

Abstract
Trypanosomes are flagellated protozoan parasites (kinetoplastids) that have a unique redox metabolism based on the small dithiol trypanothione (T(SH)2). Although GSH may still play a biological role in trypanosomatid parasites beyond being a building block of T(SH)2, most of its functions are replaced by T(SH)2 in these organisms. Consequently, trypanosomes have several enzymes adapted to using T(SH)2 instead of GSH, including the glutaredoxins (Grxs). However, the mechanistic basis of Grx specificity for T(SH)2 is unknown. Here, we combined fast-kinetic and biophysical approaches, including NMR, MS, and fluorescent tagging, to study the redox function of Grx1, the only cytosolic redox-active Grx in trypanosomes. We observed that Grx1 reduces GSH-containing disulfides (including oxidized trypanothione) in very fast reactions (k > 5 × 105 m−1 s−1). We also noted that disulfides without a GSH are much slower oxidants, suggesting a strongly selective binding of the GSH molecule. Not surprisingly, oxidized Grx1 was also reduced very fast by T(SH)2 (4.8 × 106 m−1 s−1); however, GSH-mediated reduction was extremely slow (39 m−1 s−1). This kinetic selectivity in the reduction step of the catalytic cycle suggests that Grx1 uses preferentially a dithiol mechanism, forming a disulfide on the active site during the oxidative half of the catalytic cycle and then being rapidly reduced by T(SH)2 in the reductive half. Thus, the reduction of glutathionylated substrates avoids GSSG accumulation in an organism lacking GSH reductase. These findings suggest that Grx1 has played an important adaptive role during the rewiring of the thiol-redox metabolism of kinetoplastids.
Keywords: trypanosome, glutathionylation, oxidation-reduction (redox), enzyme catalysis, disulfide, thiol, fluorescence, trypanothione
Introduction
GSH metabolism requires enzymes, because the spontaneous reaction of GSH with different targets is too slow to be compatible with cell physiology (1, 2). Thiol-disulfide exchange reactions have been studied for decades and the chemistry involved in these reactions is common to both small molecules and proteins containing thiols (3–5). In the simplest form, a thiol-disulfide exchange reaction is a bimolecular nucleophilic substitution (Sn2) in which a nucleophilic thiolate attacks an electrophilic disulfide, producing a linear transition state with a negative charge delocalized over the three sulfur atoms involved, leading to the formation of a new disulfide and a thiolate as leaving group (Scheme 1A). For small molecules, the rate of reaction and the equilibrium position depend on the nucleophilicity of the three possible (in unsymmetrical disulfides) thiolates and the pH of the reaction (5, 6). Several redox processes occurring in vivo involve thiol-disulfide exchange reactions that are catalyzed by enzymes called redoxins (e.g. thioredoxins (Trx) and glutaredoxins (Grx)).
Scheme 1.
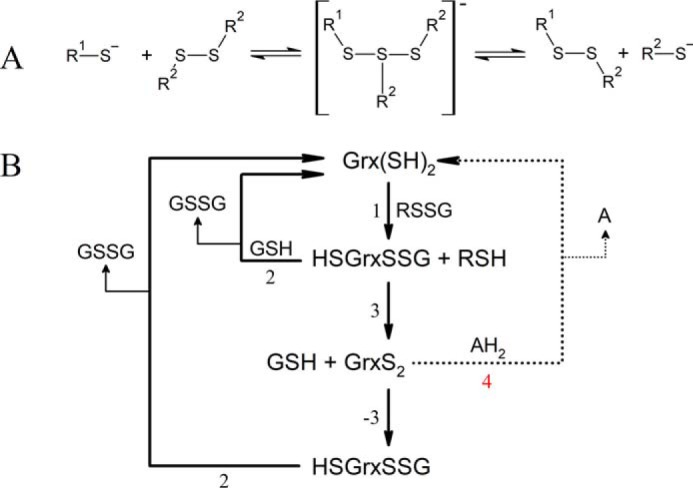
A, thiol-disulfide exchange mechanism. A nucleophilic thiolate (R1-S−) attacks a symmetrical disulfide bond forming a linear transition state that breaks apart to produce a new disulfide and a leaving thiolate (R2-S−). The whole reaction is reversible. B, reaction sequence of a GSH mixed disulfide (RSSG) being reduced by a glutaredoxin (Grx(SH)2) using GSH as reductant via a monothiol mechanism (pathway involving reactions 1 and 2), by a dithiol mechanism (pathway involving reactions 1, 3, −3, and 2); or using an alternative two-electron reductant (AH2, pathway involving reactions 1, 3, and 4). GSH GSH, GSSG GSH disulfide; Grx(SH)2, reduced glutaredoxin; GrxS2, glutaredoxin internal disulfide; HSGrxSSG, glutathionylated glutaredoxin.
Grx are a diverse group of proteins characterized by a thioredoxin-fold and a number of functions including the enzymatic reduction of protein disulfides (7–9), catalysis of thiol-disulfide exchange, preferably with GSH as co-substrate (10, 11), and iron-sulfur cluster binding (12). Grx are classified in three classes based on sequence features: class I Grx are small, single-domain and redox-active proteins carrying a conserved CXXC/S active site sequence; class II are more diverse both in sequence and domain organization, most of them contain a CGFS active site and are redox inactive; class III Grx have an unique CCXC active site (13).
The redox reactions catalyzed by class I Grx are categorized by the number of cysteine residues involved in the reaction cycle. Some substrates, such as ribonucleotide reductase (7), PAPS3 reductase (8), and OxyR (9) are reduced by Grx via a dithiol mechanism that involves both active-site cysteines cycling between a dithiol and disulfide state. However, the reduction of GSH-containing disulfides (deglutathionylation) proceeds mostly by a monothiol mechanism (Scheme 1, reactions 1 and 2) that involves only the N-terminal Cys of the Grx active site. The formation of the internal disulfide of Grx (Scheme 1, reaction 3) is considered a side reaction that can even slow down the deglutathionylation catalytic cycle (14). In any case, when formed, the internal disulfide of the Grx can be reduced back to dithiol by two consecutive reactions employing GSH as reductant (Scheme 1, reactions −3 and 2). Finally, GSSG is reduced back to GSH by GSH reductase (GR).
If GSH is the sole source of reducing equivalents, the dithiol mechanism consists of four steps involving thiol-disulfide exchange (Scheme 1, reactions 1, 3, −3, and 2), three of them being bimolecular; whereas the monothiol mechanism involves only two thiol-disulfide exchange reactions, both bimolecular (Scheme 1, reactions 1 and 2). In this context, the monothiol mechanism is kinetically faster than the dithiol mechanism. In fact, the internal disulfide formed in Grx during the dithiol mechanism can be considered a nonproductive intermediate that needs to be reintroduced in the catalytic cycle via the reaction with an additional GSH molecule. This potentially slow step may eventually be bypassed by a two-electron reductant of the Grx internal disulfide (Scheme 1, reaction 4). Worth noting, unlike Trx, which are efficiently reduced by a two-electron donor (thioredoxin reductase), Grx from most organisms lack a physiological and specific two-electron reductant, and rely on GSH as reducing agent.
The mechanism of deglutathionylation by class I Grx has been studied in detail for mammalian (15–17), yeast (18), bacterial (19, 20), and plant Grx (21). However, these studies used the hydroxyethyl disulfide (HED) assay, which couples the consumption of NADPH by GR upon reduction of GSSG generated by Grx during reduction of the mixed disulfide of GSH and 2-mercaptoethanol (GSSEtOH) (22). Hence, the HED assay yields limited information about the individual steps in the catalytic mechanism of Grx (18).
Grx are ubiquitous even in organisms equipped with redox systems that depend on low molecular weight thiols other than GSH, like actinomycetes (23), firmicutes (24), and trypanosomatids (25). African trypanosomatids, which represent an important public health problem, synthesize and use bis-glutathionyl spermidine (trypanothione, T(SH)2) as major low molecular mass redox substrate and are devoid of genes encoding for thioredoxin reductase and GR (25). They also harbor genes for two class I (named 2CGrx1 and 2CGrx2) and three class II Grx (25, 26). 2CGrx1 (hereinafter Grx1) is a cytosolic and relatively abundant protein with a canonical CPYC active site motif and a third cysteine (Cys-78) that, in vitro, is target of glutathionylation (27). Similar to human Grx2 (12), the trypanosomal Grx1 has been shown to use its active site cysteine residues either to reduce GSH-containing disulfides or to bind an iron-sulfur cluster, employing indistinctly GSH and T(SH)2 as redox cofactors or iron ligands, respectively (28, 29). Grx1, and to a minor extent 2CGrx2, display a significant T(SH)2-GSSG oxidoreductase activity (28). This, together with the fact that trypanosomatids lack GR and convert a large amount of GSH into T(SH)2 (30), has led us to propose that Grx1 surrogates for GR function (26). The biochemical and kinetic basis for trypanosomal Grx1 specificity has so far not been addressed, and this deems relevant to understand the physiological role the protein may have fulfilled during the evolution of this lineage.
With this aim, we investigated the individual steps in the catalysis of Grx1 with monothiol and dithiol substrates applying kinetic and biophysical approaches. The kinetic determinations relied on changes in fluorescence emission of a tryptophan residue (Trp18) located less than 5 Å away from the N-terminal cysteine of the Grx1 active site (PDB 2MYG (31)), and thus provided a reliable measurement of enzyme kinetics. Our data show that Grx1 evolved a remarkable capacity to sustain GSSG reduction at the expense of T(SH)2 and to catalyze thiol/disulfide-exchange reactions by a dithiol mechanism. These findings reinforce the idea that Grx1 has played an important adaptive role during the rewiring of the thiol-redox metabolism of kinetoplastids, and add a new example of functional versatility offered by the Grx domain.
Results
Fluorescence emission spectra of Grx1 and pKa of its catalytic cysteine
Oxidation, alkylation, and protonation of the catalytic Cys21 have a dramatic effect on Trp18 fluorescence, quenching over two-thirds of its intensity at 345 nm (Fig. 1). Such changes in the intrinsic fluorescence of the protein are likely associated to conformational transitions occurring at its active site upon modification of the Cys residues. This feature of Grx1 provides a sensitive and specific tool to monitor reactions of the active site, and was used herein to follow the redox (Fig. 1A) and acid-base behavior of the enzyme (Fig. 1, B and C).
Figure 1.

Changes in the emission spectrum (λexc = 280 nm) of TbGrx1 upon oxidation, alkylation, and protonation. Emission spectra of 4 μm reduced Grx1 (black) treated with: A, excess iodoacetamide (green), excess GSSG (red), or B, HCl (the arrow indicates the direction of change in emission upon acidification), and C, the corresponding titration curve at 350 nm fitted to Equation 5 (red line). Best fit pKa = 5.45 ± 0.02.
The pH titration of reduced Grx1 observed through tryptophan fluorescence (Fig. 1C), reaction with mBBr (Fig. S1) or oxidation with GSSG (Fig. 2) all yield pKa values between 5.1 and 5.5, higher than those reported for most other class I Grx that were measured by different techniques (Table 1). For instance, differences of 1 to 3 pKa units are observed when comparing the trypanosomal protein with different Grx from yeast, human, and pig. The comparatively less acidic nature of the catalytic Cys from Grx1 is to some extent shared also by some Grx from bacteria (Escherichia coli Grx1 and Grx2 pKa ∼5, and Chlamydomonas reinhardtii Grx2 pKa 4.8) and plant (Populus tremula GrxC1 and GrxC2 pKa ∼5). For most class I Grx, a basic residue (Lys ≫ Arg) located three residues upstream the active site Cys appears to contribute to the stabilization of the thiolate form (32–34). In Grx1, this position is occupied by a bulky and hydrophobic Trp residue (Fig. S2), which may explain the higher pKa of the trypanosomal protein.
Figure 2.
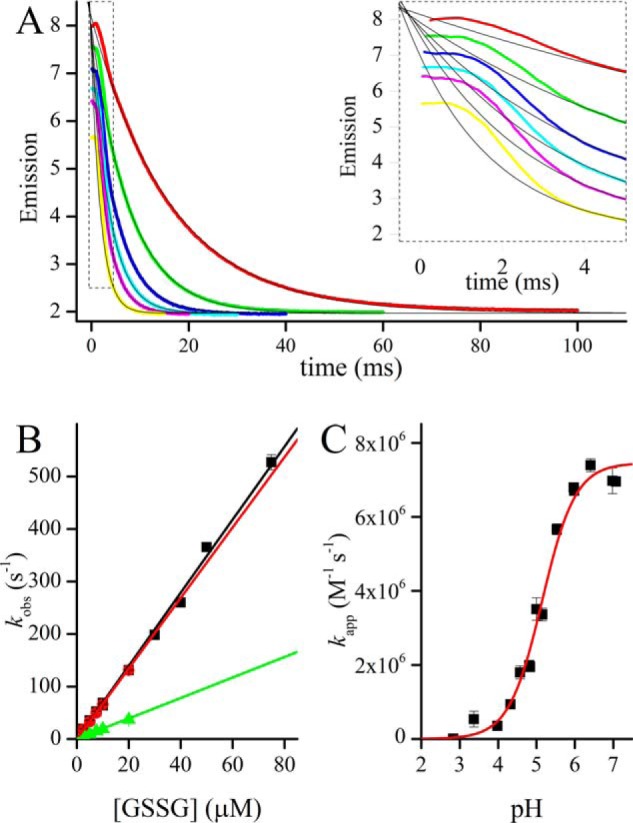
GSSG-mediated oxidation of TbGrx1. A, time courses of the oxidation of 1 μm TbGrx1(SH)2 with excess GSSG at pH 7.07 and 25 °C. From top to bottom 10, 20, 30, 40, 50, and 75 μm [GSSG]0. Inset, detail of the first 5 ms of the reaction showing a transient phase that deviates from first-order kinetics. B, second-order plots of the same reaction at pH 7.07 (■), 5.98 (red square), and 4.83 (green triangle). C, pH profile of the apparent second-order rate constant of the same reaction, the data were fitted to Equation 5 to obtain the pH-independent second order rate constant best fit values B = 7.5 ± 0.2 × 106 m−1 s−1, pKa = 5.15 ± 0.05.
Table 1.
pKa values of the catalytic cysteine of a selection of class I Grx
| Organism | Protein | Active site | Cys | pKa | Method | Reference |
|---|---|---|---|---|---|---|
| T. brucei | Grx1 | CPYC | 21 | 5.45 ± 0.02 | Intrinsic fluorescence | This work |
| 5.4 ± 0.1 | mBBr alkylation | This work | ||||
| 5.15 ± 0.05 | Oxidation by GSSG | This work | ||||
| E. coli | Grx1 | CPYC | 11 | 4.9 | Ellman reaction | 76 |
| Grx3 | CPYC | 11 | <5.5 | NMR chemical shift | 77 | |
| S. cerevisiae | Grx1 | CPYC | 27 | 3.2 | mBBr alkylation | 18 |
| 4.0 | Inactivation by iodoacetamide | |||||
| 4.5 | Alkylation with m-PEG | 78 | ||||
| Grx2 | CPYC | 61 | 3.1 | mBBr alkylation | 18 | |
| 3.5 | Inactivation by iodoacetamide | |||||
| Homo sapiens | Grx1 | CPYC | 22 | 3.5 | Inactivation by iodoacetamide | 16 |
| Grx2 | CSYC | 22 | 4.6 | Inactivation by iodoacetamide | 17 | |
| Sus scrofa | Grx1 | CPFC | 22 | 2.5 | Inactivation by iodoacetamide | 79 |
| C. reinhardtii | Grx1 | CPYC | 47 | 3.9 | Inactivation by iodoacetamide | 80 |
| Grx2 | CPYC | 47 | 4.8 | |||
| P. tremula | GrxS12 | WCSYC | 29 | 2.8 | Alkylation with bimane thioether | 81 |
| 3.8 | Inactivation by iodoacetamide | 82 | ||||
| GrxC1 | CGYC | 31 | 4.7 | NMR chemical shift | 83 | |
| 5.3 | Inactivation by iodoacetamide | 21 | ||||
| GrxC2 | CPFC | 23 | 5.0 | Inactivation by iodoacetamide | 21 | |
| GrxC3 | CPYC | 37 | 4.6 | Inactivation by iodoacetamide | 21 | |
| GrxC4 | CPYC | 27 | 4.6 | Inactivation by iodoacetamide | 21 |
Kinetics of Grx1 oxidation and reduction
Grx1 is rapidly oxidized by GSH containing disulfides
Oxidized GSH and a number of GSH-containing disulfides react with Grx1 with rate constants in the order of 106 m−1 s−1 (Table 2, Fig. 2), which is 1 order of magnitude higher than that determined previously (28). The rate of Grx1 in these reactions is comparable with that of EcGrx1 with glutathionylated RNase (kcat/Km = 3 × 106 m−1 s−1 (35)) and somewhat higher than the previously reported for Grx from yeast (3 × 105 m−1 s−1 (36), pig (7.1 × 105 m−1 s−1 (37)), or human Grx1 (1.24 × 106 m−1 s−1 (38)) with similar substrates. For comparison, the reaction of GSSG with other protein thiols is much slower, with rate constants of 600 m−1 s−1 (protein-disulfide isomerase (39)), 570 m−1 s−1 (Trx (40)), 1.2 m−1 s−1 (redox-sensitive yellow fluorescent protein (36)), and 0.1 m−1 s−1 (human serum albumin, HSA (41)). The time course of the reaction of Grx1 with excess GSSG shows a very fast phase, lasting less than 4 ms, followed by an exponential decay in emission (Fig. 2A). The slower phase fits very well to a first-order function with a kobs that depends linearly with [GSSG]0 (Fig. 2B).
Table 2.
Rate constants of TbGrx1 reactions, except for the underlined data, obtained with E. coli Grx1
| Reactant | k | pH |
|---|---|---|
| m−1 s−1 | ||
| Glutatione containing disulfides | ||
| GSSG | 7.0 ± 0.1 × 106 | 7.07 |
| 6.7 ± 0.1 × 106 | 5.98 | |
| 2.0 ± 0.1 × 106 | 4.83 | |
| 7.5 ± 0.2 × 106 | pH-independent | |
| 5.7 ± 0.2 × 106a | 7.05 | |
| TS2 | 2.5 ± 0.08 × 107 | 7.05 |
| Glutathionylspermidine disulfide | 4.2 ± 0.09 × 106 | 7.05 |
| FGSSGF | 5.7 ± 0.2 × 105 | 7.11 |
| 6.2 ± 0.05 × 105 | 6.85 | |
| 1.1 ± 0.04 × 106b | 6.85 | |
| 5.4 ± 0.1 × 105c | 6.85 | |
| Glutathionylhydroxyethyl disulfide | 6.4 ± 0.04 × 106 | 7.11 |
| Glutathionylcysteinylmethyl ester disulfide | 5.3 ± 1.1 × 106 | 7.03 |
| GlutathionylHSA | 2.6 ± 0.3 × 106 | 7.05 |
| Other disulfides | ||
| Cystine dimethyl ester | 3.7 ± 0.03 × 104 | 7.04 |
| Bishydroxyetyl disulfide | 30 ± 0.4 | 7.07 |
| Cystamine | 20 ± 0.2 | 7.10 |
| Cystine | 99 ± 5 | 7.10 |
| Thiols | ||
| T(SH)2 | 4.8 ± 0.2 × 106 | 7.05 |
| GSH | 39 ± 3 | 7.10 |
| Unspecific oxidation | ||
| H2O2 | 3.77 ± 0.06 | 7.13 |
a Reaction of E. coli Grx1 followed by changes in Trp78 intrinsic fluorescence, as described under “Experimental procedures.”
b TbGrx1C24S.
c TbGrx1C78S.
Other GSH containing disulfides, including GSSEtOH (Fig. 3A), HSA-SSG (Fig. 3B), Tb1CGrx1 Cys181–SSG, FGSSGF, and GSP disulfide all react with rate constants in the range 0.5–6 × 106 m−1 s−1 at neutral pH (Table 2). EcGrx1 was also studied by the same methodology, taking advantage of the fluorescence emission of Trp78, which changes slightly upon oxidation of the active site. The reaction of EcGrx1 with GSSG has a rate constant of 5.7 × 106 m−1 s−1.
Figure 3.
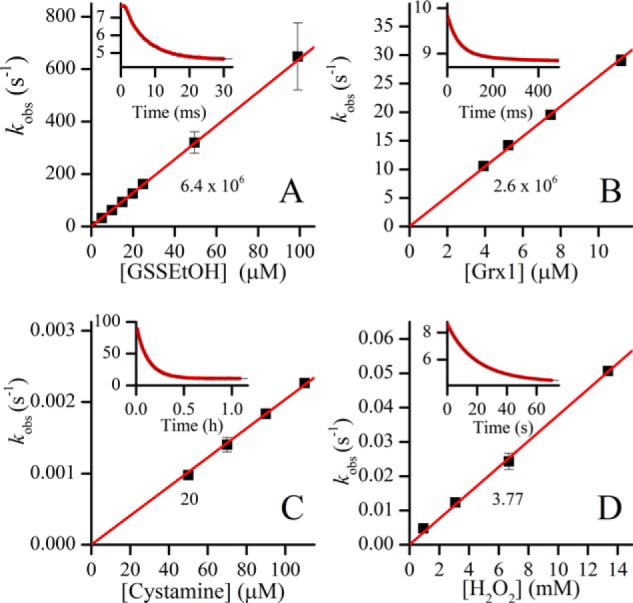
Oxidation of TbGrx1 by different oxidants. A, rate constants for 0.25 μm TbGrx1(SH)2 oxidation with different concentrations of GSH previously equilibrated with 10 mm HED for 2 h. Inset, time course of the reaction mixture containing 25 μm GSSEtOH. B, rate constants of different concentrations of TbGrx1(SH)2 with 0.4 μm HSA-SSG. Inset, time course of the reaction mixture containing 7.5 μm TbGrx1(SH)2. C, rate constants of TbGrx1(SH)2 (4 μm) oxidation with different concentrations of cystamine. Inset, time course of the reaction mixture containing 10 μm cystamine. D, rate constants of TbGrx1(SH)2 (0.4 μm) oxidation with different concentrations of H2O2. Inset, time course of the reaction mixture containing 13.4 mm H2O2. All time course plots show the fluorescence emission values in arbitrary units. Points are plotted as mean ± S.D. of n = 10 (GSSEtOH, HSA-SSG and H2O2) or n = 4 (cystamine). The numbers below the straight lines are the slopes (second-order rate constants) in units of m−1 s−1.
In contrast, disulfides lacking a GSH moiety, such as HED, cystamine (Fig. 3C), or cystine, are slow oxidants of Grx1, highlighting the selectivity for the substrate during the oxidative step of the catalysis. An important molecular factor that seems to determine the specificity exhibited by Grx is the recognition and interaction with the α-carboxylate of the γ-glutamyl of GSH (42, 43). This is supported by the fact that GSP disulfide and TS2, both having a free γ-glutamyl carboxylate but lacking the Gly carboxylate, react with similar or higher rate constant with Grx1(SH)2 than GSSG. Additionally, FGSSGF, which has the amino group of the γ-glutamyl blocked also reacts remarkably fast with Grx1(SH)2 (Table 2). Noticeably, TS2 resulted in the fastest physiological oxidant of Grx1(SH)2 with k = 2.5 × 107 m−1 s−1. In contrast, oxidation by H2O2 is extremely slow (3.77 m−1 s−1 at pH 7.4; Fig. 3D) and comparable with that of low molecular weight thiolates (44).
Reduction of Grx1S2 by GSH is extremely slow
The disulfide formed between the active site cysteines of Grx needs to be reduced to maintain the enzyme in the monothiol catalytic cycle. Synthetic dithiols (e.g. DTT) can reduce the oxidized forms of Grx. However, the reaction of Grx1S2 with DTT is not specific and occurs with a moderate rate constant (690 ± 6 m−1 s−1, Fig. S3) in the expected range for the reduction of any disulfide by this dithiol (6, 45) and is mainly driven by the entropic effect of the formation of a six-membered cyclic disulfide of DTT (trans-4,5-dihydroxy-1,2-dithiane).
Contrary to dithiol reagents, the reduction of Grx1 by GSH is very slow. Our first attempts to reduce Grx1S2 with an excess GSH were unconducive, apparently due to the rapid reoxidation of the enzyme by the contaminating GSSG present in our commercial GSH (about 0.5%). Assays conducted with a GSSG-free GSH sample yielded a rate constant for the reduction of Grx1 of 39 ± 3 m−1 s−1, identical to the previously reported value of 37 m−1 s−1 (28) (Fig. 4). Thus, under physiological conditions, the reduction of Grx1 by GSH will not be kinetically favored.
Figure 4.
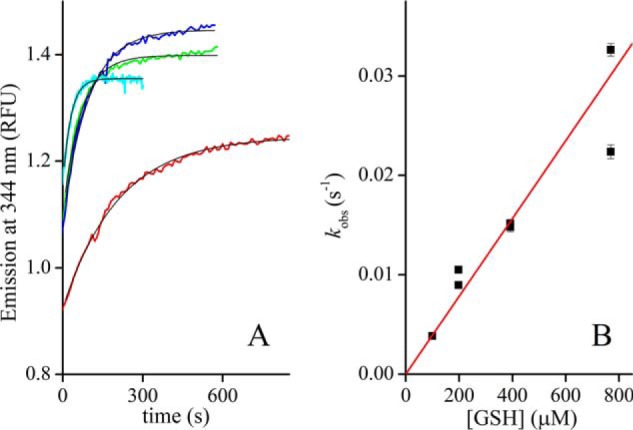
Reduction of TbGrx1S2 by GSH. A, time courses of TbGrx1S2 (4 μm) reduction by GSH at 99.5 μm (red), 198 μm (blue), 392 μm (green), and 769 μm (cyan) in the presence of GR and NADPH (see text). The black lines are the nonlinear fits to first-order exponential functions. B, dependence of the rate constant of protein reduction on GSH concentration, second-order rate constant 39 ± 3 m−1 s−1. Points are plotted as kobs ± S.E. of the fit for each replica.
The glutathionylated form of Grx1 is not particularly stable
The reactions of Grx1S2 with GSH (i.e. reaction −3) or Grx1(SH)2 with GSSG (i.e. reaction 1), produce a transient covalent intermediate, namely Grx1 glutathionylated in Cys21 (Grx1SSG). We conducted several experiments to identify the occurrence of this mixed disulfide.
First, the SOFAST-HMQC spectra of 15N-labeled Grx1S2 was monitored during titration with GSH. Upon addition of GSH, for some of the amino acids new peaks appeared at positions different from Grx1(SH)2 and at the same time the corresponding peaks of Grx1S2 disappeared. The spectra showed that even with excess GSH, up to 5 eq, Grx1(SH)2 is not formed. Grx1SSG cannot be unambiguously identified by this experiment but, compared with the spectrum of the fully reduced and fully oxidized protein, the chemical shift changes observed for several amino acids of Grx1 upon addition of GSH, indicate the formation of a distinct intermediate (Fig. 5). Interestingly, although the assignment of the oxidized protein is not available, from the assignment of Grx1(SH)2 (31) it is evident that the most prominent shifts occurred at residues that partake in the GSH-binding cleft (Fig. 5).
Figure 5.
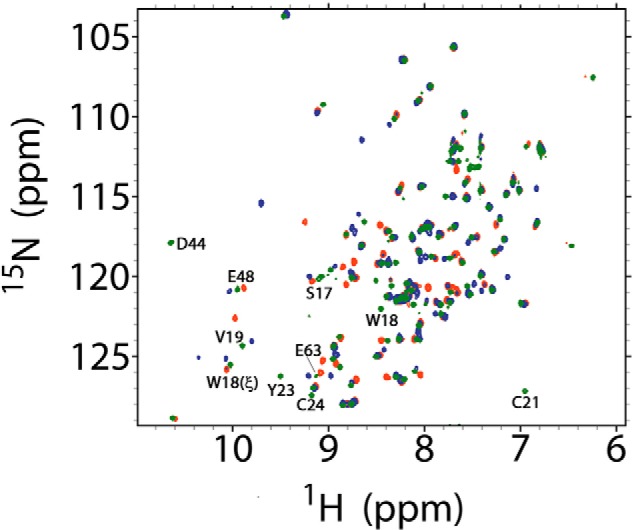
Superposition of SOFASTF-HMQC spectra of TbGrx1 under different redox states. Complete spectra of TbGrx1(SH)2 (green), TbGrx1S2 (blue), and TbGrx1S2 after the addition of 1 eq of GSH (red). Some residues belonging to the GSH binding pocket are labeled according with its assignment in the spectrum of the reduced protein (green peaks).
The formation of Grx1SSG was confirmed by MS analysis (Table 3; Fig. S4). Treatment of Grx1(SH)2 with GSSG in a 1:1 ratio resulted in the formation of the internal disulfide on Grx1 active site and a minor peak of m/z coincident with the mass estimated for the Cys21-glutathionylated form. Incubation of Grx1(SH)2 with an excess of GSSG produced Grx1S2 and a minor peak of m/z coincident with Cys78-glutathionylated Grx1S2. Reaction of Grx1S2 with 1 eq of GSH did not produce any new peaks and the Cys21-glutathionylated form was observed only when a 10-fold excess GSH was used (Table 3).
Table 3.
MS detection of glutathionylated TbGrx1
| Mass peaks | Thiol | Disulfide | Glutathionylated | |
|---|---|---|---|---|
| m/z | ||||
| TbGrx1(SH)2 (17 μm) | ||||
| No treatment | 10,833.7 | C21, C24, C78 | ||
| + 1 eq GSSG | 10,831.7 | C78 | C21–C24 | |
| 11,139.1 | C24, C78 | C21 | ||
| + 6 eq GSSG | 10,831.7 | C78 | C21–C24 | |
| 11,137.6 | C21–C24 | C78 | ||
| TbGrx1S2 (17 μm) | ||||
| No treatment | 10,831.7 | C78 | C21–C24 | |
| + 1 eq GSH | 10,831.8 | C78 | C21–C24 | |
| + 10 eq GSH | 10,832 | C78 | C21–C24 | |
| 11,139.4 | C24, C78 | C21 |
To provide further evidence on the occurrence of glutathionylated Grx1 during the reaction with GSH-mixed disulfides, Grx1(SH)2 was reacted with GSSG labeled at the amino group with fluorescein (FGSSGF) and the products were separated by gel filtration. As shown in Fig. 6A, the protein-containing fraction (Grx1 10.8 kDa) is excluded from the column and separated from the low-molecular weight components present in the reaction sample: nonreacted FGSSGF (1.34 kDa) and FGSH (0.69 kDa) released upon reduction of the corresponding disulfide by Grx1(SH)2. The peak eluting at 4.66 min did not contain protein but only fluorescein conjugated to GSH.
Figure 6.
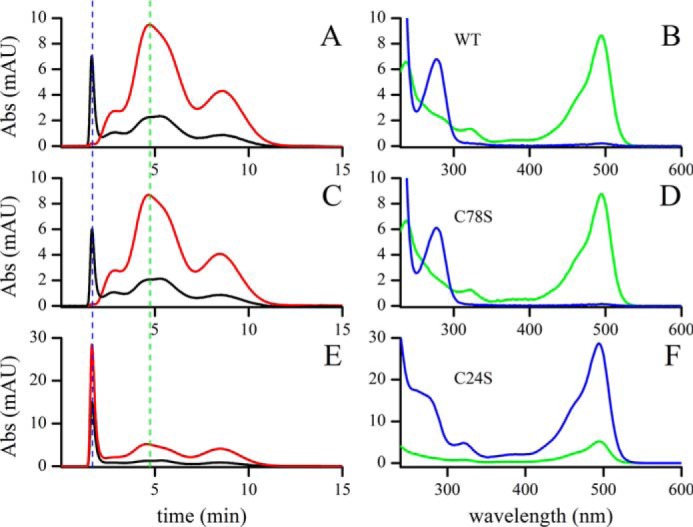
Glutathionylation of TbGrx1(SH)2 by bisfluorescein GSSG. Grx1(SH)2 (WT, C78S, and C24S) reacted with a substoichiometric amount of FGSSGF and was separated by gel filtration (two Hitrap columns in series). The gel filtration (left panels) was monitored at 280 nm (black line) and 495 nm (red line). At the times indicated by the dashed lines (1.66 min, blue, and 4.66 min, green), the UV-visible spectra (right panels) of the corresponding samples were recorded. A and B, TbGrx1(SH)2 WT; C and D, TbGrx1(SH)2 C78S; E and F, TbGrx1(SH)2 C24S.
Spectra of the first peak confirmed the presence of protein (absorbance at 280 nm) and a small amount of fluorescein (absorbance at 495 nm; Fig. 6B). Using the molar extinction coefficient of Grx1-SSGF (about 15,000 m−1 cm−1 at 280 nm) and fluorescein (70,000 m−1 cm−1 at 495 nm), we estimated that 0.7% of the Grx1 appeared glutathionylated after separation.
To confirm the finding, we also subjected two mutants of Grx1 to the same treatment. First, Grx1 C78S, lacking the cysteine outside the active site, behaved almost exactly as Grx1 WT (Fig. 6, C and D), with marginally lower glutathionylation. On the other hand, the C24S mutant, lacking the C-terminal cysteine of the active site (therefore unable of forming the internal disulfide) yielded the mixed disulfide quantitatively (Fig. 6, E and F).
To exclude potential artifacts arising from differential kinetics among the mutants we studied the reaction of FGSSGF reduction by Grx1(SH)2 (WT and the two mutants). The obtained rate constants were slightly smaller than with unlabeled GSH but still very rapid and overall comparable between the mutants and the WT protein (Table 2).
The three pieces of evidence obtained from NMR, MS, and fluorescent tagging point to a covalent intermediate consistent with Grx1SSG. At this point we cannot draw quantitative conclusions about its stability other than Grx1SSG prevalence seems to diminish if the detection is made after a dilution step (such as in MS and gel filtration), which is consistent with a relative instability of Grx1SSG with respect to Grx1S2 + GSH.
Both reduction and oxidation of Grx1 by trypanothione are extremely fast
Contrary to what we observed with GSH, the thiol-disulfide exchange between trypanothione and Grx1 is very fast in both oxidation and reduction. The kinetics are somewhat complex, both the reduction of TS2 by Grx1(SH)2 and the oxidation of T(SH)2 by Grx1S2 are biphasic when monitored through the intrinsic fluorescence of the protein (Fig. 7). In both reactions, there is a faster phase with a rate constant that is first order on trypanothione and a slower phase that appears to be zero order on trypanothione. This kinetic behavior is consistent with the expected sequence shown in reactions C and D of Table 4, which includes a mixed trypanothionylated intermediate.
Figure 7.
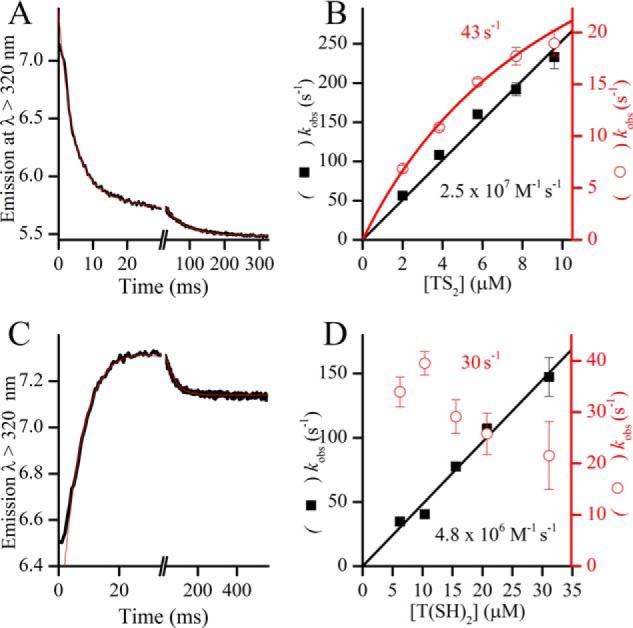
Reaction of TbGrx1 with oxidized and reduced trypanothione. A, time course of the oxidation of 0.2 μm TbGrx1(SH)2 with 9.6 μm TS2 at pH 7.05 and 25 °C. B, second-order plots of the two kobs obtained from the fit of the time courses to a double exponential function. Black squares, kfast; red circles, kslow. C, time course of the reaction of 20.8 μm T(SH)2 with 0.6 μm TbGrx1S2 at pH 7.05 and 25 °C. D, second-order plots of the kobs obtained from the fit of the time courses to a double exponential function. Black squares, kfast; red circles, kslow. The numbers in plots B and D are the second-order rate constants (black) obtained from the slope of kfast or the extrapolated value of kslow (red) at infinite concentration of trypanothione.
Table 4.
Kinetic system used to simulate the reduction of GSSG
Initial concentrations taken from Refs. 26 and 28; pH 7. Rate constants referenced to this work also appear in Table 2.

(a) kf refers to the reaction going left to right as written, kr refers to the reaction going right to left as written.
(b) KD values calculated as equilibrium constants in the direction of dissociation of the mixed disulfide.
(c) Value from Fig. 7D.
(d) Value from Fig. 7B.
(e) Calculated from the reported kf and the reported redox potentials of TXN and T(SH)2.
Both dissociation reactions of the intermediate are quite rapid with rate constants kIC = 30 s−1 and kfD = 43 s−1 and both dissociation equilibrium constants (KD) indicate that the mixed disulfide is a rather unstable species. The equilibrium constant of the global reaction.
| (Eq. 1) |
can be calculated as the following.
| (Eq. 2) |
Where KCD and KDD are the dissociation constants of reactions C and D in Table 4. Using K2 and the known reduction potential of the TS2/T(SH)2 couple (E°′ = −242 mV/NHE, (46)) and combining the Nernst equation for both couples,
| (Eq. 3) |
yields a E°′ value for the couple GrxS2/Grx(SH)2 of about −268 mV versus the normal hydrogen electrode (NHE), remarkably low for a Grx (approximately −170 mV/NHE for EcGrx1 and HsGrx1) (47, 48) but close to the −270 mV/NHE reported for EcTrx (49).
Reduction of GSSG by trypanosomal redoxins
To provide a semiquantitative idea of the relative relevance of the various reduction routes of GSSG in trypanosomatids, which are devoid of GR activity, we performed kinetic simulations with the system described in Table 4. The system was perturbed by the introduction of 10 μm GSSG with a time constant of 0.02 s, it was then allowed to equilibrate monitoring the concentration of GSSG over time in the absence or presence of Grx1 and TXN at their reported intracellular concentrations in the infective form of Trypanosoma brucei (28, 50).
In our simulated system under conditions resembling an intracellular reducing milieu, the reduction by T(SH)2 alone (reaction A) can revert the pulse of GSSG very slowly, in not less than 15 h (Fig. 8A). If the system also contains 2 μm Grx1, GSSG reduction is completed in less than 3 s, whereas in the presence of 85 μm TXN, the T(SH)2–dependent reduction of GSSG is completed in ∼5 s. In a model were both Grx1 and TXN are present, GSSG reduction is evidently faster (<2 s) with Grx1 accounting for 76% of the reductase activity and TXN contributing with the remaining 24%. Worth noting, the model assumes that Grx1 is reduced by T(SH)2 (reverse reaction D), but according to our rate constants, this reduction system is very sensitive to the presence of TS2 (reaction C). Also TXN has been reported to be susceptible to inhibition by TS2 (51). Therefore, we tested this experimental information by running a simulation of the reaction at different initial concentrations of TS2 from 0 to 50 μm, and verified that the accumulation of TS2 slows down the overall reduction of GSSG due to a drop in the steady-state concentrations of both Grx(SH)2 and TXN(SH)2 (Fig. S5). Above 15 μm TS2 ([T(SH)2]/[TS2] < 20), the reduction of GSSG by TXN becomes predominant. In principle, TXN is comparatively less sensitive than Grx1 to inhibition by TS2 due to the smaller difference in redox potentials (ETXNo′ = −249 mV/NHE (52), ETS2o′ = −242 mV/NHE (46), EGrx1o′ = −268 mV/NHE). However, previous reports indicate that the reductase activity of TXN over ribonucleotide reductase is inhibited 60% by a ([T(SH)2]/[TS2] ratio of 10 (51), which would indicate a ΔE°′ of 25 mV between TXN and trypanothione. In any case, the difference is subtle and the trend in the reduction of GSSG holds. The simulation of GSSG reduction assuming a ΔE°′ of 25 mV between TXN and trypanothione is presented in Fig. S6.
Figure 8.
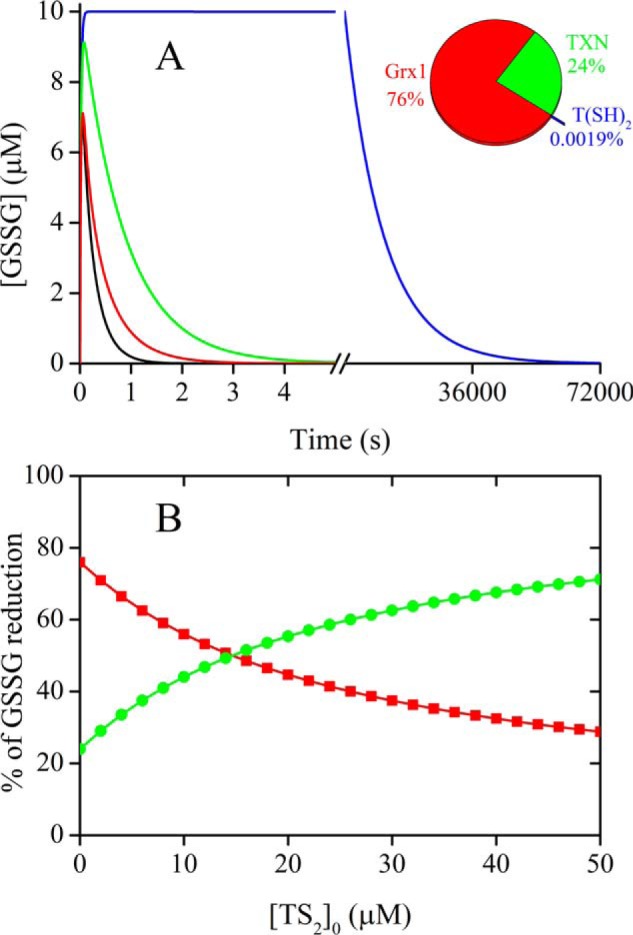
Contribution of different oxidoreductases and T(SH)2 to GSSG reduction. A, simulated reduction of a pulse of 10 μm GSSG by the kinetic system of Table 4. In addition to the complete system (black line), a system with [TXN]0 = 0 (red line), [TbGrx1]0 = 0 (green line), and [TXN]0 = [TbGrx1]0 = 0 (blue line) were simulated to assess the relative contribution of each reduction system. Inset, relative percentage of GSSG reduction by TbGrx1 (red), TXN (green), and T(SH)2 (blue) in the complete system. B, relative percentage of GSSG reduction by TbGrx1 (red) and TXN (green) in the complete system as a function of the initial concentration of TS2.
Discussion
Oxidation of Grx1(SH)2 by GSSG proceeds mainly to the internal disulfide
The oxidation of Grx1(SH)2 by GSSG consists in two elementary steps (Scheme 1, reactions 1 and 3). Reaction 1 is first-order in GSSG and Grx1(SH)2, whereas reaction 3 is 0 order in GSSG. In principle either reaction could be responsible for the change in Trp18 fluorescence as we have seen that all assayed modifications on Cys21 lead to an important decrease in emission. What we see in the time course of oxidation is a two-phase reaction consisting in a very fast transient (Fig. 2A, inset) and a first-order decay in emission that is also first order on GSSG (Fig. 2B). Therefore, reaction 1 is either rate-limiting or it accounts for most of the decrease in emission. In this regard, MS and spectrofluorometric analysis of Grx1(SH)2 treated with an excess GSSG or fluorescently-labeled GSSG, respectively, showed that the major product is Grx1S2 and only a minor fraction of Grx1 is glutathionylated at Cys21. This clearly indicates that the internal disulfide of Grx1 is more stable than its glutathionylated form and, hence, that reaction 3 is faster than reactions 1 and −3 under the conditions assayed. As will be discussed below in detail, Grx1 lacks residues involved in binding and activation of a second GSH molecule that, in canonical class I Grx, will attack the mixed disulfide with GSH releasing reduced protein and GSSG (reaction 2). This may explain the inability of the trypanosomal enzyme to operate via a monothiol mechanism and to employ the adjacent Cys24 to complete the oxidative phase of its catalytic cycle with GSH (reaction 3). The plausible biological relevance of the oxidation of Grx1 to a disulfide is provided below.
Efficient reduction of Grx1S2 requires a two-electron reductant
Reduction of Grx1S2 by GSH consists of two consecutive thiol-disulfide exchange reactions (Scheme 1, reactions −3 and 2). The lower limit for their rate constants can be estimated using the empirical Brønsted relationship obtained with low molecular weight thiols in the absence of enzyme catalysis (Equation 4) (6).
| (Eq. 4) |
Where pKanuc is the pKa of the nucleophilic attacking thiol, pKac is the pKa of the electrophilic thiol whose sulfur is being attacked in the disulfide, and pKalg is the pKa of the leaving group thiol. It is important to note that Equation 4 has been modified from the original to yield rate constants in units of m−1 s−1. The thiols involved in the reactions are GSH and the two active-site cysteines of Grx1, Cys21 and Cys24. In reaction −3, GSH act as nucleophile (pKanuc = 8.94 (44)) attacking on the electrophilic center of Cys21 sulfur (pKac = 5.15), being Cys24 (pKalg = 8.5) the leaving group. Therefore, according to Equation 4 the lower limit for this reaction is 1.4 m−1 s−1 (kapp at pH 7.0). In reaction −3, the GSH-binding site is available, therefore catalysis can be expected to be important. In fact, the rate-limiting step in the catalysis of Grx as thiol transferase is reaction 2, as evidenced by the pH profile of the activity (53).
In reaction 2, a second GSH molecule (nuc) attacks the sulfur of the bound GSH (c) and Cys21 is the leaving group. The lower kinetic limit for this reaction is kapp = 37.5 m−1 s−1, coincident with the value experimentally determined here (39 m−1 s−1, Table 2) and previously reported (28).
For human Grx1 the rate constant of reaction 2 has been measured as 3.7 × 104 m−1 s−1 at pH 9.5 (53). That value, although 3 orders of magnitude faster than the rate constant determined for Grx1, is also very similar to the value calculated using Equation 4, considering that the human enzyme has a very acidic N-terminal cysteine (pKa = 3.5, calculated kapp = 4.1 × 104 m−1 s−1 at pH 9.5 and 597 m−1 s−1 at pH 7.0). Thus, the reduction of glutathionylated Grx (reaction 2) by a second GSH molecule, the preferred reaction in most class I Grx, is relatively fast even in the absence of catalysis, because it is favored by the usually very low pKa of the N-terminal cysteine that acts as leaving group (Table 1).
For the trypanosomal Grx1, both GSH-dependent reductive steps are quite slow indicating that this monothiol is not the preferred reductant. Reduction of glutathionylated Grx by a monothiol mechanism requires binding and activation of a second GSH molecule in the active site of Grx. A recent study proposed the presence of a GSH-activator site in class I Grx that is formed by highly conserved basic residues: a Lys or Arg 3 residues upstream of the N-terminal active site Cys and an Arg and/or Lys at the N terminus of the loop containing the strictly conserved cis-Pro present in all Trx-fold proteins (34). Interestingly, in Grx1 none of these positions is occupied by positively charged residues, with Trp replacing the Lys/Arg nearby the active site of classical Grx and a Glu replacing the Lys/Arg at the P-loop of classical Grx (Fig. S2). Under such a molecular scenario, the stabilization of the glutathionylated intermediate at Cys21 as well as activation of a second GSH molecule is clearly not favored in Grx1.
In contrast to GSH, T(SH)2 proved to be an excellent reductant of Grx1S2 (k ∼5 × 106 m−1 s−1). Such performance is not strictly related to a particular selectivity of the trypanosomal protein. In fact, EcGrx1 has been shown to use T(SH)2 with higher efficiency than GSH in redox assays with diverse oxidized proteins (28). The physicochemical properties of the low molecular weight dithiol are likely responsible for making it a good reductant. The mean pKa of the Cys from T(SH)2 was estimated to be 7.4, which is significantly lower than that of GSH (pKa 8.94 (44)), likely due to the absence of one carboxylate and to the basic character of the N4 group of spermidine. Thus, in contrast to GSH, thiol-disulfide exchange using T(SH)2 as reductant requires less activation of its thiol groups because at physiological pH half of them will be in the thiolate form. Additionally, reduction of the trypanothionylated form of Grx will be solved by the intramolecular nucleophilic attack by the second Cys of T(SH)2. Nevertheless, the rate constant of T(SH)2 with Grx1 needs to involve binding and specific activation of the nucleophilic attack because the measured rate constant is nearly 6 orders of magnitude higher than the one calculated with equation 4.
In summary, the sluggishness of Grx1 reduction by GSH makes the dithiol mechanism much more efficient in the reductive as it bypasses reaction 2 when using the much faster two-electron reductant T(SH)2 (Scheme 1, reaction 4). This slow reduction of Grx1 by GSH has a physiological significance as discussed below.
Biological implications derived from Grx1 kinetic behavior and physicochemical properties
In most organisms, GR is extremely fast and efficient in the reduction of GSSG (e.g. kcat/Km is 1.2 × 107 m−1 s−1 for EcGR (54)). Organisms lacking GR evolved other possibilities, like a specialized form of thioredoxin reductase in fruit flies (55) or a Grx-domain (with GSSG reductase activity) fused to a canonical thioredoxin reductase in platyhelminth parasites (56). Indeed, any dithiol protein with E°′ < −240 mV/NHE could perform the reduction given the right concentration and conditions. Although many Trx have reduction potentials near −270 mV/NHE (47), they are too slow to reduce GSSG (rate constants ∼102 m−1 s−1 (57–60)), with the exception of the thioredoxin system of Mycobacterium tuberculosis (kcat/Km = 6.7 × 105 m−1 s−1 (59)). Trx from T. brucei is one of the slowest reported, reducing GSSG with a k = 23 m−1 s−1 (61). On the other hand, TXN from Trypanosoma cruzi was recently reported to reduce GSSG with a rate constant of 1.4 × 104 m−1 s−1 in a coupled assay with trypanothione reductase (62). We obtained the same rate constant using T. brucei TXN and measuring the rate constant by directly monitoring the reaction through TXN fluorescence (Fig. S7).
Another candidate for T(SH)2-dependent reduction of GSSG in T. cruzi and Leishmania are the GSH transferase TC52 (63) and a thiol-dependent reductase (k = 4 × 105 m−1 s−1) (64), respectively. However, because both proteins are excreted/secreted to the extracellular medium, their GSSG-reductase activity does not likely contribute to the intracellular GSH homeostasis in the parasites. Finally, the spontaneous reaction between T(SH)2 and GSSG has been proposed as responsible for maintaining the reduced pool of GSH (65). However, a rate constant of 0.31 m−1 s−1 at pH 7 was calculated using Equation 4 and it is too slow to have any biological relevance on the reduction of GSSG (Fig. 8A).
Kinetic simulations, based on rate constants obtained in this work and spanning a range of physiological concentrations of substrates and intracellular T(SH)2/TS2 ratios, suggested that Grx1 and TXN work concertedly to maintain GSH reduced at the expense of T(SH)2. According to the model, the main role of TXN is to operate as an efficient backup of Grx1 when cells face oxidative stress. The putative role of Grx1 as GSSG/T(SH)2 oxidoreductase has recently been challenged by a study showing that the overall redox state of GSH/GSSG in a T. brucei cell line lacking Grx1 (and also Grx2) is similar to that of the WT cell line (66), these observations support our experimental evidence that TXN can efficiently take over this function.
Glutaredoxins are efficient and specific catalysts of the (de)glutathionylation of proteins. A recent study shows that Grx1 does not contribute to the overall protein S-thiolation of T. brucei (i.e. the level of protein-bound GSH was almost identical in WT and Grx1-deficient cells (67)). This is likely explained by the fact that Grx1 is rapidly oxidized to its disulfide by GSSG and that the glutathionylated intermediate could only be detected at low concentrations of GSSG. Nonetheless, Grx1 may eventually participate in the glutathionylation of specific proteins when the GSH/GSSG is high. Such a mechanism may prove important to fine-tune the activity of proteins under physiological conditions. The reverse reaction, namely the reduction of mixed disulfides of GSH with proteins, is the primary function of glutaredoxins and Grx1 is not the exception (28). In the infective form of T. brucei, Grx1 accounts for about 40–50% of the deglutathionylase activity measured in cell extracts (27), which was then confirmed by the slow reversion of protein S-thiolation in Grx1-KO parasites exposed to diamide (67). The current evidence shows that Grx1 does not function as a major protein oxidoreductase in the protection against oxidative stress (66, 67) but as key redox regulator of the activity of yet unknown partner(s) involved in parasite thermotolerance (27).
The obligated dithiol mechanism used by Grx1 in thiol-disulfide exchange reactions raises interesting questions about the biochemical context in which such atypical behavior may prove useful. For a monothiol mechanism, the reduction of GSH-mixed disulfides by Grx yields GSSG, reduced target protein, and Grx(SH)2 as products (reactions 1 and 2). As shown here, this is not the preferred mechanism employed by Grx1. Instead, operating via a dithiol mechanism the products are GSH, the reduced target protein, and Grx1S2. This may prove useful for organisms lacking GR, as trypanosomatids, because GSH but not GSSG is produced by the reaction, and Grx1S2 is then efficiently reduced by T(SH)2. In contrast, in the absence of a GR, the steady accumulation of GSSG by the monothiol mechanism would gradually inactivate Grx1, and protein deglutathionylation would be compromised if the GSH/GSSG ratio is not restored. Although GSSG can be reduced by TXN, it is important to recall that this reaction is 3 orders of magnitude slower than that catalyzed by a GR. Thus the monothiol mechanism represents a dead-end path for GR-deficient cells. This led us to speculate that the dithiol mechanism evolved by the trypanosomal Grx reflects an evolutionary adaptation to overcome a kinetic bottleneck with potentially harmful consequences for the parasite. The major change undergone by Grx1 was the loss of the GSH-activator site conserved in almost all class I Grx (Fig. S2). Point mutations substituted the otherwise highly conserved basic residues of the GSH-activator site and abolished the capacity of the trypanosomal protein to operate with GSH using a monothiol mechanism. Adding value to this evolutionary hypothesis, all class I Grx from trypanosomatids lack the residues conforming the GSH-activator site and, therefore, we here propose are obligated to catalyze thiol-disulfide exchange reactions using a dithiol mechanism (Fig. S2). From an evolutionary perspective, although T. brucei can fully dispense of Grx1 activity, the protein probably played an essential role in an ancient trypanosomatid, during the establishment of a redox metabolism dependent on T(SH)2. Indeed, this process involved not only the loss of the genes encoding for GR and thioredoxin reductase but also the adaptation (mutation) of the active site of several redox proteins to use T(SH)2 as substrate (25). Thus, the nowadays superfluous GSSG reductase and protein de(glutathionylase) activity of Grx1 may have contributed to a smooth transition from a GSH to a T(SH)2-dependent metabolism.
Experimental procedures
Chemicals
Chemical reagents were of analytical grade and purchased from Sigma, Applichem, or Dorwill. Trypanothione (T(SH)2) and glutathionylspermidine (GSP) disulfide were obtained from Bachem AG, Switzerland. Unless otherwise indicated all kinetic experiments were performed in a buffer of constant ionic strength (68) consisting in Tris (30 mm), MES (15 mm), acetic acid (15 mm), 120 mm NaCl, and 0.1 mm diethylenetriamine pentaacetate (TMA buffer).
Proteins
HSA (Sigma A1653) was delipidated as previously described (69). GSH reductase from Saccharomyces cerevisiae (ScGR) was purchased from Sigma (G3664). E. coli Grx1 (EcGrx1) was a kind donation of IMCO Corp. Ltd. AB (Sweden).
Protein expression and purification
The expression vector encoding for the wildtype (WT) form of T. brucei Grx1 (accession number XP828228, TriTryp Tb927.11.1370), namely pET-trx1b TbGrx1, was kindly provided by Dr. L. Krauth-Siegel, Heidelberg University, Germany (28). The cysteine to serine mutants C24S and C78S of Grx1 were generated using the GeneArt kit (Thermo Fisher), the pET-trx1b TbGrx1 as DNA template and the following primers pairs: C24S-Forward, 5′-GTCACTTGCCCCTACAGCGTCCGAGCAGAGA-3′ and C24S-Reverse, 5′-TCTCTGCTCGGACGCTGTAGGGGCAAGTGAC-3′, and C78S-Forward, 5′-AATTTCATTGGCGGTAGCAGCGATTTGGAGG-3′ and C78S-Reverse, 5′-CCTCCAAATCGCTGCTACCGCCAATGAAATT-3′, respectively.
E. coli BL21(DE3) cells (Life Technologies) transformed with pET-trx1b TbGrx1 or the corresponding C24S or C78S mutants were grown at 37 °C and 220 rpm in 2YT medium supplemented with 50 μg/ml of kanamycin and induced with 200 μm isopropyl 1-thio-β-d-galactopyranoside when A600 ∼1.0. After incubation at 20 °C for 16 h, cells were harvested by centrifugation (4,000 × g, 10 min), resuspended in 50 mm sodium phosphate, pH 7.8, 300 mm NaCl (buffer A) plus 1 mm phenylmethylsulfonyl fluoride, 40 μg/ml of 1-chloro-3-tosylamido-7-amino-2-heptanone, 150 nm pepstatin, 4 nm cystatin, 0.1 mg/ml of aprotinin, and 1 mg/ml of lysozyme and disrupted by sonication. The lysate was treated with DNase (10 μg/ml, Life Technologies) and 10 mm MgCl2 and centrifuged at 20,000 × g for 1 h. The supernatant was filtrated (0.45 μm, Millipore) and loaded onto a HisTrap® column (GE Healthcare) equilibrated with buffer A. After washing the column with 20 mm imidazole in buffer A, the recombinant proteins were eluted with a linear gradient up to 500 mm imidazole in the same buffer. Fractions containing the fusion protein (as assessed by SDS-PAGE) were pooled and treated with 5 mm DTT, 2 mm EDTA, and His-tagged 3C-type tobacco etch virus protease (prepared as described in Ref. 29) at a 70:1 protein:protease ratio (in mg) for 2 h at room temperature. The sample was then concentrated by ultrafiltration (Vivaspin, 5 kDa molecular weight cut-off), buffer exchanged with a HiPrep® column coupled to an ÄKTA-FPLC system (GE Healthcare) equilibrated in buffer A and applied again onto a HisTrap® pre-equilibrated in buffer A. Tag-free WT Grx1 or its Cys-to-Ser mutants was collected in the flow-through, treated with 5 mm DTT and 2 mm EDTA during 30 min at room temperature, and concentrated by ultrafiltration before loading it onto a preparative size exclusion column (HiLoad 26/60 Superdex 75 prep grade column, GE) equilibrated with 100 mm sodium phosphate, pH 7.4, with 150 mm NaCl (buffer B). Fractions containing the protein of interest were collected, concentrated, and stored in aliquots at −80 °C after addition of 10% glycerol. Due to the cloning strategy, all tag-free versions of Grx1 contain an N-terminal GAME sequence instead of Met1 yielding a calculated molecular weight of 10,833.49. 15N uniformly labeled Grx1 for NMR was prepared as previously reported (31) by growing E. coli BL21(DE3)-transformed cells in M9 minimal medium containing 1 g/liter of 15NH4Cl as the only nitrogen source. The labeled protein was expressed and purified as described above and the buffer exchanged to 50 mm sodium phosphate, pH 7.2, 150 mm NaCl (buffer C) with 10 mm DTT in H2O/D2O (95:5%, v/v) and the protein concentrated by ultrafiltration. T. brucei tryparedoxin (TXN) and monothiol glutaredoxin 1 C104S mutant (Tb1CGrx1 C104S) were obtained as previously described (70, 71), respectively.
Protein reduction and oxidation
To obtain dithiol Grx1 (Grx1(SH)2), the protein was reduced with excess DTT (10 mm) for 30 min at room temperature, DTT was removed by gel filtration using a PD10 column (GE Healthcare). The active site disulfide form of Grx1 (Grx1S2) was obtained by oxidation with an excess H2O2 (1 mm) for 30 min, followed by gel filtration to remove the remaining H2O2. The product of H2O2-treated Grx1 was analyzed by MS and no further modifications were identified. Protein and thiol concentrations were assessed in Grx1(SH)2 and GrxS2. Quantitative oxidation of the active site Cys was considered when a ratio of [RSH]/[protein] = 1, meaning no oxidation of Cys78 was detected.
Thiol and protein quantitation
Thiol concentration was determined by chromogenic disulfide reduction using 4,4′-dithiodipyridine (Acros Organics), ϵ324 = 21,400 m−1 cm−1 (72). Protein concentrations were measured at 280 nm using the following molar extinction coefficients: 11,460 m−1 cm−1 for Grx1, 35,300 m−1 cm−1 for HSA, 29,500 m−1 cm−1 for TbTXN, 10,000 m−1 cm−1 for EcGrx1, and 15,470 m−1 cm−1 for Tb1CGrx1 C104S.
Preparation of mixed GSH disulfides
Different stock solutions of the heterodisulfide between GSH and 2-mercaptoethanol (GSSEtOH) were prepared by mixing excess HED (10 mm in water) with the desired concentration of GSH in Tris/MES/acetic buffer, pH 7.1, and incubating at room temperature for 2 h. Under these conditions and according to the reported rate constants for the reaction (6), the conversion of GSH to GSSEtOH is nearly quantitative (>99%) with a very small (< 0.5%) GSSG contamination.
HSA glutathionylated in Cys34 (HSA-SSG) was prepared by oxidation of the reduced protein (HSA-SH) with GSSG. Briefly, a solution of delipidated HSA (1.22 mm) was reduced with 10 mm 2-mercaptoethanol for 2 h at room temperature, excess 2-mercaptoethanol was removed by gel filtration using a PD10 column and the resulting HSA was mixed with 5 mm GSSG overnight at room temperature. Thiol concentration in the protein fraction after gel filtration was measured before and after the treatment, in a typical preparation the resulting albumin contained 41% HSA-SSG and, 48% HSA-SH and 11% nonreducible forms of HSA. The mixture was used without further purification.
Tb1CGrx1 C104S was glutathionylated at its C-terminal cysteine (Cys-181) by oxidation of the reduced protein with excess GSSG, as previously described (73). The yield of protein glutathionylation (Tb1CGrx1 Cys181–SSG) was 95–100%, as assessed by the content of protein thiols before and after oxidation and gel filtration.
Preparation of fluorescein-labeled GSH disulfide
GSH disulfide labeled with carboxyfluorescein (FGSSGF) was synthesized from 3 mm 5/6-carboxyfluorescein succinimidyl ester (NHS fluorescein, ThermoFisher) and 6 mm GSSG in borate buffer, pH 8.5. The reaction was allowed to proceed for 1 h at room temperature. The product mixture was reduced with 20 mm DTT for 30 min, acidified with TFA, and separated in a 500 mg of C18 disposable extraction column (Bakerbond spe, J.T. Baker) previously activated with 2 ml of acetonitrile and equilibrated with 0.1% TFA in water. After loading the sample, the column was washed three times with 1 ml of 0.1% TFA and the labeled GSH (FGSH) was then eluted with 50% acetonitrile. The eluate was alkalinized to pH 8 and oxidized with 5 mm H2O2 for 1 h. Finally, FGSSGF was purified with the same chromatographic protocol used for FGSH. The purity of the FGSSGF was assessed by RP HPLC (Agilent Eclipse Plus C18, 100 × 4.6 mm column) before and after reduction with 50 mm DTT to rule out contamination with FGSH.
Kinetics assays
Unless otherwise specified, all kinetic experiments were conducted using a wide-range buffer solution of constant ionic strength (I = 0.15) independent of the pH as proposed by Ellis and Morrison (68): TMA buffer, was used in the pH range of 3.5 to 9.0.
The reaction of Grx1(SH)2 (0.2–1.0 μm initial concentration) with oxidants was monitored by the change in intrinsic fluorescence of the protein under pseudo first-order conditions with the oxidant in excess. The fastest reactions (t < 60 s) were monitored in a SX20 stopped-flow spectrometer (Applied Photophysics) using an excitation wavelength (λex) of 280 nm and an emission cutoff filter λem >320 nm; intermediate reactions (300 s > t > 10 s) were followed in a Cary Eclipse spectrofluorimeter (Agilent) using a RX2000 rapid mixing stopped flow unit (Applied Photophysics) (λex, λem = 280, 350 nm, respectively); and the slowest reactions (t > 100 s) were studied using a Varioskan Flash plate reader (Thermo) (λex, λem = 280, 350 nm, respectively). In most cases, Grx(SH)2 reacted with excess oxidant. The initial concentrations of oxidants were chosen according to the reaction rate, thus GSSG and GSSEtOH and Tb1CGrx1 Cys181–SSG were used in the range from 2.5 to 100 μm, whereas cystamine, cystine methyl ester, HED, and H2O2 were in the range of 1 to 15 mm. Time courses were fitted to a first-order function and the values of kobs obtained were plotted versus the concentration of oxidant to obtain the second-order rate constant.
Exceptionally, the reactions of Grx1(SH)2 with HSA-SSG and with FGSSGF were performed under pseudo first-order conditions with Grx1(SH)2 in excess over the disulfide. This experimental design avoids interference of the higher fluorescence of HSA or fluorescein, respectively, with the fluorescent signal of Grx1. The concentrations used were 400 nm HSA-SSG (4–10 μm Grx1(SH)2) and 36 nm FGSSGF (0.4–4 μm Grx1(SH)2) and the reactions were monitored as indicated above.
Reduction of Grx1S2 by T(SH)2 was studied under pseudo first-order conditions with T(SH)2 in excess in the stopped flow. The reaction was monitored as indicated previously and the data fitted to a double exponential function to estimate the rate constants. The data were also fitted to a system of two consecutive reactions using Gepasi 3.30 (74).
Reduction of Grx1S2 by GSH was performed in the presence of ScGR and NADPH to ensure the absence of contaminating GSSG (e.g. our freshly prepared GSH stock solutions contained ∼0.5% GSSG). Thus, GSH free from GSSG was prepared by incubating different concentrations of GSH with 0.4 units/ml of GR and 10 μm NADPH during 10 min. Briefly, a mixture containing GR (0.4 units/ml), NADPH (10 μm), and GSH (0–100 μm) in buffer, pH 7.0, was incubated for 10 min at 25 °C, then Grx1S2 (2 μm) was added and fluorescence was emission monitored during 20 min in an ISS ChronosFD spectrofluorometer.
pKa measurement
The pKa of the N-terminal cysteine of Grx1(SH)2 was determined by three independent techniques: (i) pH dependence of the tryptophan fluorescence, (ii) pH dependence of the second-order rate constant of GSSG reduction, and (iii) rate of alkylation with mBBr (71). In all cases the measured variable was fitted to Equation 5 at pH < 7.5.
| (Eq. 5) |
Where A and B are the parameters characteristic of the protonated or ionized cysteine, respectively, in the protein.
NMR
The titration with GSH was performed on 550 μl of 100 μm 15N-labeled Grx1S2 solutions in 50 mm phosphate buffer, 50 mm NaCl at pH 7.0 in H2O/D2O, 10:1.
Grx1S2 was prepared by adding a stoichiometric amount of H2O2 to fully reduced Grx1. The mixture was left to react overnight at room temperature yielding Grx1 with a Cys21–Cys24 disulfide and a free thiol at Cys78.
To titrate 15N-labeled Grx1S2, GSH (6.1 mm in 50 mm phosphate buffer, pH 7.0) was added in 2-μl aliquots and a 15N-SOFAST-HMQC (75) was recorded after each addition. 15N-SOFAST-HMQC experiments were collected on a Bruker DMX 600 MHz spectrometer with a room temperature probe, at 298 K. Each experiment was acquired with 32 scans, 1024 and 128 increments 1H dimension, and 128 increments and 15N dimension, respectively. A recovery delay of 200 ms was used before each scan.
Mass spectra
The MS of Grx1(SH)2 (17 μm in 20 mm ammonium acetate) was determined before and after reaction with 1 or 6 eq of GSSG. Grx1S2 (17 μm in 20 mm ammonium acetate) was measured before and after reaction with 1 or 10 eq of GSH. The spectra were recorded on a Xevo G2 from Waters, using electrospray ionization. Prior to injection, samples were diluted in H2O:CH3CN, 50:50, with 0.1% formic acid.
Glutathionylation of Grx1 with fluorescein-labeled GSH
Grx1 WT, C24S, and C78S were reduced and total protein and thiol content was measured to verify each had the expected number of reduced cysteines. The Grx1 variants were reacted with a substoichiometric concentration of fluorescein-tagged GSH disulfide ([Grx] = 9 μm, [FGSSGF] = 7.65 μm) at pH 6.85 in TMA buffer. The reaction products were separated using two Hitrap columns in series in a HPLC (Agilent 1260 Infinity) while monitoring absorbance at 280 and 495 nm. At 1.66 and 4.66 min, coincident with the maxima at 280 and 495 nm, the UV-visible spectra were taken using the Diode Array detector of the chromatograph.
Kinetic simulation of GSSG reduction
The reduction of GSSG was simulated using Gepasi 3.30 (74) in a system containing T(SH)2, Grx1(SH)2, and TXN as possible reductants under the conditions described in Table 4.
Author contributions
B. M., M. Bellanda, M. A. C., and G. F.-S. conceptualization; B. M., M. Bellanda, and G. F.-S. resources; B. M., M. N. M., M. Bellanda, and G. F.-S. formal analysis; B. M., M. N. M., M. Bonilla, M. A. C., and G. F.-S. supervision; B. M., M. A. C., and G. F.-S. funding acquisition; B. M., M. N. M., M. Bonilla, M. Bellanda, and M. A. C. methodology; B. M., M. N. M., M. Bellanda, M. A. C., and G. F.-S. writing-original draft; B. M. and G. F.-S. project administration; B. M., M. Bonilla, M. Bellanda, M. A. C., and G. F.-S. writing-review and editing; M. Bonilla, M. D., K. G., M. Bellanda, M. A. C., and G. F.-S. investigation.
Supplementary Material
Acknowledgments
We thank Dr. Marino Bellini (Padova University) for expert assistance with MS, G. Burger (University of Montreal) for access to unpublished sequences of Diplonema papillatum, and Prof. Luise Krauth-Siegel and Natalie Dirdjaja (University of Heidelberg) for the gift of trypanothione.
This work was supported by Grant C601-348 from Comisión Sectorial de Investigación Científica Universidad de la República (to B. M. and G. F. S.), CRP/URU14-01 International Centre for Genetic Engineering and Biotechnology and Fondo para la Convergencia Estructural del Mercosur, COF 03/11 (to M. A. C.), and CPDA137397/13 from PRAT Università degli Studi di Padova, and European Comission Project number 261863 Bio-NMR (to M. Bellanda). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7.
- PAPS
- adenosine 3′-phosphate 5′-phosphosulfate
- GR
- GSH reductase
- HED
- hydroxyethyl disulfide
- HSA
- human serum albumin
- NHE
- normal hydrogen electrode
- TXN
- T. brucei tryparedoxin
- HMQC
- heteronuclear multiple quantum coherence
- GSP
- glutathionylspermidine
- mBBr
- monobromobimane.
References
- 1. Berndt C., Lillig C. H., and Flohé L. (2014) Redox regulation by glutathione needs enzymes. Front. Pharmacol. 5, 168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Flohé L. (2013) The fairytale of the GSSG/GSH redox potential. Biochim. Biophys. Acta 1830, 3139–3142 10.1016/j.bbagen.2012.10.020 [DOI] [PubMed] [Google Scholar]
- 3. Jensen E. V. (1959) Sulfhydryl-disulfide interchange. Science 130, 1319–1323 10.1126/science.130.3385.1319 [DOI] [PubMed] [Google Scholar]
- 4. Wilson J. M., Bayer R. J., and Hupe D. J. (1977) Structure-reactivity correlations for the thiol-disulfide interchange reaction. J. Am. Chem. Soc. 99, 7922–7926 10.1021/ja00466a027 [DOI] [Google Scholar]
- 5. Gilbert H. F. (1995) Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 251, 8–28 10.1016/0076-6879(95)51107-5 [DOI] [PubMed] [Google Scholar]
- 6. Szajewski R. P., and Whitesides G. M. (1980) Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J. Am. Chem. Soc. 102, 2011–2026 10.1021/ja00526a042 [DOI] [Google Scholar]
- 7. Holmgren A. (1979) Glutathione-dependent synthesis of deoxyribonucleotides: characterization of the enzymatic mechanism of Escherichia coli glutaredoxin. J. Biol. Chem. 254, 3672–3678 [PubMed] [Google Scholar]
- 8. Tsang M. L. (1981) Assimilatory sulfate reduction in Escherichia coli: identification of the alternate cofactor for adenosine 3′-phosphate 5′-phosphosulfate reductase as glutaredoxin. J. Bacteriol. 146, 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zheng M., Aslund F., and Storz G. (1998) Activation of the OxyR transcription factor by reversible disulfide bond formation. Science 279, 1718–1721 10.1126/science.279.5357.1718 [DOI] [PubMed] [Google Scholar]
- 10. Racker E. (1955) Glutathione-homocystine transhydrogenase. J. Biol. Chem. 217, 867–874 [PubMed] [Google Scholar]
- 11. Eriksson S. A., and Mannervik B. (1970) The reduction of the l-cysteine-glutathione mixed disulfide in rat liver. involvement of an enzyme catalyzing thiol-disulfide interchange. FEBS Lett. 7, 26–28 10.1016/0014-5793(70)80608-1 [DOI] [PubMed] [Google Scholar]
- 12. Lillig C. H., Berndt C., Vergnolle O., Lönn M. E., Hudemann C., Bill E., and Holmgren A. (2005) Characterization of human glutaredoxin 2 as iron-sulfur protein: a possible role as redox sensor. Proc. Natl. Acad. Sci. U.S.A. 102, 8168–8173 10.1073/pnas.0500735102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ströher E., and Millar A. H. (2012) The biological roles of glutaredoxins. Biochem. J. 446, 333–348 10.1042/BJ20112131 [DOI] [PubMed] [Google Scholar]
- 14. Yang Y., Jao Sc., Nanduri S., Starke D. W., Mieyal J. J., and Qin J. (1998) Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry 37, 17145–17156 10.1021/bi9806504 [DOI] [PubMed] [Google Scholar]
- 15. Gravina S. A., and Mieyal J. J. (1993) Thioltransferase is a specific glutathionyl mixed disulfide oxidoreductase. Biochemistry 32, 3368–3376 10.1021/bi00064a021 [DOI] [PubMed] [Google Scholar]
- 16. Mieyal J. J., Starke D. W., Gravina S. A., and Hocevar B. A. (1991) Thioltransferase in human red blood cells: kinetics and equilibrium. Biochemistry 30, 8883–8891 10.1021/bi00100a023 [DOI] [PubMed] [Google Scholar]
- 17. Gallogly M. M., Starke D. W., Leonberg A. K., Ospina S. M., and Mieyal J. J. (2008) Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry 47, 11144–11157 10.1021/bi800966v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Discola K. F., de Oliveira M. A., Rosa Cussiol J. R., Monteiro G., Bárcena J. A., Porras P., Padilla C. A., Guimarães B. G., and Netto L. E. (2009) Structural aspects of the distinct biochemical properties of glutaredoxin 1 and glutaredoxin 2 from Saccharomyces cerevisiae. J. Mol. Biol. 385, 889–901 10.1016/j.jmb.2008.10.055 [DOI] [PubMed] [Google Scholar]
- 19. Björnberg O., and Holmgren A. (1991) Characterization of homogeneous recombinant glutaredoxin from Escherichia coli: purification from an inducible λPL expression system and properties of a novel elongated form. Protein Expr. Purif. 2, 287–295 10.1016/1046-5928(91)90085-W [DOI] [PubMed] [Google Scholar]
- 20. Aslund F., Ehn B., Miranda-Vizuete A., Pueyo C., and Holmgren A. (1994) Two additional glutaredoxins exist in Escherichia coli: glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc. Natl. Acad. Sci. U.S.A. 91, 9813–9817 10.1073/pnas.91.21.9813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Couturier J., Jacquot J.-P., and Rouhier N. (2013) Toward a refined classification of class I dithiol glutaredoxins from poplar: biochemical basis for the definition of two subclasses. Front. Plant Sci. 4, 518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagai S., and Black S. (1968) A thiol-disulfide transhydrogenase from yeast. J. Biol. Chem. 243, 1942–1947 [PubMed] [Google Scholar]
- 23. Reyes A. M., Pedre B., De Armas M. I., Tossounian M. A., Radi R., Messens J., and Trujillo M. (2018) Chemistry and redox biology of mycothiol. Antioxid. Redox Signal. 28, 487–504 10.1089/ars.2017.7074 [DOI] [PubMed] [Google Scholar]
- 24. Chandrangsu P., Loi V. V., Antelmann H., and Helmann J. D. (2018) The role of Bacillithiol in Gram-positive firmicutes. Antioxid. Redox Signal. 28, 445–462 10.1089/ars.2017.7057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manta B., Bonilla M., Fiestas L., Sturlese M., Salinas G., Bellanda M., and Comini M. A. (2018) Polyamine-based thiols in trypanosomatids: evolution, protein structural adaptations, and biological functions. Antioxid. Redox Signal. 28, 463–486 10.1089/ars.2017.7133 [DOI] [PubMed] [Google Scholar]
- 26. Comini M. A., Krauth-Siegel R. L., and Bellanda M. (2013) Mono- and dithiol glutaredoxins in the trypanothione-based redox metabolism of pathogenic trypanosomes. Antioxid. Redox Signal. 19, 708–722 10.1089/ars.2012.4932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Musunda B., Benítez D., Dirdjaja N., Comini M. A., and Krauth-Siegel R. L. (2015) Glutaredoxin-deficiency confers bloodstream Trypanosoma brucei with improved thermotolerance. Mol. Biochem. Parasitol. 204, 93–105 10.1016/j.molbiopara.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 28. Ceylan S., Seidel V., Ziebart N., Berndt C., Dirdjaja N., and Krauth-Siegel R. L. (2010) The dithiol glutaredoxins of african trypanosomes have distinct roles and are closely linked to the unique trypanothione metabolism. J. Biol. Chem. 285, 35224–35237 10.1074/jbc.M110.165860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manta B., Pavan C., Sturlese M., Medeiros A., Crispo M., Berndt C., Krauth-Siegel R. L., Bellanda M., and Comini M. A. (2013) Iron-sulfur cluster binding by mitochondrial monothiol glutaredoxin-1 of Trypanosoma brucei: molecular basis of iron-sulfur cluster coordination and relevance for parasite infectivity. Antioxid. Redox Signal. 19, 665–682 10.1089/ars.2012.4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krauth-Siegel R. L., and Comini M. A. (2008) Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim. Biophys. Acta 1780, 1236–1248 10.1016/j.bbagen.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 31. Stefani M., Sturlese M., Manta B., Löhr F., Mammi S., Comini M., and Bellanda M. (2016) 1H, 13C, and 15N resonance assignment of the cytosolic dithiol glutaredoxin 1 from the pathogen Trypanosoma brucei. Biomol NMR Assign 10, 85–88 10.1007/s12104-015-9643-x [DOI] [PubMed] [Google Scholar]
- 32. Jao S. C., English Ospina S. M., Berdis A. J., Starke D. W., Post C. B., and Mieyal J. J. (2006) Computational and mutational analysis of human glutaredoxin (thioltransferase): probing the molecular basis of the low pKa of cysteine 22 and its role in catalysis. Biochemistry 45, 4785–4796 10.1021/bi0516327 [DOI] [PubMed] [Google Scholar]
- 33. Van Laer K., Oliveira M., Wahni K., and Messens J. (2014) The concerted action of a positive charge and hydrogen bonds dynamically regulates the pKa of the nucleophilic cysteine in the NrdH-redoxin family. Protein Sci. 23, 238–242 10.1002/pro.2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Begas P., Liedgens L., Moseler A., Meyer A. J., and Deponte M. (2017) Glutaredoxin catalysis requires two distinct glutathione interaction sites. Nat. Commun. 8, 14835 10.1038/ncomms14835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lundström-Ljung J., Vlamis-Gardikas A., Aslund F., and Holmgren A. (1999) Reactivity of glutaredoxins 1, 2 and 3 from Escherichia coli and protein disulfide isomerase towards glutathionyl-mixed disulfides in ribonuclease A. FEBS Lett. 443, 85–88 10.1016/S0014-5793(98)01698-6 [DOI] [PubMed] [Google Scholar]
- 36. Björnberg O., Ostergaard H., and Winther J. R. (2006) Mechanistic insight provided by glutaredoxin within a fusion to redox-sensitive yellow fluorescent protein. Biochemistry 45, 2362–2371 10.1021/bi0522495 [DOI] [PubMed] [Google Scholar]
- 37. Rabenstein D. L., and Millis K. K. (1995) Nuclear magnetic resonance study of the thioltransferase-catalyzed glutathione/glutathione disulfide interchange reaction. Biochim. Biophys. Acta 1249, 29–36 10.1016/0167-4838(95)00067-5 [DOI] [PubMed] [Google Scholar]
- 38. Jensen K. S., Winther J. R., and Teilum K. (2011) Millisecond dynamics in glutaredoxin during catalytic turnover is dependent on substrate binding and absent in the resting states. J. Am. Chem. Soc. 133, 3034–3042 10.1021/ja1096539 [DOI] [PubMed] [Google Scholar]
- 39. Darby N. J., and Creighton T. E. (1995) Characterization of the active site cysteine residues of the thioredoxin-like domains of protein disulfide isomerase. Biochemistry 34, 16770–16780 10.1021/bi00051a027 [DOI] [PubMed] [Google Scholar]
- 40. Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem. 54, 237–271 10.1146/annurev.bi.54.070185.001321 [DOI] [PubMed] [Google Scholar]
- 41. Bocedi A., Cattani G., Stella L., Massoud R., and Ricci G. (2018) Thiol disulfide exchange reactions in human serum albumin: the apparent paradox of the redox transitions of Cys34. FEBS J. 285, 3225–3237 10.1111/febs.14609 [DOI] [PubMed] [Google Scholar]
- 42. Bushweller J. H., Billeter M., Holmgren A., and Wüthrich K. (1994) The nuclear magnetic resonance solution structure of the mixed disulfide between Escherichia coli glutaredoxin(C14S) and glutathione. J. Mol. Biol. 235, 1585–1597 10.1006/jmbi.1994.1108 [DOI] [PubMed] [Google Scholar]
- 43. Nordstrand K., slund F., Holmgren A., Otting G., and Berndt K. D. (1999) NMR structure of Escherichia coli glutaredoxin 3-glutathione mixed disulfide complex: implications for the enzymatic mechanism. J. Mol. Biol. 286, 541–552 10.1006/jmbi.1998.2444 [DOI] [PubMed] [Google Scholar]
- 44. Portillo-Ledesma S., Sardi F., Manta B., Tourn M. V., Clippe A., Knoops B., Alvarez B., Coitiño E. L., and Ferrer-Sueta G. (2014) Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry 53, 6113–6125 10.1021/bi500389m [DOI] [PubMed] [Google Scholar]
- 45. Noguera M. E., Vazquez D. S., Ferrer-Sueta G., Agudelo W. A., Howard E., Rasia R. M., Manta B., Cousido-Siah A., Mitschler A., Podjarny A., and Santos J. (2017) Structural variability of E. coli thioredoxin captured in the crystal structures of single-point mutants. Sci. Rep. 7, 42343 10.1038/srep42343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fairlamb A. H., and Cerami A. (1992) Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol 46, 695–729 10.1146/annurev.mi.46.100192.003403 [DOI] [PubMed] [Google Scholar]
- 47. Aslund F., Berndt K. D., and Holmgren A. (1997) Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J. Biol. Chem. 272, 30780–30786 10.1074/jbc.272.49.30780 [DOI] [PubMed] [Google Scholar]
- 48. Sagemark J., Elgán T. H., Bürglin T. R., Johansson C., Holmgren A., and Berndt K. D. (2007) Redox properties and evolution of human glutaredoxins. Proteins 68, 879–892 10.1002/prot.21416 [DOI] [PubMed] [Google Scholar]
- 49. Krause G., Lundström J., Barea J. L., Pueyo de la Cuesta C., and Holmgren A. (1991) Mimicking the active site of protein-disulfide isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J. Biol. Chem. 266, 9494–9500 [PubMed] [Google Scholar]
- 50. Comini M. A., Krauth-Siegel R. L., and Flohé L. (2007) Depletion of the thioredoxin homologue tryparedoxin impairs antioxidative defence in African trypanosomes. Biochem. J. 402, 43–49 10.1042/BJ20061341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dormeyer M., Reckenfelderbäumer N., Ludemann H., and Krauth-Siegel R. L. (2001) Trypanothione-dependent synthesis of deoxyribonucleotides by Trypanosoma brucei ribonucleotide reductase. J. Biol. Chem. 276, 10602–10606 10.1074/jbc.M010352200 [DOI] [PubMed] [Google Scholar]
- 52. Reckenfelderbäumer N., and Krauth-Siegel R. L. (2002) Catalytic properties, thiol pK value, and redox potential of Trypanosoma brucei tryparedoxin. J. Biol. Chem. 277, 17548–17555 10.1074/jbc.M112115200 [DOI] [PubMed] [Google Scholar]
- 53. Srinivasan U., Mieyal P. A., and Mieyal J. J. (1997) pH profiles indicative of rate-limiting nucleophilic displacement in thioltransferase catalysis. Biochemistry 36, 3199–3206 10.1021/bi962017t [DOI] [PubMed] [Google Scholar]
- 54. Henderson G. B., Murgolo N. J., Kuriyan J., Osapay K., Kominos D., Berry A., Scrutton N. S., Hinchliffe N. W., Perham R. N., and Cerami A. (1991) Engineering the substrate specificity of glutathione reductase toward that of trypanothione reduction. Proc. Natl. Acad. Sci. U.S.A. 88, 8769–8773 10.1073/pnas.88.19.8769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanzok S. M., Fechner A., Bauer H., Ulschmid J. K., Müller H. M., Botella-Munoz J., Schneuwly S., Schirmer R., and Becker K. (2001) Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 291, 643–646 10.1126/science.291.5504.643 [DOI] [PubMed] [Google Scholar]
- 56. Bonilla M., Denicola A., Marino S. M., Gladyshev V. N., and Salinas G. (2011) Linked thioredoxin-glutathione systems in platyhelminth parasites: alternative pathways for glutathione reduction and deglutathionylation. J. Biol. Chem. 286, 4959–4967 10.1074/jbc.M110.170761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bao R., Zhang Y., Lou X., Zhou C. Z., and Chen Y. (2009) Structural and kinetic analysis of Saccharomyces cerevisiae thioredoxin Trx1: implications for the catalytic mechanism of GSSG reduced by the thioredoxin system. Biochim. Biophys. Acta 1794, 1218–1223 10.1016/j.bbapap.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 58. Holmgren A. (1979) Reduction of disulfides by thioredoxin: exceptional reactivity of insulin and suggested functions of thioredoxin in mechanism of hormone action. J. Biol. Chem. 254, 9113–9119 [PubMed] [Google Scholar]
- 59. Attarian R., Bennie C., Bach H., and Av-Gay Y. (2009) Glutathione disulfide and S-nitrosoglutathione detoxification by Mycobacterium tuberculosis thioredoxin system. FEBS Lett. 583, 3215–3220 10.1016/j.febslet.2009.09.007 [DOI] [PubMed] [Google Scholar]
- 60. Cheng Z., Arscott L. D., Ballou D. P., and Williams C. H. Jr. (2007) The relationship of the redox potentials of thioredoxin and thioredoxin reductase from Drosophila melanogaster to the enzymatic mechanism: reduced thioredoxin is the reductant of glutathione in Drosophila. Biochemistry 46, 7875–7885 10.1021/bi700442r [DOI] [PubMed] [Google Scholar]
- 61. Schmidt H., and Krauth-Siegel R. L. (2003) Functional and physicochemical characterization of the thioredoxin system in Trypanosoma brucei. J. Biol. Chem. 278, 46329–46336 10.1074/jbc.M305338200 [DOI] [PubMed] [Google Scholar]
- 62. Arias D. G., Marquez V. E., Chiribao M. L., Gadelha F. R., Robello C., Iglesias A. A., and Guerrero S. A. (2013) Redox metabolism in Trypanosoma cruzi: functional characterization of tryparedoxins revisited. Free Radic. Biol. Med. 63, 65–77 10.1016/j.freeradbiomed.2013.04.036 [DOI] [PubMed] [Google Scholar]
- 63. Moutiez M., Aumercier M., Schöneck R., Meziane-Cherif D., Lucas V., Aumercier P., Ouaissi A., Sergheraert C., and Tartar A. (1995) Purification and characterization of a trypanothione-glutathione thioltransferase from Trypanosoma cruzi. Biochem. J. 310, 433–437 10.1042/bj3100433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fyfe P. K., Westrop G. D., Silva A. M., Coombs G. H., and Hunter W. N. (2012) Leishmania TDR1 structure, a unique trimeric glutathione transferase capable of deglutathionylation and antimonial prodrug activation. Proc. Natl. Acad. Sci. U.S.A. 109, 11693–11698 10.1073/pnas.1202593109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moutiez M., Meziane-Cherif D., Aumercier M., Sergheraert C., and Tartar A. (1994) Compared reactivities of trypanothione and glutathione in conjugation reactions. Chem. Pharm. Bull. 42, 2641–2644 10.1248/cpb.42.2641 [DOI] [Google Scholar]
- 66. Ebersoll S., Musunda B., Schmenger T., Dirdjaja N., Bonilla M., Manta B., Ulrich K., Comini M. A., and Krauth-Siegel R. L. (2018) A glutaredoxin in the mitochondrial intermembrane space has stage-specific functions in the thermo-tolerance and proliferation of African trypanosomes. Redox Biol. 15, 532–547 10.1016/j.redox.2018.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ulrich K., Finkenzeller C., Merker S., Rojas F., Matthews K., Ruppert T., and Krauth-Siegel R. L. (2017) Stress-induced protein S-glutathionylation and S-trypanothionylation in African trypanosomes-A quantitative redox proteome and thiol analysis. Antioxid. Redox Signal. 27, 517–533 10.1089/ars.2016.6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ellis K. J., and Morrison J. F. (1982) Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 87, 405–426 10.1016/S0076-6879(82)87025-0 [DOI] [PubMed] [Google Scholar]
- 69. Alvarez M. N., Trujillo M., and Radi R. (2002) Peroxynitrite formation from biochemical and cellular fluxes of nitric oxide and superoxide. Methods Enzymol. 359, 353–366 10.1016/S0076-6879(02)59198-9 [DOI] [PubMed] [Google Scholar]
- 70. Fueller F., Jehle B., Putzker K., Lewis J. D., and Krauth-Siegel R. L. (2012) High throughput screening against the peroxidase cascade of African trypanosomes identifies antiparasitic compounds that inactivate tryparedoxin. J. Biol. Chem. 287, 8792–8802 10.1074/jbc.M111.338285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sardi F., Manta B., Portillo-Ledesma S., Knoops B., Comini M. A., and Ferrer-Sueta G. (2013) Determination of acidity and nucleophilicity in thiols by reaction with monobromobimane and fluorescence detection. Anal. Biochem. 435, 74–82 10.1016/j.ab.2012.12.017 [DOI] [PubMed] [Google Scholar]
- 72. Grassetti D. R., and Murray J. F. Jr. (1967) Determination of sulfhydryl groups with 2,2′- or 4,4′-dithiodipyridine. Arch. Biochem. Biophys. 119, 41–49 10.1016/0003-9861(67)90426-2 [DOI] [PubMed] [Google Scholar]
- 73. Melchers J., Dirdjaja N., Ruppert T., and Krauth-Siegel R. L. (2007) Glutathionylation of trypanosomal thiol redox proteins. J. Biol. Chem. 282, 8678–8694 10.1074/jbc.M608140200 [DOI] [PubMed] [Google Scholar]
- 74. Mendes P. (1993) GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 9, 563–571 [DOI] [PubMed] [Google Scholar]
- 75. Schanda P., Kupce E., and Brutscher B. (2005) SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 33, 199–211 10.1007/s10858-005-4425-x [DOI] [PubMed] [Google Scholar]
- 76. Peltoniemi M. J., Karala A. R., Jurvansuu J. K., Kinnula V. L., and Ruddock L. W. (2006) Insights into deglutathionylation reactions: different intermediates in the glutaredoxin and protein disulfide isomerase catalyzed reactions are defined by the γ-linkage present in glutathione. J. Biol. Chem. 281, 33107–33114 10.1074/jbc.M605602200 [DOI] [PubMed] [Google Scholar]
- 77. Nordstrand K., Aslund F., Meunier S., Holmgren A., Otting G., and Berndt K. D. (1999) Direct NMR observation of the Cys-14 thiol proton of reduced Escherichia coli glutaredoxin-3 supports the presence of an active site thiol-thiolate hydrogen bond. FEBS Lett. 449, 196–200 10.1016/S0014-5793(99)00401-9 [DOI] [PubMed] [Google Scholar]
- 78. Iversen R., Andersen P. A., Jensen K. S., Winther J. R., and Sigurskjold B. W. (2010) Thiol-disulfide exchange between glutaredoxin and glutathione. Biochemistry 49, 810–820 10.1021/bi9015956 [DOI] [PubMed] [Google Scholar]
- 79. Gan Z. R., and Wells W. W. (1987) Identification and reactivity of the catalytic site of pig liver thioltransferase. J. Biol. Chem. 262, 6704–6707 [PubMed] [Google Scholar]
- 80. Gao X. H., Zaffagnini M., Bedhomme M., Michelet L., Cassier-Chauvat C., Decottignies P., and Lemaire S. D. (2010) Biochemical characterization of glutaredoxins from Chlamydomonas reinhardtii: kinetics and specificity in deglutathionylation reactions. FEBS Lett. 584, 2242–2248 10.1016/j.febslet.2010.04.034 [DOI] [PubMed] [Google Scholar]
- 81. Couturier J., Koh C. S., Zaffagnini M., Winger A. M., Gualberto J. M., Corbier C., Decottignies P., Jacquot J. P., Lemaire S. D., Didierjean C., and Rouhier N. (2009) Structure-function relationship of the chloroplastic glutaredoxin S12 with an atypical WCSYS active site. J. Biol. Chem. 284, 9299–9310 10.1074/jbc.M807998200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zaffagnini M., Bedhomme M., Marchand C. H., Couturier J. R., Gao X. H., Rouhier N., Trost P., and Lemaire S. P. (2012) Glutaredoxin s12: unique properties for redox signaling. Antioxid. Redox Signal. 16, 17–32 10.1089/ars.2011.3933 [DOI] [PubMed] [Google Scholar]
- 83. Feng Y., Zhong N., Rouhier N., Hase T., Kusunoki M., Jacquot J. P., Jin C., and Xia B. (2006) Structural insight into poplar glutaredoxin C1 with a bridging iron-sulfur cluster at the active site. Biochemistry 45, 7998–8008 10.1021/bi060444t [DOI] [PubMed] [Google Scholar]
- 84. Gonzalez-Chavez Z., Olin-Sandoval V., Rodíguez-Zavala J. S., Moreno-Sánchez R., and Saavedra E. (2015) Metabolic control analysis of the Trypanosoma cruzi peroxide detoxification pathway identifies tryparedoxin as a suitable drug target. Biochim. Biophys. Acta 1850, 263–273 10.1016/j.bbagen.2014.10.029 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.