. doi: 10.1016/j.bbagen.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Baldo-Enzi G, Baiocchi MR, Vigna G, Andrian C, Mosconi C, Fellin R. Analbuminaemia: a natural model of metabolic compensatory systems. J Inherit Metab Dis. 1987;10:317–329. doi: 10.1007/BF01799973. [DOI] [PubMed] [Google Scholar]
- 8.Demirsoy E, Sirin G, Ozker E. Coronary artery bypass surgery in a patient with analbuminemia. Tex Heart Inst J. 2011;38:85–87. [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess LJ, Marais AD. The use of simvastatin in analbuminaemia. Cardiovasc Drugs Ther. 2001;15:555–558. doi: 10.1023/a:1013780007561. [DOI] [PubMed] [Google Scholar]
- 10.Del Ben M, Burattin M, Arca M, Ceci F, Violi F, Angelico F. Treatment of severe hypercholesterolemia with atorvastatin in congenital analbuminemia. Am J Med. 2004;117:803–804. doi: 10.1016/j.amjmed.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 11.Maugeais C, Braschi S, Ouguerram K, Maugeais P, Mahot P, Jacotot B, Darmaun D, Magot T, Krempf M. Lipoprotein kinetics in patients with analbuminemia. Evidence for the role of serum albumin in controlling lipoprotein metabolism. Arterioscler Thromb Vasc Biol. 1997;17:1369–1375. doi: 10.1161/01.atv.17.7.1369. [DOI] [PubMed] [Google Scholar]
- 12.Liang K, Vaziri ND. HMG-CoA reductase, cholesterol 7alpha-hydroxylase, LCAT, ACAT, LDL receptor, and SRB-1 in hereditary analbuminemia. Kidney Int. 2003;64:192–198. doi: 10.1046/j.1523-1755.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- 13.Frohlich J, Pudek MR, Cormode EJ, Sellers EM, Abel JG. Further studies on plasma proteins, lipids, and dye- and drug-binding in a child with analbuminemia. Clin Chem. 1981;27:1213–1216. [PubMed] [Google Scholar]
- 14.Koot BG, Houwen R, Pot DJ, Nauta J. Congenital analbuminaemia: biochemical and clinical implications. A case report and literature review. Eur J Pediatr. 2004;163:664–670. doi: 10.1007/s00431-004-1492-z. [DOI] [PubMed] [Google Scholar]
- 15.Toye JM, Lemire EG, Baerg KL. Perinatal and childhood morbidity and mortality in congenital analbuminemia. Paediatr Child Health. 2012;17:e20–e23. [PMC free article] [PubMed] [Google Scholar]