

Summary
Many disease‐relevant phenotypes modeled in inbred mice have been shown to be strain‐dependent, indicating the important influence of genetic background on disease phenotypes. Although C57BL/6 mice are one of the most commonly used inbred strains in laboratory research, there are multiple substrains (eg, B6J vs B6N) that have been separated for more than 50 years. Thus, understanding the substrain differences is important for scientific rigor and reproducibility. In this study, seizure susceptibility, spontaneous seizures, and survival were compared between Scn1a +/− mice on (C57BL/6J × 129S6/SvEvTac)F1 (F1J) vs (C57BL/6N × 129S6/SvEvTac)F1 (F1N) strain backgrounds. F1N.Scn1a +/− mice were more susceptible to hyperthermia‐induced seizures, yet had milder spontaneous seizures and improved survival relative to F1J.Scn1a +/− mice. Our results indicate that choice of C57BL/6 substrain may significantly alter disease phenotypes and should be considered carefully in experimental design using the Scn1a +/− Dravet mouse model, as well as other mouse models of epilepsy.
Keywords: Dravet syndrome, epilepsy, genetics, mouse model, voltage‐gated sodium channel
1. INTRODUCTION
Mouse genetic models have offered powerful insights into our understanding of gene function and disease mechanisms. C57BL/6J (B6J) mice have been the most widely used inbred strain for knockout and transgenic models, whereas the substrain C57BL/6N (B6N) is becoming increasingly popular due to large initiatives like the International Knockout Mouse Consortium.1 Separated since 1951, these substrains have accumulated significant genetic variations.2, 3, 4 Comparative genomic analysis revealed numerous coding variants between B6J (Jackson Laboratory) and B6N (National Institutes of Health) substrains, including 34 single nucleotide variants (32 missense/one nonsense/one splice site), two indels resulting in frameshifts, and 15 structural variants overlapping genes.5 There are documented substrain‐dependent phenotypic differences in diverse areas, including neurobehavioral/neurologic function, vision, metabolism, and inflammatory response.5, 6 Understanding and controlling for these differences are critical for experimental validity and reproducibility.
Mutations in the neuronal voltage‐gated sodium channel gene SCN1A most frequently result in Dravet syndrome. Mice with heterozygous deletion of Scn1a recapitulate features of Dravet syndrome, including spontaneous seizures, susceptibility to hyperthermia‐induced seizures, and premature lethality.7, 8, 9, 10, 11 Differential effects of strain background on phenotype severity in Scn1a +/− mice have been reported.7, 8, 9, 10, 11 Scn1a +/− mice on the 129S6/SvEvTac (129S6) strain have no overt phenotype. When crossed with C57BL/6J, resulting F1.Scn1a +/− mice have a severe phenotype with spontaneous seizures and premature lethality.8 Genetic analysis revealed several responsible modifier loci and genes, including Gabra2, Cacna1g, and Hlf.7, 8, 12, 13 The effect of the C57BL/6 substrain on phenotype expressivity in Scn1a +/− mice has not been systematically evaluated, although C57BL/6 substrain differences in susceptibility to induced seizures have been documented.14, 15, 16 In the current study, we compared seizure susceptibility, spontaneous seizures, and survival in Scn1a +/− mice with B6J vs B6N alleles. Several differences were detected, with B6N alleles associated with reduced severity of spontaneous seizures and improved survival. Our results suggest that these strains carry differential modifier alleles that alter phenotype severity in the context of Scn1a heterozygous deletion, and emphasize the importance of substrain selection in experimental design and interpretation.
2. MATERIALS AND METHODS
2.1. Mice
The Scn1a tm1Kea/Mmjax (MMRRC#37107‐JAX) mouse line was generated in 129S6/SvEvTac ES cells and has been maintained continuously by backcrossing to 129S6/SvEvTac mice (Taconic Biosciences, Hudson, New York), and is referred to as 129.Scn1a +/− herein.8 Experimental mice were generated by crossing 129.Scn1a +/− mice with C57BL/6J (B6J) (stock#000664; Jackson Laboratory, Bar Harbor, Maine) or C57BL/6NJ (B6N) (stock#005304, Jackson Laboratory) mice. The resulting F1 offspring were (129 × B6J)F1.Scn1a +/− or (129 × B6N)F1.Scn1a +/−, abbreviated as F1J.Scn1a +/− and F1N.Scn1a +/−, respectively. Mice were genotyped by polymerase chain reaction (PCR), as described previously.8 Mice were group‐housed with access to food and water ad libitum and maintained on a 14‐hour light/10‐hour dark schedule. All studies were approved by the Northwestern University Animal Care and Use Committee in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Principles outlined in the Animal Research: Reporting of In Vivo Experiments guideline and Basel declaration were considered when planning experiments.
2.2. Hyperthermia‐induced seizure thresholds
On P18, mice underwent a hyperthermia‐induced seizure threshold assay as described previously.9 Core body temperatures at baseline, and at onset of myoclonic and generalized tonic‐clonic seizures (GTCS) were recorded. Following a 5‐minute acclimation to the temperature probe and chamber, body temperature was gradually elevated by 0.5°C every 2 minutes until onset of a clonic convulsion with loss of posture. Once body temperature reached 42.5°C, the heat lamp was turned off and mice remained in the chamber until a GTCS occurred or 5 minutes elapsed. Mice without a GTCS during the assay were considered seizure‐free. Immediately following the GTCS, mice were rapidly cooled to 37°C on a cooling pad and then weaned into a cage for spontaneous seizure monitoring, with the exception of one F1N.Scn1a +/− mouse that did not exhibit a hyperthermia‐induced GTCS and was excluded from subsequent spontaneous seizure analysis. Rare failures to exhibit a hyperthermia‐induced GTCS have been observed in F1J.Scn1a +/− mice.9
2.3. Spontaneous seizure monitoring
All mice that exhibited a hyperthermia‐induced seizure underwent continuous video‐monitoring for 60 hours between midday on P19 through P21. Groups of 2‐3 mice were placed in a recording chamber with access to food and water ad libitum. Analysis was performed on 18 F1J.Scn1a +/− and 20 F1N.Scn1a +/− mice. Lower group size relative to the hyperthermia assay was due to technical issues (video record corruption and rapid cooling failure). GTCS events (forelimb clonus with rearing/loss‐of‐posture) and severity (with or without tonic hindlimb extension at a 180 degrees relative to torso) were scored by reviewers blinded to the groups as described.9 Following cessation of recording, mice were monitored for short‐term survival until P30.
2.4. Extended survival monitoring
A separate cohort of mice weaned at P18‐20 into standard vivarium cages with 4‐5 sex‐ and age‐matched mice was monitored for survival until P60. Daily health surveillance was conducted and any mouse exhibiting signs of unexpected morbidity was euthanized and excluded from the study.
2.5. Statistics
Statistical analyses were conducted using Prism 7 software (GraphPad Inc., La Jolla, California). Between‐group comparisons for survival and temperature thresholds for hyperthermia‐induced seizures were conducted using time‐to‐event analysis with P‐values determined with LogRank Mantel‐Cox tests. Events occurring after predetermined cutoffs were censored (P30 or P60 for survival; 42.5°C for hyperthermia GTCS threshold). Spontaneous seizure frequencies and GTCS temperature thresholds were compared by Mann‐Whitney U tests. Within‐genotype comparisons between sexes indicated no differences, therefore groups were collapsed across sex. Data are presented as median or mean ± standard error of the mean (SEM) and P < 0.05 was considered statistically significant.
3. RESULTS
3.1. F1N vs F1J strain differences in hyperthermia‐induced seizure thresholds
F1N.Scn1a +/− mice had a lower myoclonic seizure threshold with a median temperature for the first myoclonic seizure of 40.2°C in F1N.Scn1a +/− vs 40.6°C in F1J.Scn1a +/− mice (P < 0.023; LogRank Mantel‐Cox; Figure 1A). For GTCS threshold, there was no difference in the incidence curve between F1N.Scn1a +/− and F1J.Scn1a +/− (P > 0.332; LogRank Mantel‐Cox; Figure 1B). However, comparison of mice that exhibited a seizure at ≤42.5°C revealed a lower GTCS threshold temperature in F1N.Scn1a +/− (41.8 ± 0.1°C) vs F1J.Scn1a +/− (42.2 ± 0.1°C) mice (P < 0.009; Mann‐Whitney U test). Baseline body temperature was not different between F1J.Scn1a +/− (36.2 ± 0.2°C) and F1N.Scn1a +/− mice (36.4 ± 0.2°C) (P > 0.502; Unpaired t test).
Figure 1.

Temperature threshold for hyperthermia‐induced myoclonic seizures and generalized tonic‐clonic seizures (GTCS) in F1J.Scn1a +/− (F1J; n = 24) and F1N.Scn1a +/− (F1N; n = 26) mice. A, Cumulative myoclonic seizure incidence curve. The myoclonic seizure threshold was lower in F1N.Scn1a +/− (median = 40.2°C) compared to F1J.Scn1a +/− mice (median = 40.6°C) (P < 0.023, LogRank Mantel‐Cox). B, Cumulative GTCS incidence curve. GTCS incidence curves were not significantly different between F1N.Scn1a +/− and F1B.Scn1a +/− mice (P > 0.332, LogRank Mantel‐Cox). However, for mice that exhibited a GTCS at ≤42.5°C, average GTCS threshold temperature was significantly lower between F1N (41.8 ± 0.1°C) and F1J (42.2 ± 0.1°C) (P < 0.009, Mann Whitney U‐test)
3.2. F1N vs F1J strain differences in spontaneous seizure severity and survival
Spontaneous GTCS frequency was monitored for 60 hours following induction of a single hyperthermia‐induced seizure. There was no difference in average frequency of spontaneous GTCS between F1J.Scn1a +/− (1.7 ± 0.6 seizures/d) and F1N.Scn1a +/− mice (1.6 ± 0.4 seizures/) (Figure 2A; P > 0.586; Mann‐Whitney). Furthermore, the proportion of mice exhibiting GTCS did not differ between strains, with 12/20 F1N.Scn1a +/− mice and 8/18 F1J.Scn1a +/− mice exhibiting ≥1 GTCS during the 60‐hour monitoring window (P > 0.516; Fisher's exact test).
Figure 2.
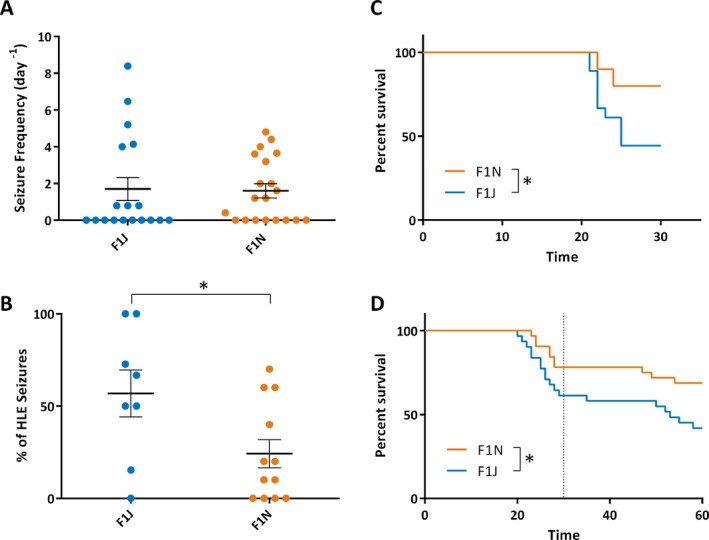
Spontaneous seizure profiles and survival of F1J.Scn1a +/− (F1J) and F1N.Scn1a +/− (F1N) mice. A, Spontaneous seizure frequencies were not significantly different between F1J.Scn1a +/− (n = 20) and F1N.Scn1a +/− (n = 18) mice (P > 0.586, Mann‐Whitney). Average seizure frequency is depicted by the thick horizontal line, and error bars represent standard error of the mean (SEM). B, A smaller proportion of generalized tonic‐clonic seizures (GTCS) advanced to tonic‐hindlimb extension in F1N.Scn1a +/− compared to F1J.Scn1a +/− mice (24 ± 8% vs 57 ± 13%, respectively; P < 0.031, Student's t test; n = 8 for F1J and n = 12 for F1N, includes only mice with GTCS). Average proportion of tonic hindlimb extension seizures is depicted by the thick horizontal line, and error bars represent standard error of the mean (SEM). C, Kaplan Meier survival curves comparing 30‐d survival following induction of a single hyperthermia‐induced seizure on P18. Survival was significantly lower in F1J.Scn1a +/− vs F1N.Scn1a +/− mice (44% vs 80%, respectively; P < 0.023 Logrank Mantel‐Cox, n = 20 per strain). D, Kaplan Meier survival curves comparing 60‐day survival in a separate cohort of naive mice that did not undergo hyperthermia induction. F1N.Scn1a +/− mice had significantly improved survival to P60 relative to F1J.Scn1a +/− mice (69% vs 42% for F1N and F1J, respectively; P < 0.032 Logrank Mantel‐Cox; n = 20 per strain). A vertical line at P30 is provided for qualitative comparison with panel C
Although there were no differences in seizure incidence or frequency, there was a difference in the proportion of GTCS advancing to the most severe stage of tonic hindlimb extension (hindlimbs at 180 degrees to the torso). In F1J.Scn1a +/− mice, 57 ± 13% of seizures advanced to tonic hindlimb extension compared to only 24 ± 8% in F1N.Scn1a +/− mice (Figure 2B; P < 0.031; unpaired t test). The proportion of GTCS advancing to tonic hindlimb extension in F1J.Scn1a +/− mice was similar to that of previous reports.9 Following video recording, this cohort was monitored for short‐term survival to 30 days of age. Only 44% of F1J.Scn1a +/− mice survived to P30 compared to 80% of F1N.Scn1a +/− mice (Figure 2C; P < 0.023; LogRank Mantel‐Cox). All recorded deaths followed a hindlimb extension seizure in video records, although not all hindlimb extension seizures led to death. There was no relationship between hyperthermia‐induced GTCS threshold and subsequent seizure frequency or survival (R 2 < 0.008 for F1J and R 2 < 0.133 for F1N).
To further evaluate the survival difference, we performed an extended survival study on a separate cohort of naive F1J.Scn1a +/− and F1N.Scn1a +/− mice that were not subjected to hyperthermia. F1N.Scn1a +/− mice had better survival, with 69% (22/32) surviving to P60, compared to 42% (13/31) of F1J.Scn1a +/− mice (Figure 2D; P < 0.032; LogRank Mantel‐Cox). Consistent with our previous observations,9, 17 induction of a GTCS by hyperthermia at P18 accelerated lethality in F1J.Scn1a +/−, with P30 survival of 44% following hyperthermia priming compared to P30 survival of 65% without hyperthermia (Figure 2C,D). There was no difference in P30 survival between F1N.Scn1a +/− mice with and without hyperthermia priming (80% and 78%, respectively; Figure 2C,D).
4. DISCUSSION
In this study, we observed strain‐dependent phenotype variability in F1N.Scn1a +/− compared to F1J.Scn1a +/− mice, including differences in hyperthermia‐induced seizure thresholds, spontaneous seizure severity, and survival.
Seizures with tonic hindlimb extension involve brainstem, a critical component of the autonomic nervous system that modulates cardiovascular and respiratory function.18 Despite unaltered seizure frequency in F1N.Scn1a +/− compared to F1J.Scn1a +/− mice, the lower proportion of GTCS advancing to tonic hindlimb extension in F1N.Scn1a +/− mice may contribute to improved survival. This is consistent with our previous observations that poor survival of Scn1a +/− mice is more strongly associated with occurrence of tonic hindlimb extension than overall GTCS frequency.9
F1N.Scn1a +/− mice had a lower temperature threshold for myoclonic and GTCS compared to F1J.Scn1a +/− mice. This is consistent with previous studies that showed lower flurothyl‐induced myoclonic and GTCS thresholds for C57BL/6N vs C57BL/6J mice, whereas repeated seizure inductions were more likely to progress rapidly from forebrain to brainstem seizures in C57BL/6J than other B6 substrains or DBA.15 The propensity of C57BL/6J mice to quickly progress to brainstem seizures may contribute to the elevated mortality risk in Scn1a +/− mice with C57BL/6J alleles.
In addition to differences in seizure susceptibility and variable expressivity of epilepsy in different C57BL/6 substrains, there are also reported differences among mice of the same substrain from different vendors. Differential phenotype severity was reported preliminarily in Scn1a +/− offspring resulting from crosses with C57BL/6 sourced from Charles River vs Jackson Laboratories.19 Similarly, differences have been reported in susceptibility to chemoconvulsant‐induced seizures in C57BL/6N mice from Harlan vs Charles River, and between different sublines from the same vendor.16, 20 Thus, it is critical for investigators to consistently source C57BL/6 substrains from the same vendor and use littermate controls to establish baseline phenotypes in their local colony.
Our results demonstrate that B6N and B6J contribute modifier alleles that differentially influence survival and severity phenotypes in Scn1a +/− mice. Although there are relatively fewer genetic differences among substrains (ie, B6J vs B6N) than between completely different strains (ie, C57BL/6J vs 129S6/SvEvTac), these differences can still influence cardiovascular, metabolic, or neurologic phenotypes.5 Preliminary examination of C57BL/6J and C57BL/6N coding sequence variation5 revealed missense variants in 2 epilepsy‐associated genes: Cyfip2 and Jmjd1c. Cyfip2 encodes cytoplasmic FMR1 interacting protein 2, which is involved in actin dynamics, axon elongation, and dendritic spine remodeling. Heterozygous de novo variants were identified in 4 unrelated individuals with epileptic encephalopathy.21 Functional studies showed gain‐of‐function effects resulting in dysregulation of actin dynamics.21 C57BL/6NJ carries an S968F missense variant (rs240617401) in Cyfip2 that results in loss‐of‐function and was associated with altered cocaine‐response phenotypes in C57BL/6NJ vs C57BL/6J.22 It is possible that S968F may also influence excitation/inhibition balance. Jmjd1c encodes a putative histone demethylase. Sáez et al23 reported heterozygous de novo variants in JMJD1C in 2 unrelated individuals with Rett syndrome and showed reduced dendritic complexity with small interfering RNA knockdown in mouse hippocampal neurons. C57BL/6J codes for a leucine at position 1715, whereas C57BL/6NJ and other strains have a proline in this position (rs13480628), although the effect on protein function is predicted to be benign.24, 25 Additional genetic and functional evidence is required for formal candidate gene consideration.
Effects of genetic background on disease phenotypes are often underestimated. This study adds to mounting evidence that C57BL/6 substrains harbor modifier alleles that must be carefully taken into account for experimental design and data interpretation.
DISCLOSURES
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
This work was supported by NIH grant R01‐NS084959 (J.A.K.). We thank Alexandra Huffman and Samantha Duarte for technical assistance.
Kang SK, Hawkins NA, Kearney JA. C57BL/6J and C57BL/6N substrains differentially influence phenotype severity in the Scn1a +/− mouse model of Dravet syndrome. Epilepsia Open. 2019;4:164–169. 10.1002/epi4.12287
REFERENCES
- 1. Brown SD, Moore MW. Towards an encyclopaedia of mammalian gene function: the International Mouse Phenotyping Consortium. Dis Model Mech. 2012;5:289–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bothe GW, Bolivar VJ, Vedder MJ, et al. Genetic and behavioral differences among five inbred mouse strains commonly used in the production of transgenic and knockout mice. Genes Brain Behav. 2004;3:149–57. [DOI] [PubMed] [Google Scholar]
- 3. Bryant CD, Zhang NN, Sokoloff G, et al. Behavioral differences among C57BL/6 substrains: implications for transgenic and knockout studies. J Neurogenet. 2008;22:315–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Matsuo N, Takao K, Nakanishi K, et al. Behavioral profiles of three C57BL/6 substrains. Front Behav Neurosci. 2010;4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simon MM, Greenaway S, White JK, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ashworth A, Bardgett ME, Fowler J, et al. Comparison of neurological function in males and females from two substrains of C57BL/6 mice. Toxics. 2015;3:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawkins NA, Zachwieja NJ, Miller AR, et al. Fine mapping of a Dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA‐Seq. PLoS Genet. 2016;12:e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller AR, Hawkins NA, McCollom CE, et al. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2014;13:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hawkins NA, Anderson LL, Gertler TS, et al. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann Clin Transl Neurol. 2017;4:326–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ogiwara I, Miyamoto H, Morita N, et al. Nav1.1 localizes to axons of parvalbumin‐positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–9. [DOI] [PubMed] [Google Scholar]
- 12. Calhoun JD, Hawkins NA, Zachwieja NJ, et al. Cacna1 g is a genetic modifier of epilepsy in a mouse model of Dravet syndrome. Epilepsia. 2017;58:e111–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hawkins NA, Kearney JA. Hlf is a genetic modifier of epilepsy caused by voltage‐gated sodium channel mutations. Epilepsy Res. 2016;119:20–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kadiyala SB, Papandrea D, Herron BJ, et al. Segregation of seizure traits in C57 black mouse substrains using the repeated‐flurothyl model. PLoS One. 2014;9:e90506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loscher W, Ferland RJ, Ferraro TN. The relevance of inter‐ and intrastrain differences in mice and rats and their implications for models of seizures and epilepsy. Epilepsy Behav. 2017;73:214–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muller CJ, Groticke I, Hoffmann K, et al. Differences in sensitivity to the convulsant pilocarpine in substrains and sublines of C57BL/6 mice. Genes Brain Behav. 2009;8:481–92. [DOI] [PubMed] [Google Scholar]
- 17. Hawkins NA, Lewis M, Hammond RS, et al. The synthetic neuroactive steroid SGE‐516 reduces seizure burden and improves survival in a Dravet syndrome mouse model. Sci Rep. 2017;7:15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Applegate CD, Samoriski GM, Burchfiel JL. Evidence for the interaction of brainstem systems mediating seizure expression in kindling and electroconvulsive shock seizure models. Epilepsy Res. 1991;10:142–7. [DOI] [PubMed] [Google Scholar]
- 19. White JL, Lee K, Tarhan B, et al. Variable phenotype in a Dravet syndrome model in Charles River versus Jackson C57BL/6 mice. Society for Neuroscience Annual Meeting Abstracts 2017;293.06/J11. http://www.abstractsonline.com/pp8/#!/4376. Accessed 2018 July 31.
- 20. Bankstahl M, Muller CJ, Wilk E, et al. Generation and characterization of pilocarpine‐sensitive C57BL/6 mice as a model of temporal lobe epilepsy. Behav Brain Res. 2012;230:182–91. [DOI] [PubMed] [Google Scholar]
- 21. Nakashima M, Kato M, Aoto K, et al. De novo hotspot variants in CYFIP2 cause early‐onset epileptic encephalopathy. Ann Neurol. 2018;83:794–806. [DOI] [PubMed] [Google Scholar]
- 22. Kumar V, Kim K, Joseph C, et al. C57BL/6N mutation in cytoplasmic FMRP interacting protein 2 regulates cocaine response. Science. 2013;342:1508–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sáez MA, Fernández‐Rodríguez J, Moutinho C, et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet Med. 2016;18:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vaser R, Adusumalli S, Leng SN, et al. SIFT missense predictions for genomes. Nat Protoc. 2016;11:1–9. [DOI] [PubMed] [Google Scholar]