

Abstract
The chemistry of DNA and its repair selectivity control the influence of genomic oxidative stress on the development of serious disorders such as cancer and heart diseases. DNA is oxidized by endogenous reactive oxygen species (ROS) in vivo or in vitro as a result of high energy radiation, non‐radiative metabolic processes, and other consequences of oxidative stress. Some oxidations of DNA and tumor suppressor gene p53 are thought to be mutagenic when not repaired. For example, site‐specific oxidations of p53 tumor suppressor gene may lead to cancer‐related mutations at the oxidation site codon. This review summarizes the research on the primary products of the most easily oxidized nucleobase guanine (G) when different oxidation methods are used. Guanine is by far the most oxidized DNA base. The primary initial oxidation product of guanine for most, but not all, pathways is 8‐oxoguanine (8‐oxoG). With an oxidation potential much lower than G, 8‐oxoG is readily susceptible to further oxidation, and the products often depend on the oxidants. Specific products may control the types of subsequent mutations, but mediated by gene repair success. Site‐specific oxidations of p53 tumor suppressor gene have been reported at known mutation hot spots, and the codon sites also depend on the type of oxidants. Modern methodologies using LC–MS/MS for codon specific detection and identification of oxidation sites are summarized. Future work aimed at understanding DNA oxidation in nucleosomes and interactions between DNA damage and repair is needed to provide a better picture of how cancer‐related mutations arise.
Keywords: DNA mutations, DNA oxidation, LC–MS/MS, oxidative products, p53 tumor suppressor gene
1. Introduction
Oxidation of DNA occurs continuously in the human body via reactions with reactive oxygen species (ROS), e. g., hydroxyl radical (.OH), singlet oxygen (1O2), peroxynitrite (ONOO−), hydrogen peroxide (H2O2), and superoxide (O2 .−). ROS are produced from endogenous processes, like metabolism and inflammation, and also by exogenous factors, like UV radiation, air pollution, and tobacco smoke.1 Unrepaired oxidative DNA damage can lead to neurodegenerative disease, cardiovascular disease, inflammation, aging and even cancers.2 Increasing oxidative DNA damage levels have been observed in human subject with increase age in coronary heart disease.3,4 Oxygen‐derived radicals are likely lesions that induce mutagenesis in hotspot codons in the ras oncogene and the p53 tumor suppressor gene leading cancers.5, 6, 7, 8 Thus, a detailed knowledge of DNA oxidation chemistry, along with knowledge of genome‐wide repair selectivity and efficiency, can lead to a complete understanding of the influence of DNA oxidation on related disease generation and progression.9,10
This review focuses on guanine, which has the lowest standard potential (1.3 V vs. NHE) of all native nucleobases11 and is by far the most frequently oxidized DNA base.12 The primary product of guanine oxidation is 8‐oxo‐7,8‐dihydroguanine (8‐oxoG),13 which is a major biomarker for oxidative DNA damage.14 Oxidations of other DNA bases can also occur and are summarized in recent reviews.2,13 8‐OxoG has a much lower oxidation potential (0.75 V vs. NHE) than guanine, and can be more easily oxidized.12 Considerable effort has been devoted to identifying the oxidation products of 8‐oxoG, and is well accepted that 8‐oxoG is not the final oxidation product of guanine in DNA.15, 16, 17 Imidazolone (Iz) was reported as a direct product of guanine oxidation and can be dehydrated to oxazolone (Ox). Other reported oxidation products of G include 5‐guanidinohydantoin (Gh), spirodihydantoin (Sp), 2,2,4‐triaminooxazolone (Oz), dehydroguanidinohydantoin (DGh), trioxo‐[1,3,5]‐triazinane‐1‐carboxamidine (CAC), cyanuric acid (CA), N‐nitro‐dehydroguanidinohydantoin (NO2‐DGh), parabanic acid (PA), oxaluric acid (OA), 4‐hydroxy‐2,5‐dioxo‐imidazolidine‐4‐carboxylic acid (HICA), with most resulting from the initial 8‐oxoG.18, 19, 20
Different oxidation products on DNA may result in different kinds of gene mutations, with the caveat that occurrence of the mutation depends on the specificity of efficiency of repair of the targeted gene. During normal DNA replication, guanine pairs with cytosine. If guanine is oxidized to 8‐oxoG, it mispairs with adenine, and thymine pairs with adenine in the next replication, which leads a G to T transversion.21,22 However, when guanine is oxidized to Iz, it mispairs with guanine, which leads to a G to C transversion.23 Ox, the hydrolysis product of Iz, gives rise to a G to T transversion.24,25 Gh causes G to C transversions, Sp stereoisomers cause both G to C and G to T transversions, depending on the isomeric forms of the oxidation product.26 If the resulting mutations are located on critical genes, such as tumor suppressor genes or oncogenes and repair is not effective, cancers can be promoted.27
Tumor suppressor genes encode tumor suppressor proteins that control critical processes, such as cell growth, cell division, autophagy, and tumor inhibition.28,29 Databases have collected vast amounts of data on mutations at specific codons show that frequently mutated “hot spots” codons and mutation patterns are correlated with specific types of cancers. For example, in p53 tumor suppressor gene mutation codon hotspots include 157, 158, 248, 249 for lung cancer, 175, 248, 273 for breast cancer, and 175, 248, 282 for liver cancer. DNA damage by carcinogens including DNA adduct formation and oxidative damage at specific sites are correlated with tissue specific cancers, although insufficient DNA repair is also an issue.7,8,30
The first part of this paper reviews the chemistry of DNA oxidation damage, mainly focusing on guanine oxidation and the oxidative products caused by different kinds of ROS. Next, a summary of findings on the selectivity of DNA oxidation on p53 tumor suppressor gene is given. The following section summarizes modern mass spectrometry and molecular modeling approaches to investigate DNA oxidations. Finally, we summarize and discuss possible future research in this area.
2. DNA Oxidation Products
2.1. Oxidation by Hydroxyl Radical (.OH)
The ROS radical . OH can be produced from γ‐rays of 60Co and by Fenton reactions of Fe(II) or Cu(II) ions with hydrogen peroxide.31,32 H2O2 exists in human cells, Fe(II) ions are physiologically necessary for iron‐containing proteins, and Cu(II) ions exist in blood plasma and cell nuclei. Redox cycling of quinone‐like metabolites with Cu(II) and NADPH can form . OH radical and other ROS. As a simple example, catechol undergoes this type of redox cycling that leads to a 2 e‐oxidation to o‐quinone, which subsequently undergoes two 1 electron enzymatic reductions back to catechol (Scheme 1). ROS including H2O2, O2 .−, and .OH have been reported and would amplified by the redox cycling.33,34 DNA adducts can also be produced in this pathway. Thus, . OH radical is generated by multiple sources within human cells, and DNA oxidation by . OH most likely happens endogenously and exogenously.
Scheme 1.

Redox cycling of quinones with M2+ utilizing electrons from NADPH produce ROS that can oxidize DNA.
7,8‐dihydro‐8‐oxo‐deoxyguanosine (8‐oxo‐dG) was reported as a minor product of deoxyguanosine (dG) oxidation induced by external radiation for single‐stranded (ss)DNA. Two major products Ox, and its precursor Iz were found after exposure of 3’,5’‐di‐O‐acetyl‐2’‐deoxyguanosine to γ‐rays of 60Co, which produced . OH radicals. Ox and Iz represented more than 80 % of the . OH‐induced oxidation products, but their formation pathway could involve 8‐oxo‐dG as a transient. For double‐stranded (ds) DNA, Ox and 8‐oxo‐dG were reported as the two major products.31 (Scheme 2)
Scheme 2.
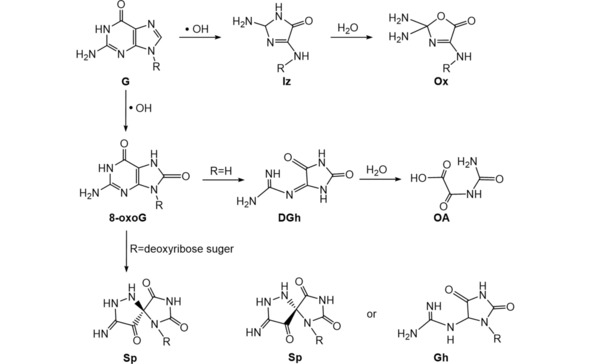
Summary of .OH radical induced oxidation products of guanine (G).31,32,50
Fenton's reagent, comprised of H2O2 and a transition metal ion (Fe2+ or Cu2+), was established as a strong oxidant of organic compounds in 1894.35 40 years later, it was found that . OH is the actual oxidant in the system.36 Fenton chemistry can happen in cells, in Wilson's disease, the abnormal increased intracellular levels of copper can stimulate production of . OH which can damage the lipids, proteins, and nucleic acids through the Fenton reaction.37 A significant amount of research has focusing on DNA oxidation by Fenton's reagent. Fenton's reagent was reported to transform DNA and also liberate bases from DNA, due to oxidation of the carbon on the deoxyribose, producing deoxyribonic acid. Both liberation and deoxyribonic acid formation resulted from breaking the sugar‐phosphate backbone.38 The changes in primary and secondary structure of DNA were investigated in the presence of H2O2 and metal ions, Fe2+ accelerated the double‐ and single‐strand breaks, cross‐linking, and decreased the rates of base destruction. Cu2+ increased the rates of base destruction. The relative magnitude of resulting DNA damage was base destruction>single‐strand break>double‐strand break>cross‐linking.39 By adding ferrous chelator o‐phenanthroline, cellular DNA strand breaks were not observed, and cells were protected from lethal injuries, showing that ferrous ions played significant roles in DNA damage by the Fenton reaction.40,41 There are also other ferrous chelators such as citrate or nitrilotriacetic acid that would enhance the DNA lesions, including strand breaks and 8‐oxo‐dG formation.42
Sequence‐specific DNA oxidation by Fenton's reagent was investigated, iron‐mediated Fenton reaction occurs preferentially at a limited number of sequences on ds‐DNA. Purine‐T−G‐purine (RTGR) is one of these, and results suggested that Fe2+ interacted with the guanine N7 prior to oxidation.43 A series of DNA cleavage agents were designed and synthesized containing sequence‐specific DNA binding molecules tethered to an iron chelator, ethylenediaminetetraacetic acid (EDTA). These molecules bound heterogeneous ds‐DNA and in the presence of O2 and dithiothreitol (DDT) cleaved the DNA backbone at specific sites.44 In this system, ROS such as hydroxyl radical, iron‐bound oxygen, and superoxide, were suspected DNA‐cleaving species.47
8‐oxo‐dG was found as the initial product from reacting deoxyguanosine with . OH produced from Fenton's reagent.48,49 Oscillating levels of 8‐oxo‐dG with the reaction time was found when oxidizing free guanine or ds‐DNA with Fenton's reagent. 8‐oxo‐dG and Gh were detected in the products by LC–MS/MS, with Gh as a further oxidation product of 8‐oxo‐dG (Scheme 2). Subsequent oxidation of 8‐oxo‐dG presumably caused the decrease of its concentration,32 but the oscillation mechanism remains unexplained. These oxidation products of 8‐oxo‐dG were formed using iron‐ and copper‐mediated Fenton oxidation. DGh was produced when incubating 8‐oxoG with Fenton's reagent, and its hydrolysis product OA was detected as the final oxidation product after 96 h. Under similar conditions, two diastereoisomeric forms of Sp were found as oxidation products of 8‐oxo‐dG 50 (Scheme 2).
2.2. Type I Photooxidation
During the long history of sensitized photooxidation studies of DNA, two different pathways have emerged. In these types of photooxidation, excited photosensitizers were exposed to UV radiation or visible light. They can return to the ground state by electron or hydrogen abstraction from the substrate DNA (type I mechanism) or by transferring energy to ground‐state oxygen to form singlet oxygen (type II mechanism).51,52 In type I oxidations of DNA, guanine residues form radical cations because of G's low oxidation potential compared to the other nucleobases. Positive “holes” initiated in the oxidation move along the DNA duplex to end up at the most easily oxidized G sites.53,54
Sp was reported as the major oxidation product of triplet‐excited 2‐hydroxyacetophenone (AP‐OH) treated monomeric dG by SELINQUATE NMR technique (Scheme 3).55 To investigate G oxidation products in ss‐DNA, a synthetic single‐stranded oligonucleotide (ATCTGTACT) containing a single G was oxidized using the photoexcited riboflavin as sensitizer. Electrospray ionization‐mass spectrometer (ESI‐MS) showed that an Iz‐containing oligonucleotide was formed immediately after irradiation and an Ox‐containing oligonucleotide was subsequently produced as the final product through spontaneous hydrolysis after 20 h (Scheme 3). Ox was confirmed by high performance liquid chromatography (HPLC) of enzymatic digestion products of the oxidized DNA fragment.56 For the ds‐oligomer (TTGGTA/ATACCAAA), an Iz‐containing oligonucleotide was found as the major product (25 % yield) of riboflavin‐sensitized photooxidation, while an 8‐oxoG‐containing oligomer was a minor product (5 % yield) (Scheme 3). The formation of Iz was confirmed by analysis of enzymatic digestion products of riboflavin‐sensitized photooxidation of calf thymus DNA.57
Scheme 3.
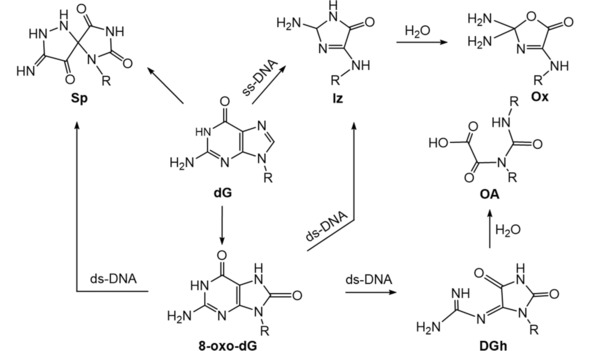
Summary of type I sensitized photooxidation of dG.19,20,55, 56, 57
The oxidation products of 8‐oxo‐dG‐containing ds‐DNA oligomers and sequence‐specific formation of those products were investigated using LC‐Quadrupole‐Time of Flight (QTOF) mass spectrometry. DGh, OA, Sp, Iz were found as the major oxidation products (Scheme 3), and yields depended on the concentration of riboflavin and the sequence context.20 Different sequence contexts (CAGAAAOCCC, COA, GOG, AOG, COT, TOC, AGC, CGA, GGG, O representing 8‐oxo‐dG) were used to make further oxidation products, then enzyme hydrolyzed to individual nucleobase or nucleoside products for LC–MS/MS analysis.
Quantitative analyses of riboflavin‐sensitized photooxidation of ds‐DNA oligomers (19 base pairs in exon 5, 25 base pairs in exon 7 and 21 base pairs in exon 8) representing fragments of the p53 tumor suppressor gene was done by HPLC‐MS/MS. Isotopically labeled guanines were placed at specific positions on the DNA duplex. After photooxidation, these DNA oligomers were hydrolyzed by enzymatic digestion to individual oxidized and unoxidized nucleosides, and relative amounts of oxidized guanines at specific positions were measured from the ratio of peak area of the isotopically labeled product divided by the sum of peak areas of labeled and unlabeled products. Results showed that both 8‐oxo‐dG and Ox were the major products and preferentially formed at guanine residues in methylated CpG dinucleotides.19
2.3. Singlet 1O2 (Type II Photooxidation)
A major source of singlet oxygen (1O2) is photosensitization in which energy is transferred from an excited triplet‐state sensitizer (Sens) to ground‐state oxygen (Eq 1). This is called type II photosensitization.51,52 1O2 can also be produced intracellularly by neutrophils during phagocytosis, and extracellularly by stimulated macrophages.58,59

Using methylene blue‐mediated type II photooxidation and analysis by LC‐mass spectrometry (LC–MS), 1H and 13C NMR,60 two 4R* and 4 S* diastereomers of 4,8‐dihydro‐4‐hydroxy‐8‐oxo 2’‐deoxyguanosine (4‐OH‐8‐oxodG) were found as the major products of guanine oxidation with free dG in aqueous solution. Later, the same group reported CA as the main type II photosensitized oxidation product of 8‐oxo‐dG. Ox and its precursor Iz were minor oxidation products.61 A thermolabile naphthalene endoperoxide derivative N,N’‐di(2,3‐dihydroxypropyl)‐1,4‐naphthalene‐dipropanamide (DHPNO2) (DHPNO2 decomposes to 1O2 at 37 °C) was used to generate [18O]‐labeled singlet oxygen for delineation of mechanistic aspects of singlet oxygen‐mediated oxidation products of dG. It was found that 8‐oxo‐dG and two diastereomers of 4‐OH‐8‐oxodG are the primary oxidation products, also two diastereomers of Sp as major products (Scheme 4).62,63
Scheme 4.
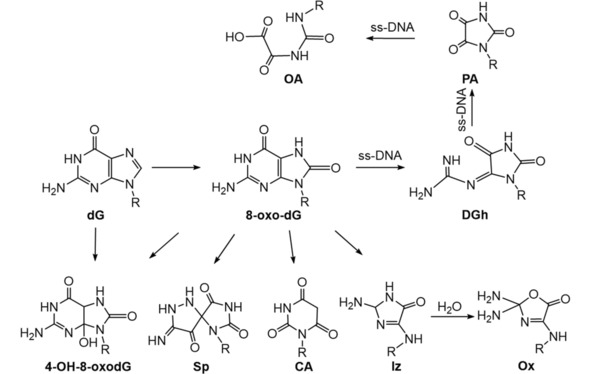
Summary of 1O2 induced (type II photooxidation) oxidation products of dG.60, 61, 62, 63, 64
Using DHPNO2 as a source of 1O2, together with single stranded oligomers, OA‐containing oligomer was found as the final stable product formed via DGh‐ and PA‐containing oligomers as intermediates (Scheme 4).64
2.4. Transition Metal Oxidants
High‐valent transition metal complex, such as manganese (III)‐bis‐aqua‐meso‐tetrakis(4‐N‐methylpyridiniumyl)‐porphyrin (Mn‐TMPyP), N,N’‐ethylenebis(salicylideneanimato) oxochromium (V), Cr (V)‐Salen, peroxo‐chromium (V) complex can oxidize guanines. The one‐electron oxidant Na2IrCl6 has a redox potential ∼0.9 V vs NHE, which allows it to oxidize 8‐oxoG (0.74 V vs. NHE) with very little oxidation of G (1.3 V vs. NHE). Na2IrCl6 has been used to investigate oxidation products of 8‐oxoG.65 Oxidation products of dG by Mn‐TMPyP/KHSO5 were found at 90 % Iz within one minute without no remaining 8‐oxo‐dG.66 Two different routes of guanine oxidation on ds‐DNA were found. First, Iz‐containing oligomers were produced via two intermediates, 5‐hydroxy‐8‐oxo‐7,8‐dihydroguanosine (5‐OH‐8‐oxodG) and DGh. In the second route, PA was the main oxidation product (Scheme 5).67 Peroxo‐chromium (V) formed from reacting bis(2‐hydroxyethylbutanato)oxochromate(V) and bis(hydroxyethyl)amino‐tris(hydroxymethyl) methaneoxochromate(V) complexes with hydrogen peroxide oxidized guanine in ds‐DNA to Gh (Scheme 5).68 One‐electron oxidants such as Na2IrCl6, K3Fe(CN)6, and CoCl2/KHSO5 oxidized free 8‐oxo‐dG to a diastereomeric mixture of Sp nucleosides in neutral aqueous solution as measured by NMR and ESI‐MS/MS.65 CoCl2/KHSO5 oxidation of 8‐oxo‐dG gave two epimers of Gh and two epimers of iminoallantoin (Ia) nucleosides as products via the intermediate 5‐OH‐8‐oxodG (Scheme 6). These products were converted to oxidized iminoallantoin (Ia°x) nucleoside by Na2IrCl6.69 For ss‐DNA oligomers, Gh‐containing DNA oligomer was the major oxidation product of 8‐oxo‐dG‐containing ss‐oligomer by IrCl6 2− (Scheme 6).70
Scheme 5.
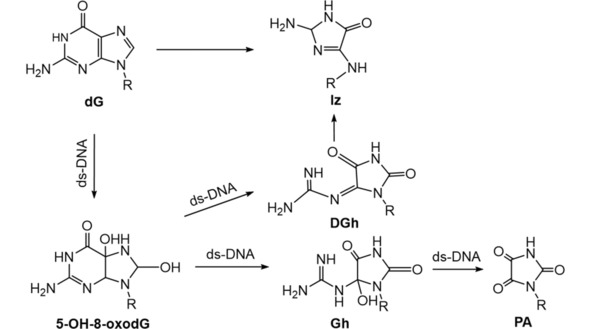
Summary of Mn‐TMPyP/KHSO5 and peroxo‐chromium (V) induced oxidation products of dG.65, 66, 67, 68
Scheme 6.
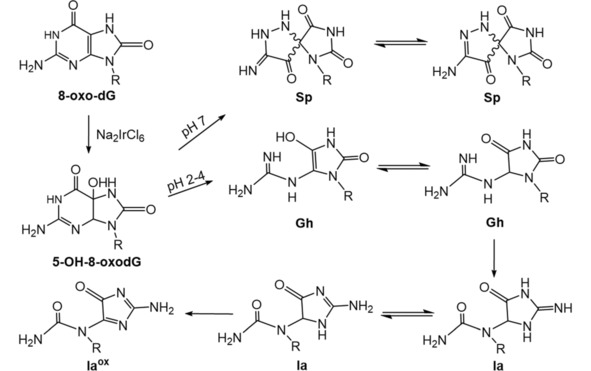
2.5. Peroxynitrite (ONOO−)
Peroxynitrite (ONOO−) is produced by nitric oxide (NO) reacting with superoxide (O2 .−) (Eq 2) in vivo, and represents a biologically important oxidant.71,72 It forms under endogenous conditions and is linked to amyotrophic lateral sclerosis, atherosclerosis, and neurodegeneration. It also forms from exogenous sources such as tobacco smoke, which contains quinone radicals that reduce oxygen to O2 .− that can react with NO in the smoke to form peroxynitrite.18,73,74

8‐oxo‐dG and Ox in a ratio 1/25 are oxidation products of free dG reacting with ONOO−.73 Major products of 3’,5’‐di‐O‐acetyl‐8‐oxo‐dG from ONOO− oxidation was OA via an 5‐iminoimidazolidine‐2,4‐dione intermediate. Minor products included CA and PA.75 Products of 8‐oxo‐dG may depend on source, DGh, NO2‐DGh, and CAC were found as the major products at high ONOO− fluxes (above 2.5 μM/s) with peroxynitrite as the oxygen source. Sp, Gh, and HICA were the major products at low ONOO− flux (0.67 μM/s), where water was the oxygen source.76
Calf thymus DNA oxidized by ONOO− gave four oxidation products, 8‐oxo‐dG, Ox, Sp, and Gh. 8‐oxodG and Ox were dose‐dependent. Amounts of Sp and Gh increased with increase of ONOO− concentration (ONOO− up to 0.2 equiv. of guanine content in DNA, 55 μM), but decreased at 550 μM ONOO− (Scheme 7).77 Ss‐oligomers containing 8‐oxo‐dG were oxidized using ratios of ONOO−/oligomer <5, with OA‐containing oligomer as the final product. At ONOO−/oligomer ratio >10, major products were CAC‐ and CA‐containing oligomers.78
Scheme 7.

Summary of ONOO− induced oxidation products of 8‐oxo‐dG.73,75, 76, 77, 78
2.6. Protein radicals
Chemical pathway details of DNA oxidation in the eukaryotic cell environment of nucleosome core particles (NCP) where DNA is associated with histone proteins is a relatively unexplored landscape. DNA‐protein crosslinks have been observed upon exposure to ROS, and other proteins associated with DNA can also form DNA crosslinks.79 Hydroperoxides generated on histones, then decomposed with Cu+ form 8‐oxo‐dG on DNA.80,81 Site selective DNA damage was found upon photolysis of NCPs containing histones modified with an azoalkane radical precursor, and the DNA damage site depends on its proximity to the protein radical.82 These studies stress the mechanistic role of coupled protein and DNA damage in systems where these macromolecules are physically bound.
3. Site‐Specific Oxidations of p53 Tumor Suppressor Gene
P53 was identified as a tumor suppressor gene in the 1980s.83 It codes for p53 protein that controls cellular stress response, and sustains impaired‐function mutations in cancers.84, 85, 86, 87 Unrepaired DNA lesions on p53 would lead to mutations. Mutations in the p53 tumor suppressor gene are found in 50–60 % of human cancers, and some of these correlate with oxidatively damaged codon sites.30,88,89 Oxidation products of guanine pair with bases other than cytosine, which leads to mutations. DNA oxidation by ROS is non‐random, and may precede mutations at the same sites.20
Fenton's reagent induced base pair changes at mutation hot spot p53 codons 248, 249, 250 in human fibroblasts investigated by restriction fragment length polymorphism/polymerase chain reaction (RFLP/PCR).5 The DNA sequence containing mutated sites was amplified by PCR. The second G (CGG→CCG) and third G (CGG→CGA) in codons 248 and (AGG→ATG, AGG→AGT) in codon 249, and the first C (CCC→ACC) in codon 250 was mutated after the Fenton oxidation.
A yeast reporter system was used to detect change‐in‐function mutations in p53 gene, which were caused by DNA oxidation with ROS generated from polycyclic aromatic hydrocarbons (PAH) o‐quinones.8 P53 DNA fragment was incubated with PAH o‐quinone and redox cycling system (NADPH and CuCl2). The yIG397 yeast strain contains an adenine reporter gene under the control of p21 promoter. A p53‐p21 reporter binding assay was used to measure the incidence of mutation. Then, p53 plasmid DNA was recovered from the yeast, and sequenced with S6 (5’‐dCTGGGACAGCCAAGTCTGT3’), R6 (5’‐dCCTCATTCAGCTCTCGGAA3’) primers to reveal the mutations. 53 yeast colonies were examined, in which 63 mutations were sequenced, 29 were G to T transversions, and 16 occurred at hot spots of lung cancer such as codon 157 (GTC→ATC), 244 (GGC→GAC), 245 (GGC→TGC), 249 (AGG→ATG, AGG→AGC), and 273 (CGT→CTT).
Co‐generation of nitric oxide and superoxide induced mutation at codon 248 in the p53 gene.90 Human bronchial epithelial cells (BEAS‐2B) were treated with nitric oxide donor systems and harvested 72 h later. The genomic DNA was extracted, purified and subjected to fish‐RFLP/PCR to detect mutations. Mutations in codon 248 were observed as CGG→TGG, CGG→CAG, and CGG→CCG.
Codon specific mutations in the p53 exon 7 were investigated by ultraviolet B light radiation in human skin fibroblasts.89 Fibroblasts were treated with UVB radiation grown for 72 h. Then, DNA was extracted and mutations in p53 codons 247–250 were found by the genotypic RFLP‐PCR assay. Mutations occurred in codon 250 (CCC→ACC), codon 249 (AGG→AGT), codon 248 (CGG→AGG), and codon 247 (AAC→AAA, AAC→AAT). These base pair changes are most likely due to pyrimidine photodimers as premutagenic lesions. Isotopically labeled DNA oligonucleotides at specific guanine were irradiated using riboflavin photosensitizer, then enzymatic hydrolysis process to yield 2’‐deoxynucleosides analyzed by tandem MS in multiple reaction monitoring mode.19 Amounts of 8‐oxo‐dG lesion at the first G in codon 245 (GGC) (19 %), the second G in codon 248 (CGG) (14 %) and the Ox lesion originating from the first G in codon 245 (GGC) (16 %), the second G in codon 248 (CGG) (18 %) were much higher than those for other positions. Also, methylated cytosines increased the reactivity of the G in the CpG dinucleotides. Methylation of cytosine opposite the second G in codon 248 led to a 9‐fold increase of yield of Ox, while the 5’ neighboring methylated cytosine caused a 4‐fold increase. When both these cytosine were methylated, a 6‐fold increase was found. In contrast, 8‐oxo‐dG yields were only increased by 20–30 % relative to the unmethylated p53 codon 248.
We compared oxidation of a 32 base pair double‐stranded oligonucleotide representing exon 7 of the p53 gene by catechol/Cu2+/NADPH and Fenton's reagent.91 Oxidized oligonucleotides were cut by a restriction endonuclease to provide smaller fragments to enable to determine the positions of 8‐oxo‐dG by LC–MS/MS sequencing (Scheme 8). G's in codons 243, 244, 245 and 248 were most frequently oxidized by catechol/Cu2+/NADPH, while G in codons 243 and 248 were most frequently oxidized by Fenton's reagent.
Scheme 8.

Sample workup procedure for oxidative site determination by LC–MS/MS sequencing.91
P53 oxidation by different ROS sources is nonrandom and mutations occur at specific codons. Mutations in codon 245 and 248 are the most frequent and are “hot spots” of many cancers based on the p53 database.30 Also, guanine reactivity towards oxidants may be affected by DNA modifications, e. g. cytosine methylation to 5‐methylcytosine (MeC). MeC bases are formed by enzymatic methylation of the C5 position of cytosine in cytosine‐phosphate‐guanine (CpG) dinucleotide sites. MeC is thought to exist at every CpG site (including 46 different sites on both DNA strands) along exon 5–8 in the human p53 tumor suppressor gene.92 The introduction of opposite and 5’‐neighboring MeC increases the reactivity of the target G.19 Experimental results thus far suggest that oxidative damage of p53 may play an important role along with DNA adduction in the initiation of cancers.19,91 (Adduction denotes a nucleophilic addition reaction of a base on DNA with an electrophilic metabolite to form a nucleobase adduct).
4. LC–MS/MS Approaches to Study Codon‐Specific Oxidation
Mass spectrometry (MS) has emerged as a powerful modern tool to investigate structurally damaged DNA, and is the subject of a recent comprehensive review.93 DNA oxidation can occur on any human gene. Here we discuss mainly codon‐specific oxidations on p53 tumor suppressor gene that have been widely investigated. Stable isotope labeling of DNA has been widely used along with MS to examine the reactive sites and cytosine methylation on the formation of 8‐oxo‐dG and downstream oxidation products of reactions with nitrosoperoxycarbonate and riboflavin‐mediated photolysis using MRM.19 Experiments were done on 15N3,13C1‐labeled DNA oligodeoxynucleotides synthesized by standard phophoramidite chemistry using a DNA synthesizer. 15N3,13C1‐dG phosphoramidite and 15N3,13C1‐dG phosphoramidite were also synthesized and used. Percent oxidation at labeled guanine equals the LC MRM peak area of the labeled oxidation product divided by the sum of LC MRM peak areas of labeled and unlabeled oxidation products. In this way, the amounts and locations of 8‐oxo‐dG and other products formed on the DNA strands are revealed. While the approach is powerful and reliable, the need for synthesis of isotopically labeled oligonucleotides is a serious limitation, and enzymatic oligonucleotide digestion is required.94, 95, 96
Over the past decade, LC–MS/MS methods have emerged to sequence ds‐oligonucleotides 20 bp or smaller.93,97 We recently developed a restriction‐enzyme assisted version of MS/MS sequencing that enables reactive codons to be identified on ds‐oligonucleotides longer than 20 bp.101 This is important because DNA conformation in longer p53 gene strands can influence secondary structure reactivity.102 We used this direct LC–MS/MS methodology to locate primary oxidation sites in ds‐oligonucleotides of >20 bp without labeling or hydrolysis.91 The 32 bp ds‐oligonucleotide representing exon 7 of the p53 tumor suppressor gene was oxidized by catechol/Cu2+/NADPH or Fenton's reagent, then cut at known specific sites by a restriction endonuclease to provide smaller strands for LC–MS/MS sequencing (Scheme 8). Multiple oxidation sites were identified and compared by looking for fragments with m/z that differ from the unreacted fragment m/z. For example, m/z 1002.4 (z=−4) was observed for unoxidized Fragment 1 (Scheme 8) and this fragment when oxidized gave four peaks with retention times 13.6 min, 14.9 min, 16.2 min, and 17.9 min. The CID spectrum of ion 1006.4 reflecting m/z for dG→8‐oxodG conversion for peak 1 is in Figure 1B and for peak 2 in Figure 1 C. Collision‐induced dissociation (CID) in MS provides an–bn and wn ions by fragmentation of the phosphodiester backbone.103 The position of 8‐oxo‐dG was determined from m/z differences in an–bn and wn ions of corresponding unoxidized and oxidized oligonucleotide fragments. MS/MS for Peak 1 of singly oxidized Fragment 1 (Figure 1B) shows increases in m/z from a6–b6 to a8–b8 compared to the unoxidized Fragment 1. This reveals that the fifth G was oxidized to 8‐oxo‐dG, CATGOGCGGCATG (O=8‐oxo‐dG). The MS/MS spectrum for peak 2 of singly oxidized Fragment 1 (Figure 1 C) shows an increase in mass of all ions from a9–b9 compared with that of the unoxidized Fragment 1. This shows that the eighth G was oxidized to 8‐oxo‐dG, CATGGGCOGCATG. Similar analysis of the third and the fourth peaks, revealed that peak 3 represents oxidation of the sixth G (CATGGOCGGCATG) and peak 4 represents oxidation of the fourth G (CATOGGCGGCATG). This restriction enzyme assisted sequencing is simple, reliable, relatively rapid, and facilitates direct detection and mapping of modified sites.
Figure 1.
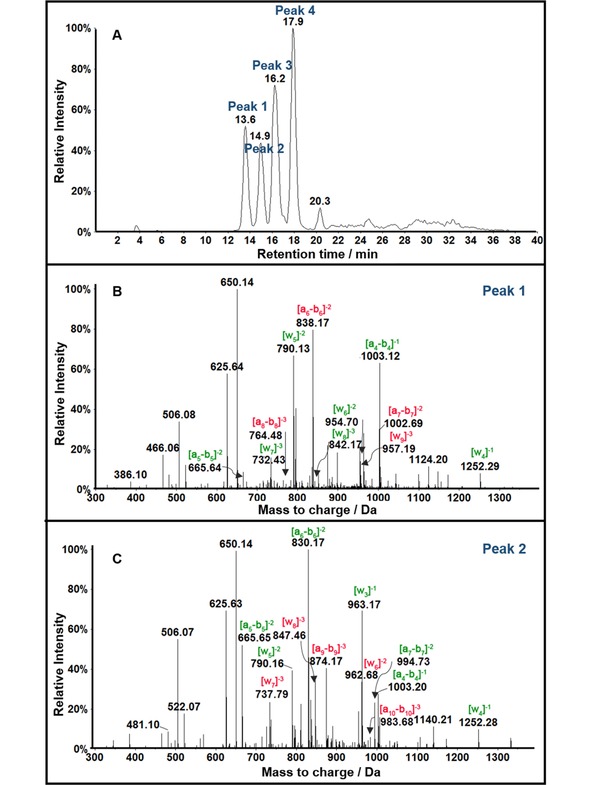
LC‐QTOF mass spectrometry of Fragment 1 (Scheme 8) from Exon 7 oxidized by Catechol/Cu2+/NADPH. (A) Extracted ion chromatogram for m/z 1006.4 representing z=−4 of singly oxidized products. (B) CID analysis of m/z 1006.4 for peak 1 eluting at 13.6 min and (C) CID analysis of peak 2 eluting at 14.9 min. Reproduced with permission from ref.91, copyright American Chemical Society, 2017.
5. Molecular modeling of DNA Oxidation
Molecular modeling is generally useful to gain better insight into experimental reactivity and specificity of DNA oxidation and adduction.104 In order help reveal to how codon specificity of the p53 oxidations arises, we used the molecular modeling program Autodock 4.2.6105 and investigated the favored docking position of suspected oxidant species in Cu2+/NADPH/catechol oxidation of the B‐DNA form of the exon 7 ds‐oligonucleotide. The standard B‐DNA form of 32 bp p53 exon 7 ds‐oligonucleotide was modeled using make‐na106 and solvated with water using CHIMERA software.107 An Amber solvation model was used with appropriate box size to accommodate water molecules. Each possible species involved (H2O2, .OH, catechol, benzoquinone, Cu(I)OOH) were tested individually to find the best energy‐minimized binding sites to the hydrated ds‐oligonucleotide. A Lamarckian genetic algorithm (LGA) was used in Autodock 4.2.6 to find the binding energy between the oligonucleotides and the ligands. Parameters were set with 25,000,000 evaluations and 50 docked conformations for an individual docking computation.
Figure 2 shows preferred most probable binding sites for catechol on p53 exon 7. It was found that catechol bound at multiple positions, i. e., the fourth, fifth, sixth, eighth, and thirteenth guanines, correlating with the multiple positional isomers (CATOGGCGGCATG, CATGOGCGGCATG, CATGGOCGGCATG, CATGGGCOGCATG) found experimentally for p53 exon 7 singly oxidized by Cu2+/NADPH/catechol (Figure 1A).91
Figure 2.

Models of catechol in yellow docking near reactive guanines in the B form of the 32 bp exon 7 p53 fragment showing catechol docked (arrows) (A) with the fourth and fifth guanines; (B) with the sixth guanine; (C) with the eighth guanine; (D) with the thirteenth guanine. Reproduced with permission from ref.91, copyright American Chemical Society, 2017.
Thus, the model predicted binding sites in studies of exon oxidation91 and adduction102,108 lead to the view that there may be a common pathway in which reactants first bind to specific reaction sites on the ds‐oligonucleotide followed by the chemical reaction step.
6. Summary
The results summarized above make clear that 8‐oxo‐dG is a primary, but not final product of dG oxidation by ROS and other oxidants in vitro. A rich pathway‐dependent oxidation chemistry of 8‐oxo‐dG leads to myriad downstream products that depend on mode of oxidation. These including DGh, OA, Sp and Gh for .OH radical oxidation, Sp, DGh, PA, OA, CA, and Iz from photooxidation of 8‐oxo‐dG, and Sp, Iz, DGh, PA, Gh, Ia, Ia°x products by transition metal oxidation. In ONOO− oxidation, Sp, Gh, DGH, CAC, PA, OA, CA, and HICA are found as 8‐oxo‐dG oxidation products with composition depending on ONOO− concentration. Only the oxidation product Iz, its hydrolysis product Ox and 4‐OH‐8‐oxodG are produced directly without 8‐oxo‐dG as an intermediate. LC–MS/MS is a powerful tool to investigate DNA oxidation as well as adduction, and can help to predict mutational hotspots within the human genome.
The individual oxidation products of DNA may influence the type of mutation that occurs. For example, 8‐oxo‐dG and Ox cause G to T transversions, Iz and Gh cause G to C transversions, and Sp stereoisomers lead to both G to C and G to T transversions. Mutations in the p53 gene caused by oxidation occur at known mutation hot spots identified by analysis of numerous tumors and cancer cell cultures and are available in extensive databases.30 This information makes it possible to correlate exogenous metabolite‐directed and other oxidation reactions with different types of cancers. Adduction reactions of DNA bases with electrophilic metabolites (e. g. SN2 reactions) can contribute to these mutations, and must be considered along with gene oxidations to arrive at an accurate understanding of the chemical pathways.109,110 More frequent mutations in human p53 may also arise due to the increased reactivity of its C‐methylated MeCpG sites in hot spot‐containing exons 5 to 8.
While the chemistry of the various reaction types is fairly well understood, it is not totally clear which types reactions on DNA bases in humans are the most important in leading to the ultimate mutations. The influence of DNA‐histone binding on DNA oxidation in nucleosomes is a complicating factor that has yet to be fully elucidated. Yet, oxidation and adduction sites on DNA and p53 gene fragments in solution without histones correlate with downstream mutations. Clearly the situation is very complicated and must also involve specificity and efficiency of gene repair processes.9,27 Investigations of the chemistry of DNA oxidation and DNA repair have remained somewhat separated. Great progress in understanding the role of DNA chemical modification in mutations might be expected if we can find new ways to study combined interactions of these two important processes.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Di Jiang received her B.Sc. (2011) and M.Sc. (2014) in Chemistry from Jilin University, China. She is currently a Ph.D. student under the supervision of Professor James F. Rusling in Chemistry, University of Connecticut, USA. Her research is mainly focused on DNA damage detection of p53 tumor suppressor gene and protein damage by LC–MS/MS.

Biographical Information
James F. Rusling was awarded a B.Sc. in Chemistry from Drexel University in 1969, and Ph. D. from Clarkson University in 1979. He is Professor of Chemistry at University of Connecticut, and Professor of Surgery and member of the Neag Cancer Center at UConn Health Center, as well as adjunct Professor of Physical Chemistry at National Univ. of Ireland. Galway. Current research includes developing new cancer diagnostic devices for detection of biomarker proteins and peptides, low‐cost 3D printed immunoarrays for point‐of‐care diagnostics, electrochemical and mass spectrometric arrays for toxicity screening, tumor suppressor gene damage, and fundamental bioelectrochemistry. He has authored over 400 research papers and several books, and is a also musician interested in Irish and American folk styles.

Acknowledgements
The authors thank the National Institute of Environmental Health Sciences (NIEHS), NIH, USA, Grant No. ES03154 for financial support. They also thank Prof. Emeritus John B. Schenkman for insightful contributions and discussions during the research on DNA damage.
D. Jiang, J. F. Rusling, ChemistryOpen 2019, 8, 252.
References
- 1. Loft S., Poulsen H. E., J. Mol. Med. 1996, 74, 297–312. [DOI] [PubMed] [Google Scholar]
- 2. Cooke M. S., Evans M. D., Dizdaroglu M., Lunec J., FASEB J. 2003, 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- 3. Rattan S., Siboska G. E., Wikmar F. P., Clark B. F. C., Woolley P., Med. Sci. Res. 1995, 23, 469–470. [Google Scholar]
- 4. Collins A. R., Gedik C. M., Olmedilla B., Southon S., Bellizzi M., FASEB J. 1998, 12, 1397–1400. [PubMed] [Google Scholar]
- 5. Hussain S. P., Aguilar F., Amstad P., Cerutti P., Oncogene. 1994, 9, 2277–2281. [PubMed] [Google Scholar]
- 6. Du M. Q., Carmichael P. L., Philips D. H., Mol. Carcinog. 1994, 11, 170–175. [DOI] [PubMed] [Google Scholar]
- 7. Kang D. H., Oxidative Stress, 2002, 13, 540–549. [DOI] [PubMed] [Google Scholar]
- 8. Yu D., Berlin J. A., Penning T. M., Field J., Chem. Res. Toxicol. 2002, 15, 832–842. [DOI] [PubMed] [Google Scholar]
- 9. Gillingham D., Sauter B., ChemBioChem 2017, 18, 2368–2375. [DOI] [PubMed] [Google Scholar]
- 10. Greenberg M. M., Biochem. Soc. Trans. 2004, 32, 46–50. [DOI] [PubMed] [Google Scholar]
- 11. Steenken S., Jovanovic S. V., J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar]
- 12. Kanvah S., Joseph J., Schuster G. B., Barnett R. N., Cleveland C. L., Landman U., Acc. Chem. Res. 2010, 43, 280–287. [DOI] [PubMed] [Google Scholar]
- 13. Burrows C. J., Muller J. G., Chem. Rev. 1998, 98, 1109–1151. [DOI] [PubMed] [Google Scholar]
- 14. Cadet J., Loft S., Olinski R., Evans M. D., Bialkowski K., Wagner J. R., Dedon P. C., Moller P., Greenberg M. M., Cooke M. S., Free Radical Res. 2012, 46, 367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Matter B., Malejka-Giganti D., Csallany A. S., Tretyakova N., Nucleic Acids Res. 2006, 34, 5449–5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cadet J., Douki T., Ravanat J. L., Acc. Chem. Res. 2008, 41, 1075–1083. [DOI] [PubMed] [Google Scholar]
- 17. Hickerson R. P., Prat F., Muller J. G., Foote C. S., Burrows C. J., J. Am. Chem. Soc. 1999, 121, 9423–9428. [Google Scholar]
- 18. Niles J. C., Wishnok J. S., Tannenbaum S. R., Nitric Oxide 2006, 14, 109–121. [DOI] [PubMed] [Google Scholar]
- 19. Ming X., Matter B., Song M., Veliath E., Shanley R., Jones R., Tretyakova N., J. Am. Chem. Soc. 2014, 136, 4223–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lim K. S., Cui L., Taghizadeh K., Wishnok J. S., Chan W., DeMott M. S., Babu I. R., Tannenbaum S. R., Dedon P. C., J. Am. Chem. Soc. 2012, 134, 18053–18064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng K. C., Cahill D. S., Kasai H., Nishimura S., Loeb L. A., J. Biol. Chem. 1992, 267, 166–172. [PubMed] [Google Scholar]
- 22. Shibutani S., Takeshita M., Grollman A. P., Nature 1991, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 23. Maor-Shoshani A., Ben-Ari V., Livneh Z., Proc. Natl. Acad. Sci. India 2003, 100, 14760–14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Henderson P. T., Delaney J. C., Gu F., Tannenbaum S. R., Essigmann J. M., Biochem. 2002, 41, 914–921. [DOI] [PubMed] [Google Scholar]
- 25. Duarte V., Gasparutto D., Jaquinod M., Cadet J., Nucleic Acids Res. 2000, 28, 1555–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Henderson P. T., Delaney J. C., Muller J. G., Neeley W. L., Tannenbaum S. R., Burrows C. J., Essigmann J. M., Biochem. 2003, 42, 9257–9262. [DOI] [PubMed] [Google Scholar]
- 27. Donigan K., Sweasy J. B., Mol. Carcinog. 2009, 48, 362–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maiuri M. C., Tasdemir E., Criollo A., Morselli E., Vicencio J. M., Carnuccio R., Kroemer G., Cell Death Differ. 2009, 16, 87–93. [DOI] [PubMed] [Google Scholar]
- 29. Jones R. G., Thompson C. B., Genes Dev. 2009, 23, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.L. Hjortsberg, J. M. Rubio-Nevado, D. Hamroun, C. B'eroud, M. Claustre, T. Soussi, The p53 Mutation handbook 2.0, available online, updated October, 2017. http://p53.fr/tp5; last accessed 27 June 2018.
- 31. Cadet J., Berger M., Buchko G. W., Joshi P. C., Raoul S., Ravanat J. L., J. Am. Chem. Soc. 1994, 116, 7403–7404. [Google Scholar]
- 32. White B., Smyth M. R., Stuart J. D., Rusling J. F., J. Am. Chem. Soc. 2003, 125, 6604–6605. [DOI] [PubMed] [Google Scholar]
- 33. Song B., Shen M., Jiang D., Malla S., Mosa I. M., Choudhary D., Rusling J. F., Analyst 2016, 141, 5722–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bist I., Song B., Mosa I. M., Keyes T. E., Martin A., Forster R. J., Rusling J. F., ACS Sens. 2016, 1, 272–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fenton H. J. H., J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar]
- 36. Haber F., Weiss J., Proc. R. Soc. London Ser. A 1934, 147, 332–351. [Google Scholar]
- 37. Wu F., Wang J., Pu C., Qiao L., Jiang C., Int. J. Mol. Sci. 2015, 16, 6419–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rhaese H. J., Freese E., Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1968, 155, 476–490. [PubMed] [Google Scholar]
- 39. Massie H. R., Samis H. V., Baird M. B., Biochim. Biophys. Acta Nucleic Acids Protein Synth. 1972, 272, 539–548. [DOI] [PubMed] [Google Scholar]
- 40. de Mello Filho A. C., Meneghini R., Biochim. Biophys. Acta Mol. Cell Res. 1985, 847, 82–89. [DOI] [PubMed] [Google Scholar]
- 41. Mello-Filho A. C., Meneghini R., Mutat. Res. 1991, 251, 109–113. [DOI] [PubMed] [Google Scholar]
- 42. Toyokuni S., Sagripanti J. L., Free Radical Res. 1999, 31, 123–128. [DOI] [PubMed] [Google Scholar]
- 43. Rai P., Cole T. D., Wemmer D. E., Linn S., J. Mol. Biol. 2001, 312, 1089–1101. [DOI] [PubMed] [Google Scholar]
- 44. Schultz P. G., Taylor J. S., Dervan P. B., J. Am. Chem. Soc. 1982, 104, 6861–6863. [Google Scholar]
- 45. Schultz P. G., Dervan P. B., J. Am. Chem. Soc. 1983, 105, 7748–7750. [Google Scholar]
- 46. Dreyer G. B., Dervan P. B., Proc. Natl. Acad. Sci. USA 1985, 82, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hertzberg R. P., Dervan P. B., J. Am. Chem. Soc. 1982, 104, 313–315. [Google Scholar]
- 48. Kasai H., Nishimura S., Nucleic Acids Res. 1984, 12, 2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Floyd R. A., Watson J. J., Wong P. K., Altmiller D. H., Rickard R. C., Free Radical Res. Commun. 1986, 1, 163–172. [DOI] [PubMed] [Google Scholar]
- 50. White B., Tarun M. C., Gathergood N., Rusling J. F., Smyth M. R., Mol. BioSyst. 2005, 1, 373–381. [DOI] [PubMed] [Google Scholar]
- 51. Foote C. S., Photochem. Photobiol. 1991, 54, 659. [DOI] [PubMed] [Google Scholar]
- 52. Baptista M. S., Cadet J., Di Mascio P., Ghogare A. A., Greer A., Hamblin M. R., Lorente C., Nunez S. C., Ribeiro M. S., Thomas A. H., Vignoni M., Yoshimura T. M., Photochem. Photobiol. 2017, 93, 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Candeias L. P., Steenken S., J. Am. Chem. Soc. 1993, 115, 2437–2440. [Google Scholar]
- 54. Hall D. B., Holmlin R. E., Barton J. K., Nature 1996, 382, 731–735. [DOI] [PubMed] [Google Scholar]
- 55. Adam W., Arnold M. A., Grüne M., Nau W. M., Pischel U., Saha-Möller C. R., Org. Lett. 2002, 4, 537–540. [DOI] [PubMed] [Google Scholar]
- 56. Gasparutto D., Ravanat J. L., Gérot O., Cadet J., J. Am. Chem. Soc. 1998, 120, 10283–10286. [Google Scholar]
- 57. Kino K., Saito I., Sugiyama H., J. Am. Chem. Soc. 1998, 120, 7373–7374. [Google Scholar]
- 58. Steinbeck M. J., Khan A. U., Karnovsky M. J., J. Biol. Chem. 1992, 267, 13425–13433. [PubMed] [Google Scholar]
- 59. Steinbeck M. J., Khan A. U., Karnovsky M. J., J. Biol. Chem. 1993, 268, 15649–15654. [PubMed] [Google Scholar]
- 60. Ravanat J. L., Cadet J., Chem. Res. Toxicol. 1995, 8, 379–388. [DOI] [PubMed] [Google Scholar]
- 61. Raoul S., Cadet J., J. Am. Chem. Soc. 1996, 118, 1892–1898. [Google Scholar]
- 62. Ravanat J. L., Martinez G. R., Medeiros M. H. G., Mascio P. D., Cadet J., Tetrahedron 2006, 62, 10709–10715. [Google Scholar]
- 63. Dumont E., Gruber R., Bignon E., Morell C., Moreau Y., Monari A., Ravanat J. L., Nucleic Acids Res. 2016, 44, 56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Duarte V., Gasparutto D., Yamaguchi L. F., Ravanat J. L., Martinez G. R., Medeiros M. H. G., Mascio P. D., Cadet J., J. Am. Chem. Soc. 2000, 122, 12622–12628. [Google Scholar]
- 65. Luo W., Muller J. G., Rachlin E. M., Burrows C. J., Org. Lett. 2000, 2, 613–616. [DOI] [PubMed] [Google Scholar]
- 66. Vialas C., Pratviel G., Claparols C., Meunier B., J. Am. Chem. Soc. 1998, 120, 11548–11553. [Google Scholar]
- 67. Vialas C., Claparols C., Pratviel G., Meunier B., J. Am. Chem. Soc. 2000, 122, 2157–2167. [Google Scholar]
- 68. Joudah L., Moghaddas S., Bose R. N., Chem. Commun. 2002, 16, 1742–1743. [DOI] [PubMed] [Google Scholar]
- 69. Luo W., Muller J. G., Rachlin E. M., Burrows C. J., Chem. Res. Toxicol. 2001, 14, 927–938. [DOI] [PubMed] [Google Scholar]
- 70. Duarte V., Muller J. G., Burrows C. J., Nucleic Acids Res. 1999, 27, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Szabó C., Ischiropoulos H., Radi R., Nat. Rev. Drug Discovery 2007, 6, 662–680. [DOI] [PubMed] [Google Scholar]
- 72. Pacher P., Beckman J. S., Liaudet L., Physiol. Rev. 2007, 87, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Douki T., Cadet J., Free Radical Res. 1996, 24, 369–380. [DOI] [PubMed] [Google Scholar]
- 74. Pryor W. A., Stone K., Ann. N. Y. Acad. Sci. 1993, 686, 12–27. [DOI] [PubMed] [Google Scholar]
- 75. Niles J. C., Burney S., Singh S. P., Wishnok J. S., Tannenbaum S. R., Proc. Natl. Acad. Sci. USA 1999, 96, 11729–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Niles J. C., Wishnok J. S., Tannenbaum S. R., Chem. Res. Toxicol. 2004, 17, 1510–1519. [DOI] [PubMed] [Google Scholar]
- 77. Yu H., Venkatarangan L., Wishnok J. S., Tannenbaum S. R., Chem. Res. Toxicol. 2005, 18, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tretyakova N. Y., Niles J. C., Burney S., Wishnok J. S., Tannenbaum S. R., Chem. Res. Toxicol. 1999, 12, 459–466. [DOI] [PubMed] [Google Scholar]
- 79. Tretyakova N. Y., Groehler A., Ji S., Acc. Chem. Res. 2015, 48, 1631–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Luxford C., Dean R. T., Davies M. J., Chem. Res. Toxicol. 2000, 13, 665–672. [DOI] [PubMed] [Google Scholar]
- 81. Furukawa A., Hiraku Y., Oikawa S., Luxford C., Davies M. J., Kawanishi S., Biochem. J. 2005, 388, 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhou C., Greenberg M. M., J. Am. Chem. Soc. 2014, 136, 6562–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. May P., May E., Oncogene, 1999, 18, 7621–7636. [DOI] [PubMed] [Google Scholar]
- 84. Soussi T., Oncogene, 2007, 26, 2145–2156. [DOI] [PubMed] [Google Scholar]
- 85. Pfeifer G. P., Besaratinia A., Hum. Genet. 2009, 125, 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ozaki T., Nakagawara A., J. Biomed. Biotechnol. 2011, 2011, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Turgeon M. O., Perry N. J. S., Poulogiannis G., Front. Oncol. 2018. 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Soussi T., Kato S., Levy P., Ishioka C., Hum. Mutat. 2005, 25, 6–17. [DOI] [PubMed] [Google Scholar]
- 89. Amstad P., Hussain S. P., Cerutti P., Mol. Carcinog. 1994, 10, 181–188. [DOI] [PubMed] [Google Scholar]
- 90. Souici A. C., Mirkovitch J., Hausel P., Keefer L. K., Felley-Bosco E., Carcinogenesis 2000, 21, 281–287. [DOI] [PubMed] [Google Scholar]
- 91. Jiang D., Malla S., Fu Y. J., Choudhary D., Rusling J. F. , Anal. Chem. 2017, 89, 12872–12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tornaletti S., Pfeifer G. P., Oncogene 1995, 10, 1493–1499. [PubMed] [Google Scholar]
- 93. Tretyakova N., Villalta P. W., Kotapati S., Chem. Rev. 2013, 113, 2395–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Tretyakova N., Matter B., Jones R., Shallop A., Biochemistry 2002, 41, 9535–9544. [DOI] [PubMed] [Google Scholar]
- 95. Rajesh M., Wang G., Jones R., Tretyakova N., Biochemistry 2005, 44, 2197–2207. [DOI] [PubMed] [Google Scholar]
- 96. Tretyakova N., Goggin M., Sangaraju D., Janis G., Chem. Res. Toxicol. 2012, 25, 2007–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Xiong W., Glick J., Lin Y., Vouros P., Anal. Chem., 2007, 79, 5312–5321. [DOI] [PubMed] [Google Scholar]
- 98. Liao Q., Shen C., Vouros P., J. Mass Spectrom. 2009, 44, 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chowdhury G., Guengerich F. P., Chem. Res. Toxicol. 2009, 22, 1310–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chowdhury G., Guengerich F. P., Curr. Protoc. Nucleic Acid Chem. 2011, 1, 716.1-7.16.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Malla S., Kadimisetty K., Fu Y. J., Choudhary D., Jansson I., Schenkman J. B., Rusling J. F., Chem. Sci. 2015, 6, 5554–5563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Malla S., Kadimisetty K., Fu Y. J., Choudhary D., Schenkman J. B., Rusling J. F., Sci. Rep. 2017, 7, 1–7, DOI:10.1038/srep40890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. McLuckey S. A., Van Berkel G. J., Glish G. L., J. Am. Soc. Mass Spectrom. 1992, 3, 60–70. [DOI] [PubMed] [Google Scholar]
- 104. Dumont E., Monari A., Front. Chem. 2015, 3 : 43. doi: 10.3389/fchem.2015.00043 [Google Scholar]
- 105.
- 105a.AutoDock 4.2 is distributed free of charge as open source software, http://autodock.scripps.edu (accessed November 2018);
- 105b.ADT is distributed free of charge as part of the MGL Tools package, http://mgltools.scripps.edu/downloads. (last accessed November 2018).
- 106.J. Stroud, Make nucleic acid server. http://structure.usc.edu/make-na/server.html (last accessed November 2018).
- 107. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 108. Malla S., Kadimisetty K., Jiang D., Choudhary D., Rusling J. F., Biochemistry, 2018, 57, 3883–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rusling J. F., Hvastkovs E. G., Schenkman J. B., Drug Metabolism Handbook, (Eds.: A. F. Nassar, P. F. Hollenburg, J. Scatina) Wiley: Trenton, NJ, 2009, pp. 307–340. [Google Scholar]
- 110. Hvastkovs E. G., Schenkman J. B., Rusling J. F., Annu. Rev. Anal. Chem. 2012, 5, 79–105. [DOI] [PMC free article] [PubMed] [Google Scholar]