

Summary
Objective
Although the cystine/glutamate antiporter System xc − (Sxc −) plays a permissive role in glioma‐associated seizures, its contribution to other acquired epilepsies has not been determined. As such, the present study investigates whether and how Sxc − contributes to the pentylenetetrazole (PTZ) chemical kindling model of epileptogenesis.
Methods
Male Sxc − null (sut/sut) mice and their wild‐type littermates were administered PTZ (i.p.) daily for up to 21 days (kindling paradigm). Seizure severity was scored on a 5‐point behavioral scale. Mossy fiber sprouting, cellular degeneration, and Sxc − light chain (xCT) messenger RNA (mRNA) were explored using Timm staining, thionin staining, and real‐time quantitative polymerase chain reaction (qPCR), respectively. Levels of reduced and oxidized glutathione and cysteine were determined via high‐performance liquid chromatography (HPLC). Plasma membrane protein levels of glutamate and γ‐aminobutyric acid (GABA) receptor subunits as well as the K+/Cl− co‐transporter KCC2 were quantified via western blot analysis.
Results
Repeated administration of PTZ produced chemical kindling in only 50% of Sxc − null mice as compared to 82% of wild‐type littermate control mice. Kindling did not result in any changes in xCT mRNA levels assessed in wild‐type mice. No cellular degeneration or mossy fiber sprouting was discernible in either genotype. Except for a small, but significant, decrease in oxidized cysteine in the hippocampus, no other change in measured redox couples was determined in Sxc − null mice. Cortical levels of the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptor subunit GluA1 were decreased in Sxc − null mice as compared to wild‐type littermates, whereas all other proteins tested showed no difference between genotypes.
Significance
This study provides the first evidence that Sxc − signaling contributes to epileptogenesis in the PTZ kindling model of acquired epilepsy. Further data indicate that a reduction in AMPA receptor signaling could underlie the resistance to PTZ kindling uncovered in Sxc − null mice.
Keywords: astrocytes, GluA1, kindling, pentylenetetrazole, xCT
1.
Key Points.
Mice lacking the System xc − (Sxc −) transporter are resistant to pentylenetetrazole (PTZ) kindling as compared to wild‐type littermates
Basal levels of Sxc − pression appear to be sufficient to facilitate kindling in wild‐type mice
Sxcnull mice have reduced expression of the GluA1 AMPA receptor subunit in cortical plasma membrane
2. INTRODUCTION
System xc − (Sxc −) is a sodium‐independent anionic amino acid antiporter comprising 2 protein components linked via a disulfide bridge: xCT (encoded by SLC7A11), the light chain that confers substrate specificity, and 4f2hc (encoded by SLC3A2), the associated glycoprotein heavy chain that traffics xCT to the plasma membrane.1, 2 Although xCT expression is inducible in a number of tissues, its constitutive expression is limited to the central nervous system (CNS) and lymphoid organs, including the thymus and spleen.3, 4 Within the CNS, xCT is detected in most major brain regions, with studies investigating its cellular source demonstrating that it is predominantly of astrocytic origin.5, 6
Sxc − imports cystine (CySS) and exports glutamate (Glu) with 1:1 stoichiometry across the cellular plasma membrane.7 Early functional characterization of Sxc − established its fundamental physiologic role as a cellular CySS supplier. Following Sxc −‐mediated uptake, CySS is rapidly reduced to cysteine (Cys), a critical component of many structural, catalytic, and regulatory domains of proteins and a precursor to the essential thiol antioxidant, glutathione (GSH).8 Cys is also directly exported from the cell via neutral amino acid transporters.9 Thus, Sxc − regulates intracellular and extracellular thiol redox systems.8, 9 In addition, several studies have determined that Sxc − contributes significantly to the ambient extracellular Glu that bathes the CNS in vivo.10, 11, 12
Alterations in redox homeostasis adversely affect neuronal synaptic plasticity,13, 14 stressing the importance of redox balance in the normal function of brain network activity. In addition, Glu released from astrocytes is known to modulate neuronal excitability and enhance synaptic strength (for review see Ref. 15). Furthermore, multiple studies demonstrate that changes in redox balance and/or extracellular Glu levels could be permissive in generating ictal activity. For example, mice under chronic oxidative stress show increased incidence of spontaneous and handling‐induced seizures, which occurs in association with decreased expression of the glial Glu transporters glutamate transporter 1 (GLT‐1) and glutamate aspartate transporter (GLAST).16 Diminished Glu uptake in mice null for the Glu transporters GLT‐1 or GLAST occurs in association with spontaneous seizures or prolonged seizure duration in an amygdaloid kindling model, respectively.17, 18 Moreover, increased Sxc −‐mediated cystine/glutamate exchange in glioma19 occurs in association with elevated Glu levels,20 and pharmacologic inhibition of Sxc − reduces peritumoral Glu levels in patients with human glioblastoma21 as well as seizure frequency in glioma‐bearing mice.22 Whether Sxc − is involved in non–tumor‐associated seizure generation is not known, although xCT levels are upregulated in hippocampi resected from patients with temporal lobe epilepsy.23 Finally, transgenic xCT null mice require an elevated dose of pilocarpine or kainic acid to elicit behavioral seizures—in addition to demonstrating decreased seizure severity and mortality in response to an acute dose of N‐methyl‐d‐aspartate (NMDA)—suggesting that Sxc − signaling is permissive in generating acute seizure activity.10
The dual regulation of redox systems and Glu homeostasis by Sxc − and the potential for alterations in such to regulate brain ictal activity prompted our interest in the possibility that this antiporter may also contribute to the aberrant changes that predispose the CNS to develop epilepsy.
Kindling, a model of epileptogenesis, is the time‐dependent sensitization of neuronal circuitry that results in a decrease in convulsive seizure threshold such that a normally nonconvulsive stimulus through repeated administration eventually elicits behavioral convulsions. In the seminal kindling studies performed by Goddard, the stimulus was a subconvulsive electrical current, delivered daily via depth electrodes implanted in rat amygdala.24 Later, studies pioneered the use of systemic administration of subconvulsive doses of pentylenetetrazole (PTZ).25, 26 Thus, the overall goal of this study was to explore the contribution of Sxc − to epileptogenesis by comparing PTZ kindling in mice wild‐type or null for the SLC7A11 gene.
3. METHODS
3.1. Animal husbandry
Mice were maintained in the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) accredited laboratory animal resource facility of Syracuse University on a 12‐hour light/dark schedule (7 am/7 pm). Standard mouse chow and water were provided ad libitum. Experiments were carried out using male mice (8‐12 weeks at start of experimentation) in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals as approved by the Syracuse University Institutional Animal Care and Use Committee. Purchased C57BL/6J mice (Jackson Laboratories [JAX] Stock #000664) were allowed to acclimatize to the facility for at least 1 week before any manipulations. Wild‐type (SLC7A11 +/+) and xCT mutant (SLC7A11 sut/sut) mice were bred in‐house from heterozygous (SLC7A11 +/sut) breeding units (F1) that were obtained by crossing SLC7A11 sut/sut male mice (JAX, Stock #001310) with SLC7A11 +/+ female C3H/HeSnJ mice (JAX, Stock #000661). F2 SLC7A11 +/sut progeny were also used as breeding units for studies. Genotyping was performed via polymerase chain reaction (PCR) analysis of tail genomic DNA samples: +/+ primers, 5′‐GAA GTG CTC CGT GAA GG‐3′ (forward), 5′‐ATC TCA ATC CTG GGC AGA TG‐3′ (reverse); sut/sut primers, 5′‐CCA CTG TTG TAG GTC AGC TTA GG‐3′ (forward), 5′‐CAG GAC CTG TGA ATA TGA TAG GG‐3′ (reverse). Mice were segregated by sex at weaning and placed 2 to 3 per cage such that at least one mouse of each genotype was represented. These breeding and housing strategies were employed to control for environmental differences, genetic background influences, and genetic drift. Gross morphologic analysis of SLC7A11 sut/sut and SLC7A11 +/+ male mouse brains at 12 weeks of age revealed no significant differences (not shown).
3.2. Pentylenetetrazole kindling
Five days before each study, mice were acclimated to handling by performing mock daily intraperitoneal (i.p.) injections, which consisted of inverting the mouse and rubbing its abdomen. They were also acclimatized to the procedure room for at least 1 hour on each day of injection. PTZ (Sigma Chemical Co., St. Louis, MO), made fresh daily, was dissolved in saline and filter sterilized, and it was administered intraperitoneally (i.p.) in a volume of 10 mL/kg body weight. C3H/HeSnJ or C57BL/6J mice were treated with a dose of 35 or 39 mg/kg, respectively, once daily for up to 21 days. These doses were chosen for each strain because in initial dose‐ranging studies performed in wild‐type mice only (this study and Ref. 27 respectively), they produced a mean maximal acute seizure score of 2 (see scoring criteria described below). Following each injection, mice were monitored for 30 minutes and the time and severity of behavioral seizures scored and recorded by an observer blinded to genotype using a 5‐point modified Racine scale (0‐4: 0 = no behavioral change; 1 = hypoactivity; 2 = myoclonus; 3 = generalized convulsion with righting reflex; 4 = generalized convulsion with loss of righting reflex).28 Six of 24 SLC7A11 sut/sut vs 0/27 SLC7A11 +/+ (P = 0.01, Fisher exact test)—were a priori excluded from the study because they responded to the first injection with a convulsive seizure (stage 3 or greater) making any further reduction in seizure threshold elicited by the kindling protocol indeterminable. Mice were deemed kindled after exhibiting convulsive seizures (stage 3 or greater) on 3 consecutive days, after which PTZ injections were stopped. Ten days later, the permanence of the kindled state was assessed by rechallenging with PTZ. A schematic of this experimental design is depicted in Figure S1. The percentage of permanently kindled mice was determined by dividing the number of animals with a maximum seizure score ≥3 by the total number of animals injected.
3.3. Real‐time quantitative polymerase chain reaction
The neocortex and dorsal hippocampus were dissected from PTZ‐kindled, PTZ‐nonkindled, or saline‐injected C57BL/6J mice 1 day following the final injection with PTZ or saline, placed in TRIzol reagent (Invitrogen, Carlsbad, CA), and stored at −80°C. Following synthesis of first‐strand complementary DNA (cDNA) from total RNA, real‐time quantitative PCR (qPCR) was performed using mouse‐specific primers for xCT (SLC7A11, Mm01292531_m1, TaqMan Gene Expression Assays, Applied Biosystems, Foster City, CA) and the reference gene hypoxanthine guanine phosphoribosyl transferase (HPRT, Mm01545399_m1, TaqMan Gene Expression Assays, Applied Biosystems) along with TaqMan Universal PCR MasterMix (Applied Biosystems). HPRT expression levels were stable under our experimental conditions. A serial dilution of cortical or hippocampal cDNA demonstrated that the calculated slope of the line comparing ∆C T (C T value of xCT − C T value of HPRT) vs input cDNA was −0.007606 or −0.000905, respectively, and that the efficiency of both primers was >90% in either tissue. Reactions were performed in duplicate or triplicate using an Eppendorf Realplex2 under the following conditions: 50°C for 2 minutes and 95°C for 10 minutes followed by 40 amplification cycles (95°C for 15 seconds and 60°C for 1 minute). Data analysis was performed using the comparative cycle threshold method (∆∆C T), where C T values of xCT were normalized to HPRT C T values from the same sample and compared to the calibrator C T values (saline controls) to determine the relative fold increase in xCT mRNA.
3.4. Timm and thionin staining
Mossy fiber sprouting, elucidated by Timm staining, was quantified in the supragranular layer of the dentate gyrus (DG) by an observer blinded to the experimental condition. Brain slices from PTZ kindled SLC7A11 +/+ and SLC7A11 sut/sut mice and saline‐injected SLC7A11 +/+ mice were sacrificed 16‐17 days after kindling acquisition or cessation of saline injections by transcardial perfusion with a proprietary (FD Rapid TimmStain Kit; FD Neuro Technologies, Inc., Baltimore, MD) sodium sulfide‐containing perfusate, followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB). Brains were postfixed in 4% PFA for 24 hours (4°C), transferred to a 30% sucrose solution in 0.1 M PB for 72 hours (4°C), and then snap‐frozen on dry ice in Optimal Cutting Temperature (O.C.T.) compound (Tissue‐Tek, Torrance, CA) prior to storage at −80°C. Frozen brains were cut serially (≈−0.94 to −2.46 mm posterior to bregma) into 40‐μm coronal sections, mounted on gelatin‐coated slides, air dried at room temperature for 24 hours, and then stored in a light‐protected box at −20°C. Timm staining was carried out per manufacturer's instructions (FD Rapid TimmStain Kit), with the exception that the time in the silver nitrate developing solution was extended to 55‐70 minutes. Mossy fiber sprouting was quantified using a 6‐point rating scale (0‐5) developed by Cavazos et al29: 0 = no Timm granules; 1 = patchy distribution of sparse granules in supragranular layer; 2 = continuous distribution of granules in supragranular layer; 3 = continuous distribution of granules with patches of confluency in supragranular layer; 4 = prominent granules that form a dense, confluent laminar band in supragranular layer; and 5 = prominent granules that form a dense, confluent laminar band in the supragranular layer that extend into the inner molecular layer. A Timm score for each mouse was determined by calculating the median of the scores assigned to the left and right dentate gyrus (DG) at ≈−1.94 mm posterior to bregma. Tissue sections within 80 μm of those processed for Timm staining were thionin‐stained (Sigma Chemical Co.) as we described previously to assess for any neurodegeneration. All tissue analyses were performed on identically processed photomicrographs acquired using a DP73 digital color camera (Olympus, Tokyo, Japan) mounted on an Olympus IX50 inverted microscope (Olympus).
3.5. Reduced/Oxidized glutathione and cysteine measurements
Fully anesthetized naive SLC7A11 +/+ or SLC7A11 sut/sut mice were perfused transcardially with ice‐cold phosphate buffered saline (PBS). The dorsal hippocampus and neocortex were rapidly dissected and snap‐frozen separately in liquid nitrogen. The concentrations of reduced and oxidized glutathione (GSH and GSSG) and cysteine (Cys and CySS) were determined via high‐performance liquid chromatography (HPLC) by the Emory‐Children's Pediatric Biomarkers Core facility.
3.6. Immunoblotting
3.6.1. Plasma membrane protein isolation
Naive SLC7A11 +/+ or SLC7A11 sut/sut mice were perfused transcardially with ice‐cold 1× PBS under full anesthesia. Bilateral dorsal hippocampi and neocortices were dissected, snap‐frozen in liquid nitrogen separately, and stored at −80°C prior to use. Plasma membrane proteins were isolated from pooled hippocampi (2‐5 mice/sample) or bilateral cortices (one mouse/sample) using an aqueous 2‐phase separation method as per manufacturer's instructions (Plasma Membrane Protein Extraction Kit; Abcam, Cambridge, UK). Isolated proteins were suspended in 0.5% Triton X‐100 in PBS. Samples were stored at −80°C until immunoblotting. Protein concentrations were quantified using the BCA assay kit (Pierce, Rockford, IL).
3.6.2. Gel electrophoresis and protein detection
Protein samples (7.5 μg) were separated by 8% Sodium Dodecyl Sulfate Polyacrylamide gel electrophoresis (SDS‐PAGE) under reducing (10 mM dithiothreitol) and chaotropic (8 M urea) conditions followed by electrophoretic transfer to a polyvinylidene difluoride (PVDF) membrane (Bio‐Rad, Hercules, CA). Membranes were blocked for 1 hour at room temperature (Odyssey blocking buffer, LI‐COR Biosciences; Lincoln, NE) and then probed overnight (4°C) with the mouse monoclonal loading control anti‐Na+/K+ ATPase α1 antibody (1:750; Abcam; RRID: AB_306023) in addition to one of the following antibodies: anti‐GluA1 rabbit polyclonal antibody (1:750; Abcam; RRID: AB_2113447); anti‐GluA2 rabbit polyclonal antibody (1:750; Abcam; RRID: AB_2232655); anti‐GluN1 rabbit monoclonal antibody (1:750; Cell Signaling; RRID: AB_1904067); anti‐GluN2A rabbit polyclonal antibody (1:750; Cell Signaling; RRID: AB_2112295); anti‐GluN2B rabbit polyclonal antibody (1:750; Cell Signaling; RRID: AB_1264223); anti‐GABAARα1 rabbit polyclonal antibody (1:3000; Abcam; RRID: AB_732498); or anti‐K+/Cl− co‐transporter (KCC2) rabbit polyclonal antibody (1:750; Abcam; RRID: AB_881571). Species‐specific secondary antibodies labeled with spectrally distinct IRDye fluorescent dyes (LI‐COR Biosciences) were used to detect primary antibodies (1 hour at 25°C) (1:10 000 dilution). Results were recorded on a LI‐COR ODYSSEY Fc Imaging system (LI‐COR Biosciences) and analyzed using Image Studio 3.1 (LI‐COR Biosciences). Na+/K+ ATPase is a protein constitutively expressed in plasma membrane. Thus, plasma membrane protein levels were normalized to their own respective Na+/K+ ATPase levels. The signal intensity pertaining to the amount of hippocampal or cortical protein per lane (7.5 μg) was determined to be in the linear range for each antibody at their respective dilution (data not shown).
3.7. Statistical analysis
All statistical analyses were performed using GraphPad Prism (Version 6.0.1; GraphPad Software, Inc., La Jolla, CA). Curves depicting kindling acquisition as a function of time were compared using a log‐rank test. Two‐way analysis of variance (ANOVA) or unpaired t test was used to compare seizure latencies, mouse weight, and the concentrations of reductants/oxidants. Proportions indicating the percent of mice convulsing were compared using a Fisher exact test. The Mann‐Whitney U test was used to compare Timm scores. Prior to parametric analysis, qPCR data were transformed to the geometric means, whereas immunoblot data was log transformed (y = log[y + 1]) and compared using 1‐way ANOVA or an unpaired t test, respectively. In all cases, significance was set at P < 0.05.
4. RESULTS
4.1. Kindling acquisition, maintenance, and mortality
Irrespective of the mouse's genotype, the number of kindled mice increased steadily over the 21‐day dosing paradigm (Figure 1A), with the mean latency to kindle being 10.3 ± 1.1 and 11.4 ± 1.2 days for SLC7a11 +/+ and SLC7a11 sut/sut mice, respectively (P = 0.54, unpaired t test). Although the latency to convulsive seizure on each of the 3 consecutive days leading up to and inclusive of the kindled state was similar between SLC7a11 +/+ and SLC7a11 sut/sut littermates (Figure 1B; P = 0.40, 2‐way ANOVA), kindling acquisition was significantly reduced in SLC7a11 sut/sut (9/18; 50%) as compared to SLC7a11 +/+ (22/27; 81.5%; Figure 1A; P = 0.02, log‐rank test).
Figure 1.
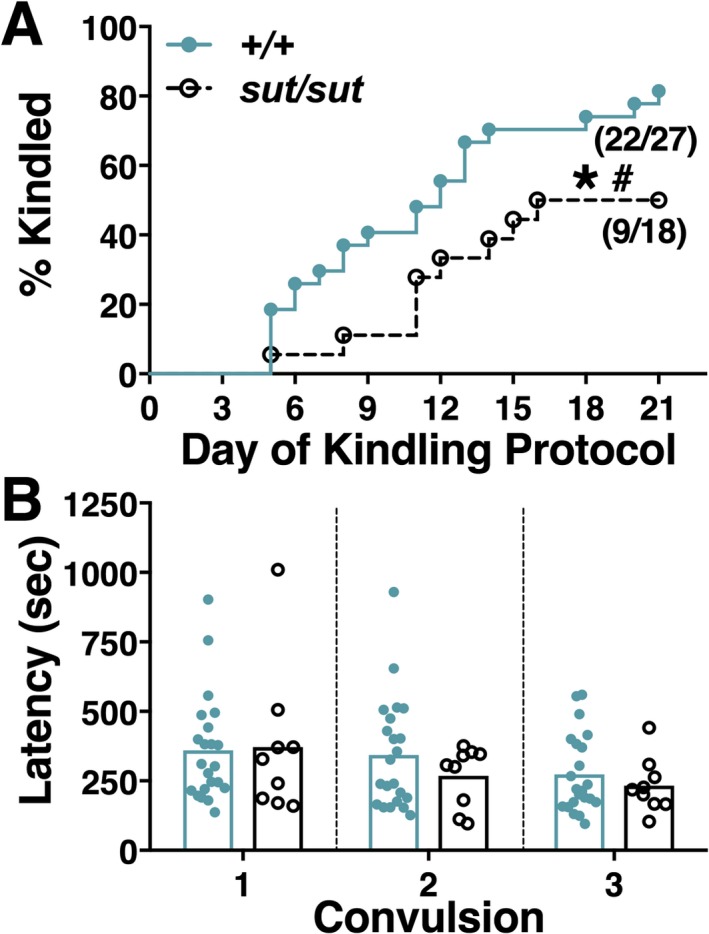
Comparison of pentylenetetrazole (PTZ) kindling acquisition and convulsive seizure latency between SLC7A11+/+ and SLC7A11sut/sut mice. SLC7A11 +/+ (+/+; closed blue circles, n = 27) and SLC7A11 sut/sut (sut/sut; open black circles, n = 18) mice were administered 35 mg/kg PTZ (i.p.) once daily for 21 days or until the mouse became kindled. Five separate experiments were performed over 24 months. A, Kindling acquisition: Each data point represents the percentage of mice kindled each day over the 21 day paradigm, which was determined by dividing the number of animals defined as kindled (see Methods) by the total number of animals injected. Fractions represent the proportion of mice kindled at the end of the protocol. The rate and proportion of SLC7A11 sut/sut mice that kindled are significantly decreased as compared to SLC7A11 +/+ littermate controls as denoted by the asterisk and pound sign, respectively (*P = 0.02, log‐rank test; # P < 0.05, Fisher exact test). B, Convulsive seizure latency: Each data point [closed blue circles (+/+) or open circles (sut/sut)] represents the latency to convulsion (behavioral score ≥3) of a single mouse for each of the 3 consecutive convulsions (depicted as Convulsion 1‐3) that led to its kindled state. Bars represent the mean latency to convulsive seizure for each genotype. Kindled SLC7A11 +/+ and SLC7A11 sut/sut mice have similar convulsive seizure latencies (P = 0.40, 2‐way analysis of variance [ANOVA])
Permanency of the kindled state, determined by rechallenge with PTZ 10 days after the third convulsion, was neither 100% for either genotype nor statistically significant between genotypes, with only 82% of the SLC7a11 +/+ (18/22) and 78% of SLC7a11 sut/sut (7/9) responding to the PTZ challenge with a seizure score of ≥3 (P = 1.00, Fisher exact test). The latency to convulsion upon rechallenge also did not differ between the genotypes (431 ± 45 vs 416 ± 51 s for SLC7a11 +/+ vs SLC7a11 sut/sut, respectively; P = 0.85, unpaired t test).
Of note, no mortality occurred in either group during the kindling paradigm. Mice that completed the paradigm exhibited an average loss of 1% body weight. However, the final weight of kindled mice did not differ between genotypes (25.2 ± 0.4 vs 23.9 ± 0.5 g for SLC7a11 +/+ and SLC7a11 sut/sut, respectively), nor did these differ from that of nonkindled mice (23.2 ± 1.1 vs 24.4 ± 0.5 g for SLC7a11 +/+ and SLC7a11 sut/sut, respectively, 2‐way ANOVA).
4.2. xCT mRNA levels
To determine whether the kindling phenotype was associated with an increase in xCT expression, we measured hippocampal and cortical xCT mRNA levels in kindled, nonkindled, and saline‐injected SLC7a11 +/+ mice via quantitative PCR, 1 day following the final PTZ or saline injection. Neither hippocampal nor cortical xCT mRNA expression levels of kindled mice differed from nonkindled or saline‐administered control mice (Figure 2), suggesting that basal levels of Sxc − are sufficient to facilitate kindling.
Figure 2.
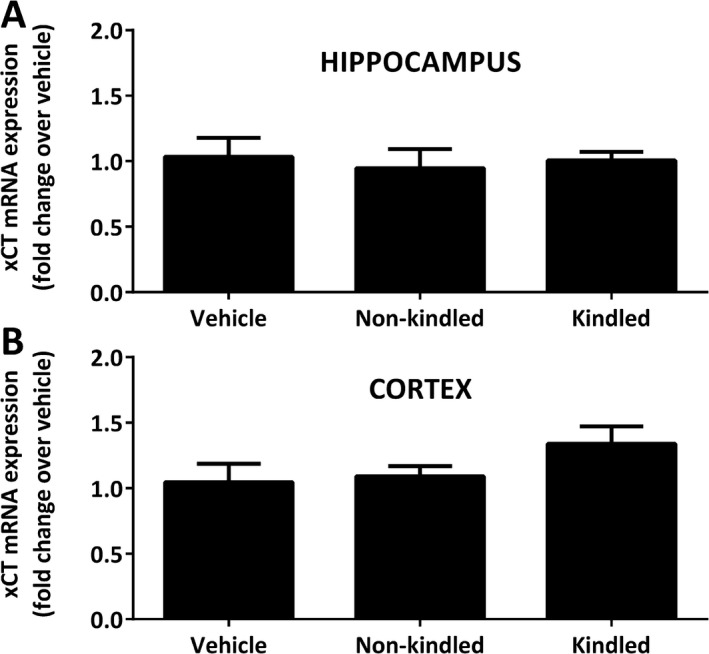
Pentylenetetrazole (PTZ) kindling phenotype is not associated with alterations in Sxc− light chain (xCT) messenger RNA (mRNA). SLC7A11 +/+ mice on a C57BL/6J background were administered 39 mg/kg PTZ once daily for 21 days or until kindled. Mice receiving vehicle (saline) were injected in parallel. The left hippocampus (A) and left cortex (B) were harvested 24 h following the final injection of PTZ or vehicle and xCT mRNA was assessed via quantitative polymerase chain reaction (qPCR). Data are expressed as mean ± standard error of the mean (SEM) fold‐change in xCT mRNA compared with vehicle‐injected controls. No significant between‐group differences in xCT mRNA expression in hippocampus (P = 0.84) or cortex (P = 0.26) was found as determined by 1‐way analysis of variance (ANOVA). N = 4‐5 each per treatment group
4.3. Histologic analysis of injury and axonal sprouting
Sixteen to 17 days following PTZ kindling, brains were examined for signs of overt cellular degeneration. Thionin‐staining of brain slices revealed that the cortex (not shown) and principal layers of the hippocampal formation (Figure 3A,E,I), including the CA1 (Figure 3B,F,J), the CA3 (Figure 3C,G,K), and the dentate gyrus (DG; Figure 3D,H,L) of kindled mice appeared grossly normal and intact irrespective of genotype. The lack of degeneration was confirmed in adjacent sections using Fluoro‐Jade C and a 4′,6‐diamidino‐2‐phenylindole (DAPI) counterstain (data not shown). Aberrant sprouting of DG granule cell axons, known as mossy fiber sprouting, is a common feature in temporal lobe epilepsy and is found in some, but not all, rodent models of epileptogenesis. PTZ‐kindled SLC7a11 +/+ mice, maintained on a C3H/HeSnJ background, had few mossy fiber synaptic terminals, as evidenced by little to no Timm granules in the supragranular layer of the DG that were comparable in number to those quantified in saline‐treated control mice (Figure 4; median Timm score = 0.75 vs 1, respectively; P = 0.69, Mann‐Whitney U test). SLC7a11 sut/sut kindled mice also showed little to no change in mossy fiber sprouting (Figure 4; median Timm score = 1). Thus, alterations in mossy fiber sprouting cannot explain the reduction in kindling acquisition in SLC7a11 sut/sut as compared to SLC7a11 +/+ mice.
Figure 3.
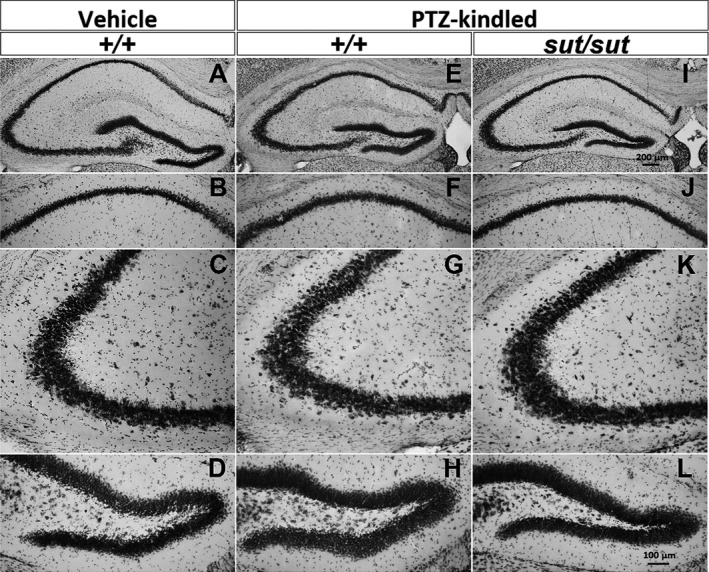
Pentylenetetrazole (PTZ) kindling phenotype is not associated with cellular degeneration. Representative photomicrographs of thionin‐stained coronal brain sections ≈−1.82 mm posterior to bregma: (A‐D) vehicle (saline)‐injected SLC7A11 +/+ (+/+; n = 3), (E‐H) kindled SLC7A11 +/+ (+/+; n = 4), and (I‐L) kindled SLC7A11 sut/sut (sut/sut; n = 2) brain sections. Images represent the hippocampal formation (A, E, I; 8×), CA1 (B, F, J; 20×), CA3 (C, G, K; 20×), or the dentate gyrus (DG) (D, H, L; 20×)
Figure 4.
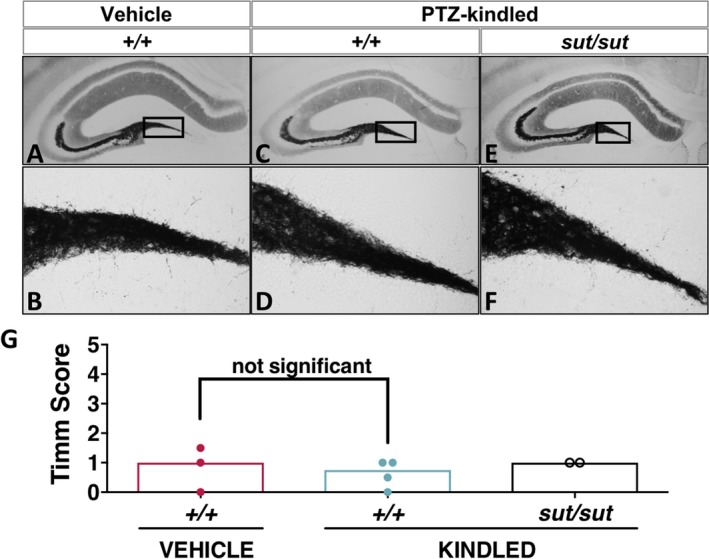
Pentylenetetrazol e (PTZ) kindling phenotype is not associated with increased mossy fiber sprouting. A‐F, Representative photomicrographs of Timm‐stained hippocampal formation ≈ −1.94 mm posterior to bregma: (A‐B) vehicle (saline)‐injected control SLC7A11 +/+ (+/+; n = 3, red bar/circles), (C‐D) kindled SLC7A11 +/+ (+/+; n = 4, blue bar/circle) (C‐D), and (E‐F) kindled SLC7A11 sut/sut (sut/sut; n = 2, black bar/circles). Images represent the hippocampal formation (A, C, E; 8×) and the DG (B, D, F; 40×). Boxes in A, C, and E depict the region of the DG assessed for mossy fiber sprouting. G, Each circle represents the Timm score (see Methods) of a single mouse determined by calculating the median of the scores assigned to the right and left dentate gyrus (DG) (40×) at ≈ −1.94 mm bregma. Open bars represent the median Timm score for each group. There was no significant difference in mossy fiber sprouting between vehicle‐injected SLC7A11 +/+ and kindled SLC7A11 +/+ mice as determined by the Mann‐Whitney U test (P = 0.69)
4.4. Redox signaling
Cys derived from Sxc − import of CySS is the rate‐limiting substrate for the production of the low‐molecular‐weight thiol GSH and contributes to the Cys/CySS redox couple across the cell plasma membrane.9, 30, 31 To determine whether loss of Sxc − resulted in a redox imbalance in SLC7a11 sut/sut brains, the hippocampal and cortical levels of reduced and oxidized GSH (GSH and GSSG) and Cys (Cys and CySS) were measured and compared to levels measured from SLC7a11 +/+ tissues. Neither GSH (Figure 5A left; P = 0.74; Figure 5C left; P = 0.28, unpaired t test) nor GSSG levels measured in either the hippocampus or cortex (Figure 5A right; P = 0.34; Figure 5C right; P = 0.12, unpaired t test) levels were different between SLC7a11 +/+ and SLC7a11 sut/sut mice. Cortical levels of Cys were also similar between the genotypes (Figure 5D left; P = 0.80, unpaired t test), whereas there was a small, but nonsignificant increase in CySS (Figure 5D right; P = 0.12, unpaired t test). Notably, in the hippocampus, the level of CySS was significantly decreased in SLC7a11 sut/sut mice as compared to SLC7a11 +/+ littermate controls (Figure 5B right; P = 0.002, unpaired t test), whereas Cys levels were unchanged (Figure 5B left; P = 1.00, unpaired t test).
Figure 5.
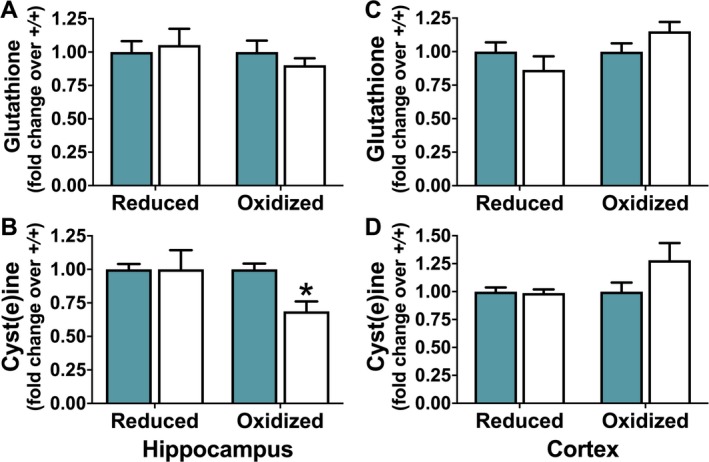
Comparison of redox couples in SLC7A11 +/+ and SLC7A11 sut/sut mice. The concentration of reduced and oxidized glutathione(GSH and GSSG) or cysteine (Cys and CySS), respectively, was determined by high‐performance liquid chromatography (HPLC) in hippocampus (A‐B) or cortex (C‐D) of naive SLC7A11 +/+ (+/+; blue bars; n = 8‐10) and SLC7A11 sut/sut (sut/sut; white bars; n = 10) littermates. Bars represent the mean ± standard error of the mean (SEM) fold‐change over control (+/+, set to one) of the concentration of hippocampal or cortical GSH (A, C; left bars), GSSG (A, C; right bars), Cys (B, D; left bars), or CySS (B, D; right bars). Comparisons between individual levels of reduced or oxidized GSH or Cys were made using an unpaired t test on raw data. An asterisk (*) represents a significant between group difference (P = 0.002)
4.5. Plasma membrane protein expression levels
Finally, we assessed cortical and hippocampal surface expression of AMPA (GluA1, GluA2), NMDA (GluN1, GluN2A, and GluN2B), and GABA (GABAARα1) receptor subunits, as well as, the K+/Cl− co‐transporter (KCC2) (Figure 6), as aberrations in plasma membrane levels of each has been associated with abnormal neuronal synchronization.32, 33, 34 Western blot analysis of this suite of plasma membrane proteins revealed no change in expression levels between SLC7a11 +/+ and SLC7a11 sut/sut littermates in the hippocampus (Figure 6A,B). Of note, cortical levels of the AMPA receptor subunit GluA1 were significantly decreased in SLC7a11 sut/sut mice as compared to SLC7a11 +/+ littermate controls (Figure 6C; P = 0.01, unpaired t test on log transformed data). No other cortical plasma membrane proteins were altered in SLC7a11 sut/sut as compared to SLC7a11 +/+ littermate controls (Figure 6C).
Figure 6.
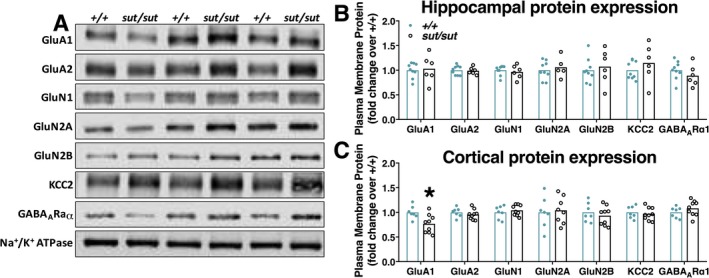
Comparison of plasma membrane protein expression in SLC7A11 +/+ and SLC7A11 sut/sut mice. Plasma membrane protein levels in hippocampus or cortex derived from SLC7A11 +/+ (+/+) or SLC7A11 sut/sut (sut/sut) littermates were compared using western blot analysis (see methods). Representative western blots of +/+ and sut/sut hippocampal plasma membrane proteins are shown in A. Each data point (closed blue circles [+/+; n = 7‐9] or open black circles [sut/sut; n = 5‐9]) represents the level of hippocampal (B) or cortical (C) protein following normalization to their own respective loading control (i.e., Na+/K+ ATPase levels). Bars indicate the mean fold‐change over control (+/+), which was set to one. Hippocampal or cortical plasma membrane protein expression levels were compared using an unpaired t test on log‐transformed data. An asterisk (*) represents a significant between group difference (P = 0.01)
5. DISCUSSION
By virtue of its localization to astrocytes and its role in regulating thiol redox systems as well as ambient extracellular Glu levels, Sxc − (cystine/glutamate antiporter) contributes importantly to CNS homeostasis. In fact, astrocytic transporters, in general, regulate brain excitatory/inhibitory (E/I) balance by providing neurons with energy substrates, by maintaining ion homeostasis, and by removing excess neurotransmitters from the extracellular space. Dysfunction in any one of these activities can facilitate epileptogenesis (reviewed in Ref. 35). In this study, we provide evidence that Sxc − signaling contributes to epileptogenesis in the PTZ‐kindling model of acquired epilepsy (Figure 1). These findings complement studies demonstrating an incontrovertible role for Sxc − in glioma‐associated epilepsy in both mice and man.21, 22
Although Sxc − has been found to be upregulated in glioma,36 we do not find any changes in xCT mRNA expression in PTZ‐kindled mice as compared to vehicle‐injected controls (Figure 2), suggesting that basal levels of Sxc − are sufficient to facilitate PTZ‐kindling. Given the lack of reliable commercial antibody for xCT (personal observations and Ref. 37), we cannot rule out the possibility that enhanced transporter trafficking (as has been described in human glioma cells in vitro38) or increased transporter kinetics (as demonstrated in rat striatum in vivo39) occurs in wild‐type mice. However, present data suggest that in vivo glutamatergic tone—maintained in large part by nonvesicular release of Glu by Sxc − 10, 11, 12—may be enough to dysregulate excitatory signaling in wild‐type mice, as we previously documented in an in vitro paradigm of hypoglycemic neuronal cell death.40 Lending credence to this interpretation are the findings that a reduction in GABAergic inhibition41, 42 and an increase in extracellular Glu and Glu receptor levels43, 44 are associated with PTZ‐kindling in rodents. Conversely, a decrease in ambient, extracellular Glu levels has been reported in mice lacking Sxc −.10, 11, 12 This, together with the reduction in cortical GluA1 AMPA‐receptor subunit protein expression found herein (Figure 6), suggests that glutamatergic signaling may be hypofunctional in the SLC7A11 sut/sut mouse brain in a manner sufficient to reduce PTZ kindling. In keeping with this idea, hippocampal long‐term potentiation (LTP) is also reduced in SLC7A11 sut/sut mice.45 However, a subcohort of SLC7A11 sut/sut mice in this study were a priori excluded because they responded to the first injection with a convulsive seizure (Figure S1). This could simply reflect normal population biologic variability in seizure threshold or represent a true phenotypic difference. Using an acute PTZ paradigm, we hope to differentiate between these 2 possibilities. Moreover, it should be noted that enhanced synaptic GluA1 expression has been reported in the CA1 region of the hippocampus of transgenic xCT null mice,46 although we found no change in global hippocampal GluA1 levels in SLC7A11 sut/sut mice (Figure 6). It is difficult to compare these 2 studies as our plasma membrane measurements include all hippocampal subregions and would capture receptors of both synaptic and extrasynaptic origin. Nevertheless, a global plasma membrane reduction in the cortical GluA1 AMPA receptor subunit is interesting given evidence that block of AMPA receptor signaling is anti‐epileptogenic in animal models.47, 48
Sxc −‐mediated CySS import has been demonstrated to be fundamental in maintaining the extracellular and intracellular redox balance (eg, GSH/GSSG and Cys/CySS), at least in vitro where growth of xCT‐deficient cells is dependent on the addition of a reducing agent.49 Of interest, dysregulation of glutathione homeostasis is associated with impairment of synaptic strength13, 14 and thus represents another possible explanation for the resistance of SLC7A11 sut/sut mice to PTZ kindling. However, SLC7A11 sut/sut mice have normal Cys, GSH, and GSSG levels in hippocampal and cortical tissue, although hippocampal CySS levels were found to be significantly decreased (Figure 4). Our data are in agreement with those of other studies in transgenic xCT nulls showing normal hippocampal GSH levels as compared to wild‐type mice.10 To our knowledge, this study is the first to measure the concentration of GSH in cortex as well as brain cyst(e)ine levels in a mouse null for Sxc −. Taken in toto, current and previous data in both transgenic xCT nulls and SLC7A11 sut/sut mice suggest these animals must employ compensatory mechanisms (eg, EAAT3 or the alanine‐cysteine‐serine transporter [ASCT1/2] as alternative Cys transporters) to sustain GSH levels in vivo.
Finally, epileptogenic network sensitization and its resultant E/I imbalance is associated with synaptic reorganization, particularly when hilar neurons in the dentate gyrus are lost.50, 51 This reorganization includes reactive synaptogenesis of granule cell mossy fibers, whose targets have been demonstrated to include both excitatory52 and inhibitory53 neurons. However, we found no cellular loss in the hippocampus or cortex of PTZ‐kindled mice of either genotype (Figure 3). We also did not observe any significant hippocampal mossy fiber sprouting in any group tested, indicating that this structural alteration was not necessary for kindling development. These results are not unprecedented, as previous studies show that rats54 and guinea pigs55 kindle in the absence of mossy fiber sprouting. Moreover, suppression of reactive sprouting in rats56 and mice57 has been demonstrated to be ineffective in preventing epileptogenic neuronal sensitization following chemically induced status epilepticus.
In sum, our results demonstrate that SLC7A11 sut/sut mice have a reduction in PTZ‐kindling acquisition that occurs in association with decreased GluA1 levels. Given the permissiveness of Sxc − signaling to epileptogenesis—both glioma‐21, 22 and PTZ‐induced (this study)—we postulate that inhibition of astrocyte Sxc − may represent an alternative therapeutic strategy to modulate excessive glutamatergic signaling in individuals predisposed to developing epilepsy.
DISCLOSURE
None of the authors have any conflicts of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and confirm that this report is consistent with those guidelines.
Supporting information
ACKNOWLEDGMENTS
This work was supported by grants from the US National Institutes of Health (NIH)—R01 NS051445 and R01 NS105767 to SJH and R15 NS082982 to JAH.
Sears SMS, Hewett JA, Hewett SJ. Decreased epileptogenesis in mice lacking the System xc − transporter occurs in association with a reduction in AMPA receptor subunit GluA1. Epilepsia Open. 2019;4:133–143. 10.1002/epi4.12307
REFERENCES
- 1. Sato H, Tamba M, Ishii T, et al. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–8. [DOI] [PubMed] [Google Scholar]
- 2. Bassi M, Gasol E, Manzoni M, et al. Identification and characterisation of human xCT that co‐expresses, with 4F2 heavy chain, the amino acid transport activity system x c. Pflügers Arch. 2001;442:286–96. [DOI] [PubMed] [Google Scholar]
- 3. Sato H, Tamba M, Okuno S, et al. Distribution of cystine/glutamate exchange transporter, system x(c)(‐), in the mouse brain. J Neurosci. 2002;22:8028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Taguchi K, Tamba M, Bannai S, et al. Induction of cystine/glutamate transporter in bacterial lipopolysaccharide induced endotoxemia in mice. J Inflamm. 2007;4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pow DV. Visualising the activity of the cystine‐glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. [DOI] [PubMed] [Google Scholar]
- 6. Ottestad‐Hansen S, Hu QX, Follin‐Arbelet VV, et al. The cystine‐glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia. 2018;66:951–70. [DOI] [PubMed] [Google Scholar]
- 7. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256–63. [PubMed] [Google Scholar]
- 8. Sato H, Kuriyama‐Matsumura K, Siow R, et al. Induction of cystine transport via system xc− and maintenance of intracellular glutathione levels in pancreatic acinar and islet cell lines. Biochim Biophys Acta. 1998;1414:85–94. [DOI] [PubMed] [Google Scholar]
- 9. Banjac A, Perisic T, Sato H, et al. The cystine/cysteine cycle: a redox cycle regulating susceptibility versus resistance to cell death. Oncogene. 2008;27:1618–28. [DOI] [PubMed] [Google Scholar]
- 10. De Bundel D, Schallier A, Loyens E, et al. Loss of system x(c)(‐) does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. 2011;31:5792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Massie A, Schallier A, Kim SW, et al. Dopaminergic neurons of system xc−‐deficient mice are highly protected against 6‐hydroxydopamine‐induced toxicity. FASEB J. 2011;25:1359–69. [DOI] [PubMed] [Google Scholar]
- 12. McCullagh EA, Featherstone DE. Behavioral characterization of system xc‐mutant mice. Behav Brain Res. 2014;265:1–11. [DOI] [PubMed] [Google Scholar]
- 13. Robillard JM, Gordon GR, Choi HB, et al. Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS ONE. 2011;6:e20676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Almaguer‐Melian W, Cruz‐Aguado R, Bergado JA. Synaptic plasticity is impaired in rats with a low glutathione content. Synapse. 2000;38:369–74. [DOI] [PubMed] [Google Scholar]
- 15. De Pitta M, Brunel N, Volterra A. Astrocytes: orchestrating synaptic plasticity? Neuroscience. 2016;323:43–61. [DOI] [PubMed] [Google Scholar]
- 16. Liang L‐P, Patel M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic Biol Med. 2004;36:542–54. [DOI] [PubMed] [Google Scholar]
- 17. Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT‐1. Science. 1997;276:1699–702. [DOI] [PubMed] [Google Scholar]
- 18. Watanabe T, Morimoto K, Hirao T, et al. Amygdala‐kindled and pentylenetetrazole‐induced seizures in glutamate transporter GLAST‐deficient mice. Brain Res. 1999;845:92–6. [DOI] [PubMed] [Google Scholar]
- 19. Ye Z‐C, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction–mislocalization of sodium‐dependent glutamate transporters and enhanced activity of cystine–glutamate exchange. J Neurosci. 1999;19:10767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye Z‐C, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–91. [PubMed] [Google Scholar]
- 21. Robert SM, Buckingham SC, Campbell SL, et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med. 2015;7:289ra286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buckingham SC, Campbell SL, Haas BR, et al. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17:1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lewerenz J, Baxter P, Kassubek R, et al. Phosphoinositide 3‐kinases upregulate system xc(‐) via eukaryotic initiation factor 2alpha and activating transcription factor 4 – a pathway active in glioblastomas and epilepsy. Antioxid Redox Signal. 2014;20:2907–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goddard GV. Development of epileptic seizures through brain stimulation at low intensity. Nature. 1967;214:1020–1. [DOI] [PubMed] [Google Scholar]
- 25. Mason CR, Cooper RM. A permanent change in convulsive threshold in normal and brain‐damaged rats with repeated small doses of pentylenetetrazol. Epilepsia. 1972;13:663–74. [DOI] [PubMed] [Google Scholar]
- 26. Ito T, Hori M, Yoshida K, et al. Effect of anticonvulsants on seizures developing in the course of daily administration of pentetrazol to rats. Eur J Pharmacol. 1977;45:165–72. [DOI] [PubMed] [Google Scholar]
- 27. Claycomb RJ, Hewett SJ, Hewett JA. Prophylactic, prandial rofecoxib treatment lacks efficacy against acute PTZ‐induced seizure generation and kindling acquisition. Epilepsia. 2011;52:273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–94. [DOI] [PubMed] [Google Scholar]
- 29. Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–71. [DOI] [PubMed] [Google Scholar]
- 32. Kelley MR, Deeb TZ, Brandon NJ, et al. Compromising KCC2 transporter activity enhances the development of continuous seizure activity. Neuropharmacology. 2016;108:103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mathern GW, Pretorius JK, Mendoza D, et al. Increased hippocampal AMPA and NMDA receptor subunit immunoreactivity in temporal lobe epilepsy patients. J Neuropathol Exp Neurol. 1998;57:615–34. [DOI] [PubMed] [Google Scholar]
- 34. Raol YH, Lund IV, Bandyopadhyay S, et al. Enhancing GABAA receptor α1 subunit levels in hippocampal dentate gyrus inhibits epilepsy development in an animal model of temporal lobe epilepsy. J Neurosci. 2006;26:11342–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eid T, Lee TSW, Patrylo P, et al. Astrocytes and glutamine synthetase in epileptogenesis. J Neurosci Res. 2018:1–18. 10.1002/jnr.24267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yuen TI, Morokoff AP, Bjorksten A, et al. Glutamate is associated with a higher risk of seizures in patients with gliomas. Neurology. 2012;79:883–9. [DOI] [PubMed] [Google Scholar]
- 37. Van Liefferinge J, Bentea E, Demuyser T, et al. Comparative analysis of antibodies to xCT (Slc7a11): forewarned is forearmed. J Comp Neurol. 2016;524:1015–32. [DOI] [PubMed] [Google Scholar]
- 38. Chase LA, Smith D, Schiller N. Cell surface expression of xCT is regulated by the Akt signaling pathway. FASEB J. 2013;27(1017):10. [Google Scholar]
- 39. Baker DA, Xi Z‐X, Shen H, et al. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thorn TL, He Y, Jackman NA, et al. A cytotoxic, co‐operative interaction between energy deprivation and glutamate release from system xc− mediates aglycemic neuronal cell death. ASN Neuro. 2015;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kapur J, Bennett JP Jr, Wooten GF, et al. Evidence for a chronic loss of inhibition in the hippocampus after kindling: biochemical studies. Epilepsy Res. 1989;4:100–8. [DOI] [PubMed] [Google Scholar]
- 42. Kapur J, Michelson HB, Buterbaugh GG, et al. Evidence for a chronic loss of inhibition in the hippocampus after kindling: electrophysiological studies. Epilepsy Res. 1989;4:90–9. [DOI] [PubMed] [Google Scholar]
- 43. Li Z‐q, Yamamoto Y, Morimoto T, et al. The effect of pentylenetetrazole‐kindling on the extracellular glutamate and taurine levels in the frontal cortex of rats. Neurosci Lett. 2000;282:117–9. [DOI] [PubMed] [Google Scholar]
- 44. Schröder H, Becker A, Lössner B. Glutamate binding to brain membranes is increased in pentylenetetrazole‐kindled rats. J Neurochem. 1993;60:1007–11. [DOI] [PubMed] [Google Scholar]
- 45. Li Y, Tan Z, Li Z, et al. Impaired long‐term potentiation and long‐term memory deficits in xCT‐deficient sut mice. Biosci Rep. 2012;32:315–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Williams LE, Featherstone DE. Regulation of hippocampal synaptic strength by glial xCT. J Neurosci. 2014;34:16093–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kodama M, Yamada N, Sato K, et al. Effects of YM90K, a selective AMPA receptor antagonist, on amygdala‐kindling and long‐term hippocampal potentiation in the rat. Eur J Pharmacol. 1999;374:11–9. [DOI] [PubMed] [Google Scholar]
- 48. Namba T, Morimoto K, Sato K, et al. Antiepileptogenic and anticonvulsant effects of NBQX, a selective AMPA receptor antagonist, in the rat kindling model of epilepsy. Brain Res. 1994;638:36–44. [DOI] [PubMed] [Google Scholar]
- 49. Sato H, Shiiya A, Kimata M, et al. Redox imbalance in cystine/glutamate transporter‐deficient mice. J Biol Chem. 2005;280:37423–9. [DOI] [PubMed] [Google Scholar]
- 50. Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate‐treated rats. J Comp Neurol. 1997;385:385–404. [PubMed] [Google Scholar]
- 51. Cavazos JE, Sutula TP. Progressive neuronal loss induced by kindling: a possible mechanism for mossy fiber synaptic reorganization and hippocampal sclerosis. Brain Res. 1990;527:1–6. [DOI] [PubMed] [Google Scholar]
- 52. Scharfman HE, Sollas AL, Berger RE, et al. Electrophysiological evidence of monosynaptic excitatory transmission between granule cells after seizure‐induced mossy fiber sprouting. J Neurophysiol. 2003;90:2536–47. [DOI] [PubMed] [Google Scholar]
- 53. Sloviter RS, Zappone CA, Harvey BD, et al. Kainic acid‐induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyperinhibition in chronically epileptic rats. J Comp Neurol. 2006;494:944–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Osawa M, Uemura S, Kimura H, et al. Amygdala kindling develops without mossy fiber sprouting and hippocampal neuronal degeneration in rats. Psychiatry Clin Neurosci. 2001;55:549–57. [DOI] [PubMed] [Google Scholar]
- 55. Mohapel P, Armitage LL, Gilbert TH, et al. Mossy fiber sprouting is dissociated from kindling of generalized seizures in the guinea‐pig. NeuroReport. 2000;11:2897–901. [DOI] [PubMed] [Google Scholar]
- 56. Longo BM, Mello LE. Blockade of pilocarpine‐or kainate‐induced mossy fiber sprouting by cycloheximide does not prevent subsequent epileptogenesis in rats. Neurosci Lett. 1997;226:163–6. [DOI] [PubMed] [Google Scholar]
- 57. Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials