

Abstract
Betaglycan (BG) is a membrane-bound co-receptor of the TGF-β family that selectively binds transforming growth factor-β (TGF-β) isoforms and inhibin A (InhA) to enable temporal-spatial patterns of signaling essential for their functions in vivo. Here, using NMR titrations of methyl-labeled TGF-β2 with BG's C-terminal binding domain, BGZP-C, and surface plasmon resonance binding measurements with TGF-β2 variants, we found that the BGZP-C–binding site on TGF-β2 is located on the inner surface of its extended finger region. Included in this binding site are Ile-92, Lys-97, and Glu-99, which are entirely or mostly specific to the TGF-β isoforms and the InhA α-subunit, but they are unconserved in other TGF-β family growth factors (GFs). In accord with the proposed specificity-determining role of these residues, BG bound bone morphogenetic protein 2 (BMP-2) weakly or not at all, and TGF-β2 variants with the corresponding residues from BMP-2 bound BGZP-C more weakly than corresponding alanine variants. The BGZP-C–binding site on InhA previously was reported to be located on the outside of the extended finger region, yet at the same time to include Ser-112 and Lys-119, homologous to TGF-β2 Ile-92 and Lys-97, on the inside of the fingers. Therefore, it is likely that both TGF-β2 and InhA bind BGZP-C through a site on the inside of their extended finger regions. Overall, these results identify the BGZP-C–binding site on TGF-β2 and shed light on the specificity of BG for select TGF-β–type GFs and the mechanisms by which BG influences their signaling.
Keywords: transforming growth factor β (TGF-B), cell surface receptor, cell signaling, cardiac development, endocrinology, nuclear magnetic resonance (NMR), betaglycan, finger region, ILV methyl labeling
Introduction
Betaglycan (BG)3 is a co-receptor of the highly diversified transforming growth factor β family (TGF-β) of signaling proteins, which have essential roles in diverse processes, ranging from patterning of embryos and organs in embryogenesis to regulation of the endocrine and adaptive immune systems in adults (1–3). BG is expressed by epithelial, mesenchymal, and other cell types, but unlike the type I and type II signaling receptors (4–6) that bind TGF-β family growth factors (GFs) to transduce the signal by phosphorylating Smads and other effectors (7–9), it is not required for signal transduction (10). BG is nonetheless essential in vertebrates, such as mice, where knockout of the BG gene is embryonic lethal (11).
BG is a membrane-anchored proteoglycan composed of a large extracellular domain (766 amino acids in humans), a single transmembrane-spanning helix, and a short (43 amino acids in humans) highly-conserved cytoplasmic tail (Fig. 1A). BG's extracellular domain is composed of two subdomains, each roughly equal in size: the membrane-distal orphan domain (BGO) and the C-terminal membrane-proximal zona pellucida domain (BGZP). The structure of BG's orphan domain is not known, but it is likely two tandem β-sandwiches, designated BGOR1 and BGOR2, similar to that of endoglin (12), a homologous co-receptor of the TGF-β family with the same overall domain structure (13). BGZP is likely composed of tandem immunoglobulin-like domains, designated BGZP-N and BGZP-C, based on homology to endoglin, as well as available structures of the BGZP C-terminal subdomain (BGZP-C) from rat (14) and mouse (15).
Figure 1.

Overall domain structure of betaglycan and proposed mechanisms for potentiation of TGF-β signaling and inhibition of activin signaling. A, overall domain structure of betaglycan. B, previously proposed mechanism for potentiation of TGF-β signaling by betaglycan (29) in which BG binds TGF-β dimers asymmetrically with a 1:1 stoichiometry in a manner that blocks one of the TβRII-binding sites but not the other (step i). Sequestration of TGF-β2 on the membrane by BG is proposed to promote binding of TβRII (step ii), which in turn promotes the concerted recruitment of TβRI and displacement of the BG orphan domain, and by an unknown mechanism, binding of a second TβRI:TβRII pair (step iii). C, previously proposed mechanism for inhibition of activin signaling by InhA (21, 23) in which BG binds InhA through its α-subunit (step i). Sequestration of InhA on the membrane by BG is proposed to promote binding of ActRII, which in turn sequesters ActRII away from activin A in a complex that is incapable of recruiting ActRIb and signaling (step ii).
BG binds and forms stable binary complexes with TGF-β1, -β2, and -β3 homodimers (16, 17) and potentiates type II receptor (TβRII) binding and signaling (Fig. 1B) (10, 18). BG's effects are greatest for TGF-β2, which binds TβRII 200–500-fold more weakly than TGF-β1 and -β3 (10, 19, 20) and only signals at supraphysiological concentrations in the absence of BG (10, 18, 19). BG also binds and forms stable complexes with inhibin A (InhA) (21), a TGF-β family heterodimer formed by an α-subunit (Inh α) and a βΑ-subunit (Inh βA). Binding of InhA by BG, which occurs through the α-subunit, potentiates binding of the activin type II receptor, ActRII, or the closely related activin type IIB receptor, ActRIIB, to the β-subunit (Fig. 1C) (21). BG-potentiated binding of ActRII, or ActRIIB, to the inhibin β-subunit has been proposed to underlie its ability to antagonize activins, homo- or heterodimers of Inh β-subunits, by sequestering activin's type II receptors, ActRII and ActRIIB, in a ternary complex incapable of recruiting a type I receptor (21–23).
BG is expressed at high levels in developing and adult hearts and livers, as well as by gonadotropic cell types in the anterior pituitary, and thus its expression overlaps with that of the GFs whose functions it influences, including TGF-β2, which is expressed at high levels in developing and adult hearts and livers (24), as well inhibins, which are produced in the gonads and antagonize activin signaling in the anterior pituitary (25). TGF-β2 knockout mice are embryonic lethal (24), similar to the BG knockout mice (11), and both have many of the same characteristics, including severe heart and liver defects. BG therefore appears to function not by directly altering signaling outcomes, but instead by mediating TGF-β2, and likely inhibin signaling, in a restricted spatial-temporal manner. BG is nonetheless likely to have other functions, as suggested by its expression in other tissues (11, 26) and by reports that it binds and effects the function of several other TGF-β family GFs, including BMP-2, BMP-4, BMP-7, and GDF-5 (27).
Biochemical and biophysical studies have begun to shed light on how this essential co-receptor functions. Esparza-López et al. (28) and Mendoza et al. (17) showed that both BGO and BGZP-C bind TGF-βs, whereas Wiater et al. (23) showed that only BGZP-C binds inhibins. Villarreal et al. (29), who used surface plasmon resonance (SPR), isothermal titration calorimetry, and other measurements, showed that the full-length BG extracellular domain binds TGF-β homodimers asymmetrically with 1:1 stoichiometry and is positioned such that one of the binding sites for TβRII on the GF fingertip is occluded by BGZP-C (Fig. 1B). Makanji et al. (30), who used site-directed mutagenesis and functional analysis of secreted but not purified InhA variants, concluded that the binding site for BGZP-C on the InhA α-subunit lies on the convex outer surface of the fingers (Fig. S1).
The objective of this study was to identify the binding site for BGZP-C on TGF-β2. One motivation for this was homology models of InhA, which showed that three of the seven residues identified by Makanji et al. (30) lie on the concave inner surface of the fingers and not the outer surface (Fig. S1). Another motivation was that among these three residues on the inner surface, two (Inh α Lys-119 and Inh α Ser-112) were either entirely or mostly specific to the TGF-βs and Inh α, but unconserved in other family members (Fig. S1). Among the remaining four residues, one other, Inh α Tyr-50 on the outside of the fingers, is unique to Inh α but different in the TGF-βs and other family members (Fig. S1). All other residues are conserved, not just among the TGF-βs and Inh α but among most other GFs of the family (Fig. S1). Hence, it is unclear whether the binding site on Inh α resides on the outside of the fingers, the inside, or both. It is also unclear whether TGF-β and Inh α utilize the same or distinct sites to bind BGZP-C.
Our studies, which included NMR titrations of sparsely labeled TGF-β2 with unlabeled BGZP-C, as well as accompanying analysis of TGF-β2 variants by SPR-based binding studies, showed that the BGZP-C–binding site lies exclusively on the inner surface of the fingers. Included in this site are three residues, Ile-92, Lys-97, and Glu-99, that are either completely (Lys-97) or mostly (Ile-92 and Glu-99) specific to the three TGF-β isoforms and Inh α and of a totally different character (hydrophobic versus charged, negatively versus positively charged, etc.) in other family GFs. Our results further show that purified BG binds BMP-2 weakly or not at all and that substitution of TGF-β2 with the corresponding residues from BMP-2 weakens binding to BGZP-C even more than substitution with neutral alanine residues.
Results
Overview
Our overall strategy to identify the BGZP-C–binding site on TGF-β2 was to assign the methyl resonances of its Ile, Leu, and Val (ILV) residues and to use these to map the BGZP-C–binding site. One reason for choosing methyls, instead of amides, was our prior finding that in isolation BGZP-C binds TGF-β homodimers with 2:1 stoichiometry (29), thus forming a 66-kDa complex (26-kDa GF homodimer plus two 20-kDa molecules of BGZP-C), which would be difficult to study by amide-based approaches, even with deuteration. One other reason is the poor solubility of the TGF-β2/BGZP-C complex, which for reasons described below could only be studied at pH values higher than 10.0, which would be impossible with amides because of rapid solvent-exchange rates. Protonated, 13C–labeled methyl groups, in the context of otherwise fully deuterated proteins, provide a viable alternative approach because of their much more favorable transverse spin-relaxation properties and their insensitivity to pH (31).
Methyl groups, while proven probes for mapping binding sites in the context of large macromolecular complexes, must be assigned to be useful. Many approaches have been proposed for this, including assignment of the backbone resonances and extension to the side chains using triple-resonance–based approaches (32), assignment by conservative substitution, such as Ile → Leu or Val or Leu → Ile (33), and identification by characteristic patterns of NOESY cross-peaks (34) or chemical shifts and residual dipolar couplings (35). Our approach was to use direct assignment, the rationale being that assignment of the backbone, and extension to the side-chain methyls, should be possible for the unbound form of the 26-kDa TGF-β2 homodimer. One of the caveats, however, was the necessity to perform these assignments under acidic conditions, far away from the basic conditions where we planned to perform the binding studies with BGZP-C. One of the consequences of this was the necessity to complement our direct assignment approach at low pH using a combination of pH titrations and conservative substitutions to obtain the necessary methyl assignments at high pH. Our presentation of the results therefore begins with assignment of the backbone and side-chain methyl resonances of TGF-β2 at low pH and then follows with a description of how we transferred these assignments to high pH where we studied the 66-kDa TGF-β(BGZP-C)2 complex.
Assignment and secondary structure of TGF-β2
TGF-βs, and most other GFs of the TGF-β family, have micromolar or lower solubilities from about pH 4.5 to about 10 because of the hydrophobic nature of their surfaces and their close to neutral charge over this range (36, 37). The poor solubility is known to be further aggravated by the presence of salts, such as NaCl (37). Thus, the previous NMR studies of TGF-β1 and TGF-β3 were carried out under acidic conditions in the absence of salts, 95% H2O, 5% 2H2O, pH 4.2, 45 °C for TGF-β1 and 87% H2O, 5% 2H2O, 8% [2H]dioxane, 3% [2H]methanol, pH 2.9, 45 °C for TGF-β3, where they were both highly soluble and structured (38, 39). To identify the ideal conditions for TGF-β2, we collected spectra of 15N TGF-β2 as the pH was varied from 2 to 5 and the temperature was varied from 25 to 50 °C. We found the conditions that led to the most dispersed 1H–15N shift correlation (HSQC) spectrum and the least accumulation of precipitate over time was 95% H2O, 5% 2H2O, pH 2.9, and 37 °C. Through analysis of standard triple resonance data sets with 0.2–0.3 mm 13C,15N samples under these conditions, we were able to assign the backbone of all residues, except Ile-105 (Fig. 2A).
Figure 2.

Assignment and secondary structure of human TGF-β2. A, assigned 1H-15N HSQC spectrum of TGF-β2 recorded in 5% 2H2O, pH 2.7, 37 °C, 700 MHz. Assigned backbone amide signals are indicated by their residue number and one-letter code. Horizontal bars identify the side-chain amide resonances of Asn and Gln. B, secondary structure probabilities of TGF-β2 calculated using the program PECAN (67), with helical and strand probabilities plotted as positive and negative values, respectively. Secondary structure shown along the top was calculated from the crystal structure of TGF-β2 (PDB 2TGI) (68) using the program DSSP (69).
The secondary structure propensities, calculated from the assigned shifts using the program PECAN, showed that with the exception of the heel helix (α3), the protein had the expected secondary structure (Fig. 2B). The absence of a detectable heel helix suggests that rather than adopting a canonical closed form in which the two monomers pack extensively against one another, as in the two reported TGF-β2 crystal structures (40, 41), the protein instead adopts the open form, i.e. an alternative conformation, previously reported for TGF-β3 (42), in which the monomers rotate away from one another and the heel helix unfolds (Fig. S2).
TGF-β2/BGZP-C complex at high pH
To investigate possible conditions for studying the TGF-β2/BGZP-C complex, samples of 10 μm TGF-β2 with a slight molar excess of BGZP-C (2.25 eq for each 1.0 eq of TGF-β2 homodimer) were prepared between pH 3.0 and 11.0 in 1.0 pH increments. This analysis showed that all samples had extensive precipitate, except for the pH 10.0 sample, which was slightly precipitated, and for the pH 11.0 sample, which had no precipitate.
To determine whether the TGF-β2/(BGZP-C)2 complex was stably formed at pH 11.0, Ile, Leu, Val-methyl–protonated but otherwise fully deuterated TGF-β2 and BGZP-C samples (2H–ILV–TGF-β2 and 2H-ILV-BGZP-C, respectively) were prepared by expression in Escherichia coli on the appropriately isotopically labeled medium, refolded, and examined alone by NMR at high pH, 25 mm [2H]glycine, pH 11.0, and at lower pH values, where we expected them to be soluble and natively folded (Fig. S3, A and B). The methyl spectra revealed some shift perturbations between the two pH values, especially for TGF-β2, yet regardless of the pH, the spectra were generally well-dispersed, suggesting that both TGF-β2 and BGZP-C are mostly, or entirely, natively folded at pH 11. To determine whether TGF-β2 and BGZP-C could still bind under these conditions, SPR sensorgrams were recorded as BGZP-C was injected over minimally-biotinylated TGF-β2 captured on a high-density streptavidin surface at pH 7.4 and pH 11.0 (Fig. S3, C and D). The KD values at the two pH values, derived by fits of the equilibrium response as a function of concentration to a standard binding isotherm (Fig. S3, C and D), showed that there was only a 3-fold weakening of the affinity at pH 11 compared with pH 7.4 (Table 1). Thus, both TGF-β2 and BGZP-C appear to retain most or all of their native structure at pH 11.0 and in turn bind one another with an affinity that is only slightly weakened relative to that at neutral pH.
Table 1.
Binding affinities of mouse TGF-β2 variants for BGZP-C and BGO
All measurements, with the exception of those indicated in footnotes a and d, were made in HBS-EP buffer at pH 7.4 with TGF-βs minimally carboxy-biotinylated and captured on a high-density streptavidin surface.
| TGF-β2 variant | BGZP-C binding |
BGO binding |
||
|---|---|---|---|---|
| Kd | Rmax | Kd | Rmax | |
| μm | RU | μm | RU | |
| TGF-β2 WT | 0.20 ± 0.12 | 315 ± 6 | 0.085 ± 0.023 | 398 ± 6 |
| TGF-β2 WTa | 0.611 ± 0.06 | 148 ± 5 | NDb | NDb |
| TGF-β2 I33A | 8.22 ± 0.14 | 250c | 0.076 ± 0.001 | 315 ± 4 |
| TGF-β2 E84A | 0.17 ± 0.01 | 274 ± 4 | 0.065 ± 0.007 | 426 ± 5 |
| TGF-β2 T87A | 0.26 ± 0.01 | 268 ± 2 | 0.060 ± 0.001 | 379 ± 3 |
| TGF-β2 L89A | 0.27 ± 0.02 | 236 ± 5 | 0.083 ± 0.007 | 350 ± 10 |
| TGF-β2 Y91A | 0.40 ± 0.02 | 86 ± 2 | 0.22 ± 0.04 | 189 ± 9 |
| TGF-β2 I92A | 1.8 ± 0.2 | 159 ± 8 | 0.080 ± 0.01 | 286 ± 6 |
| TGF-β2 I92D | 12.6 ± 0.3 | 98c | 0.36 ± 0.03 | 129 ± 4 |
| TGF-β2 K97A | 0.96 ± 0.07 | 110 ± 4 | 0.065 ± 0.001 | 182 ± 3 |
| TGF-β2 K97V | 1.97 ± 0.02 | 78c | 0.11 ± 0.01 | 103 ± 3 |
| TGF-β2 I98A | 0.32 ± 0.02 | 267 ± 6 | 0.14 ± 0.01 | 336 ± 8 |
| TGF-β2 E99A | 1.5 ± 0.1 | 297 ± 12 | 0.12 ± 0.01 | 411 ± 12 |
| TGF-β2 E99R | 8.8 ± 0.6 | 79c | 0.31 ± 0.07 | 114 ± 9 |
| avi-mmTGF-β2–7 md | 2.1 ± 0.1 | 76 ± 1 | NDb | NDb |
a This was measured in 25 mm glycine, 150 mm NaCl, pH 11.0, with minimally carboxy-biotinylated TGF-β2 WT captured on a high-density streptavidin surface.
b ND means not determined.
cRmax value was constrained to the value shown based on the Rmax obtained for binding of BGO.
d This was measured in HBS-EP buffer at pH 7.4 with biotinylated avi-tagged mmTGF-β2–7 m captured on a high-density streptavidin surface.
TGF-β2 pH titration
To obtain the TGF-β2 Ile, Leu, and Val (ILV) methyl signal assignments at pH 11.0 where we intended to map the BGZP-C–binding site, standard NMR experiments, such as C(CO)NH, H(CCO)NH, and HCCH-TOCSY, were applied to samples of 13C,15N TGF-β2 at pH 2.9 to extend the assignments from the backbone to the ILV side-chain methyls. This enabled the identification of the methyl signals of all the ILV residues in TGF-β2. To verify the assignments, a 1H-13C shift correlation spectrum of 2H–ILV–TGF-β2 was recorded under the same conditions and overlaid onto the constant time 1H-13C shift correlation spectrum of uniformly 13C,15N-labeled TGF-β2 (Fig. 3A). The 22 signals apparent in the spectrum of 2H–ILV–TGF-β2, which was expected based on the 7 Ile, 11 Leu, and 4 Val residues in TGF-β2 and the methyl precursors used, 3,3′-2H, 4-13C α-ketobutyrate for Ile (43) and 2-13C-methyl-2-hydroxy-3-oxobutanoate for Leu and Val (44), overlaid perfectly with the seven assigned Ile-δ1 signals and one of two methyls for the 11 assigned Leu-δ1/δ2 and four assigned Val γ1/γ2 signals.
Figure 3.
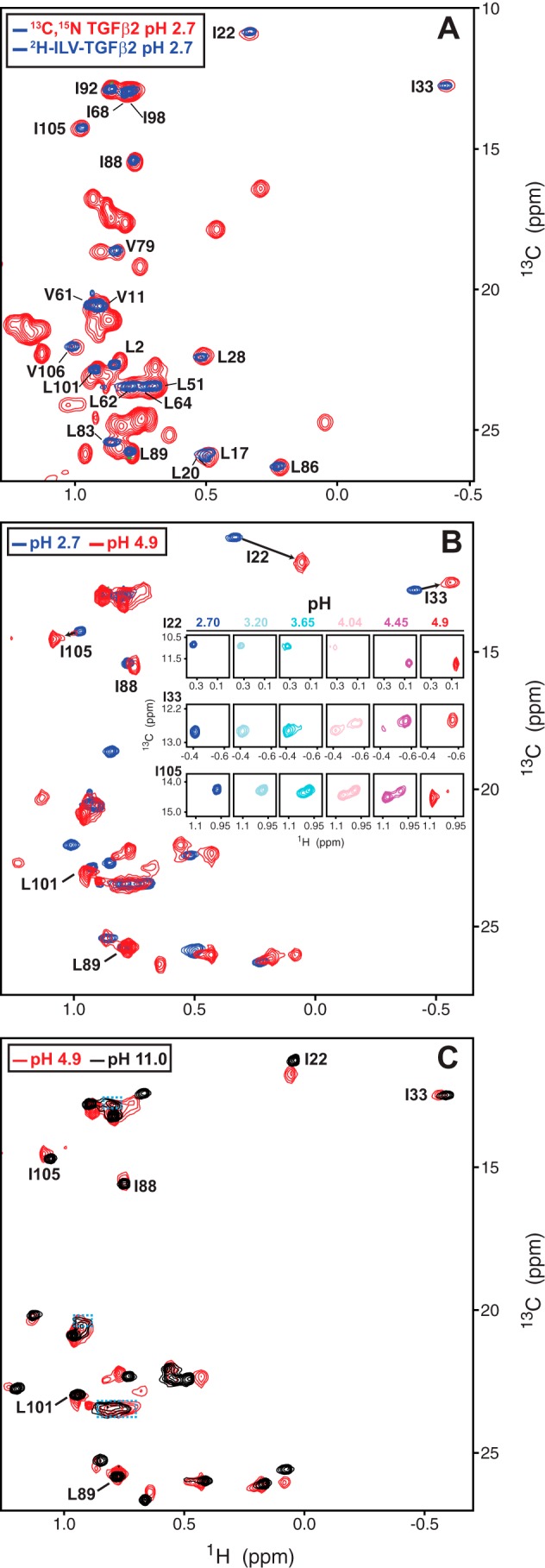
pH titration of human TGF-β2. A, overlay of the methyl region of the CT-HSQC spectrum of 13C,15N-TGF-β2 (red contours) or HSQC spectrum of 2H-MP-TGF-β2 (blue contours). Spectra were recorded at pH 2.7 and 37 °C. Assigned methyls are indicated by their residue number and one-letter code. B, HSQC spectra of 2H-MP-TGF-β2, recorded between pH 2.7 and 4.9. Overlay of the end points of the titration at pH 2.7 (blue) and pH 4.9 (red) is shown in the main panel. Peaks assignments shown correspond to those that could be confidently inferred from the assigned signals at pH 2.7. Expansions of three representative peaks, Ile-22, Ile-33, and Ile-105, at all points along the titration, are shown in the inset. C, overlay of the methyl region of the HSQC spectrum of 2H-MP-TGF-β2 at pH 4.9 (red contours) and 11.0 (black contours). Peaks assignments shown correspond to those that could be confidently inferred from the assigned signals at pH 2.7.
To transfer the methyl assignments from pH 2.7 to pH 11.0, 1H-13C shift correlation spectra of 2H–ILV–TGF-β2 were collected under the limited range of pH values, 2.7–4.9 and 11.0, where 2 μm or higher samples could be prepared without significant precipitation. This showed unexpectedly large shift changes between pH 2.7 and 4.9 but surprisingly small changes between pH 4.9 and 11.0 (Fig. 3, B and C). Although the spectra in the low pH range were collected in relatively fine increments, it nonetheless proved difficult to follow all but the most resolved signals as the majority of signals exhibited slow exchange behavior, with signals corresponding to low and high pH forms of the protein being simultaneously observed at intermediate pH values in this range (Fig. 3B, insets). This behavior suggested that the large shifts observed as the pH was changed from 2.7 to 4.9 were not due to ionization of nearby side-chain carboxylic acid groups but instead a large rearrangement of the protein. This slow exchange behavior is also accompanied by an apparent shift of several methyl signals from the random coil methyl position (∼0.85 ppm 1H for Ile-δ1 and Leu-δ methyls and ∼0.90 ppm 1H for Val-γ methyls) to a nonrandom coil position as the pH was raised from 2.7 to 4.9. One process that could account for these observations is a large-scale conformational rearrangement from the open form at pH 2.7 to the closed form at pH 4.9. The slow exchange behavior is presumably caused by the opening and closing process, which would be expected to be relatively slow (millisecond time scale) owing to the large number of atoms that must simultaneously rearrange. The decrease in the number of random coil methyls is presumably due to the 1 Val (Val-61), 2 Leu (Leu-62 and Leu-64), and 1 Ile (Ile-68) residues in the heel helix that are structurally disordered in the open form but not the closed form.
Although the slow exchange behavior prevents reliable assignment of most of the methyl signals, it is nonetheless possible to tentatively assign a few of the peaks at pH 11.0, such as Leu-89 or Leu-101, which did not appear to shift at all, or Ile-33, Ile-22, Ile-88, and Ile-105, which shifted as the pH was varied, but were well-resolved (Fig. 3, A–C). Thus, due to the unexpected large-scale conformational rearrangement and slow exchange behavior, it was possible to transfer only about a third of the assigned methyl signals at pH 2.7 to pH 11.0.
Titration of TGF-β2 with BGZP-C
To identify the BGZP-C–binding site on TGF-β2, 1H-13C shift correlation spectra of ILV–TGF-β2 were collected at pH 11 with sub- to super-stoichiometric amounts of unlabeled BGZP-C added (Fig. 4, A and B). Through spectral overlays, it was evident that three of the tentatively assigned signals, Ile-33, Ile-88, and Leu-101, underwent sizable shifts. These three residues are each located on the concave inner surface of the extended finger region and are spatially quite close to one another, with the γ2 methyl of Ile-33 packing against the Ile-88 δ1 methyl and the Leu-101 δ2 methyl packing against the Ile-88 δ2 methyl (Fig. 5A). The other three tentatively assigned methyls that did not shift, Ile-22, Ile-105, and Leu-89 (Fig. 4A), were not spatially close to one another, with two them, Leu-89 and Ile-105, lying far apart on the convex outer surface of the extended finger region, and one of them, Ile-22, buried in the hydrophobic interface between monomers (Fig. 5A). This strongly suggested that the BGZP-C–binding site on TGF-β2 lies principally on the concave inner surface of the fingers.
Figure 4.

Titration of 2H-MP human TGF-β2 with unlabeled BGZP-C. A, overlay of the methyl region of the HSQC spectrum of 2H-MP-TGF-β2 in either the unbound state (blue contours) or bound state (red contours) with an excess of unlabeled BGZP-C (1 eq of TGF-β2 homodimer with 2.25 eq of BGZP-C). Peak assignments shown correspond to those that could be confidently inferred from the assigned signals at pH 2.7. B, expansions of six representative peaks, Ile-33, Ile-88, and Leu-101, and three other unassigned peaks, designated peaks a–c, at all points along the titration. C, overlay of the HSQC spectrum of WT 2H-MP-TGF-β2 (blue contours) and the I33L variant (red contours). Inferred assignments for Ile-33 in the WT protein and Leu-33 in the variant are shown. D, overlay of the HSQC spectrum of WT 2H-MP-TGF-β2 (blue contours) and the L101I variant (red contours). Inferred assignments for Leu-101 in the WT protein and Ile-101 in the variant are shown.
Figure 5.

Structure of human TGF-β2 homodimer and identification of residues in the BGZP-C–binding site. A, structure of the TGF-β2 homodimer (PDB 2TGI), highlighting the Ile, Leu, and Val residues found on either the inside or outside of the extended finger region (red and blue text, respectively). Same residues are shown on only one of the two monomers to simplify the presentation. B, structure of the TGF-β2 homodimer (PDB 2TGI), highlighting the residues that lie on either the inside or outside of the fingers that were substituted in this study (red and blue text, respectively). Same residues are shown on only one of the two monomers to simplify the presentation. C, structure of the engineered TGF-β mini-monomer, mmTGF-β2-7M (PDB 5TX6), highlighting the Ile, Leu, and Val residues found on the inside of the extended finger region (red text). Engineered loop that replaced the heel helix is highlighted in green. D, alignment of the residues from the finger region of all TGF-β family growth factors in humans; positions highlighted in color were either shown to shift upon titration of 2H-MP-TGF-β2 with unlabeled BGZP-C or to be affected in their binding affinity for BGZP-C upon substitution. Hydrophobic residues are colored green; acidic residues are colored red; basic residues are colored blue, and neutral residues are colored purple. Residues highlighted in yellow correspond to those that are either entirely (Lys-97) or mostly unique (Val-92/Ile-92 and Glu-99) to TGF-βs and Inh α.
To ascertain whether this was correct, two separate 2H–ILV–TGF-β2 samples were prepared in which Ile-33 and Leu-101 were conservatively substituted with leucine and isoleucine, respectively (I33L and L101I). The 1H-13C shift correlation spectra of these variants were then recorded at pH 11.0 and compared with the spectrum of WT 2H–ILV–TGF-β2 (Fig. 4, C and D). The spectra of I33L 2H–ILV–TGF-β2 and L101I 2H–ILV–TGF-β2 each had no peak corresponding to the assigned Ile-33 and Leu-101 peaks, respectively, while at the same time they both had an extra peak, located in the Leu/Val region of the spectrum for the I33L variant and in the Ile region of the spectrum for the L101I variant. This indicates the assignments for the Ile-33 and Leu-101 methyls were correct. One further point to note is that the peak tentatively assigned to Ile-88 in the spectra of both the I33L and L101I variants also shifted. This is expected even with these conservative Ile → Leu and Leu → Ile substitutions, owing to the close spatial proximity of these residues, and thus it provides further, albeit indirect, evidence that all three of these assignments are correct.
The overlay of the spectrum of ILV–TGF-β2 bound to an excess of BGZP-C at pH 11 relative to ILV–TGF-β2 alone revealed that several other as yet unassigned methyls also underwent sizable shifts upon addition of BGZP-C (Fig. 4A). If the binding site indeed lies on the concave inner surface of the fingers, then it is reasonable to expect that the unassigned peaks that underwent the largest shifts (Fig. 4A, peaks a–d) correspond to other methyl-bearing residues, such as Leu-28, Val-61, Leu-64, Leu-86, and Ile-92, that are spatially close to Ile-33, Ile-88, and Leu-101 (Fig. 5A). To determine whether this was correct, four additional conservatively substituted ILV–TGF-β2 variants were prepared, L28I, V61I, L86I, and I92L. The 1H-13C shift correlation spectra of these variants, relative to that of WT ILV–TGF-β2, confirms this, with peaks a–d in the WT spectrum corresponding to Ile-92, Val-61, Leu-28, and Leu-86, respectively (Fig. S4).
Together, these results show that the BGZP-C–binding site lies on the concave inner surface of the fingers and extends horizontally from the near the loops connecting the fingertips toward the edge of the heel helix and vertically from finger 1 at the bottom to finger 4 at the top (Fig. 5A). The binding site overlaps in part with the TβRII-binding site, which includes the loops connecting fingers 1/2 and 3/4, but it does not appear to extend to the convex outer surface of the fingers since the signals of two assigned residues in this region, Leu-89 and Ile-105, did not shift upon addition of BGZP-C (Fig. 4A). Another observation in accord with this is that Ile-98, which lies on the outside of the fingers (Fig. 5A) and by a process of elimination must correspond to one of the two unassigned Ile resonances between 0.65–0.75 1H and 13.0–13.5 ppm 13C (Fig. 4A), does not shift upon addition of BGZP-C (Fig. 4A).
Site-directed mutagenesis of TGF-β2 and effects on BGZP-C and BGO binding
To further validate the BGZP-C–binding site and to identify potential interaction hot spots, several residues both inside the binding site on the concave surface of the fingers and outside on the convex surface were substituted with alanine. The residues on the inside that were substituted include the three residues whose methyl groups shifted the most upon BGZP-C binding, Ile-33, Ile-92, and Leu-101, and three other prominent nonmethyl-bearing residues, Tyr-90, Lys-97, and Glu-99 (Fig. 5B). The residues on the outside that were substituted include two residues whose methyl groups did not appreciably shift, Leu-89 and Ile-98, and three other prominent residues, Glu-84, Thr-87, and Tyr-91 (Fig. 5B).
Although it was possible to isolate each of these variants in high yield from inclusion bodies, the amount of dimer that could be isolated following the folding reaction varied significantly, with some variants, such as I33A, leading to reduced but still significant dimer formation, whereas others, such as I98A and Y90A, led to little to no dimer (Fig. S5). The criterion whether a variant could be purified was whether subsequent fractionation of the dimeric form of the protein on a C18 reverse-phase column resulted in a suitable single peak with LC-ESI-MS intact mass within 0.5 Da of that expected. Those variants that did not meet this criterion, Y90A and L101A, were excluded from further analysis. The SPR measurements were performed by capturing minimally biotinylated TGF-β2 dimers onto a streptavidin surface, followed by duplicate or triplicate injections of either BGZP-C or BGO until equilibrium was reached. Typical sensorgrams, and accompanying fits of the equilibrium response as a function of concentration, are presented in Fig. 6. The measured binding constants are presented in Table 1.
Figure 6.

SPR-based binding studies of mouse TGF-β2 and variants. A, SPR sensorgrams for injection of a 2-fold dilution series of BGZP-C (2 μm to 3.9 nm) or BGO (2 μm to 3.9 nm) over immobilized WT TGF-β2 (left and right panels, respectively). Injections were performed in duplicate over the time period shown by the solid gray bar. B, saturation plots in which the equilibrium response from the sensorgrams in A were fit as a function of the injected concentration to a standard binding isotherm to derive the KD and the Rmax. Fitted values and the estimated errors are provided in Table 1. C and E, sensorgrams as in A, but for BGZP-C (2 μm to 3.9 nm) or BGO (2 μm to 3.9 nm) injected over immobilized TGF-β2 I33A or TGF-β2 T87A, respectively. D and F, saturation plots as in B, but for TGF-β2 I33A or TGF-β2 T87A, respectively.
The substitution of the four residues on the inside of the fingers with alanine, Ile-33, Ile-92, Lys-97, and Glu-99, each led to a 5-fold or greater decrease in the binding affinity for BGZP-C, with substitution of Ile-33, whose methyl group was strongly perturbed, leading to the largest alteration, 41-fold. The substitution of the five residues on the outside of the fingers with alanine, Glu-84, Thr-87, Leu-89, Tyr-91, and Ile-98, led in contrast to at most a 2-fold weakening of the binding affinity for BGZP-C. To ascertain whether the substitutions that led to a large disruption of BGZP-C–binding affinity did so indirectly, the binding affinity of all nine alanine variants for BGO, which has been shown previously to bind at a site distinct from BGZP-C (17), were also measured. The BGO–binding affinities of all the TGF-β2 alanine variants were within 1.5-fold that of WT TGF-β2, except TGF-β2 Y91A, which was within 2.6-fold. These results show that the large decrease in the binding affinity for BGZP-C by the four variants on the inside of the fingers cannot be explained by indirect effects, such as the presence of non-native protein or an overall perturbation of the structure, but instead it must be due to loss of specific contacts that contribute favorably to binding.
Binding of BGZP-C to mini-monomeric TGF-β
To further confirm and narrow down the possible BGZP-C–binding site, we took advantage of an engineered TGF-β monomer previously reported (36), designated mmTGF-β2-7M, that included the entire finger region but had a flexible loop in place of the heel helix (Fig. 5C). This form of TGF-β, which was based on the backbone of TGF-β2, also included several substitutions from TGF-β1, including I92V, to confer high-affinity TβRII binding. To investigate whether mmTGF-β2-7M might bind BGZP-C, we captured a biotinylated avi-tagged form of the protein on a high-density SPR streptavidin surface, measured its affinity to BGZP-C by equilibrium analysis, and found that the KD values were weakened by roughly 10-fold relative to WT TGF-β2 dimer at pH 7.4 (KD 2.1 μm versus 0.20 μm, Table 1). This indicated that mmTGF-β2-7M indeed possessed most, but not all, of the residues that form the BGZP-C–binding site.
To further investigate the binding, we prepared deuterated ILV-methyl–protonated mmTGF-β2-7M and extended the assignments from the backbone to the side-chain methyl groups under conditions where the protein was assigned (10 mm sodium phosphate, 10 mm CHAPS, pH 4.7). To transfer the methyl assignments to the conditions for a titration experiment, pH 11 in the absence of CHAPS, we collected spectra at a constant concentration of CHAPS (10 mm) as the pH was raised to 11, and at pH 11, with a descending concentration of CHAPS. These titrations led to small shift perturbations for most residues and were in the fast exchange regime, thus enabling assignment of all 18 expected ILV methyl resonances at pH 11 in the absence of CHAPS (Fig. S6A).
The addition of BGZP-C to ILV-mmTGF-β2-7M at pH 11.0 led to sizable shifts and slow exchange binding for several residues, including Ile-33, Ile-88, Leu-28, and Leu-86 (Fig. S6A). Val-92 and Leu-101 also shifted based on the disappearance of their unbound signals when more than 1.0 eq of BGZP-C had been added (Fig. S6, A and B). The extent of these shifts was, however, unclear, as it was impossible to unambiguously identify the corresponding bound signals due to the relatively close proximity of the unbound and bound signals to one another. The signals that were perturbed in mmTGF-β2-7M by BGZP-C were all the same (or homologous) to those in TGF-β2, except Val-61, which was located in the heel helix and was absent in mmTGF-β2-7M. Together, the SPR and NMR results show that mmTGF-β2-7M binds BGZP-C in the same overall manner as TGF-β2, albeit with an affinity that is about 10-fold weaker. The weakening is probably due to lack of contact with Val-61 and other surrounding residues in the heel helix, which are absent in mmTGF-β2-7M.
BMP binding
Previously, it was claimed based on SPR measurements that the full-length betaglycan extracellular domain (BGO-ZP) bound amine-coupled BMP-2 with an affinity comparable with that of amine-coupled TGF-β1 (KD 10 and 5 μm, respectively) (27). Because the affinity for binding of BGO-ZP to TGF-β1 differed markedly from what our group reported for binding of BGO-ZP to amine-coupled TGF-β1, -β2, and -β3 (KD 0.095, 0.036, and 0.143 nm) or streptavidin-captured biotinylated TGF-β2 (KD 4.2 nm) (17, 29), and because the studies here identified three residues that contributed to binding and were mostly unique to the three TGF-βs and Inh α (Fig. 5D), we revisited whether betaglycan can bind BMP-2.
Increasing concentrations of BGO-ZP were injected over TGF-β2 and BMP-2 immobilized by amine coupling at densities of 180 and 380 RU, respectively, on the surface of a carboxymethyl dextran sensor chip. There was a robust concentration-dependent response that approached the expected Rmax (550 and 1140 RU, respectively) when BGO-ZP (80 kDa, assumed 1:1 binding) was injected over the TGF-β2 surface, but not the BMP-2 surface (Fig. 7, A and C, and Table 2). To determine whether the reduced response from the BMP-2 surface was due to an over estimation of the BMP-2 surface density, noggin, a BMP antagonist shown previously to bind BMP-2 with sub-nanomolar affinity (45), was injected. This resulted in a robust concentration-dependent response that approached the expected Rmax (530 RU, calculated based on binding of 36-kDa noggin with 1:1 stoichiometry) (Fig. 7B). Although it proved impossible to reliably fit the noggin/BMP-2 response due to near-zero disassociation rates, it was possible to fit the BGO-ZP/TGF-β2 responses using a simple 1:1 kinetic model. This yielded a KD of 2.2 nm, which was comparable to that measured for BGO-ZP binding to biotinylated TGF-β2 (4.2 nm) (29) and more than an order of magnitude less than that measured for amine-coupled TGF-β2 (36 nm) (17). The response for BGO-ZP binding to BMP-2 was too weak to enable reliable derivation of a binding constant, but it was nonetheless clear that the affinity of BGO-ZP for BMP-2 was orders of magnitude weaker than that of BGO-ZP for TGF-β2 due to the minimal response relative to the expected Rmax (1140 RU).
Figure 7.

SPR-based binding studies of BMP-2. A and C, SPR sensorgrams for injection of BGO-ZP (BG) over immobilized BMP-2 (A) or TGF-β2 (C) in HBS-EP buffer adjusted to pH 7.4. Raw sensorgrams, recorded in duplicate, are shown in gray, and the fitted curves (TGF-β2 only) for the 1:1 binding model are shown in orange. Concentrations of BG injected over the BMP-2 and TGF-β2 surfaces were 12.5, 25, 50, and 100 nm and 25, 50, and 100 nm, respectively. B, SPR sensorgrams for injection of noggin over immobilized BMP-2 in HBS-EP buffer adjusted to pH 7.4. Concentrations of noggin injected were 1.95, 3.9, 7.8, 15.6, 31.2, 62.5, 125, and 250 nm.
Table 2.
Rates and binding affinity of BGO-ZP with TGF-β2
| Buffer | kon | koff | KD | Rmax |
|---|---|---|---|---|
| m−1 s−1 | s−1 | nm | RU | |
| HBS-EP | 5.8 × 104 | 1.3 × 10−4 | 2.2 | 375 |
To determine whether it might be possible to engender BMP-2 with increased affinity for BGO-ZP, two variants of BMP-2 were generated in which either two or three of the proposed specificity–determining residues for BGO-ZP were substituted with the corresponding residues from TGF-β2 (Fig. S8). Although we were able to refold and purify WT BMP-2, we were unable to refold and purify these two variants. Therefore, as an alternative, the proposed specificity-determining residues in TGF-β2, Ile-92, Lys-97, and Glu-99, were substituted with the corresponding residues in BMP-2, Asp, Val, and Lys, respectively, to determine whether this would weaken binding to BGO-ZP even more than substitution with neutral alanine residues. These variants could be readily purified and were found to have binding affinities that were further weakened relative to the corresponding alanine variants. The decreases were largest for Ile-92 and Glu-99 where the I92D and I92A variant and E99R and E99A variant differed by 7- and 6-fold, respectively, and the K97V and K97A variants differed by 2-fold (Table 1). This suggested that TGF-β2 Ile-92 and Glu-99 have a more important role in dictating specificity compared with TGF-β2 Lys-97.
Discussion
The selectivity with which betaglycan recognizes TGF-β2 and inhibin is vital in enabling temporal-spatial patterns of signaling that underlie the unique functions of these GFs in vivo- for TGF-β2, morphogenetic transformation of endothelial progenitors in the heart (11, 46), and for inhibins, antagonism of activins in the anterior pituitary (21, 47). Through a previous study of InhA, it was reported that betaglycan's ZP-C domain, which is solely responsible for recognition of inhibins (23) and partly responsible for recognition of TGF-βs (17, 28), likely binds to a patch of residues that reside on the convex outer surface of the extended finger region of the Inh α subunit (30).
The binding site for BGZP-C on TGF-β2 identified in this study, while falling in the same general area of the molecule, differs in that it lies on the concave inner surface of the fingers (Fig. 5). The data supporting the identification of this site include BGZP-C-induced chemical shift perturbations (CSPs) of Ile, Leu, and Val methyl groups and a large set of single amino acid alanine variants, both of ILV residues that did or did not undergo CSPs and of several nearby nonmethyl-bearing amino acids. The binding affinities of the variants were fully consistent with the NMR CSPs, with the largest perturbations in affinity found for residues on the concave inner surface that underwent the largest CSPs, Ile-33 and Ile-92, as well as two charged residues in close spatial proximity, Lys-97 and Glu-99.
The binding site delineated by the residue substitutions spans from the top of finger 4 to the bottom of finger 1 and includes two tryptophan residues, Trp-30 and Trp-32, conserved in all TGF-β family members. Through previous studies, it is known that substitutions in this region can impair folding (20), and indeed many of the variants in this study folded poorly (Fig. S5). Three steps were taken to ensure that perturbations of the measured binding affinities were not influenced by non-native protein or a perturbation of the structure. First, only those variants that could be purified to a single species and had the correct mass (to within 0.5 Da) were analyzed. Second, the binding studies were carried out by immobilizing the TGF-β variants on the surface of an SPR sensor chip, ensuring that even if non-native protein were present, it would not alter the binding constant derived from the SPR response. Third, binding of all variants to the BG orphan domain (BGO) was carried out in parallel with BGZP-C, thus serving as an internal control that the variants did not perturb the structure.
The binding site for BGZP-C on TGF-β2 identified in this study appears to be mostly composed of residues from a single monomer, although there is a minor contribution from residues in the heel helix of the other monomer. The residues found in the binding site, which may include other residues such as Tyr-90 whose contribution to binding could not be assessed, include a large number of hydrophobic residues, as well two charged residues Lys-97 and Glu-99. The composition of this binding site is reminiscent of the binding site between the TGF-βs and TβRII, which includes a large number of hydrophobic residues, surrounded by two conserved charged residues (Arg-25 and Arg-94 in TGF-β1 and -β3 and Lys-25 and Lys-94 in TGF-β2) that form hydrogen-bonded ion pairs with oppositely charged residues on TβRII (Glu-119 and Asp-32, respectively) (19, 20, 48, 49). This suggests that Lys-97 and Glu-99 in the TGF-βs might be important specificity determinants for BGZP-C. Consistent with this, it is observed that Lys-97 and Glu-99 are strictly conserved in TGF-β1, -β2, -β3, and inhibin α but are unconserved in most other family members (Fig. 5D). For Glu-99, most family members have a basic residue, such as arginine or lysine, but a few have an uncharged polar residue, and one, nodal, has an acidic residue. For Lys-97, most family members have a hydrophobic residue, although a few others have a neutral polar residue. Ile-92, also within the binding site, has been shown to contribute very significantly to binding but is substituted in most other family members with an acidic residue. Ile-92 might therefore also contribute significantly to specificity by creating an unfavorable interaction with noncognate GFs, instead of forming an attractive interaction with cognate GFs.
The identification of three residues within the binding site unique to the TGF-βs and the inhibin α subunit seemed at odds with the prior published report that BG binds BMP-2 with comparable affinity as TGF-β1 (27). This prompted us to re-measure the binding of the full-length BG extracellular domain to BMP-2 and TGF-β2. Consistent with expectations of our model, BG bound BMP-2 with an affinity that was orders of magnitude weaker than that of TGF-β2. The inability to refold BMP-2 variants precluded us from testing whether we could engender BMP-2 with the ability to bind BG with high affinity, but we were able to show that transferring individual residues from BMP-2 into the three specificity-determining residues identified in TGF-β2, Ile-92, Lys-97, and Glu-99, led to further decreases in affinity relative to the corresponding alanine variants. These results lend further support to the idea that these residues are important for determining specificity and further suggest that BG's effects on BMP signaling might not be due to direct binding to BMP GFs, but indirectly through other mechanisms.
The homology model of InhA showed that three residues in the TGF-β–binding site that contribute significantly to binding, Ile-92, Lys-97, and Ile-88, are structurally homologous to three residues identified in the InhA-binding site, Ser-112, Lys-119, and Val-108 (Fig. S1). Thus, there is a high likelihood that these three InhA residues reside on the concave inner surface of the fingers, rather than the convex outer surface. The residues identified in the InhA site that lie on the convex outer surface, Tyr-50, Thr-111, Phe-118, and Tyr-120, are structurally homologous to TGF-β2 residues Glu-35, Tyr-91, Pro-96, and Ile-98, respectively. Although not all four of the homologous residues were examined in TGF-β2, it was found that the Ile-98 δ1 did not shift upon binding BGZP-C (Fig. 5) and that substitution of either Tyr-91 or Ile-98 with alanine did not lead to a significant perturbation in the BGZP-C–binding affinity (Table 1). Thus, among the residues in TGF-β2 that are homologous to those in the InhA α-subunit–binding site, they either corresponded to residues in the TGF-β2-binding site on the inside of the fingers or were on the outside of the fingers but did not contribute to binding.
Through a previous deletion study, it has been shown that betaglycan uses a common region, spanning residues 607–620, to bind and affect the signaling activity of both the TGF-βs and inhibins (23). Thus, the interfaces on the TGF-βs and the Inh α subunit for BGZP-C are also likely the same. This raises the possibility that the four residues on the outer surface of the Inh α subunit previously suggested to mediate binding might have been mis-identified. To this end, it is worth noting that only about one-third of the more than 40 variants in the InhA study were studied as single amino acid variants. Thus, among the four residues noted above, only Tyr-50 and Tyr-120 were studied as single amino acid variants. The inferences about the involvement of the other two residues, Thr-111 and Phe-118, were derived from measurements with double and triple mutants and are therefore indirect. It is further worth noting that only two of the more than 40 InhA variants were fully purified, while the remainder were used in crude form from the conditioned medium. In addition, the two variants that were purified, K119A/Y120A and T111A/S112A/Y120A, were examined by binding to ActRIIB, which does not provide a fully rigorous check for the presence of non-native protein, as it binds to the nonsubstituted β-subunit, not the substituted α-subunit. Finally, the luciferase assays used were conducted by measuring the response as a function of increasing concentrations of the InhA variant, which has the drawback that the EC50 will be artificially increased if any non-native protein is present. The indirect manner in which some of the residues were identified, together with the less stringent procedures to minimize against the possibility that non-native protein might influence the measurements, might therefore explain why some residues on the outside of the fingers were mis-identified in the InhA study.
The binding site on the concave inner surface, in addition to providing insight as to the potential mechanism by which betaglycan selectively recognizes TGF-βs and inhibins, also provides insights into the mechanism by which betaglycan promotes TGF-β receptor complex assembly and inhibits activin receptor complex assembly. Through previously purified component binding studies, our laboratory showed that the full-length BG extracellular domain binds TGF-β homodimers asymmetrically with 1:1 stoichiometry and that the ZP-C subdomain competes to block binding of one molecule of TβRII, but not both. The binding site for BGZP-C that we identified here fully accounts for this as it shares several residues in the loops connecting fingers 1/2 and 3/4 that are used to bind TβRII, including Leu-28 and Ile-33 in the finger 1/2 loop and Ile-92 in the finger 3/4 loop (42). Through previous studies, it has been further shown that after binding TGF-βs and promoting TβRII binding, BG must be displaced to allow TβRI to bind (10, 28, 29). Through competition binding studies, it was previously shown that at the concentrations tested, the full-length BG extracellular domain completely blocked binding of TβRI by the TGF-β/TβRII complex, whereas the orphan domain mostly blocked binding (29). The incomplete blocking of the TβRI binding by the orphan domain alone was attributed to the lower affinity of the orphan domain for TGF-β compared with the full-length BG extracellular domain. However, based on extensive overlap with the TβRI-binding site revealed in this study, it is likely that the BGZP-C domain also blocks TβRI. The revised mechanism suggested by these findings thereby has the orphan domain of BG bound such that it blocks one of the TβRI-binding sites but leaves both of the TβRII-binding sites unoccupied, whereas the ZP-C domain of BG is bound such that it blocks not only TβRII but also TβRI (Fig. S9). This manner of binding may be responsible for directing a hierarchical manner of assembly, whereby BG first promotes the binding of one molecule of TβRII, followed by the paired TβRI, and in turn assembly, by an unknown series of steps, of the second TβRII/TβRI pair.
The proposed mechanism for inhibin-mediated antagonism of activin signaling states that through its ZP-C domain, BG binds to the inhibin α subunit and promotes binding of the activin type II receptors, ActRII and ActRIIB, to the inhibin β subunit by membrane localization effects (21). This has been proposed to result in the sequestration of ActRII and ActRIIB away from activins in a ternary complex incapable of recruiting a type I receptor, such as ActRIB. The details by which inhibin is prevented from recruiting a type I receptor, however, is not fully understood. The identification of the betaglycan ZP-C–binding site on the underside of the fingers provides a rational explanation for this as it overlaps directly in the site where type I receptors of the TGF-β family bind. This, however, cannot completely account for the inability of inhibin to bind and recruit a type I receptor as the BGZP-C domain would be expected to block binding of the type I receptor on one side of the dimer, but not on the other. Thus, other mechanisms, such as the one suggested by Zhu et al. (50), might also be operative.
Overall, the results reported here have led to the precise identification of the BGZP-C–binding site on TGF-β2, which has begun to shed light on the specificity of BG for a select few members of the TGF-β family and the potential mechanisms by which it influences their signaling.
Experimental procedures
Synthesis of ethyl 2-hydroxy-2-13C-methyl-3-oxobutanoate
Ethyl 2-13C-methyl-3-oxobutanoate was formed from ethyl 3-oxobutanoate (Sigma) (4.55 g, 35 mmol) and 13C-methyl iodide (Sigma) (5.0 g, 35 mmol) in the presence of K2CO3 as described previously (44). The reaction mixture was extracted with diethyl ether, dried over Na2SO4, concentrated under reduced pressure, and purified by silica gel column chromatography (15% EtOAc/hexanes). The purified product, ethyl 2-13C-methyl 3-oxobutanoate (2.01 g), along with P(OEt)3 (4.61 g, 27.74 mmol) and Cs2CO3 (0.904 g, 2.77 mmol) was added to a flame-dried 100-ml Schlenck flask. The flask was sealed, backfilled with O2 (three times), and after adding DMSO (30 ml) via syringe, the reaction was stirred at 25 °C overnight as described previously (51). The reaction was diluted with EtOAc (50 ml) and transferred to a separatory funnel. The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The resulting crude oil was purified by column chromatography on silica gel (20% ethyl acetate:hexanes) to give ethyl 2-hydroxy-2-13C-methyl-3-oxobutanoate (1.09 g, 6.81 mmol). 1H NMR (400 MHz, CDCl3) 4.27 ppm (q, J = 7.1 Hz, 2H), 4.19 ppm (d, J = 3.4 Hz, 1H), 2.29 ppm (s, 3H), 1.60 ppm (d, J1H-13C = 130 Hz, 3H), and 1.31 ppm (t, J = 7.1 Hz, 3H).
Expression and purification of TGF-β growth factor domains
The growth factor domains of human TGF-β2, mouse TGF-β2, and accompanying variants, the previously reported TGF-β2 mini-monomer, mmTGF-β2–7M (36), and N-terminally avi-tagged mmTGF-β2-7M were each expressed in Escherichia coli at 37 °C in the form of insoluble inclusion bodies (sequences of the expressed proteins are provided in Fig. S7). The cells were disrupted by sonication, and after washing, the overexpressed protein was reconstituted in 8 m urea and refolded by dilution into nondenaturing buffer at 4 °C containing 30 mm CHAPS (Gold Biotechnology, Denver, CO) at pH 9.5. The natively folded growth factor domains were isolated by high-resolution cation–exchange chromatography (Source S, GE Healthcare), followed by reverse-phase chromatography (C18 Jupiter, Phenomenex, Torrance, CA). Intact masses of the purified proteins were measured by LC electrospray ionization TOF MS (LC-ESI-TOF-MS, Bruker Micro TOF, Billerica, MA) and were found in all cases to be within 0.5 Da of their calculated masses. Other details can be found in the previously described protocol for expressing, refolding, and purifying TGF-β2 (52).
Expression and purification of mammalian-expressed rBGO-ZP, rBGO, and rBGZP-C
Rat betaglycan extracellular domain, rBGO-ZP, and its two component TGF-β–binding domains, the orphan domain, rBGO, and the C-terminal zona pellucida domain, rBGZP-C, were each expressed as secreted proteins in suspension-cultured HEK-293 expi cells and HEK-293 expi expression medium (GE Healthcare), as described previously (29). Secreted proteins were purified from the conditioned medium by performing a 2-fold dilution into PBS, followed by sequential purification of the C-terminally His-tagged proteins by nickel metal-affinity chromatography (chelating Sepharose, GE Healthcare) and size-exclusion chromatography (Superdex 200, GE Healthcare), as described previously (29).
Expression and purification of bacterially-expressed TβRII and rBGZP-C
Unlabeled TβRII ectodomain, for use in the biotinylation reactions, was produced as an insoluble protein in E. coli and refolded and purified as described previously (52). Isotopically labeled samples of rBGZP-C for NMR were produced in E. coli by inserting the coding region for the C-terminal portion of the rat betaglycan zona pellucida domain (residues 590–756 of rat betaglycan, with residue numbering beginning at the N-terminal methionine) downstream of the coding region for thioredoxin (Trx), a hexahistidine tag, and a thrombin cleavage site, LVPRGS, in the bacterial T7 expression vector pET32a (EMD Millipore, Billerica, MA). The Trx–BGZP-C fusion protein was overexpressed in BL21(DE3) cells cultured in LB medium at 37 °C containing 50 μg/ml carbenicillin. Cells were resuspended in 100 mm Tris, 10 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, pH 8.0, and lysed by sonication. Inclusion bodies were isolated by washing the insoluble fraction with lysis buffer with 500 mm NaCl, followed by lysis buffer with 1% Triton X-100. Inclusion bodies were solubilized by stirring for 12 h in 8 m urea, 25 mm Tris, 50 mm DTT, pH 8, and after removal of the remaining insoluble material by centrifugation, the supernatant was buffer exchanged into 8 m urea, 25 mm Tris, pH 4. Urea solubilized Trx–BGZP-C was then slowly diluted into folding buffer (100 mm Tris, 0.25 m guanidine hydrochloride, 0.5 m arginine, 2 mm GSH, 0.5 mm GSSG, pH 9.0) to a final protein concentration no higher than 0.1 mg/ml. After stirring 14 h at 4 °C, the folding mixture was concentrated and dialyzed into purification buffer (2 m urea, 10 mm Tris, 15 mm CHES, 1 μm pepstatin, 1 μm leupeptin, 1 mm benzamidine, pH 9.2). After centrifugation and filtration, Trx–BGZP-c was fractionated by loading it onto a Source Q anion-exchange column (GE Healthcare) equilibrated with purification buffer and eluted with a linear gradient from 0 to 0.3 m NaCl. After overnight digestion with thrombin (3 units/mg fusion protein), the cleavage mixture was loaded onto a nickel-affinity column (chelating Sepharose, GE Healthcare) equilibrated in 25 mm sodium phosphate, 200 mm NaCl, pH 8.0, which enabled separation of rBGZP-C away from His-tagged thioredoxin. Intact mass of the purified protein was measured by electrospray ionization TOF MS (Bruker Micro TOF, Billerica, MA) and was found to be within 1.0 Da of its calculated mass.
Expression of deuterated, methyl-protonated proteins
Deuterated Ile, Leu, and Val methyl-protonated human TGF-β2 and mmTGF-β2–7M (2H-MP-TGF-β2 and 2H-MP-mmTGF-β2-7M) were produced by inoculating colonies from a freshly transformed plate of BL21(DE3) cells into water-based M9 medium followed by incubation at 37 °C. Once the cell density rose to 0.25, the cells were pelleted in a sterile centrifuge bottle and inoculated into the same volume of 2H2O-based M9 medium containing 99.9% 2H2O and 3 g/liter [2H]glucose (Cambridge Isotope Laboratories, Andover, MA) and incubated at 37 °C. Once the cell density reached 0.55, 75 mg of 4-13C-3,3′-2H-α-ketobutyric acid (Cambridge Isotope Laboratories, Andover, MA) and 200 mg of saponified ethyl 2-hydroxy-2-13C-methyl-3-oxobutanoate were added (saponification was achieved by incubating 200 mg of ethyl 2-hydroxy-2-13C-methyl-3-oxobutanoate for 1 h at 25 °C in 10 ml of 2H2O containing 0.1 m sodium hydroxide). Protein expression was induced 1 h later by the addition of 200 mg IPTG.
Backbone assignment of TGF-β2
Samples of 13C,15N human TGF-β2 for backbone assignment were prepared in 5% 2H2O at pH 2.7 at a protein concentration of 0.25 mm - 0.35 mm in a 5 mm Shigemi tube (Shigemi, Allison Park, PA). All NMR data were acquired at 37 °C using a Bruker AVI 700 MHz spectrometer (Bruker, Billerica, MA) equipped with 5 mm 1H{13C,15N} TCI cryogenically cooled probe. Backbone resonance assignments of TGF-β2 were obtained by collecting and analyzing sensitivity-enhanced triple-resonance data sets, including HNCACB (53), CBCA(CO)NH (54), HNCA (55), HN(CO)CA (55), HNCO (56), and HN(CA)CO (57). All spectra were processed with NMRPipe (58) and analyzed using NMRViewJ (59).
Methyl spectra
Samples of 2H–ILV–TGF-β2 or 2H-ILV-mmTGF-β2-7M for the binding titrations were prepared in 25 mm 2H-glycine at pH 11.0 in either straight 3 mm tubes (Wilmad, Vineland, NJ) or a 5 mm susceptibility matched microcells (Shigemi, Allison Park, PA). Separate samples, each with the labeled protein being 15 - 25 μm, were used for each point in the titration. Spectra were recorded at 37 °C using a 1H-13C HSQC pulse sequence with a WATERGATE solvent suppression scheme (60) as implemented on a Bruker AVII 800 MHz spectrometer (Bruker, Billerica, MA) equipped with a 5 mm 1H{13C,15N} TCI cryogenically cooled probe. Spectra were acquired with 128 complex points and between 64 and 128 scans per time increment. Spectra were processed with NMRPipe (58) and analyzed using NMRFAM-SPARKY (61) and CcpNmr (62). Spectra of 2H-MP TGF-β2 for the pH titrations were acquired in the same manner, except samples were prepared in 5% 2H2O with the pH being adjusted by the addition of small aliquots of 0.1 m NaOH or 0.1 m HCl prior to NMR data collection.
GF biotinylation for SPR
Minimally biotinylated mouse TGF-β2, and single amino variants, were prepared by overnight dialysis of a 0.5 ml solution containing 125 μg growth factor homodimer (5 nmol) and 200 μg TβRII (15 nmol) into 25 mm MES, pH 6.0. DMSO was added to a final concentration of 40% v/v, followed by 0.0115 mg of ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC, Pierce) (60 nmol), 0.026 mg of sulfo-N-hydroxysulfosuccinimide (120 nmol, Pierce), and 0.20 mg (+)-biotinyl-3,6,9-trioxaundecanediamine (EZ-Link Amine-PEG3-Biotin) (480 nmol, Pierce). The reaction was allowed to proceed for 1 h at 25 °C and then quenched by the addition of 0.4 ml 100 mm acetic acid. The growth factor was separated away from TβRII and the reagents by loading it onto a Source S high resolution cation exchange column equilibrated in 20 mm sodium acetate, 30% isopropanol pH 5.2 and eluting it with a linear gradient from 0 - 70% B over 10 column volumes. LC-ESI-TOF MS showed that roughly 10% of the growth factor was singly modified, while the remaining 90% was unmodified, indicating that at most the growth factor was singly biotinylated.
Biotinylated avi-tagged mmTGF-β2–7m was prepared by overnight dialysis of 0.5 mg of avi-tagged mmTGF-β2–7m and 1.0 mg TβRII into 25 mm Bicine, pH 8.3. Biotinylation was achieved by incubation at 37 °C for 4 h in the presence of 40 μm biotin, 10 mm ATP, 10 mm Mg(OAc)2, and 20 μg of BirA biotin ligase (63). Biotinylated avi-tagged mmTGF-β2–7m was isolated from the reaction mixture as above for chemically biotinylated TGF-β2 homodimers.
SPR measurements
SPR binding studies were performed with a BIAcore 3000 instrument (GE Healthcare, Piscataway, NJ) and were analyzed using the software package Scrubber2 (Biologic Software, Canberra, Australia). Streptavidin-coated sensor chips for capture of biotinylated ligands were made by activating the surface of a CM-5 sensor chip (GE Healthcare) with EDC and N-hydroxysulfosuccinimide (NHS) (Pierce) followed by injection of streptavidin (Pierce) diluted into sodium acetate at pH 4.5 until the surface density reached 6000 - 8000 RUs. Biotinylated ligands were captured onto the streptavidin surface to a surface density between 50–300 RU. Equilibrium binding assays were performed by injecting the analytes, BGZP-C or BGO, in 10 mm HEPES, 150 mm NaCl, 0.1% surfactant P20 (Pierce) at pH 7.4 (HBS-EP buffer) at a rate of 10 μl min−1 for 720 s, followed by a dissociation period of 600 s. Regeneration of the surface was achieved by a 10 s injection at 100 μl min−1 of 4 m guanidine hydrochloride in 4 mm HCl solution. Baseline correction was performed by double referencing. Equilibrium analyses were performed by fitting the equilibrium to the equation RU = RUmax·Conc/(KD + Conc). Binding studies of BGO-ZP and noggin were performed as described above, but with human BMP-2 and human TGF-β2 directly attached to the surface of a CM5 sensor chip by amine coupling with NHS and EDC. Human BMP-2 and noggin for these measurements were purchased from PeproTech (Rocky Hill, NJ).
Inhibin A homology model
The program HHPred (64) was used to find the best alignment of the human Inh α sequence to other TGF-β family GFs whose structure are known. The resulting alignment, which identified human BMP-6 (PDB 2R53) as the best match (Z-score 9.2e−33; next best match human GDF-5 (PDB 3EVS), Z-score 3.4e−32), was in turn used to generate a homology model of the Inh α monomer using the program SwissModel (65). A model of the Inh Α heterodimer was generated by superimposing the model for the Inh α monomer onto one of the inhibin β-subunits in the structure of the closed form of activin A from its structure bound to follistatin (66).
Author contributions
M. A. H., P. M., and A. P. H. conceptualization; M. A. H., R. D. H., T. C. K., G. H., M. V., M. M., and A. P. H. resources; M. A. H., P. M., C. Z., R. B. K., R. D. H., and A. P. H. data curation; M. A. H., P. M., C. Z., R. B. K., R. D. H., T. C. K., and A. P. H. formal analysis; M. A. H., R. B. K., C. S. H., R. D. H., U. I., K. E. C., and A. P. H. supervision; M. A. H. and A. P. H. funding acquisition; M. A. H., P. M., C. Z., R. B. K., and A. P. H. validation; M. A. H., P. M., C. Z., R. B. K., C. S. H., R. D. H., T. C. K., U. I., K. E. C., G. H., M. V., M. M., and A. P. H. investigation; M. A. H. and A. P. H. visualization; M. A. H., P. M., T. C. K., M. M., and A. P. H. methodology; M. A. H. and A. P. H. writing-original draft; M. A. H. and A. P. H. project administration; M. A. H., P. M., C. Z., R. B. K., R. D. H., T. C. K., G. H., and A. P. H. writing-review and editing.
Supplementary Material
Acknowledgments
David Brinkman is thanked for the initial efforts to generate and characterize several TGF-β2 variants; Erich Hellemann for measuring the mass of the TGF-β2 variants; Mike Delk for maintaining the NMR instruments, and Profs. Craig Harrison and Daniel Bernard for providing valuable comments on the manuscript. Molecular graphics and analyses were performed with UCSF Chimera. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health NIGMS Grant P41-GM103311).
This work was supported by National Institutes of Health Grants GM58670 and CA172886 and the University of Pittsburgh Vascular Medicine Institute. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S9.
NMR signal assignments for TGF-β2 have been deposited to BioMagResBank under accession number 27574.
- BG
- betaglycan
- TGF-β
- transforming growth factor-β
- InhA
- inhibin A
- TGF-β
- transforming growth factor-β
- GF
- growth factor
- PDB
- Protein Data Bank
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- SPR
- surface plasmon resonance
- BMP
- bone morphogenetic protein
- EDC
- ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- NHS
- N-hydroxysulfosuccinimide
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- HSQC
- heteronuclear single quantum coherence
- CSP
- chemical shift perturbation
- Trx
- thioredoxin
- RU
- response unit
- MP
- methyl-protonated
- ILV
- isoleucine, leucine, and valine.
References
- 1. Katagiri T., and Watabe T. (2016) Bone morphogenetic proteins. Cold Spring Harb. Perspect. Biol. 8, a21899 10.1101/cshperspect.a021899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morikawa M., Derynck R., and Miyazono K. (2016) TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 8, a021873 10.1101/cshperspect.a021873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Namwanje M., and Brown C. W. (2016) Activins and inhibins: roles in development, physiology, and disease. Cold Spring Harb. Perspect. Biol. 8, a021881 10.1101/cshperspect.a021881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boyd F. T., and Massagué J. (1989) Transforming growth factor-β inhibition of epithelial cell proliferation linked to the expression of a 53-kDa membrane receptor. J. Biol. Chem. 264, 2272–2278 [PubMed] [Google Scholar]
- 5. Laiho M., Weis F. M., Boyd F. T., Ignotz R. A., and Massagué J. (1991) Responsiveness to transforming growth factor-β (TGF-β) restored by genetic complementation between cells defective in TGF-β receptors I and II. J. Biol. Chem. 266, 9108–9112 [PubMed] [Google Scholar]
- 6. Laiho M., Weis M. B., and Massagué J. (1990) Concomitant loss of transforming growth factor (TGF)- β receptor types I and II in TGF-β-resistant cell mutants implicates both receptor types in signal transduction. J. Biol. Chem. 265, 18518–18524 [PubMed] [Google Scholar]
- 7. Heldin C. H., and Moustakas A. (2016) Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 8, a022053 10.1101/cshperspect.a022053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hata A., and Chen Y. G. (2016) TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061 10.1101/cshperspect.a022061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y. E. (2017) Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 9, a022129 10.1101/cshperspect.a022129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. López-Casillas F., Wrana J. L., and Massagué J. (1993) Betaglycan presents ligand to the TGF β signaling receptor. Cell 73, 1435–1444 10.1016/0092-8674(93)90368-Z [DOI] [PubMed] [Google Scholar]
- 11. Stenvers K. L., Tursky M. L., Harder K. W., Kountouri N., Amatayakul-Chantler S., Grail D., Small C., Weinberg R. A., Sizeland A. M., and Zhu H. J. (2003) Heart and liver defects and reduced transforming growth factor β2 sensitivity in transforming growth factor β type III receptor-deficient embryos. Mol. Cell. Biol. 23, 4371–4385 10.1128/MCB.23.12.4371-4385.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saito T., Bokhove M., Croci R., Zamora-Caballero S., Han L., Letarte M., de Sanctis D., and Jovine L. (2017) Structural basis of the human endoglin-BMP9 interaction: insights into BMP signaling and HHT1. Cell Rep. 19, 1917–1928 10.1016/j.celrep.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheifetz S., Bellón T., Calés C., Vera S., Bernabeu C., Massagué J., and Letarte M. (1992) Endoglin is a component of the transforming growth factor-β receptor system in human endothelial cells. J. Biol. Chem. 267, 19027–19030 [PubMed] [Google Scholar]
- 14. Lin S. J., Hu Y., Zhu J., Woodruff T. K., and Jardetzky T. S. (2011) Structure of betaglycan zona pellucida (ZP)-C domain provides insights into ZP-mediated protein polymerization and TGF-β binding. Proc. Natl. Acad. Sci. U.S.A. 108, 5232–5236 10.1073/pnas.1010689108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diestel U., Resch M., Meinhardt K., Weiler S., Hellmann T. V., Mueller T. D., Nickel J., Eichler J., and Muller Y. A. (2013) Identification of a novel TGF-β-binding site in the zona pellucida C-terminal (ZP-C) domain of TGF-β-receptor-3 (TGFR-3). PLoS ONE 8, e67214 10.1371/journal.pone.0067214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. López-Casillas F., Payne H. M., Andres J. L., and Massagué J. (1994) Betaglycan can act as a dual modulator of TGF-β access to signaling receptors: mapping of ligand binding and GAG attachment sites. J. Cell Biol. 124, 557–568 10.1083/jcb.124.4.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mendoza V., Vilchis-Landeros M. M., Mendoza-Hernández G., Huang T., Villarreal M. M., Hinck A. P., López-Casillas F., and Montiel J. L. (2009) Betaglycan has two independent domains required for high affinity TGF-β binding: proteolytic cleavage separates the domains and inactivates the neutralizing activity of the soluble receptor. Biochemistry 48, 11755–11765 10.1021/bi901528w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheifetz S., Hernandez H., Laiho M., ten Dijke P., Iwata K. K., and Massagué J. (1990) Distinct transforming growth factor-β (TGF-β) receptor subsets as determinants of cellular responsiveness to three TGF-β isoforms. J. Biol. Chem. 265, 20533–20538 [PubMed] [Google Scholar]
- 19. De Crescenzo G., Hinck C. S., Shu Z., Zúñiga J., Yang J., Tang Y., Baardsnes J., Mendoza V., Sun L., López-Casillas F., O'Connor-McCourt M., and Hinck A. P. (2006) Three key residues underlie the differential affinity of the TGFβ isoforms for the TGFβ type II receptor. J. Mol. Biol. 355, 47–62 10.1016/j.jmb.2005.10.022 [DOI] [PubMed] [Google Scholar]
- 20. Baardsnes J., Hinck C. S., Hinck A. P., and O'Connor-McCourt M. D. (2009) TβR-II discriminates the high- and low-affinity TGF-β isoforms via two hydrogen-bonded ion pairs. Biochemistry 48, 2146–2155 10.1021/bi8019004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lewis K. A., Gray P. C., Blount A. L., MacConell L. A., Wiater E., Bilezikjian L. M., and Vale W. (2000) Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 404, 411–414 10.1038/35006129 [DOI] [PubMed] [Google Scholar]
- 22. Wiater E., and Vale W. (2003) Inhibin is an antagonist of bone morphogenetic protein signaling. J. Biol. Chem. 278, 7934–7941 10.1074/jbc.M209710200 [DOI] [PubMed] [Google Scholar]
- 23. Wiater E., Harrison C. A., Lewis K. A., Gray P. C., and Vale W. W. (2006) Identification of distinct inhibin and transforming growth factor β-binding sites on betaglycan: functional separation of betaglycan co-receptor actions. J. Biol. Chem. 281, 17011–17022 10.1074/jbc.M601459200 [DOI] [PubMed] [Google Scholar]
- 24. Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., and Doetschman T. (1997) TGFβ knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Woodruff T. K., Krummen L., McCray G., and Mather J. P. (1993) In situ ligand binding of recombinant human [125I]activin-A and recombinant human [125I]inhibin-A to the adult rat ovary. Endocrinology 133, 2998–3006 10.1210/endo.133.6.8243328 [DOI] [PubMed] [Google Scholar]
- 26. MacConell L. A., Leal A. M., and Vale W. W. (2002) The distribution of betaglycan protein and mRNA in rat brain, pituitary, and gonads: implications for a role for betaglycan in inhibin-mediated reproductive functions. Endocrinology 143, 1066–1075 10.1210/endo.143.3.8707 [DOI] [PubMed] [Google Scholar]
- 27. Kirkbride K. C., Townsend T. A., Bruinsma M. W., Barnett J. V., and Blobe G. C. (2008) Bone morphogenetic proteins signal through the transforming growth factor-β type III receptor. J. Biol. Chem. 283, 7628–7637 10.1074/jbc.M704883200 [DOI] [PubMed] [Google Scholar]
- 28. Esparza-Lopez J., Montiel J. L., Vilchis-Landeros M. M., Okadome T., Miyazono K., and López-Casillas F. (2001) Ligand binding and functional properties of betaglycan, a co-receptor of the transforming growth factor-β superfamily. Specialized binding regions for transforming growth factor-β and inhibin A. J. Biol. Chem. 276, 14588–14596 10.1074/jbc.M008866200 [DOI] [PubMed] [Google Scholar]
- 29. Villarreal M. M., Kim S. K., Barron L., Kodali R., Baardsnes J., Hinck C. S., Krzysiak T. C., Henen M. A., Pakhomova O., Mendoza V., O'Connor-McCourt M. D., Lafer E. M., López-Casillas F., and Hinck A. P. (2016) Binding properties of the transforming growth factor-β coreceptor beta-glycan: proposed mechanism for potentiation of receptor complex assembly and signaling. Biochemistry 55, 6880–6896 10.1021/acs.biochem.6b00566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Makanji Y., Walton K. L., Wilce M. C., Chan K. L., Robertson D. M., and Harrison C. A. (2008) Suppression of inhibin A biological activity by alterations in the binding site for betaglycan. J. Biol. Chem. 283, 16743–16751 10.1074/jbc.M801045200 [DOI] [PubMed] [Google Scholar]
- 31. Kerfah R., Plevin M. J., Sounier R., Gans P., and Boisbouvier J. (2015) Methyl-specific isotopic labeling: a molecular tool box for solution NMR studies of large proteins. Curr. Opin. Struct. Biol. 32, 113–122 10.1016/j.sbi.2015.03.009 [DOI] [PubMed] [Google Scholar]
- 32. Tugarinov V., and Kay L. E. (2003) Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 125, 13868–13878 10.1021/ja030345s [DOI] [PubMed] [Google Scholar]
- 33. Amero C., Asunción Durá M., Noirclerc-Savoye M., Perollier A., Gallet B., Plevin M. J., Vernet T., Franzetti B., and Boisbouvier J. (2011) A systematic mutagenesis-driven strategy for site-resolved NMR studies of supramolecular assemblies. J. Biomol. NMR 50, 229–236 10.1007/s10858-011-9513-5 [DOI] [PubMed] [Google Scholar]
- 34. Kerfah R., Plevin M. J., Pessey O., Hamelin O., Gans P., and Boisbouvier J. (2015) Scrambling free combinatorial labeling of alanine-β, isoleucine-δ1, leucine-proS and valine-proS methyl groups for the detection of long range NOEs. J. Biomol. NMR 61, 73–82 10.1007/s10858-014-9887-2 [DOI] [PubMed] [Google Scholar]
- 35. Pederson K., Chalmers G. R., Gao Q., Elnatan D., Ramelot T. A., Ma L. C., Montelione G. T., Kennedy M. A., Agard D. A., and Prestegard J. H. (2017) NMR characterization of HtpG, the E. coli Hsp90, using sparse labeling with 13C-methyl alanine. J. Biomol. NMR 68, 225–236 10.1007/s10858-017-0123-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim S. K., Barron L., Hinck C. S., Petrunak E. M., Cano K. E., Thangirala A., Iskra B., Brothers M., Vonberg M., Leal B., Richter B., Kodali R., Taylor A. B., Du S., Barnes C. O., et al. (2017) An engineered transforming growth factor β (TGF-β) monomer that functions as a dominant negative to block TGF-β signaling. J. Biol. Chem. 292, 7173–7188 10.1074/jbc.M116.768754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pellaud J., Schote U., Arvinte T., and Seelig J. (1999) Conformation and self-association of human recombinant transforming growth factor-β3 in aqueous solutions. J. Biol. Chem. 274, 7699–7704 10.1074/jbc.274.12.7699 [DOI] [PubMed] [Google Scholar]
- 38. Bocharov E. V., Blommers M. J., Kuhla J., Arvinte T., Bürgi R., and Arseniev A. S. (2000) Sequence-specific 1H and 15N assignment and secondary structure of transforming growth factor β3. J. Biomol. NMR 16, 179–180 10.1023/A:1008315600134 [DOI] [PubMed] [Google Scholar]
- 39. Hinck A. P., Archer S. J., Qian S. W., Roberts A. B., Sporn M. B., Weatherbee J. A., Tsang M. L., Lucas R., Zhang B. L., Wenker J., and Torchia D. A. (1996) Transforming growth factor β 1: three-dimensional structure in solution and comparison with the X-ray structure of transforming growth factor β2. Biochemistry 35, 8517–8534 10.1021/bi9604946 [DOI] [PubMed] [Google Scholar]
- 40. Daopin S., Piez K. A., Ogawa Y., and Davies D. R. (1992) Crystal structure of transforming growth factor-β2: an unusual fold for the superfamily. Science 257, 369–373 10.1126/science.1631557 [DOI] [PubMed] [Google Scholar]
- 41. Schlunegger M. P., and Grütter M. G. (1992) An unusual feature revealed by the crystal structure at 2.2 A resolution of human transforming growth factor-β2. Nature 358, 430–434 10.1038/358430a0 [DOI] [PubMed] [Google Scholar]
- 42. Hart P. J., Deep S., Taylor A. B., Shu Z., Hinck C. S., and Hinck A. P. (2002) Crystal structure of the human TβR2 ectodomain–TGF-β3 complex. Nat. Struct. Biol. 9, 203–208 [DOI] [PubMed] [Google Scholar]
- 43. Goto N. K., Gardner K. H., Mueller G. A., Willis R. C., and Kay L. E. (1999) A robust and cost-effective method for the production of Val, Leu, Ile (δ1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR 13, 369–374 10.1023/A:1008393201236 [DOI] [PubMed] [Google Scholar]
- 44. Gans P., Hamelin O., Sounier R., Ayala I., Durá M. A., Amero C. D., Noirclerc-Savoye M., Franzetti B., Plevin M. J., and Boisbouvier J. (2010) Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins. Angew. Chem. Int. Ed. Engl. 49, 1958–1962 10.1002/anie.200905660 [DOI] [PubMed] [Google Scholar]
- 45. Groppe J., Greenwald J., Wiater E., Rodriguez-Leon J., Economides A. N., Kwiatkowski W., Affolter M., Vale W. W., Izpisua Belmonte J. C., and Choe S. (2002) Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 420, 636–642 10.1038/nature01245 [DOI] [PubMed] [Google Scholar]
- 46. Brown C. B., Boyer A. S., Runyan R. B., and Barnett J. V. (1999) Requirement of type III TGF-β receptor for endocardial cell transformation in the heart. Science 283, 2080–2082 10.1126/science.283.5410.2080 [DOI] [PubMed] [Google Scholar]
- 47. Bernard D. J., Chapman S. C., and Woodruff T. K. (2002) Inhibin binding protein (InhBP/p120), betaglycan, and the continuing search for the inhibin receptor. Mol. Endocrinol. 16, 207–212 10.1210/mend.16.2.0783 [DOI] [PubMed] [Google Scholar]
- 48. Groppe J., Hinck C. S., Samavarchi-Tehrani P., Zubieta C., Schuermann J. P., Taylor A. B., Schwarz P. M., Wrana J. L., and Hinck A. P. (2008) Cooperative assembly of TGF-β superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 29, 157–168 10.1016/j.molcel.2007.11.039 [DOI] [PubMed] [Google Scholar]
- 49. Radaev S., Zou Z., Huang T., Lafer E. M., Hinck A. P., and Sun P. D. (2010) Ternary complex of transforming growth factor-β1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 285, 14806–14814 10.1074/jbc.M109.079921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhu J., Lin S. J., Zou C., Makanji Y., Jardetzky T. S., and Woodruff T. K. (2012) Inhibin α-subunit N terminus interacts with activin type IB receptor to disrupt activin signaling. J. Biol. Chem. 287, 8060–8070 10.1074/jbc.M111.293381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liang Y. F., and Jiao N. (2014) Highly efficient C-H hydroxylation of carbonyl compounds with oxygen under mild conditions. Angew. Chem. Int. Ed. Engl. 53, 548–552 10.1002/anie.201308698 [DOI] [PubMed] [Google Scholar]
- 52. Huang T., and Hinck A. P. (2016) Production, isolation, and structural analysis of ligands and receptors of the TGF-β superfamily. Methods Mol. Biol. 1344, 63–92 10.1007/978-1-4939-2966-5_4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wittekind M., and Mueller L. (1993) Hncacb, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α-carbon and β-carbon resonances in proteins. J. Magn. Reson. Series B. 101, 201–205 10.1006/jmrb.1993.1033 [DOI] [Google Scholar]
- 54. Grzesiek S., and Bax A. (1993) Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR 3, 185–204 [DOI] [PubMed] [Google Scholar]
- 55. Grzesiek S., and Bax A. (1992) Improved 3D triple-resonance NMR techniques applied to a 31-kDa protein. J. Magn. Reson. 96, 432–440 [Google Scholar]
- 56. Kay L. E., Ikura M., Tschudin R., and Bax A. (1990) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J. Magn. Reson. 89, 496–514 [DOI] [PubMed] [Google Scholar]
- 57. Clubb R. T., Thanabal V., and Wagner G. (1992) A constant-time 3-dimensional triple-resonance pulse scheme to correlate intraresidue H-1(N), N-15, and C-13(′) chemical-shifts in N-15-C-13-labeled proteins. J. Magn. Reson. 97, 213–217 [Google Scholar]
- 58. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 59. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 60. Piotto M., Saudek V., and Sklenár V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665 10.1007/BF02192855 [DOI] [PubMed] [Google Scholar]
- 61. Lee W., Tonelli M., and Markley J. L. (2015) NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–1327 10.1093/bioinformatics/btu830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 63. Cull M. G., and Schatz P. J. (2000) Biotinylation of proteins in vivo and in vitro using small peptide tags. Methods Enzymol. 326, 430–440 10.1016/S0076-6879(00)26068-0 [DOI] [PubMed] [Google Scholar]
- 64. Alva V., Nam S. Z., Söding J., and Lupas A. N. (2016) The MPI Bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 44, W410–W415 10.1093/nar/gkw348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thompson T. B., Lerch T. F., Cook R. W., Woodruff T. K., and Jardetzky T. S. (2005) The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 9, 535–543 10.1016/j.devcel.2005.09.008 [DOI] [PubMed] [Google Scholar]
- 67. Eghbalnia H. R., Wang L., Bahrami A., Assadi A., and Markley J. L. (2005) Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements. J. Biomol. NMR 32, 71–81 10.1007/s10858-005-5705-1 [DOI] [PubMed] [Google Scholar]
- 68. Daopin S., Li M., and Davies D. R. (1993) Crystal structure of TGF-β2 refined at 1.8 A resolution. Proteins 17, 176–192 10.1002/prot.340170207 [DOI] [PubMed] [Google Scholar]
- 69. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.