

Summary
Objective
To present the baseline data of the international TuberOus SClerosis registry to increase disease Awareness (TOSCA) with emphasis on the characteristics of epilepsies associated with tuberous sclerosis complex (TSC).
Methods
Retrospective and prospective patients’ data on all aspects of TSC were collected from multiple countries worldwide. Epilepsy variables included seizure type, age at onset, type of treatment, and treatment outcomes and association with genotype, seizures control, and intellectual disability. As for noninterventional registries, the study protocol did not specify any particular clinical instruments, laboratory investigations, or intervention. Evaluations included those required for diagnosis and management following local best practice.
Results
Epilepsy was reported in 83.6% of patients (1852/2216) at baseline; 38.9% presented with infantile spasms and 67.5% with focal seizures. The mean age at diagnosis of infantile spasms was 0.4 year (median <1 year; range <1‐30 years) and at diagnosis of focal seizures was 2.7 years (median 1 year; range <1‐66 years). A total of 1469 patients (79.3%) were diagnosed with epilepsy <2 years. The rate of infantile spasms was higher in patients with a TSC 2 mutation than in patients with a TSC1 mutation (47.3% vs 23%). ɣ‐aminobutyric acid (GABA)ergic drugs were the most common treatment modality for both infantile spasms (78.7%) and focal seizures (65.5%). Infantile spasms and focal seizures were controlled in 76.3% and 58.2% of patients, respectively. Control of seizures was associated with lower rates of intellectual disability in both groups.
Significance
This registry reports the largest international cohort of patients with TSC. Findings confirmed the typical onset pattern of infantile spasms and other focal seizures in the first 2 years of life, and the high rates of infantile spasms in patients with TSC2 mutation. Our results underscored the occurrence of focal seizures at all ages, including an onset that preceded emergence of infantile spasms. Seizure control was shown to be associated with lower rates of intellectual disability but did not preclude the presence of intellectual disability.
Keywords: epilepsy, registry, TOSCA, tuberous sclerosis complex
1.
Key Points.
Epilepsy was reported in 1852 patients (83.6%) at baseline; of these, there were focal seizures in 67.5% and infantile spasms in 38.9% patients
Epilepsy was diagnosed before 2 years of age in approximately 79% of patients
The rate of infantile spasms was higher in patients with TSC2 mutation than in patients with TSC1 mutation
2. INTRODUCTION
Epilepsy is one of the most common neurologic symptoms in patients with tuberous sclerosis complex (TSC), with reported prevalence from 62% to 93%.1, 2, 3 It is also a significant cause of morbidity and mortality in patients with TSC.2, 4 Epilepsy usually begins during the first months of life and in the majority before the first year.5 Early onset epilepsy often presents as focal seizures initially and can precede, coexist with, or evolve into infantile spasms.5, 6 However, patients with TSC can present with almost all seizure types such as tonic, atonic, or tonic‐clonic seizures.6 There are several therapeutic options available for the treatment of focal seizures and infantile spasms associated with TSC including antiepileptic drugs (AEDs), hormonal therapy, epilepsy surgery, ketogenic diet, and vagus nerve stimulation. However, about two‐thirds of patients develop treatment refractory epilepsies, associated with increased rates of intellectual disability and other TSC‐associated neuropsychiatric disorders (TAND).6, 7
The natural history of epilepsy in TSC has been evaluated in only a handful of studies.1, 2, 8, 9 Most of these studies were retrospective in nature, reported a single‐center cohort, and had relatively small sample size. Only one large cohort was reported by Jeong et al, who evaluated the natural history of epilepsy in a cohort of patients with TSC enrolled from the United States and Belgium (n = 1816; 81.8% had history of focal seizures and 49.2% had infantile spasms).2 The TuberOus SClerosis registry to increase disease Awareness (TOSCA) is an international study that enrolled patients from 170 centers across 31 countries worldwide. The baseline core data of TOSCA provided understanding of the overall TSC manifestations. The study showed epilepsy in 83.5% of patients, subependymal giant cell astrocytoma in 24.4%, and renal angiomyolipomas in 47.2%.10 Herein we report baseline data from TOSCA with the aim of describing the characteristics of epilepsy among this large cohort of patients with TSC.
3. METHODS
The methods of the TOSCA study have been described in detail previously.11 In short, TOSCA is a multicenter, international disease study designed to collect data, retrospectively and prospectively, on patients with TSC from several countries worldwide. Sites with specialists in managing one or more aspects of TSC (in children and adults) were included in the registry. Centers were dedicated mainly to epilepsy care but with almost 30% of patients (mainly adult) enrolled from other specialties as well.
The registry consists of a “core” section and 6 subsections (“petals”). In the “core” section, general information on patient background such as demographic data, family history, genotype, vital signs, prenatal history, clinical features of TSC across all organ systems, comorbidities, and rare manifestations were reported. Data were collected retrospectively and prospectively at baseline (first inclusion visit) and interim analysis was performed on this data collection. A prospective follow‐up observation period was up to 5 years, with regular visits scheduled at a minimum interval of 1 year to ensure an ongoing data stream. These follow‐up data were not included in this article. Subsections (“petals”) represent specific research projects to record in‐depth data related to specific disease manifestations. Given that this is an international noninterventional study, evaluations included were those required for disease diagnosis and management according to the local best practice. The study protocol, therefore, did not specify any particular clinical instruments or laboratory investigations.
Patients of any age who fulfilled clinical criteria for TSC diagnosis were eligible if they had at least 1 documented visit for TSC within the previous 12 months or were newly diagnosed with TSC before participating in the registry. Variables were obtained on the basis of the most recent data collected during the last visit and included seizure type (focal seizures, infantile spasms, other seizures), age at onset of epilepsy, type of epilepsy treatment (grouped as γ‐aminobutyric acid (GABA)ergics, hormonal therapy, ketogenic diet, fructose derivatives, vagus nerve stimulator, mammalian target of rapamycin (mTOR) inhibitors, surgery, other modalities) and treatment outcome (eg, epilepsy resolved spontaneously, was controlled with treatment, or was not controlled with treatment). In addition, we compared the characteristics of epilepsy between the overall epilepsy cohort and those with epilepsy diagnosed before 2 years of age (early onset seizure group). The association between seizure type and genotype as well as between seizure control and intellectual ability was evaluated. Intellectual ability was evaluated by clinician or by formal neuropsychological test and categorized as normal (IQ > 70), mild intellectual disability (ID) (IQ 51–70), moderate ID (IQ 36–50), severe ID (IQ 20‐35), and profound ID (IQ < 20).
All eligible patients enrolled in the TOSCA study were considered in the analysis. Continuous variables were analyzed in terms of value (number of patients, mean, standard deviation, median, minimum and maximum), whereas categorical variables (eg, presence/absence of a condition or manifestation) were analyzed in terms of frequency distribution at baseline. Missing data were not imputed.
This study was designed, implemented, and reported in accordance with the Good Clinical Practice and the ethical principles specified in the Declaration of Helsinki.12 The protocol was approved by the local ethics committee at each center before patient enrollment.
4. RESULTS
4.1. Demographics and clinical characteristics
As of September 30, 2015 (data cutoff date for the third interim analysis), 2216 eligible patients from 170 sites across 31 countries worldwide were enrolled in the TOSCA study (Table 1). The third interim analysis included baseline data for all the patients enrolled in the study.
Table 1.
Patients enrolled from different countries in TOSCA (N = 2216)
| Countries | Number of patients, n (%) |
|---|---|
| Europe | |
| France | 228 (10.3) |
| The Netherlands | 224 (10.1) |
| Germany | 162 (7.3) |
| Spain | 119 (5.4) |
| Belgium | 110 (5.0) |
| Italy | 97 (4.4) |
| Portugal | 54 (2.4) |
| Austria | 52 (2.3) |
| Poland | 52 (2.3) |
| United Kingdom | 32 (1.4) |
| Greece | 30 (1.4) |
| Slovakia | 26 (1.2) |
| Norway | 24 (1.1) |
| Sweden | 23 (1.0) |
| Romania | 21 (0.9) |
| Latvia | 18 (0.8) |
| Estonia | 12 (0.5) |
| Lithuania | 11 (0.5) |
| Slovenia | 8 (0.4) |
| Czech Republic | 7 (0.3) |
| Denmark | 4 (0.2) |
| Outside Europe | |
| China | 252 (11.4) |
| Taiwan | 140 (6.3) |
| Australia | 101 (4.6) |
| Japan | 98 (4.4) |
| Turkey | 91 (4.1) |
| Russia | 60 (2.7) |
| Israel | 59 (2.7) |
| Thailand | 50 (2.3) |
| South Africa | 31 (1.4) |
| Korea | 20 (0.9) |
Baseline patient demographics and clinical characteristics are summarized in Table 2. Baseline data were available for 1154 female (52.1%) and 1,062 male (47.9%) patients; 806 patients (36.4%) were adult (>18 years) and 1410 (63.6%) were children or adolescents. The median age at consent was 13 years (range < 1‐71). The mean age at TSC diagnosis was 7.0 years (median age 1 year; range < 1‐69). Molecular testing for genetic mutations was performed for 1000 patients who met clinical criteria for TSC (45.1%). Of these, 638 patients (63.8%) had a TSC2 mutation, 191 patients (19.1%) had a TSC1 mutation, and 6 patients (0.6%) had both TSC1 and TSC2 mutations. No further molecular details were requested for this study. Of patients who had genetic molecular testing performed, no TSC mutation was identified in 144 patients (14.4%), whereas test results were not available for 9 patients (0.9%). Prenatal diagnosis of TSC was reported in 144 patients (6.5%) and 500 patients (22.6%) had relatives affected with TSC.
Table 2.
Baseline patient demographics and clinical characteristics (N = 2216)
| Characteristics | Baseline data |
|---|---|
| Age at diagnosis of TSCa, years, median (range) | 1 (<1‐69) |
| Gender, n (%) | |
| Male | 1062 (47.9) |
| Female | 1154 (52.1) |
| Patients with molecular testing, n (%) | 1000 (45.1%) |
| Genetic testing, n (%)b | |
| No mutation identified | 144 (14.4) |
| TSC1 mutationc | 197 (19.7) |
| TSC2 mutationc | 644 (64.4) |
| Variation type, n (%)d | |
| Pathogenic mutation | 678 (67.8) |
| Variant of unknown significance | 66 (6.6) |
| Time from first TSC clinical diagnosis to first molecular testing, months | |
| Mean (SD) | 80.8 (116.5) |
| Median (range) | 23 (<1‐721) |
| Patients with prenatal diagnosis, n (%) | 144 (6.5) |
| Biologic mother/father evaluated for TSC, n | |
| Mother | 936 |
| Father | 820 |
| TSC inherited from one parent, n | |
| Total | 51 |
| Mother | 30 |
| Father | 21 |
| Patients with affected relatives, n (%)e | |
| Total | 500 (22.6) |
| 1 | 275 (12.4) |
| 2 | 138 (6.2) |
| 3 | 50 (2.3) |
| >3 | 54 (2.4) |
| Patients with at least one blood relative participating in TOSCA, n (%) | 230 (10.4) |
SD, standard deviation; TSC, tuberous sclerosis complex; TOSCA, TuberOus SClerosis registry to increase disease Awareness.
Data available for 2179 patients.
Information on the type of mutation was missing for 9 patients.
The count (n) includes 6 patients who had both TSC1 and TSC2 mutations.
The count (n) includes 23 patients who had both variation types.
Patients switching from one category to the other during the study visits were counted in each category.
4.2. Characterization of epilepsy
Epilepsy was reported at the baseline visit in 1852 patients (83.6%) (overall epilepsy cohort). In this overall epilepsy cohort, a history of focal seizures was reported in 1250 patients (67.5%) and infantile spasms in 720 patients (38.9%). The co‐occurrence of focal seizures and epileptic spasms was reported in 380 patients (20.5%; Table 3). Of these, epileptic spasms occurred before focal seizures in 242 patients (13.1%) and focal seizures occurred first in 63 patients (3.4%). In 75 patients (4%), focal seizures and epileptic spasms were reported as starting concomitantly. The mean age at diagnosis of focal seizures was 2.7 years (median age 1 year; range < 1‐66 years), whereas mean age at diagnosis of spasms was 0.4 years (median age < 1 year; range < 1‐30 years). In 691 patients (95.6%), infantile spasms were reported within the typical age range (before 2 years). In 22 patients (3%), it occurred between 2 and 5 years and in 10 patients (1.4%) at an older age. The differences in the occurrence rate and age at diagnosis of focal seizures and epileptic spasms (alone or in combination with other types) among patients with TSC1 mutation and TSC2 mutation are shown in Table 4. Infantile spasms were more frequent in patients with a TSC2 mutation compared to those with a TSC1 mutation (47.3% vs 23%).
Table 3.
Type of epilepsy and treatment outcomes in overall epilepsy cohort and in patients diagnosed at <2 years at baseline
| Characteristics | Overall epilepsy cohort (N = 1852), n (%) | Early onset seizure group, (N = 1461), n (%) |
|---|---|---|
| Epilepsy type | ||
| Focal seizuresa | 1250 (67.5) | 984 (67.4) |
| Infantile spasmsa | 720 (38.9) | 684 (46.8) |
| Focal seizures only | 765 (41.3) | 530 (36.3) |
| Infantile spasms only | 246 (13.3) | 221 (15.1) |
| Co‐occurrence of infantile spasms and focal seizures | 380 (20.5) | 375 (25.7) |
| Treatmentb for infantile spasm | ||
| No. of patients who received treatment | 696 (96.7) | 663 (96.9) |
| Type of treatment | ||
| GABAergics | 548 (78.7) | 527 (79.5) |
| ACTH | 122 (17.5) | 121 (18.3) |
| mTOR inhibitors | 38 (5.5) | ‐ |
| Surgery | 29 (4.2) | ‐ |
| Ketogenic diet | 27 (3.9) | ‐ |
| Vagus nerve stimulator | 15 (2.2) | ‐ |
| Fructose derivatives | 9 (1.3) | ‐ |
| Treatment outcomes for infantile spasm | ||
| Resolved spontaneously | 23 (3.3) | 34 (5.0) |
| Controlled with treatment | 530 (76.3) | 506 (74.5) |
| Not controlled with treatment | 108 (15.5) | 106 (15.6) |
| Unknown | 34 (4 9) | 33 (4.9) |
| Treatmentb for focal seizures | ||
| No. of patients who received treatment | 1226 (98.1) | 969 (98.5) |
| Type of treatment | ||
| GABAergics | 803 (65.5) | 683 (70.5) |
| mTOR inhibitors | 95 (7.7) | ‐ |
| Surgery | 85 (6.9) | ‐ |
| Ketogenic diet | 58 (4.7) | ‐ |
| Vagus nerve stimulator | 47 (3.8) | ‐ |
| Fructose derivatives | 43 (3.5) | ‐ |
| ACTH | 35 (2.9) | 32 (3.3) |
| Treatment outcomes for focal seizures | ||
| Resolved spontaneously | 9 (0.7) | 10 (1.0) |
| Controlled with treatment | 713 (58.2) | 552 (56.6) |
| Not controlled with treatment | 466 (38.0) | 384 (39.3) |
| Unknown | 38 (3.1) | 30 (3.1) |
ACTH, adrenocorticotropic hormone; mTOR, mammalian target of rapamycin.
Alone or with other seizures.
As single therapy and in combination with other modalities.
Table 4.
Characteristics of epilepsy according to mutation type
| Characteristics | Overall epilepsy cohort with molecular testing | Early onset seizure group with molecular testing | ||
|---|---|---|---|---|
| TSC1 mutation (N = 152), n (%) | TSC2 mutation (N = 569), n (%) | TSC1 mutation (N = 98), n (%) | TSC2 mutation (N = 489), n (%) | |
| Epilepsy type | ||||
| Focal seizuresa | 113 (74.3) | 409 (71.9) | 75 (76.5) | 350 (71.6) |
| Infantile spasmsa | 35 (23) | 269 (47.3) | 34 (34.7) | 260 (53.2) |
| Infantile spasms only | 12 (7.9) | 67 (11.8) | 11 (11.2) | 61 (12.5) |
| Focal seizures only | 88 (57.9) | 220 (38.7) | 52 (53.1) | 168 (34.4) |
| Concomitant infantile spasms and focal seizures | 21 (13.8) | 163 (28.6) | 21 (21.4) | 161 (32.9) |
| Age at diagnosis, years | ||||
| Focal seizures | ||||
| Mean | 3.7 | 2.2 | 1.1 | 0.9 |
| Median | 2.0 | <1 | 1 | <1 |
| Range | <1‐47 | <1‐59 | <1‐14 | <1‐16 |
| Infantile Spasms | ||||
| Mean | 0.3 | 0.3 | 0.3 | 0.2 |
| Median | <1 | <1 | <1 | <1 |
| Range | <1‐6 | <1‐5 | <1‐6 | <1‐4 |
| Treatmentb for infantile spasm | ||||
| No. of patients who received treatment | 33 (94.3) | 264 (98.1) | 32 (94.1) | 256 (98.5) |
| Type of treatment | ||||
| GABAergics | 22 (66.7) | 223 (84.5) | 22 (68.8) | 216 (84.4) |
| ACTH | 4 (12.1) | 43 (16.3) | 4 (12.5) | 40 (15.6) |
| Surgery | 2 (6.1) | 15 (5.7) | ‐ | ‐ |
| mTOR inhibitors | 2 (6.1) | 16 (6.1) | ‐ | ‐ |
| Vagus nerve stimulator | 1 (3) | 9 (3.4) | ‐ | ‐ |
| Fructose derivatives | 0 | 2 (0.8) | ‐ | ‐ |
| Ketogenic diet | 0 | 14 (5.3) | ‐ | ‐ |
| Treatment outcomes for infantile spasm | ||||
| Resolved spontaneously | 1 (3) | 6 (2.3) | 3 (8.8) | 9 (3.5) |
| Controlled with treatment | 21 (63.6) | 206 (78) | 20 (58.8) | 199 (76.8) |
| Not controlled with treatment | 10 (30.3) | 40 (15.2) | 10 (29.4) | 40 (15.4) |
| Unknown | 1 (3) | 12 (4.5) | 1 (2.9) | 11 (4.2) |
| Treatmentb for focal seizures | ||||
| No. of patients who received treatment | 113 (100) | 402 (98.3) | 75 (100) | 348 (99.4) |
| Type of treatment | ||||
| GABAergics | 67 (59.3) | 316 (78.6) | 51(68) | 280 (80.5) |
| ACTH | 2 (1.8) | 7 (1.7) | 2 (2.7) | 7 (2.0) |
| Surgery | 8 (7.1) | 37 (9.2) | ‐ | ‐ |
| mTOR inhibitors | 6 (5.3) | 34 (8.5) | ‐ | ‐ |
| Vagus nerve stimulator | 4 (3.5) | 16 (4) | ‐ | ‐ |
| Fructose derivatives | 6 (5.3) | 14 (3.5) | ‐ | ‐ |
| Ketogenic diet | 2 (1.8) | 33 (8.2) | ‐ | ‐ |
| Treatment outcomes for focal seizures | ||||
| Resolved spontaneously | 0 | 3 (0.7) | 0 | 4 (1.1) |
| Controlled with treatment | 67(59.3) | 229 (57.0) | 44 (58.7) | 195 (55.9) |
| Not controlled with treatment | 42 (37.2) | 163 (40.5) | 30 (40) | 145 (41.5) |
| Unknown | 4 (3.5) | 7 (1.7) | 1 (1.3) | 5 (1.4) |
ACTH, adrenocorticotropic hormone; mTOR, mammalian target of rapamycin.
Alone or with other seizures.
As single therapy and in combination.
4.3. Treatment
At baseline, a total of 1226 patients (98.1%) with focal seizures in the overall epilepsy cohort received treatment. The majority received GABAergics as a single agent or in combination with other treatment modalities (803, 65.5%). Additional treatment modalities included mTOR inhibitors (95, 7.7%), surgery (85, 6.9%), ketogenic diet (58, 4.7%), vagus nerve stimulation (47, 3.8%), fructose derivatives (43, 3.5%), and corticotropin (ACTH; 35, 2.9%). Focal seizures were controlled by treatment in 713 patients (58.2%), resolved spontaneously in 9 (0.7%), and were not controlled in 466 patients (38%). Outcome data were not available for 38 patients (3.1%).
A total of 696 patients (96.7%) in the overall epilepsy cohort received treatment for infantile spasms. The most frequent treatment modalities (as single agents or in combination with other treatment modalities) included GABAergics (548, 78.7%) and ACTH (122, 17.5%). Additional treatment modalities included mTOR inhibitors (38, 5.5%), surgery (29, 4.2%), ketogenic diet (27, 3.9%), vagus nerve stimulator (15, 2.2%), and fructose derivatives (9, 1.3%). Infantile spasms were controlled with treatment in 530 (76.3%), resolved spontaneously in 23 (3.3%), and were not controlled in 108 patients (15.5%). Outcome data were not available for 34 patients (4.9%). The type of treatment and overall outcome of focal seizures and infantile spasms in the patients diagnosed with epilepsy before the age of 2 years (early onset seizure group) were similar to those in the overall epilepsy cohort (Table 3). Type of treatment and overall outcome of focal seizures and infantile spasms in relation to mutation type are shown in Table 4.
4.4. Association between seizure control and intellectual ability
In the overall epilepsy cohort, a total of 563 of 1250 patients with focal seizures reported at baseline (45%) had received an evaluation of their intellectual abilities assessed by the clinician or by standardized tests depending on the local best practice. Of these, 229 patients (40.7%) had normal intellectual ability, whereas the degree of intellectual disability was recorded as mild in 178 (31.6%), moderate in 58 (10.3%), severe in 86 (15.3%), and profound in 12 patients (2.1%). Of 720 patients in the overall epilepsy cohort with a history of infantile spasms, 279 patients (38.8%) had been evaluated by formal tests for IQ. Of these, 61 patients (21.9%) had normal intellectual ability, whereas mild, moderate, severe, and profound degrees of intellectual disability were observed in 82 (29.4%), 73 (26.2%), 46 (16.5%) and 17 patients (6.1%), respectively. The proportion of patients with normal intellectual ability was higher in patients controlled with treatment than in those not controlled with treatment (23.5% vs 7.9%, patients with infantile spasms, and 47.5% vs 26.8%, patients with focal seizures; Figure 1).
Figure 1.
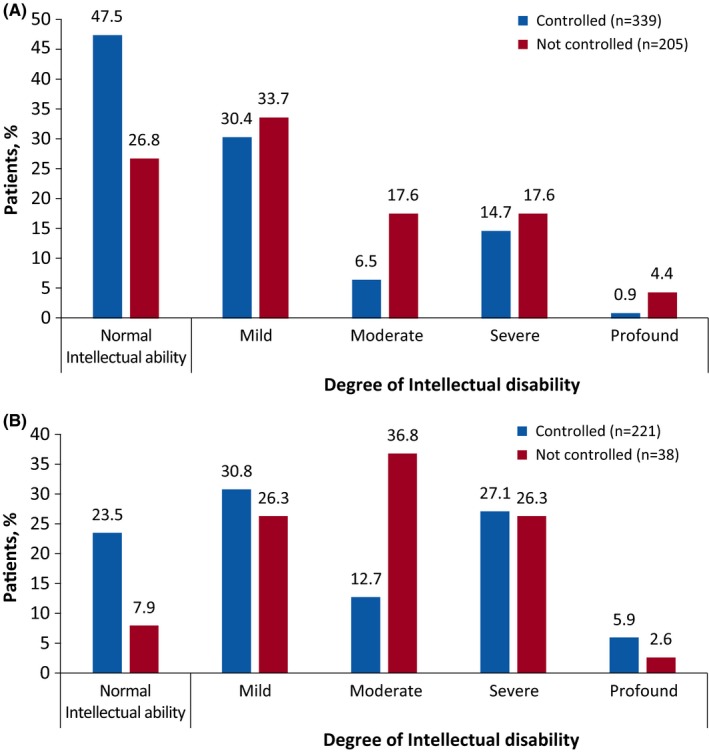
Effect of seizure control on intellectual ability. A, Patients with focal seizures. B, Patients with infantile spasms
In the early onset seizure group, 459 of 984 patients (46.6%) diagnosed with focal seizures had been evaluated for IQ. Of these, 158 patients (34.4%) had normal intellectual ability, whereas mild, moderate, severe, and profound degrees of intellectual disability were observed in 151 (32.9%), 81 (17.6%), 57 (12.4%), and 12 (2.6%) patients, respectively. Among the 684 patients diagnosed with infantile spasms in the early onset seizure group, 273 (39.9%) were evaluated for IQ. A total of 59 patients (21.6%) had normal intellectual ability, whereas mild, moderate, severe, and profound degrees of intellectual disability was observed in 82 (30%), 71 (26%), 44 (16.1%), and 17 (6.2%) patients, respectively. Similar to the overall epilepsy cohort, the proportion of patients with normal intellectual ability was higher in the early onset seizure group that had been controlled than the group with uncontrolled focal seizure (41.2% vs 21.4%) and infantile spasms (23.3% vs 7.9%) after treatment.
5. DISCUSSION
The TOSCA study, which represents the largest cohort of patients with TSC described to date, enrolled patients from 31 countries and from various specialty clinics. The third interim analysis results showed an occurrence at baseline of epilepsy in about 83.6% of the of 2216 enrolled patients, which was in line with both the baseline “core” data of the TOSCA registry10 and other previous reports,1, 8, 13 confirming epilepsy to be the most common clinical presentation of TSC.
A database of 1816 individuals with TSC showed that focal seizures were present in 81.8% of the patients and infantile spasms in 49.2% of the patients included.2 In agreement with this report, focal seizures were also the most common seizure observed in our cohort, followed by infantile spasms. A history of infantile spasms was reported in about 38.9% patients with TSC in the TOSCA study, which was similar to that observed in a single center clinical case series of 291 patients by Chu‐Shore et al1, which reported infantile spasms in approximately 37% of patients with TSC. However, Jeong et al.2 reported a slightly higher rate of infantile spasms (about 49.2%). This could be because patients with TSC in the series of Jeong et al were self‐enrolled by families that might have biased recruitment to more severe cases.
It has been reported that epilepsies associated with TSC most often have their onset during infancy or early childhood, although they may occur at any age, with focal seizures and infantile spasms being the most common seizure types.5, 14 In our study, epilepsy was diagnosed before 2 years of age in about 79% of patients, showing this early onset in the majority of patients. However, seizure onset occurred later in 27% of the cohort, and even in patients older than 40 years of age (about 7 patients [0.6%] diagnosed with focal seizures at age >40 years). This emphasizes that patients with TSC remain at increased risk of epilepsy (mainly focal epilepsies) throughout their lifetime. The occurrence of infantile spasms was higher in the first 2 years (46.8% vs 38.9%), but the occurrence rate of focal seizures was similar (67.4% vs 67.5%) between the whole cohort and the early onset seizure group, respectively. Late‐onset epileptic spasms occurred in 2%‐6% of patients.15 This late occurrence of epileptic spasms was reported in structural epilepsies without specifying TSC.15, 16 Our data suggest that TSC can be considered as a cause of late‐onset epileptic spasms. These findings mainly highlight that infantile spasms and focal seizures occur at an early age in the majority of patients with TSC. Infantile spasms can precede, co‐occur, or follow focal seizures, with this co‐occurrence being a characteristic of TSC.17
This emphasizes the importance of parental education and the potential role of serial electroencephalography (EEG) recordings to detect possible subclinical seizures. This has been suggested in patients with antenatal diagnosis of TSC or patients diagnosed with TSC before the onset of clinical seizures The question on the usefulness to initiate the treatment for subclinical seizures (seizures on EEG without clinical manifestations), or even paroxysmal EEG activity without subclinical seizures, is still under debate.18, 19 Two projects (EPISTOP20 and PREVENT21) are ongoing with the aim of evaluating the impact of an early presymptomatic treatment to be administered at identification of EEG abnormalities without any clinical seizures reported or recorded in patients with TSC.
With respect to genotype, infantile spasms were more frequently seen in patients with a TSC2 mutation compared to those with a TSC1 mutation (47.3% vs 23%). Furthermore, patients with a TSC2 mutation had an earlier onset of infantile spasms and focal seizures (Table 4). These findings reinforced the observations seen in previous studies evaluating the genotype‐phenotype relationships in patients with TSC,1, 22and could suggest a better efficacy of earlier treatments.
GABAergic drugs were the most frequent therapy used in patients with focal seizures and patients with infantile spasms. The term GABAergic drugs was used for vigabatrin and did not include other GABAergic drugs. This finding is in line with the current recommendations that vigabatrin should be used as a first‐line AED treatment for infantile spasms with TSC and for focal seizures occurring before the age of 1 year.5, 23 A better outcome with vigabatrin initiation in association with hormonal therapy was reported in a cohort of patients with infantile spasms.24 However, patients with TSC were excluded from this study and the potential additional benefit from this inaugural bi‐therapy might need additional investigation. The number of patients resistant to treatment was higher for focal seizure (38%) than for infantile spasms (15.5%) and was independent of the mutation type. This emphasizes the better control of infantile spasms with a less efficacy on focal seizures control as they might persist or newly occur after the control of spasms. This finding is important for the development of new therapies for epilepsy in TSC, especially for focal seizures that were not controlled as well by available treatments. The recent EXIST 3 study showing higher efficacy of adjunctive everolimus therapy in patients with treatment‐refractory seizures associated with TSC compared to placebo, is promising in this regard.25
The number of patients not controlled on treatment at baseline was much lower in our study (infantile spasms, 15.5%; focal seizures; 38%) than the 62.5% reported in a previous study by Chu‐Shore et al.1 This lower number of treatment‐resistant patients could be explained by the earlier diagnosis age shown by the high number of young patients enrolled in our study and by the fact that patients were diagnosed and managed in specialized reference centers that follow established recommended guidelines.23 There has been substantial progress in understanding of diagnosis and treatment of epilepsy in patients with TSC during the last decade. The recommendations of serial EEG studies before clinical seizures have allowed the detection of some patients with subtle or infraclinical seizures. In addition, current recommendation on the use of vigabatrin for both infantile spasms and focal seizures for infants might have contributed to this lower rate of pharmacoresistance.5, 23
Studies exploring the efficacy of the ketogenic diet and vagus nerve stimulation in patients with TSC‐associated epilepsy showed that these nonpharmacologic therapies were effective in reducing seizure frequency.26, 27, 28, 29 Surprisingly, the ketogenic diet and vagus nerve stimulation were not commonly used in this cohort with a large proportion of patients with pharmacoresistant epilepsy. This cannot be totally due to lack of availability of such therapies, as many of the TOSCA sites were tertiary centers for TSC and epilepsy treatment. Some explanation can emerge explaining this lower use, as vagus nerve stimulation devices create difficulties for routine use of magnetic resonance imaging (MRI) of the brain or kidney, and the ketogenic diet is more difficult to achieve in patients with psychiatric and behavior disorders that are frequent in the patients with TSC presenting epilepsy. In addition, many of the severely delayed patients can be in specialized institutions where the ketogenic diet is not available. The number of patients that underwent epilepsy surgery was also relatively low in this cohort. Epilepsy surgery for patients with TSC needs specific expertise, as there is often more than a single tuber focus and surgery may need invasive monitoring of seizures to determine the resection area.30, 31 This might be the main limiting factor for epilepsy surgery but should be a reminder to refer patients with pharmacoresistant epilepsy early on to expert epilepsy surgery centers to define the possibility of such therapy that is showing fair results, even in patients with multiple foci.32
The correlation between intellectual ability and seizure control in TSC has been reported.33, 34, 35, 36, 37 Patients with uncontrolled seizures had a higher rate of intellectual disability compared to those with controlled seizures. This association is known primarily for infantile spasms,33, 34, 35, 36, 37but our cohort showed that pharmacoresistant focal seizures equally impact intellectual development in patients with TSC. Similar to these findings, the present study showed that a smaller proportion of patients who were controlled with treatment had intellectual disability compared to those uncontrolled in both groups of infantile spasms and focal seizures.
We acknowledge selection bias as one of the limitations of our study, given that recruitment was achieved through clinical centers with expertise in TSC. Milder cases, or those without seizure disorders in childhood, may not have been included. This study was recruited mainly from pediatric and adult centers for TSC dedicated to epilepsy care, but with almost 30% of patients (mainly adult) enrolled from other specialties as well. Despite the involvement of several specialty clinics, the rate of epilepsy observed in our study was very high (>80%). Furthermore, due to the observational nature of the registry, only data collected from routine clinical practice were reported. This may in part explain the incomplete genetic and neuropsychological scores data, given the widespread differences in access to clinical evaluation of patients with TSC across the globe even in specialized centers, and the lack of access to neuropsychological evaluation. Nevertheless, participation of a large number of centers (170 sites in 31 countries) with complementary expertise has helped in the inclusion of a significant number of patients with TSC, which is likely to be representative of tertiary hospital clinical practice. The lower number of unknown data for epilepsy in this registry with a very large cohort reflects good quality data collection. Moreover, medical data reported in the registry were collected directly by the patients’ physician and not provided by patients and families, ensuring a high level of medical accuracy. Finally, therapies were recorded in groups of AEDs, as the purpose of this registry was to increase disease awareness and to report epilepsy characteristics, and not to evaluate the efficacy of specific therapies.
In conclusion, TOSCA provides valuable insights into the characteristics of epilepsy in patients with TSC. Our aim was to increase awareness on the disease and to present a picture of patients’ characteristics and interventions in a very large number of centers dedicated for TSC care around the world. The findings in this study support previous studies highlighting the higher prevalence of epilepsy in patients with TSC with onset during infancy or early childhood, the correlation between seizure control and intellectual outcome, and the ongoing need for therapies for both infantile spasms and focal seizures as well as for even closer developmental observations correlated with these. The findings also emphasize the better seizure control compared to previous studies that might be multifactorial encompassing earlier diagnosis and a widespread use of the international guidelines for therapies. However, standardized neuropsychological and psychiatric assessment is still lacking in many countries, even in reference centers, and this should be better addressed and implemented.10, 38 Finally, the surgery as a rare recourse in the therapy arsenal should be further investigated and a closer interaction with expert centers for epilepsy surgery should be encouraged.
Further analysis of collected data of the TOSCA study will provide more details in understanding the treatment interventions and outcomes and might inform on the development of our knowledge on a rare disease exploring time‐related changes through these data regarding different aspects as age of diagnosis, therapies, and therapy responses.
6. DISCLOSURE OF CONFLICTS OF INTEREST
J.C.K., E.I.B., T.C., V.C., P.C., G.B.d'A., P.J.dV., J.C.F., M.F., C.F., C.H., S.J., R.N., F.O'C., J.Q., M.S., R.T., M.D., J.A.L., A.M., S.Y., M.P.B., B.Z., and A.C.J. received honoraria and travel support from Novartis. V.C. received personal fees for consulting, lecture fees, and travel from Actelion, Bayer, Biogen Idec, Boehringer Ingelheim, Gilead, GSK, MSD, Novartis, Pfizer, Roche, and Sanofi; grants from Actelion, Boehringer Ingelheim, GSK, Pfizer, and Roche; and personal fees for developing educational material from Boehringer Ingelheim and Roche. P.J.dV. has been on the study steering group of the EXIST‐1, ‐2, and ‐3 studies sponsored by Novartis, and co‐PI on 2 investigator‐initiated studies partly funded by Novartis. R.N. received grant support paid to her institution from Eisai, and lectures fees from Nutricia, Eisai, Advicenne, and GW Pharma. Y.T. received personal fees from Novartis for lecture and received a grant from the Japanese government for intractable epilepsy research. S.J. was partly financed by the EC Seventh Framework Programme (FP7/2007‐2013; EPISTOP, grant agreement no. 602391), the Polish Ministerial funds for science (years 2013‐2018) for the implementation of international cofinanced project, and the grant EPIMARKER of the Polish National Center for Research and Development No STRATEGMED3/306306/4/2016. J.C.K., P.C., C.H., J.A.L., and J.Q. received research grant from Novartis. R.M., L.D′A., and S.S. are employees of Novartis. V.S. reported no conflict of interest. This study was funded by Novartis Pharma AG. The study participants have not received funds for their participation in the study. The results presented in this manuscript have not been published previously except as a poster presentation at the Tuberous Sclerosis Complex International Congress on November 4, 2016. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
This study was funded by Novartis Pharma AG. We thank the patients and their families, investigators, and staff from all the participating sites. We thank Manojkumar Patel, Novartis Healthcare Pvt. Ltd., for providing medical editorial assistance with this manuscript.
Appendix 1. Tosca investigators
1.1.
Japan: Nobuo Shinohara, Shigeo Horie, Masaya Kubota, Jun Tohyama, Katsumi Imai, Mari Kaneda, Hideo Kaneko, Yasushi Uchida, Shoichi Endo, Yoshikazu Inoue, Katsuhisa Uruno; Turkey: Ayse Serdaroglu, Zuhal Yapici, Banu Anlar, Sakir Altunbasak; Russia: Olga Lvova, Oleg Valeryevich Belyaev, Oleg Agranovich, Elena Vladislavovna Levitina, Yulia Vladimirovna Maksimova, Antonina Karas; China: Yuwu Jiang, Liping Zou, Kaifeng Xu, Yushi Zhang, Guoming Luan, Yuqin Zhang, Yi Wang, Meiling Jin, Dingwei Ye, Weiping Liao, Liemin Zhou, Jie Liu, Jianxiang Liao, Bo YAN, Yanchun Deng, Li Jiang, Zhisheng Liu, Shaoping Huang, Hua Li; Korea: Kijoong Kim; Taiwan: Pei‐Lung Chen, Hsiu‐Fen Lee, Jeng‐Dau Tsai, Ching‐Shiang Chi, Chao‐Ching Huang; Australia: Kate Riney, Deborah Yates, Patrick Kwan; Thailand: Surachai Likasitwattanakul, Charcrin Nabangchang, Lunliya Thampratankul Krisnachai Chomtho, Kamornwan Katanyuwong, Somjit Sriudomkajorn; South Africa: Jo Wilmshurst; Israel: Reeval Segel, Tal Gilboa, Michal Tzadok, Aviva Fattal‐Valevski; Greece: Panagiotis Papathanasopoulos, Antigone Syrigou Papavasiliou, Stylianos Giannakodimos, Stylianos Gatzonis, Evangelos Pavlou, Meropi Tzoufi; Belgium: Marc Dhooghe, Hélène Verhelst, Filip Roelens, Marie Cecile Nassogne, Pierre Defresne, Liesbeth De Waele, Patricia Leroy, Nathalie Demonceau, Patrick Van Bogaert, Berten Ceulemans, Lina Dom; France: Pierre Castelnau, Anne De Saint Martin, Audrey Riquet, Mathieu Milh, Claude Cances, Jean‐Michel Pedespan, Dorothee Ville, Agathe Roubertie, Stéphane Auvin, Patrick Berquin, Christian Richelme, Catherine Allaire, Sophie Gueden, Sylvie Nguyen The Tich, Bertrand Godet; Spain: Maria Luz Ruiz Falco Rojas, Jaume Campistol Planas, Antonio Martinez Bermejo, Patricia Smeyers Dura, Susana Roldan Aparicio, Maria Jesus Martinez Gonzalez, Javier Lopez Pison, Manuel Oscar Blanco Barca, Eduardo Lopez Laso, Olga Alonso Luengo, Francisco Javier Aguirre Rodriguez, Ignacio Malaga Dieguez, Ana Camacho Salas, Itxaso Marti Carrera, Eduardo Martinez Salcedo, Maria Eugenia Yoldi Petri, Ramon Cancho Candela; Portugal: Ines da Conceicao Carrilho, Jose Pedro Vieira, José Paulo da Silva Oliveira Monteiro, Miguel Jorge Santos de Oliveira Ferreira Leao, Catarina Sofia Marceano Ribeiro Luis, Carla Pires Mendonca; Lithuania: Milda Endziniene; Latvia: Jurgis Strautmanis; Estonia: Inga Talvik; Italy: Maria Paola Canevini, Antonio Gambardella, Dario Pruna, Salvatore Buono, Elena Fontana, Bernardo Dalla Bernardina; Romania: Carmen Burloiu, Iuliu Stefan Bacos Cosma, Mihaela Adela Vintan, Laura Popescu; Czech Republic: Karel Zitterbart; Slovakia: Jaroslava Payerova, Ladislav Bratsky, Zuzana Zilinska; Austria: Ursula Gruber‐Sedlmayr, Edda Haberlandt, Kevin Rostasy, Ekaterina Pataraia; United Kingdom: Frances Elmslie, Clare Ann Johnston, Pamela Crawford; Denmark: Peter Uldall; Sweden: Paul Uvebrant, Olof Rask; Norway: Marit Bjoernvold, Andreas Sloerdahl, Ragnar Solhoff, Martine Sofie Gilje Jaatun; Poland: Marek Mandera, Elzbieta Janina Radzikowska, Mariusz Wysocki; Germany: Michael Fischereder, Gerhard Kurlemann, Bernd Wilken, Adelheid Wiemer‐Kruel, Klemens Budde, Klaus Marquard, Markus Knuf, Andreas Hahn, Hans Hartmann, Andreas Merkenschlager, Regina Trollmann
Nabbout R, Belousova E, Benedik MP, et al. on behalf of the TOSCA Consortium and TOSCA Investigators . Epilepsy in tuberous sclerosis complex: Findings from the TOSCA Study. Epilepsia Open. 2019;4:73–84. 10.1002/epi4.12286
TOSCA Investigators are listed in Appendix 1.
Contributor Information
Rima Nabbout, Email: rimanabbout@yahoo.com.
the TOSCA Consortium and TOSCA Investigators:
Nobuo Shinohara, Shigeo Horie, Masaya Kubota, Jun Tohyama, Katsumi Imai, Mari Kaneda, Hideo Kaneko, Yasushi Uchida, Shoichi Endo, Yoshikazu Inoue, Katsuhisa Uruno, Ayse Serdaroglu, Zuhal Yapici, Banu Anlar, Sakir Altunbasak, Olga Lvova, Oleg Valeryevich Belyaev, Oleg Agranovich, Elena Vladislavovna Levitina, Yulia Vladimirovna Maksimova, Antonina Karas, Yuwu Jiang, Liping Zou, Kaifeng Xu, Yushi Zhang, Guoming Luan, Yuqin Zhang, Yi Wang, Meiling Jin, Dingwei Ye, Weiping Liao, Liemin Zhou, Jie Liu, Jianxiang Liao, Bo YAN, Yanchun Deng, Li Jiang, Zhisheng Liu, Shaoping Huang, Hua Li, Kijoong Kim, Pei‐Lung Chen, Hsiu‐Fen Lee, Jeng‐Dau Tsai, Ching‐Shiang Chi, Chao‐Ching Huang, Kate Riney, Deborah Yates, Patrick Kwan, Surachai Likasitwattanakul, Charcrin Nabangchang, Lunliya Thampratankul Krisnachai Chomtho, Kamornwan Katanyuwong, Somjit Sriudomkajorn, Jo Wilmshurst, Reeval Segel, Tal Gilboa, Michal Tzadok, Aviva Fattal‐Valevski, Panagiotis Papathanasopoulos, Antigone Syrigou Papavasiliou, Stylianos Giannakodimos, Stylianos Gatzonis, Evangelos Pavlou, Meropi Tzoufi, Marc Dhooghe, Hélène Verhelst, Filip Roelens, Marie Cecile Nassogne, Pierre Defresne, Liesbeth De Waele, Patricia Leroy, Nathalie Demonceau, Patrick Van Bogaert, Berten Ceulemans, Lina Dom, Pierre Castelnau, Anne De Saint Martin, Audrey Riquet, Mathieu Milh, Claude Cances, Jean‐Michel Pedespan, Dorothee Ville, Agathe Roubertie, Stéphane Auvin, Patrick Berquin, Christian Richelme, Catherine Allaire, Sophie Gueden, Sylvie Nguyen The Tich, Bertrand Godet, Maria Luz Ruiz Falco Rojas, Jaume Campistol Planas, Antonio Martinez Bermejo, Patricia Smeyers Dura, Susana Roldan Aparicio, Maria Jesus Martinez Gonzalez, Javier Lopez Pison, Manuel Oscar Blanco Barca, Eduardo Lopez Laso, Olga Alonso Luengo, Francisco Javier Aguirre Rodriguez, Ignacio Malaga Dieguez, Ana Camacho Salas, Itxaso Marti Carrera, Eduardo Martinez Salcedo, Maria Eugenia Yoldi Petri, Ramon Cancho Candela, Ines da Conceicao Carrilho, Jose Pedro Vieira, José Paulo da Silva Oliveira Monteiro, Miguel Jorge Santos de Oliveira Ferreira Leao, Catarina Sofia Marceano Ribeiro Luis, Carla Pires Mendonca, Milda Endziniene, Jurgis Strautmanis, Inga Talvik, Maria Paola Canevini, Antonio Gambardella, Dario Pruna, Salvatore Buono, Elena Fontana, Bernardo Dalla Bernardina, Carmen Burloiu, Iuliu Stefan Bacos Cosma, Mihaela Adela Vintan, Laura Popescu, Karel Zitterbart, Jaroslava Payerova, Ladislav Bratsky, Zuzana Zilinska, Ursula Gruber‐Sedlmayr, Edda Haberlandt, Kevin Rostasy, Ekaterina Pataraia, Frances Elmslie, Clare Ann Johnston, Pamela Crawford, Peter Uldall, Paul Uvebrant, Olof Rask, Marit Bjoernvold, Andreas Sloerdahl, Ragnar Solhoff, Martine Sofie Gilje Jaatun, Marek Mandera, Elzbieta Janina Radzikowska, Mariusz Wysocki, Michael Fischereder, Gerhard Kurlemann, Bernd Wilken, Adelheid Wiemer‐Kruel, Klemens Budde, Klaus Marquard, Markus Knuf, Andreas Hahn, Hans Hartmann, Andreas Merkenschlager, and Regina Trollmann
REFERENCES
- 1. Chu‐Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jeong A, Wong M. Systemic disease manifestations associated with epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57:1443–9. [DOI] [PubMed] [Google Scholar]
- 3. Krueger DA. Management of CNS‐related disease manifestations in patients with tuberous sclerosis complex. Curr Treat Options Neurol. 2013;15:618–33. [DOI] [PubMed] [Google Scholar]
- 4. Webb DW, Fryer AE, Osborne JP. Morbidity associated with tuberous sclerosis: a population study. Dev Med Child Neurol. 1996;38:146–55. [DOI] [PubMed] [Google Scholar]
- 5. Curatolo P, Jozwiak S, Nabbout R. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16:582–6. [DOI] [PubMed] [Google Scholar]
- 6. Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015;14:733–45. [DOI] [PubMed] [Google Scholar]
- 7. de Vries PJ, Whittemore VH, Leclezio L, et al. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr Neurol. 2015;52:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Devlin LA, Shepherd CH, Crawford H, et al. Tuberous sclerosis complex: clinical features, diagnosis, and prevalence within Northern Ireland. Dev Med Child Neurol. 2006;48:495–9. [DOI] [PubMed] [Google Scholar]
- 9. Vignoli A, La Briola F, Turner K, et al. Epilepsy in TSC: certain etiology does not mean certain prognosis. Epilepsia. 2013;54:2134–42. [DOI] [PubMed] [Google Scholar]
- 10. Kingswood JC, d'Augeres GB, Belousova E, et al. TuberOus SClerosis registry to increase disease Awareness (TOSCA) ‐ baseline data on 2093 patients. Orphanet J Rare Dis. 2017;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kingswood JC, Bruzzi P, Curatolo P, et al. TOSCA ‐ first international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet J Rare Dis. 2014;9:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) adopts Consolidated Guideline on Good Clinical Practice in the Conduct of Clinical Trials on Medicinal Products for Human Use. Int Dig Health Legis 1997;48:231–4. [PubMed] [Google Scholar]
- 13. Jozwiak S, Schwartz RA, Janniger CK, et al. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. J Child Neurol. 2000;15:652–9. [DOI] [PubMed] [Google Scholar]
- 14. Rosser T, Panigrahy A, McClintock W. The diverse clinical manifestations of tuberous sclerosis complex: a review. Semin Pediatr Neurol. 2006;13:27–36. [DOI] [PubMed] [Google Scholar]
- 15. Eisermann MM, Ville D, Soufflet C, et al. Cryptogenic late‐onset epileptic spasms: an overlooked syndrome of early childhood? Epilepsia. 2006;47:1035–42. [DOI] [PubMed] [Google Scholar]
- 16. Auvin S, Lamblin MD, Pandit F, et al. Infantile epileptic encephalopathy with late‐onset spasms: report of 19 patients. Epilepsia. 2010;51:1290–6. [DOI] [PubMed] [Google Scholar]
- 17. Barba C, Mai R, Grisotto L, et al. Unilobar surgery for symptomatic epileptic spasms. Ann Clin Transl Neurol. 2017;4:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jozwiak S, Kotulska K, Domanska‐Pakiela D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol. 2011;15:424–31. [DOI] [PubMed] [Google Scholar]
- 19. Wu JY, Peters JM, Goyal M, et al. Clinical electroencephalographic biomarker for impending epilepsy in asymptomatic tuberous sclerosis complex infants. Pediatr Neurol. 2016;54:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Long‐term, Prospective Study Evaluating Clinical and Molecular Biomarkers of Epileptogenesis in a Genetic Model of Epilepsy ‐ Tuberous Sclerosis Complex (EPISTOP). Available at: https://clinicaltrials.gov/ct2/show/NCT02098759
- 21. Preventing Epilepsy Using Vigabatrin In Infants With Tuberous Sclerosis Complex (PREVeNT Trial) A Randomized, Double‐blind, Placebo‐controlled Seizure Prevention Clinical Trial for Infants With TSC. Available at: https://clinicaltrials.gov/ct2/show/NCT02849457
- 22. Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krueger DA, Northrup H. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O'Callaghan FJ, Edwards SW, Alber FD, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open‐label trial. Lancet Neurol. 2017;16:33–42. [DOI] [PubMed] [Google Scholar]
- 25. French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment‐resistant focal‐onset seizures associated with tuberous sclerosis (EXIST‐3): a phase 3, randomised, double‐blind, placebo‐controlled study. Lancet. 2016;388:2153–63. [DOI] [PubMed] [Google Scholar]
- 26. Overwater IE, Bindels‐de Heus K, Rietman AB, et al. Epilepsy in children with tuberous sclerosis complex: chance of remission and response to antiepileptic drugs. Epilepsia. 2015;56:1239–45. [DOI] [PubMed] [Google Scholar]
- 27. Parain D, Penniello MJ, Berquen P, et al. Vagal nerve stimulation in tuberous sclerosis complex patients. Pediatr Neurol. 2001;25:213–6. [DOI] [PubMed] [Google Scholar]
- 28. Major P, Thiele EA. Vagus nerve stimulation for intractable epilepsy in tuberous sclerosis complex. Epilepsy Behav. 2008;13:357–60. [DOI] [PubMed] [Google Scholar]
- 29. Zamponi N, Petrelli C, Passamonti C, et al. Vagus nerve stimulation for refractory epilepsy in tuberous sclerosis. Pediatr Neurol. 2010;43:29–34. [DOI] [PubMed] [Google Scholar]
- 30. Fallah A, Rodgers SD, Weil AG, et al. Resective epilepsy surgery for tuberous sclerosis in children: determining predictors of seizure outcomes in a multicenter retrospective cohort study. Neurosurgery. 2015;77:517–524; discussion 524. [DOI] [PubMed] [Google Scholar]
- 31. Arya R, Tenney JR, Horn PS, et al. Long‐term outcomes of resective epilepsy surgery after invasive presurgical evaluation in children with tuberous sclerosis complex and bilateral multiple lesions. J Neurosurg Pediatr. 2015;15:26–33. [DOI] [PubMed] [Google Scholar]
- 32. Liang S, Zhang J, Yang Z, et al. Long‐term outcomes of epilepsy surgery in tuberous sclerosis complex. J Neurol. 2017;264:1146–54. [DOI] [PubMed] [Google Scholar]
- 33. Eisermann MM, DeLaRaillere A, Dellatolas G, et al. Infantile spasms in Down syndrome–effects of delayed anticonvulsive treatment. Epilepsy Res. 2003;55:21–7. [DOI] [PubMed] [Google Scholar]
- 34. Winterkorn EB, Pulsifer MB, Thiele EA. Cognitive prognosis of patients with tuberous sclerosis complex. Neurology. 2007;68:62–4. [DOI] [PubMed] [Google Scholar]
- 35. Askalan R, Mackay M, Brian J, et al. Prospective preliminary analysis of the development of autism and epilepsy in children with infantile spasms. J Child Neurol. 2003;18:165–70. [DOI] [PubMed] [Google Scholar]
- 36. Koo B, Hwang PA, Logan WJ. Infantile spasms: outcome and prognostic factors of cryptogenic and symptomatic groups. Neurology. 1993;43:2322–7. [DOI] [PubMed] [Google Scholar]
- 37. Jambaque I, Chiron C, Dumas C, et al. Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res. 2000;38:151–60. [DOI] [PubMed] [Google Scholar]
- 38. de Vries PJ, Belousova E, Benedik MP, et al. TSC‐associated neuropsychiatric disorders (TAND): findings from the TOSCA natural history study. Orphanet J Rare Dis. 2018;13:157. [DOI] [PMC free article] [PubMed] [Google Scholar]