

Abstract
Breast cancer screening and new precision therapies have led to improved patient outcomes. Yet, a positive prognosis is less certain when primary tumors metastasize. Metastasis requires a coordinated program of cellular changes that promote increased survival, migration, and energy consumption. These pathways converge on mitochondrial function, where distinct signaling networks of kinases, phosphatases, and metabolic enzymes regulate these processes. The protein kinase A–anchoring protein dAKAP1 compartmentalizes protein kinase A (PKA) and other signaling enzymes at the outer mitochondrial membrane and thereby controls mitochondrial function and dynamics. Modulation of these processes occurs in part through regulation of dynamin-related protein 1 (Drp1). Here, we report an inverse relationship between the expression of dAKAP1 and mesenchymal markers in breast cancer. Molecular, cellular, and in silico analyses of breast cancer cell lines confirmed that dAKAP1 depletion is associated with impaired mitochondrial function and dynamics, as well as with increased glycolytic potential and invasiveness. Furthermore, disruption of dAKAP1–PKA complexes affected cell motility and mitochondrial movement toward the leading edge in invasive breast cancer cells. We therefore propose that depletion of dAKAP1–PKA “signaling islands” from the outer mitochondrial membrane augments progression toward metastatic breast cancer.
Keywords: A-kinase–anchoring protein (AKAP), protein kinase A (PKA), cell signaling, signal transduction, breast cancer, cancer biology, cell migration, mitochondria, signaling islands
Introduction
Approximately one in eight women are diagnosed with breast cancer. Recent advances in detection and diagnosis, when combined with precision therapies that tailor drug treatment to the genetic profile of patients increase the likelihood of good prognoses (1). Nonetheless, 5-year survival rates plummet to 22% for patients with metastatic (stage IV) disease (2). Elucidating the molecular mechanisms that underlie the transition to metastatic breast cancers remains challenging, yet it is a necessary prelude to the development of new therapies. The onset of metastasis is often marked by a cellular reprogramming paradigm that alters gene expression patterns, migration competency, and metabolism (3). This paradigm shares many features with the developmental process known as the epithelial-to-mesenchymal transition (EMT)6 (4–6). Furthermore, highly proliferative cancer cells often exhibit enhanced glycolytic capacity (3, 7). Recent studies show that both glycolysis and oxidative metabolism at mitochondria are critical for the progression to metastasis and the survival of cancer cells as they establish distant tumors (8–10). Such multifaceted control of tumor cell metabolism highlights the importance of mitochondrial signaling events during metastatic tumor progression.
Bioinformatic analyses of mRNA and protein data sets have identified predictive elements of tumor progression that change during EMT and metastasis (11, 12). Many of these altered proteins regulate processes such as cytoskeletal remodeling, cell adhesion, and cellular metabolism, which are critical to tumor cell migration and survival (11). One class of proteins that are frequently altered in cancers are A-kinase–anchoring proteins (AKAPs), a family of scaffolding proteins that tether protein kinase A (PKA) and other signaling enzymes to intracellular membranes and organelles (13–15). These “AKAP signaling islands” locally control the phosphorylation state and activity of target substrates within precisely defined microenvironments (13, 16). This provides a means to not only efficiently control the relay of information within cells but also to insulate signals at defined subcellular locations to prevent indiscriminate cross-talk (13, 17). Recent evidence suggests that PKA activity facilitates the acquisition and maintenance of epithelial-like properties in breast cancer cells (18). Consequently, the loss of anchored PKA signaling at organelles including mitochondria may contribute to phenotypic changes that precede metastasis.
The mitochondrial anchoring protein dAKAP1 (also called sAKAP84, AKAP-121, and AKAP-149; gene is AKAP1) has been implicated in the establishment and growth of primary tumors in some cancers (19, 20). Using a combination of in silico and experimental approaches, we discovered that differential expression of dAKAP1 in breast tumors accompanies molecular and cellular changes that promote metastasis. This mitochondrial anchoring protein, originally identified in male germ cells, is a dual function AKAP that sequesters both the type I and type II PKA holoenzymes (21–23). Subsequent studies have shown that this versatile anchoring protein has the capacity to confer bidirectional control of protein phosphorylation by localizing both PKA and protein phosphatase 1 (PP1) to the outer mitochondrial membrane (13). Mitochondrial dAKAP1-anchored PKA phosphorylates and inhibits the mitochondrial fission enzyme dynamin-related protein 1 (Drp1) to alter mitochondrial morphology (24, 25). In this report, we show that the loss of dAKAP1 signaling islands from the outer mitochondrial membrane occurs as breast cancer cells acquire a more mesenchymal phenotype. Classification of dAKAP1 expression levels as “high” or “low” segregates a panel of breast cancer cell lines into functionally distinct groups that differ both in their metabolism and cell motility. Functionally, we show that dAKAP1-associated PKA represses mitochondrial fission and mitochondrial movement toward the leading edge. These findings support the notion that low dAKAP1 promotes motility in breast cancer cells. This infers that therapeutically regulating these mitochondrial signaling complexes may be applicable to the management of tumor metabolism and invasiveness.
Results
dAKAP1 levels are lower in distant metastases than in primary tumors
The in vivo tumor microenvironment contains two key compartments: tumor cells and the surrounding stroma (20, 26, 27). In some cancers, stromal cells utilize glycolytic metabolism to “feed” the tumor cells and thereby support cell survival (20, 26, 27). This promotes altered tumor metabolism that is associated with metastasis and cell proliferation (9). Because dAKAP1 may be involved in the establishment and growth of certain tumors, we sought to establish whether changes in the expression pattern of this anchoring protein could serve as a cellular index of metastatic potential (19, 20). A panel of 45 paired primary and metastatic breast cancer tumors was screened immunohistochemically for dAKAP1 levels. Analysis of a representative tissue pair is shown in Fig. 1, A–F. Staining in tumors (Fig. 1, A and D, shaded regions) was compared with staining in neighboring stromal tissue. Immunofluorescent detection of dAKAP1 (green) and counterstaining with the nuclear marker DAPI (blue) revealed the tissue distribution of this anchoring protein (Fig. 1, B and E). Samples were classified based on fluorescent intensity, and pseudocolor signal intensity gradients were used to visualize dAKAP1 protein levels in these compartments (Fig. 1, C and F). As predicted, strong dAKAP1 staining in tumors was evident in 60% of the primary tumor samples (Fig. 1G, yellow). In contrast, only 42% of the corresponding metastatic tumor sections exhibited robust expression of this anchoring protein (Fig. 1H, yellow). Thus, dAKAP1 expression is suppressed at secondary metastatic sites as compared with primary tumors. In keeping with this notion, only 4% of the primary breast tumors were scored as weakly expressing dAKAP1 as compared with 22% of the metastatic tumors (Fig. 1, G and H, teal). Paired analyses of these 45 matched tissue samples provide further evidence of reduced dAKAP1 tumor staining after metastasis (Fig. 1I). This supports the hypothesis that low dAKAP1 expression is associated with a molecular environment that promotes metastasis.
Figure 1.

dAKAP1 expression is decreased after metastasis. A–H, immunohistochemical evaluation of 45 paired primary and metastatic breast cancer tissue sections. Representative images from either the primary tumor (A–C) or the site of metastasis (D–F). A and D, bright field images of tissue sections. Tumor (brown) regions are denoted. B and E, immunofluorescent detection of dAKAP1 (green) and nuclei (blue). C and F, signal intensity of dAKAP1 staining is specified by pseudocolor gradient. Amalgamated data of 45 primary (G) and metastatic (H) tumor sections quantifying signal intensity enrichment of dAKAP1 staining in tumor. Strong (yellow), intermediate (gray), and weak (teal) signals are categorized. I, paired analysis of dAKAP1 staining intensity in tumors between 45 matched primary and metastatic samples. Cases of decreases (red lines) or increases (gray lines) in expression between samples are denoted.
dAKAP1 and mesenchymal gene expression are inversely related
Tumor cells are thought to transition through a mesenchymal-like state as they invade surrounding tissue and establish distant tumors (28). We therefore explored the relationship between the expression of dAKAP1, as well as 10 other AKAPs, and a set of mesenchymal markers across a large panel of breast cancer cell lines. mRNA expression data were obtained from the Cancer Cell Line Encyclopedia (CCLE) for AKAP genes and an in-house curated set of 36 mesenchymal markers (Fig. 2, A and B) (11, 12, 29). We observed a strong inverse correlation between the expression of mesenchymal genes and the mitochondrial anchoring protein dAKAP1 (Fig. 2, A and B). This inverse correlation was unique among not only the AKAPs but also other genes involved in mitochondrial morphology and function (Fig. 2, C and D). Furthermore, a similar inverse association with dAKAP1 expression was also apparent across a variety of cancer etiologies, suggesting that this relationship to mesenchymal markers is broadly conserved across multiple cancer types (Fig. S1, A and B). Correlative analyses in both breast cancer cell lines and tissues further support the notion that dAKAP1 expression is inversely related to a mesenchymal-like phenotype (Fig. S1, C–E). Although some AKAPs were positively correlated with mesenchymal markers in our analyses, we chose to focus on dAKAP1 to study how the regulation of outer mitochondrially anchored PKA signaling might influence breast cancer cell physiology. This negative correlation between dAKAP1 mRNA levels and average mesenchymal marker expression persisted across the 59 breast cancer cell lines represented in the CCLE (Pearson's r = −0.74 (Figs. 2, E and F, and S1, F and G). Moreover, an expanded analysis of metastasis-related genes revealed that reduced dAKAP1 expression is particularly coincident with the up-regulation of adhesion molecules and extracellular matrix components (Fig. S1H).
Figure 2.

dAKAP1 expression is inversely related to mesenchymal markers. A, Pearson's r values quantifying the correlation of AKAPs mRNA and 36 mesenchymal markers from gene array analysis data of CCLE breast cancer cell lines (n = 59) (29). Median and interquartile ranges (red bars) are indicated. B, heat map displaying r values of correlation of AKAP and mesenchymal gene mRNA expression. The rows (mesenchymal genes) are organized with hierarchical clustering, and columns (AKAPs) are organized by mean r value. Intensity scale indicates r values ranging from high (yellow) to low (teal). C, Pearson's r values as in A but with mitochondrion-related proteins as columns. D, heat map of r values as in B with mitochondrial proteins as columns. Note that repeated genes from previous panels display the same data with new clustering applied. E, heat map of dAKAP1 mRNA intensity and mean mesenchymal marker mRNA across each individual breast cancer cell line (of 59 total) used in this data set, organized by ascending dAKAP1 mRNA expression. F, scatter plot of dAKAP1 mRNA against mean mesenchymal marker mRNA. Error bars represent S.E. Breast cancer cell lines MCF7, BT474, MDA231 (also called MDA-MB-231), and HS578T used later in this study are highlighted in magenta. G and H, heat maps depicting correlation of mesenchymal marker expression with dAKAP1 mRNA (G) and dAKAP1 protein expression (H) in patient samples (32) from four clinically defined breast cancer subtypes (indicated above each lane). I, immunoblot analysis of patient samples from breast cancer tumors monitoring dAKAP1 expression (top), E-cadherin epithelial marker (second from top), N-cadherin (middle) and Snail mesenchymal markers (second from bottom), and Eif2α loading control (bottom). Individual patients are designated A–D at the bottom of the panel (see Table S1 for patient sample details).
Next, it was important to determine whether this relationship was conserved in the context of clinical patient samples. Primary breast cancer tumors can be classified in several ways but are clinically resolved into four subtypes: Basal, Her2, Luminal A, and Luminal B (30–32). With this in mind, we analyzed mRNA and protein levels in data sets generated from patient samples (Fig. 2, G and H). Again, dAKAP1 expression was negatively correlated with the appearance of mesenchymal markers. A similar negative correlation was evident in each breast cancer subtype, suggesting that reduction in this anchoring protein is a general phenomenon in tumor cells (Fig. 2, G and H). The inverse correlation was particularly evident in basal and Her2 classifications. This analysis provides evidence that decreased dAKAP1 levels are associated with the appearance of mesenchymal proteins in clinical samples from breast cancer patients.
Biochemical validation initially involved immunoblot analysis of primary tumor lysates obtained from four patients (Fig. 2I; samples are detailed in Table S1). Samples were arranged in descending order of dAKAP1 protein levels (as determined in exploratory analyses) (Fig. 2I, top). E-cadherin served as a marker of epithelial identity (Fig. 2I, second from top), whereas N-cadherin and the transcription factor Snail were used as mesenchymal markers (Fig. 2I, middle and second from bottom) (11). Detection of Eif2α served as a loading control (Fig. 2I, bottom). Strong immunoblot detection of dAKAP1 expression correlated with high E-cadherin and lower levels of N-cadherin (Fig. 2I, top, middle, and second from bottom, patients A and B) (9, 28, 33). Similarly, reduced detection of dAKAP1 was linked to increases in N-cadherin, consistent with the characteristic cadherin switch observed in EMT (Fig. 2I, top, middle, and second from bottom, patients A and B). Low expression of dAKAP1 was also linked to increases in the transcription factor and mesenchymal marker Snail (Fig. 2I, top, middle, and second from bottom, patients C and D). Based on these cumulative analyses, we predicted that dAKAP1 levels drop as cells adopt a mesenchymal phenotype.
dAKAP1 links to changes in mitochondrial function and cell motility
To systematically examine the role of dAKAP1 in breast cancer cell survival and metastasis, we turned to well-characterized model cell lines. Altered metabolism and increased cell motility are hallmarks of tumor progression (9, 28, 33). We queried published cell line data sets to investigate whether changes in dAKAP1 are linked to either process. Data from a large-scale proteomic analysis on a panel of breast cancer cell lines showed that dAKAP1 abundance varies across molecular subtypes (34). Our analysis shows that luminal subtypes express higher levels of dAKAP1, whereas basal/claudin-low types show minimal expression (Fig. 3A). Immunoblot analysis of lysates from four representative cell line confirmed that expression levels of dAKAP1 protein followed this predicted trend (Fig. 3B, top). Quantification of data from three independent experiments reinforced the notion that dAKAP1 protein is low in basal/claudin-low cells (Fig. 3C, gold columns). Independent analysis by RII overlay shows the distribution of AKAPs in each breast cancer cell line (Fig. S1I). However, this technique cannot be used to delineate the identity of individual anchoring proteins. Immunofluorescent detection of dAKAP1 (green) in fixed cells revealed a reticular staining pattern consistent with the mitochondrial compartment (Fig. 3D). Moreover, the signal intensity in each cell line mirrored the relative expression differences observed in the previous experiments. To assess the functional significance of these changes in dAKAP1 expression, we compared metabolic state across seven cell lines and found that less dAKAP1 protein corresponded to an enhanced glycolytic phenotype (Fig. 3E, gold) (34, 35). In parallel, we equated dAKAP1 expression to the invasive potential of nine cell lines evaluated by Transwell assay (Fig. 3F) (34, 36). Again, dAKAP1 was markedly reduced in the most invasive cancer cell lines (Fig. 3F, gold). This classification was also apparent at the level of mRNA expression (Fig. 3, G and H). On the basis of these cumulative analyses, breast cancer cell lines were segregated into two groups: “dAKAP1-high” lines (gray), including MCF7 and BT474, which are metabolically oxidative and noninvasive; and “dAKAP1-low” lines (gold), including MDA231 and HS578T, which are glycolytic and motile. These four representative breast cancer cell lines (also marked in magenta in Fig. 2F) were selected for further molecular and mechanistic analyses to investigate how dAKAP1 signaling at the mitochondria impacts cell motility.
Figure 3.

Classification of dAKAP1-high and dAKAP1-low breast cancer cell lines. A, average dAKAP1 iBAQ intensities across 20 breast cancer cell lines (data extracted from Table S1 and reported in Ref. 34). The subtype classification is indicated above each shaded region. Error bars indicate standard deviation between quantified dAKAP1 protein identifiers. B, immunoblot analysis of dAKAP1 levels (top) and Eif2α (bottom) loading control in representative breast cancer cell lines. C, quantification of amalgamated data (n = 3 independent blots) by densitometry. Error bars represent S.E. Statistical significance was determined by ordinary one-way ANOVA (p = 0.0013; F(3, 8) = 14.76). MCF7 and BT474 cells (gray) were designated dAKAP1-high. MDA231 and HS578T (gold) cells were designated dAKAP1-low. (See also magenta circles in Fig. 2F.) D, immunofluorescent detection of dAKAP1 (green) and nuclei (blue) in each cell line. Scale bars (10 μm) are indicated. E and F, scatter plots comparing dAKAP1 protein expression (34) with metabolic analysis of aerobic respiration/glycolysis (OCR/ECAR) (35) (E). Cell lines are designated as predominantly glycolytic (gold) or aerobic (gray). F, comparison with invasive potential (36) assessed by Transwell assay. G and H, mRNA expression for the same breast cancer cell lines (29) to metabolic analysis (35) (G) or invasive potential (H) (36). * denotes cell lines used experimentally.
dAKAP1 sequesters PKA and other second messenger–responsive enzymes to coordinate local signaling events at the outer mitochondrial membrane, including modulation of mitochondrial membrane potential (ΔΨm) (37–39). Measurement of ΔΨm serves as a gauge of respiratory chain function and an index of mitochondrial health (40, 41). Mitochondrial membrane potentials were assessed in our representative breast cancer cell lines by calculating the ratio of tetramethylrhodamine (TMRM) (Fig. 4A, top row) to MitoTracker Green FM signal intensity (Fig. 4A, middle row). The amalgamated data reveal that resting ΔΨm values are low in both the dAKAP1-low cell lines MDA231 and HS578T (Fig. 4, A, bottom row, and B). Furthermore, siRNA silencing of dAKAP1 in the dAKAP1-high cell line MCF7 decreased ΔΨm compared with nonsilenced control cells (Fig. S2, A–C). This suggests that expression of this anchoring protein directly influences ΔΨm. Parallel experiments conducted on MDA231 cells showed a similar trend (Fig. S2G). Thus, we propose that dAKAP1 supports high mitochondrial membrane potential and decreased expression of this anchoring protein may diminish respiratory chain function in breast cancer.
Figure 4.

Mitochondrial membrane potential is low in dAKAP1-low cell lines. A, mitochondrial membrane potentials were calculated by comparing the ratio of TMRM (top row) to MitoTracker Green FM fluorescence (middle row) in each cell line. Composite images (bottom row) reveal elevated mitochondrial membrane potentials in dAKAP1-high cell lines. Scale bars (10 μm) are indicated. B, quantification of amalgamated data (n = 4 independent experiments). The number of cells used in analyses are listed below each column. Error bars represent S.E. Statistical significance was determined by one-way ANOVA (p = 0.0043; F(3, 12) = 7.523).
Mitochondrial morphology is altered by dAKAP1 expression in breast cancer cells
Mitochondrial morphology reflects a dynamic equilibrium between the opposing actions of mitochondrial fission and fusion (42, 43). The size and distribution of mitochondria within the cell can be indicative of localized energy demand (44–47). Consequently, motile breast cancer cells often contain smaller, fragmented mitochondria (48). This is thought to promote mitochondrial trafficking during the energetically taxing process of cell migration (48, 49). The dynamin-related GTPase Drp1 is a recognized PKA substrate that is responsible for mitochondrial fission under normal growth conditions (50–52). Nutrient starvation is a metabolic stressor that suppresses fission through the phosphorylation of Drp1 at Ser-637 (53–55). Furthermore, dAKAP1-associated PKA is responsible for the phosphorylation of this site, which inhibits Drp1 fission activity and is important for cell survival in neurons (25, 56). It has been established previously that dAKAP1-associated PKA phosphorylates Drp1 at Ser-637 in neurons (24, 25, 56). We propose that this dAKAP1-mediated event is also physiologically relevant in breast cancer cells, particularly because cell survival under harsh conditions is an important feature of tumor cells (Fig. 5A). Gene silencing in dAKAP1-high MCF7 cells confirmed this notion (Fig. 5, B–G). Cells were treated with control- or dAKAP1-selective siRNA, and depletion of the anchoring protein from cell lysates was confirmed by immunoblot (Figs. 5B, top, and S2D, top). Phosphopeptide antibodies were used to evaluate phosphorylation status of Ser-637 on Drp1. In control cells, long-term serum starvation enhanced the pSer-637 signal (Fig. 5B, second from top, lane 2). This effect was lost upon siRNA depletion of the anchoring protein (Fig. 5B, second from top, lane 4). Immunoblot detection of total Drp1 and Eif2α served as loading controls for these experiments (Fig. 5B, second from bottom and bottom). Thus, gene silencing of dAKAP1 attenuates local phosphorylation of Ser-637 on Drp1. Densitometric analysis of immunoblots from five independent experiments corroborated these results (Fig. 5C).
Figure 5.
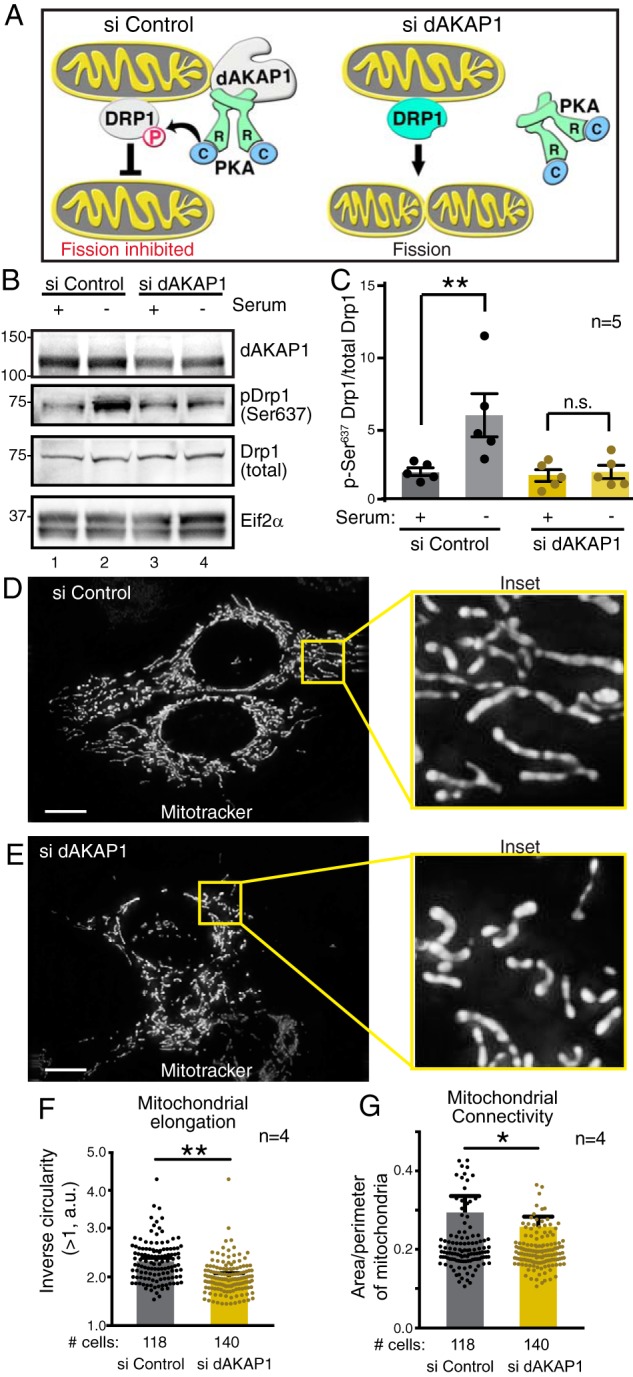
dAKAP1 facilitates phosphorylation of Drp1 to modulate mitochondrial morphology. A, schematic of dAKAP1-anchored PKA phosphorylation of the dynamin-related GTPase Drp1. Left, anchored PKA favors phosphorylation of Drp1 to inhibit mitochondrial fission. Right, gene silencing of dAKAP1 abolishes this effect. B, siRNA depletion of dAKAP1 suppresses phosphorylation of Ser-637 on Drp1. Immunoblot detection of dAKAP1 (top), pDrp1–Ser-637 (second from top), total Drp1 (second from bottom), and Eif2α loading control (bottom) is shown. Analysis of MCF7 cells treated with siControl (lanes 1 and 2) and siRNA against dAKAP1 (lanes 3 and 4) is shown. Samples in lanes 2 and 4 are starved. C, quantification of amalgamated data (n = 5 independent blots) by densitometry. Error bars represent S.E. Statistical significance was determined by two-tailed Student's t test. D–G, fluorescence detection of MitoTracker Green FM to monitor mitochondrial morphologies of MCF7 cells treated with siControl (D) or siRNA (E) against dAKAP1. Scale bars (10 μm) are indicated. Insets reveal selected 10-μm regions at higher magnification. Quantification of mitochondrial elongation (F) and mitochondrial connectivity (G) in siControl (gray) and si-dAKAP1 (gold)–treated cells (n = 4 independent experiments). The number of cells used in each analysis is indicated below each column. Error bars represent S.E. Statistical significance was determined by two-tailed Student's t test.
Concomitant changes in mitochondrial morphology were evaluated by live-cell imaging of serum-starved MCF7 cells loaded with MitoTracker Green FM (Fig. 5, D–G). Mitochondrial networks were more interconnected and elongated in cells treated with the control siRNA, particularly when visualized in enlarged images (Fig. 5D and inset). In contrast, mitochondria were fragmented and disorganized in cells treated with dAKAP1 siRNA (Fig. 5E and inset). Analysis of >100 cells under each condition confirmed that mitochondrial elongation (Fig. 5F, gold) and mitochondrial connectivity (Fig. 5G, gold) were reduced upon depletion of this anchoring protein.
Mitochondrial fission/fusion dynamics are hindered with dAKAP1 with depletion
To assess the state of mitochondrial dynamics in real time, we monitored the dispersal of mitochondrially targeted photoactivatable GFP (mito-paGFP) through the organellar networks. This offers a quantifiable index of mitochondrial dynamics. MCF7 cells (dAKAP1-high) were co-transfected with photoactivatable mito-paGFP and photostable mito-dsRed markers (Fig. 6, A–C, top panels, and Movies S1–S3). Photoactivation at 405 nm within a 1-μm diameter region allowed selective visualization and tracking of mito-paGFP over a time course of 24 min (Fig. 6, A–C, lower panels). Simultaneous imaging of mito-dsRed illuminated the entire mitochondrial network (Fig. 6, A–C, upper panels). In control cells, mito-paGFP rapidly diffused out of the region of activation, indicative of mitochondrial membrane fusion (Fig. 6, A, E, and F, charcoal). In contrast, siRNA depletion of dAKAP1 restricted the diffusion of mito-paGFP, suggesting a decrease in mitochondrial fusion and interconnectedness compared with control cells (Fig. 6, B, E, and F, gold). Rescue upon expression of a SNAP-tagged murine dAKAP1 ortholog that is resistant to the human siRNA restored the free movement of the photoactivated probe (Fig. 6, C, E, and F, gray). Delivery of the SNAP-targeting fluorophore 647-SiR was used to visualize dAKAP1 expression in “rescued” cells (Figs. 6D and S2E). Measuring the area of mito-paGFP distribution immediately after photoactivation provided an independent means of assessing mitochondrial morphology. These measurements confirmed our findings in Fig. 5 that gene silencing of dAKAP1 corresponds to a more fragmented mitochondrial network (Fig. 6G, gold), whereas rescue with the murine-anchoring protein reverses this effect (Fig. 6G, gray). Collectively, the data in Figs. 5 and 6 indicate that depletion of the anchoring protein in dAKAP1-high cells shifts the balance of fission and fusion to favor fragmented mitochondrial networks.
Figure 6.

Live-cell imaging analyses of mitochondrial dynamics. A–C, tracking of mitochondrially targeted paGFP (yellow) and whole-cell detection of photostable mitochondrially targeted dsRed (teal) monitored organellar dynamics over a time course of 24 min. Analysis was conducted on MCF7 cells treated with siControl (A), si-dAKAP1 (B), and rescue (C) upon expression of murine SNAP-tagged dAKAP1. Upper rows, composite images of both fluorescent channels. White circles denotes site of photoactivation. Lower rows, gray-scale images of paGFP signal. D, detection of SNAP-tagged dAKAP1 upon conjugation of 647-SiR fluorescent dye. Scale bars (5 μm) are indicated. E, diffusion of fluorescent paGFP signal was calculated. Amalgamated data for siControl (charcoal), si-dAKAP1 (gold), and rescued cells (gray). The numbers of cells in each analysis are indicated. Error bars represent S.E. Statistical significance was determined individually by two-tailed Student's t test. The rates of dispersal (F) and degrees of mitochondrial connectivity (G) were calculated for the same experimental conditions. The number of cells used in each analysis is indicated below each column. Data were collected over n ≥ 4 independent experiments. Error bars represent S.E. Statistical significance was determined by two-tailed Student's t test. Welch's correction was used in F to account for uneven variances.
Cell motility and mitochondrial positioning are impeded with dAKAP1 expression
Tumor cell motility and the invasion of other organs are defining features of metastasis (3, 7). Remodeling of mitochondrial networks and PKA phosphorylation events at the leading edge of migrating cells contribute to tumor cell movement (48, 49, 57). Because dAKAP1-associated PKA prevents mitochondrial fragmentation via the phosphorylation of Drp1, we reasoned that this local signaling event would also decrease cell migration. To test this hypothesis, we investigated the motility of dAKAP1-low MDA231 cells. Cells were induced to express WT dAKAP1 or a PKA-anchoring defective point mutant (dAKAP1ΔPKA, Figs. 7A and S2F) (17). Transwell migration assays monitored cell movement through a permeable membrane toward chemoattractant (Fig. 7, B and C). The results were normalized to WT MDA231 cells (Fig. 7, B, right panels, and C). Doxycycline induction of dAKAP1 expression blocked MDA231 cell migration as compared with controls (Fig. 7, A, top panel, lane 2; B, top right panel; and C, charcoal squares). Interestingly, induction of the dAKAP1ΔPKA mutant modestly enhanced Transwell migration (Fig. 7, A, top panel, lane 4; B, bottom right panel; and C, red triangles). Similar results were obtained when these experiments were repeated using a Transwell invasion assay that measures migration through a 3D matrix (Matrigel) into the bottom chamber (Fig. 7, D and E). Two important pieces of information were gleaned from these experiments: 1) conversion of MDA231 cells to dAKAP-high–expressing cells blocks migration, and 2) dAKAP1-anchored PKA is necessary for this process.
Figure 7.

dAKAP1-anchored PKA represses cell motility and modulates mitochondrial positioning. A, MDA231 cells were infected with doxycycline-inducible dAKAP1 (lanes 1 and 2) and a PKA anchoring–deficient mutant dAKAP1ΔPKA (lanes 3 and 4). Shown are immunoblot detection of dAKAP1 (top), PKA anchoring assessed by RII overlay (17) (middle), and Eif2α loading control (bottom). B and C, Transwell migration monitors cell movement across a porous membrane. B, representative images of crystal violet–stained migrating cells under each experimental condition. Scale bars (100 μm) are indicated; experimental are conditions indicated. C, amalgamated data (n = 5 independent experiments) normalized to uninduced WT dAKAP1 (gray circles) or dAKAP1ΔPKA mutant (pink triangles). Transwell migration of cells overexpressing dAKAP1 (charcoal squares) or dAKAP1ΔPKA (red inverted triangles) is indicated. D and E, Transwell invasion evaluates cell movement through a Matrigel barrier. D, representative image of crystal violet–stained invading cells under each experimental condition. Scale bars (100 μm) are indicated. E, quantification of data (n = 5 independent experiments) as defined above. F–I, migration of MitoTracker-loaded MDA231 cells into scratch wounds (yellow lines) over a time course of 18 h. Montages of control (F) and MDA231 cells overexpressing dAKAP1 (G) or dAKAP1ΔPKA (H) are shown. I, migration velocities (μm/min) of control (gray) and cells overexpressing dAKAP1 (charcoal) or dAKAP1ΔPKA (red) (n = 3 independent experiments). The numbers of cells analyzed from three independent experiments are indicated. Scale bars (100 μm) are indicated. J–L, fluorescent detection revealed the location of mitochondrial networks in migrating control cells (J) and cells overexpressing dAKAP1 (K) or dAKAP1ΔPKA mutant (L). Representative images depicting composite bright field and MitoTracker signals (top), MitoTracker alone (middle), and posterior–anterior line plots (bottom) reveal the intracellular position of fluorescent mitochondria. The cell body is outlined. Line plots are traced (top, white line, and middle, green line). The centroid (black circles) and anterior (shaded regions) of migrating cells are indicated. The scale is defined by the x axis of the line plot (75 μm). M, localization indexes (58) mark the position of mitochondria in control (gray), dAKAP1 (charcoal), or dAKAP1ΔPKA (red) cells (n = 3 independent experiments). The number of cells analyzed from three experiments is indicated. N and O, schematic highlighting the conclusions of this study. N, high dAKAP1 expression favors nonmotile breast cancer cells with elongated mitochondrial networks. O, reducing dAKAP1 fragments the mitochondria and enhances cell motility. Error bars presented in C, E, L, and M signify S.E. In each case statistical significance was determined by two-tailed Student's t test.
Live-cell imaging of MDA231 cells loaded with MitoTracker Green FM migrating into a scratch wound consolidated both findings (Fig. 7, F–H, and Movies S4–S6). The migration velocity of individual control cells into the wound area was calculated as 33.5 ± 2.8 μm/h over a time course of 18 h (n = 16; Fig. 7, F and I, gray column). Doxycycline induction of dAKAP1 retarded cell migration as evidenced by a reduced cell velocity of 22.4 ± 1.6 μm/h over the same time course (n = 15; Fig. 7, G and I, charcoal column). Overexpression of the dAKAP1ΔPKA mutant restored rates of cell migration to control levels (34.9 ± 1.7 μm/h (n = 15); Fig. 7, H and I, red column). Importantly, dAKAP1 expression also decreased the number of cells observed entering the wound area, an effect that was reversed with the dAKAP1ΔPKA mutant (Fig. 7, F–H).
Anterior positioning of mitochondria toward the leading edge increases velocity and the directional persistence of migrating breast cancer cells (58). Accordingly, we used time lapse imaging to monitor the intracellular location of mitochondrial labeled with MitoTracker (Fig. 7, J–L). Mitochondrial localization indexes (MLI) were defined as the ratio of the integrated fluorescent signal in front of the centroid (black dots) to the entire cell (white lines) (Fig. 7, J–L, top panels) (58). Line plots monitored the distribution of fluorescent mitochondria over the length of representative cells (Fig. 7, J–L, bottom). In control cells, the majority of the mitochondria were positioned toward the anterior of the cells (Fig. 7, J and M, gray column). As anticipated, the induction of dAKAP1 reduced mitochondrial mass at the leading edge (Fig. 7, K and M, charcoal column). This effect was lost in cells expressing dAKAP1ΔPKA (Fig. 7, L and M, red column). Thus, we concluded that the loss of dAKAP1 signaling islands biases mitochondrial morphology and positioning toward a migratory phenotype.
Discussion
We report that reduced expression of the mitochondrial PKA-anchoring protein dAKAP1 in breast cancers contributes to tumor progression toward a malignant state. Three lines of investigation support this model. First, an immunohistochemical screening of 45 matched tissue biopsies from primary and metastatic sites in patients with advanced breast cancer confirmed that dAKAP1 expression decreases upon disease progression (Fig. 1A). This pattern was observed across the spectrum of breast cancer classifications represented in the tissue microarray (denoted by receptor status in Table S1). Second, analysis of clinical samples showed that dAKAP1 levels decrease as tumors adopt a mesenchymal profile. Immunoblot analyses of patient samples in Fig. 2I support this view, showing that loss of the anchoring protein is concomitant with enhanced detection of mesenchymal markers. Similarly, dAKAP1 abundance is nominal in “claudin-low” cell lines that maintain a mesenchymal-like phenotype (Fig. 3A) (59). Although the availability of sufficient clinical material limited the scope of our analysis, this biochemical switch is consistent with a prevailing view that transcriptional remodeling and changes in cell motility are hallmarks of a mesenchymal profile (4, 11). Third, indirect support for this notion comes from the bioinformatic data presented in Fig. 2, G and H. With the possible exception of luminal A breast cancers, dAKAP1 expression inversely correlates with mesenchymal markers across the clinical subtypes. Thus, our integrated biochemical, histochemical, and bioinformatic strategy offers compelling support for the depletion of dAKAP1 mRNA and protein levels as breast cancer cells progress toward metastasis.
Although the molecular mechanisms that attenuate dAKAP1 expression in breast cancer tumors are poorly understood, one plausible explanation is ubiquitin-mediated protein degradation. Accordingly, hypoxic signaling to the E3 ubiquitin ligase Siah2 has been implicated in the targeted destruction of dAKAP1 with concurrent effects on mitochondrial morphology and function (24, 60). This may be particularly relevant in the hypoxic and nutrient-deprived core of solid tumors (61), prompting us to evaluate how pathological changes in dAKAP1 levels impact mitochondrial signaling. Our starting point was to assess the levels of anchoring protein in a range of breast cancer cell lines classified on the basis of their metabolic and invasive properties (35, 36). The data in Fig. 4 indicate that cell lines designated as dAKAP1-high display robust basal mitochondrial membrane potentials. This feature is often associated with enhanced oxidative phosphorylation, a metabolic process that exploits the transfer of electrons across inner mitochondrial membranes to promote ATP synthesis (40, 62). Paradoxically these findings imply that although dAKAP1 signaling islands reside on the outer mitochondrial membrane, they somehow seem to influence metabolic processes that occur inside these organelles. One explanation is that molecular flexibility within AKAP–PKA assemblies bestows a sufficient range of motion on anchored PKA to engage distal substrates (13, 16). Such a mechanism is supported by recent evidence that anchored PKA holoenzymes remain active and intact upon exposure to physiological levels of cAMP (14). However, we do not know how anchored PKA action is transduced across the outer mitochondrial membrane or whether other dAKAP1-binding partners are effectors of oxidative phosphorylation. Nonetheless, these findings add to the literature implicating dAKAP1 and other mitochondrial anchoring proteins such as Rab32, OPA1, and SKIP as local effectors of mitochondrial physiology (63–67).
Dynamic changes in mitochondrial morphology are indicative of different cellular and metabolic states (42, 53–55). One well-defined PKA target is the mitochondrial fission effector Drp1 (50, 51). This dynamin-related GTPase mechanically constricts the outer mitochondrial membrane to create the site of organellar fission (42, 52). Although changes in the phosphorylation status of Drp1 have been observed in neurons, this AKAP-mediated event may also be pertinent to the core of breast cancer tumors where nutrient deprivation is linked to elongated mitochondrial networks (25, 56, 61). To recapitulate this scenario in situ we exposed dAKAP1-high MCF7 cells to chronic serum starvation and were able to block fission. Biochemical and mitochondrial imaging experiments (in Fig. 5) show that gene silencing of dAKAP1 abolishes the inhibitory phosphorylation event at Ser-637 of Drp1 allowing mitochondrial fission to proceed unopposed. This tallies with live-cell imaging experiments presented in Fig. 6 showing that depletion of dAKAP1 impedes the diffusion of fluorophore upon photoactivation of a mitochondrially targeted paGFP. As diffusion into surrounding mitochondria is a quantifiable index of organelle fusion, our cumulated data imply that dAKAP1 modulates these dynamic changes in mitochondrial morphology. Importantly, these changes can be reversed upon rescue with the murine dAKAP1 ortholog. Nevertheless, we recognize that other factors also augment mitochondrial fragmentation. For example, the PTEN-induced putative kinase (PINK), enhances mitochondrial fission by displacing PKA from dAKAP1 (68, 69). Likewise, fission effector proteins, including Mff and Fis1, sequester Drp1 at the outer mitochondrial membrane where it is proximal to dAKAP1 signaling islands (42). Irrespective of which mechanism predominates, we concluded that dAKAP1 signaling islands influence dynamic changes in mitochondrial morphology in a variety of cellular and pathophysiological contexts.
Cell motility is an energy-intensive process defined as the movement of a cell from one location to another and is often considered an experimental surrogate for tumor metastasis (33, 36). A growing body of literature argues that a fragmented mitochondrial morphology drives cell migration (44, 45, 48, 49). Mitochondrial transport to the leading edge of motile cells supports the local production of ATP to supply cytoskeletal elements that participate in this type of cell movement (47, 57, 58). Anterior positioning of mitochondrial networks at the leading edge distinguishes motile cell lines with high metastatic potential from those with low metastatic potential (58). In keeping with this notion, the experiments represented in Fig. 7, J–L, detect fluorescently labeled mitochondrial networks toward the anterior of dAKAP1-low MDA231 cells. Conversion to dAKAP1-high upon overexpression of the anchoring protein not only diminished mitochondrial mass at the leading edge but also reduced the migration velocity of individual cells. Parallel studies recorded similar effects in Transwell migration and invasion assays. Mechanistically, dAKAP1-induced attenuation of cell migration proceeds through anchored PKA, as overexpression of the dAKAP1ΔPKA mutant had no effect. However, displacement of other dAKAP1-binding partners, including protein phosphatase PP1, microtubule-associated Rho guanine exchange factor and oncoprotein Lfc, as well as a protein tyrosine phosphatase D1-src module may further accentuate these effects (40, 70–73). Thus, dAKAP1 may serve as a nexus for the integration and relay of diverse signals from the mitochondria to the microtubule network. Finally, we propose a model where dAKAP1 levels are high in primary tumors to facilitate robust mitochondrial function that sustains growth and proliferation (Fig. 7N). As the cell becomes migratory, dAKAP1 levels drop and the mitochondrial signaling environment is altered to facilitate the changing needs of the cell (Fig. 7O). In migratory dAKAP1-low cell types, fragmented mitochondria may facilitate the trafficking of these energetic organelles to the leading edge, where local energy requirements are elevated. In a broader context, this study highlights the importance of mitochondrial AKAP signaling islands in the maintenance of organelle organization and function. Therefore, we propose that the elimination of dAKAP1 is a pathological development that alters the local signaling terrain as breast cancers progress toward metastasis.
Experimental procedures
Correlation and heat map generation
Cancer Cell Line Encyclopedia (CCLE) data (29)
RMA (robust multiarray average)–normalized mRNA expression data were downloaded from the Broad Institute (https://portals.broadinstitute.org/ccle).7 Pearson's correlation, calculated between each AKAP with quantified mRNA expression values and individual mesenchymal markers (36 total) in either breast cancer (Fig. 2, A–D) or any cancer etiology with >3 quantified cell lines (Fig. S1, A and B), was represented in a heat map using GraphPad Prism7 (GraphPad Software, La Jolla, CA).
Clinical sample RNA microarray, RNAseq, and proteomics data
Microarray, RNAseq, and protein expression data in tissue samples (Figs. 2, G and H, and S1, C and D) were downloaded from the Broad Institute–associated browser (http://prot-shiny-vm.broadinstitute.org:3838/BC2016/)7 (32). Pearson's correlation was calculated between dAKAP1 and expression of mesenchymal markers (36 total) in individual samples and averaged across each of the four clinical subtypes defined in the study (32). Protein expression data in breast cancer cell lines (Figs. 3, A, E, and F, and S1E) were downloaded from published data sets (34). The resulting data were represented in a heat map using GraphPad Prism7 software.
Clustering
Unsupervised hierarchical clustering was performed on mesenchymal genes (rows in Figs. 2, B, D, G, and H, and S1, B and D) using the Perseus software package (V1.6.1.3, Max Plank Institute of Biochemistry) (74) with the following settings: distance = Euclidean; linkage = average; processed with K-means enabled; number of clusters = 300; maximal number of iterations = 10; and number of restarts = 1.
Determination of metabolic and invasive potential correlations
Data for oxygen consumption rate (OCR)/extracellular acidification rate (ECAR) was determined computationally from the reported data (35) (Fig. 3, E and G, y axis). Similarly, the invasive potential was also determined computationally from published data (36) (Fig. 3, F and H, y axis). These were compared with dAKAP1 protein expression values determined using iBAQ proteomics (Fig. 3, E and F, x axis) (34) or mRNA expression values in the CCLE (Fig. 3, G and H, x axis) (29). Pearson's r was used to determine the linear relationship between OCR/ECAR and dAKAP1 expression, whereas Spearman's r was used for the nonlinear variable of invasive potential. “Glycolytic” cells were defined as an OCR/ECAR ratio < 10. “Invasive” cell lines were defined as having an invasive potential of >250. See respective studies for relevant methods for determining OCR/ECAR (35) and invasive potential (36).
Antibodies
The following antibodies were used in this study for Western blotting: AKAP149 (610720, BD Biosciences, 1:1000), AKAP1 (HPA008691, Human Protein Atlas/Sigma Aldrich, 1:1000), pDrp1 Ser-637 (PA5-37534, Invitrogen, 1:1000), total Drp1 (611112, BD Biosciences, 1:1000), Eif2α (5324S, Cell Signaling Technologies, Danvers, MA, 1:1000), V5 (R960CUS, Invitrogen, 1:5000), N-cadherin (13116S, Cell Signaling Technologies, 1:1000), Snail (3879S, Cell Signaling Technologies, 1:500), E-cadherin (610181, BD Biosciences, 1:1000), and mouse and rabbit horseradish peroxidase-conjugated secondary antibodies (NA9310 and NA9340, GE Healthcare Life Sciences, Marlborough, MA, 1:10,000). The following antibodies and stains were used in this study for immunofluorescent imaging: AKAP1 (HPA008691, Human Protein Atlas/Sigma Aldrich, 1:500) and DAPI (62248, Life Technologies, Inc., 1:1000).
Plasmids
The human dAKAP1 ORF was purchased from OriGene (RC200506, Rockville, MD). The pLIX403 plasmid was a gift from David Root (Addgene plasmid 41395). Mito-paGFP was a gift from Richard Youle (Addgene plasmid 23348). Plasmid encoding mito-dsRed was a gift from the laboratory of Suzanne Hoppins (University of Washington).
Cell lines and culture
Breast cancer cell lines MDA231 (MDA-MB-231, ATCC catalog No. HTB-26), BT474 (ATCC catalog No. HTB-20), and HS578T (ATCC catalog No. HTB-126) were purchased from ATCC (Manassas, VA). MCF7 cells were graciously provided by the laboratory of Judit Villén (University of Washington). Cell lines were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco). Cell lines with stably incorporated dAKAP1 variants were grown in DMEM supplemented with 10% defined, tetracycline-low FBS (Hyclone, Pittsburgh, PA) with 1 μg/ml puromycin (Sigma Aldrich) unless otherwise noted. All cell lines were maintained in a 5% CO2 incubator at 37 °C.
Starvation and siRNA
siRNA knockdown was achieved in MCF7 cells with 50 nm AKAP1 siGENOME SmartPool (pool of four siRNAs) or 50 nm siGENOME nontargeting siRNA (M-011426-02 and D-001210-03, respectively Dharmacon, Lafayette, CO), transfected with Lipofectamine 2000 (Invitrogen), and incubated in a 5% CO2 incubator at 37 °C for 24 h. The transfection medium was replaced with FBS-free DMEM after 24 h to achieve 48 h of starvation prior to the assay.
Cell line generation
Doxycycline-inducible cells were generated using lentiviral transduction to stably incorporate dAKAP1 variants in a pLIX403 backbone. Lentiviral particles were incubated with MDA231 cells (ATCC) for 24 h. Cells were recovered in normal media for 24 h before undergoing 4 μg/ml puromycin selection for 48 h. After selection, cells were dissociated using 0.25% trypsin-EDTA (Gibco); 200 cells were evenly distributed into a 10-cm2 dish and incubated for 48–96 h or until single-cell–derived colonies were visible. Single-cell–derived colonies were hand-picked with a pipette tip and plated into single wells of a 96-well plate. Cells were expanded and tested for induction with 1 μg/ml doxycycline for 72 h. Cell lines with moderate and comparably induced expression of V5-tagged dAKAP1 were selected for use in this study.
Tissue sample lysis
Tumor samples from breast cancer patients were obtained (Fig. 2I, patients A–C (Northwest BioSpecimens, Seattle, WA), and Table S1) as flash-frozen tissue. The tissue was powdered in a mortar on dry ice and lysed in RIPA buffer (150 mm NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mm Tris pH 8.0) with protease and phosphatase inhibitors by vortexing and incubating on a rocker at 4 °C for 30 min. Lysates were cleared at 10,000 × g for 10 min at 4 °C. The lysate shown in Fig. 2I, patient D, was obtained from OriGene (catalog No. 543487) (Table S1) in modified RIPA buffer.
Cell lysis
For cultured cells, the cell line was grown to the desired confluence and rinsed once in cold PBS. Cold lysis buffer (20 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm EDTA, and 0.5% Triton/Nonidet P-40 in water) with protease and phosphatase inhibitors was added to the dish and gently rocked at 4 °C for 20 min. Cell lysate was scraped into a chilled tube and cleared at 10,000 × g for 10 min at 4 °C.
Immunoblot analysis
A BCA assay (Pierce) was used to determine protein concentrations, and 15 μg of protein/sample was loaded onto a Bolt 4–12% bis-Tris gel (Life Technologies). The cleared lysate was boiled in 1× LDS loading buffer for 10 min before loading. Proteins were transferred to nitrocellulose membrane and blocked in either 5% milk or 5% BSA in TBST (50 mm Tris, pH 7.6, 150 mm NaCl, 0.05% Tween-20), as determined by the manufacturer of the relevant antibody. The primary antibodies were incubated at the dilutions described above overnight at 4 °C. Immunoblots were thoroughly washed in TBST before incubation in a 1:10,000 secondary antibody for 1 h at room temperature. Immunoblots were washed again in TBST before imaging on an iBright FL1000 (Thermo Fisher Scientific) with SuperSignal Dura ECL reagent (Thermo Fisher Scientific). Densitometry for blot quantification was done using ImageJ software (NIH).
Transwell migration and invasion
Transwell assays were performed by dissociating the relevant cell lines in 1 mm EDTA in PBS for 5 min at 37 °C. Cells were agitated, fully resuspended in serum-free medium, and counted. A total of 1.0 × 105 cells were seeded onto the upper chamber of the Transwell. Transwells with an 8.0-μm pore size were used uncoated for migration (catalog No. 07-200-150, Pittsburgh Corning Corp., Pittsburgh, PA) or were Matrigel-coated for invasion (catalog No. 354480, Corning BioCoat). For Matrigel invasion assays, Matrigel-coated Transwells were rehydrated in serum-free medium at 37 °C, 5% CO2 for 2 h prior to seeding. Whole-serum medium (10% FBS in DMEM) was used as the chemoattractant in the bottom chamber. Migration assays were incubated at 37 °C, 5% CO2 for 18 h, and invasion assays were incubated for 20 h. After incubation, both the chamber and the membrane were rinsed twice in PBS, fixed in 70% ethanol for 10 min, and stained in 0.05% crystal violet (w/v in water) for 30 min. Membranes and chambers were rinsed well in PBS, and total cells in the bottom chamber were counted. Representative images were acquired at 10× magnification with a DMI8 automated inverted microscope (Leica, Wetzlar, Germany) with an EL6000 component (light source, filter wheel, and ultra-fast shutter, Leica) and a CoolSnap HQ camera (Photometrics, Tucson, AZ).
Tissue staining and imaging
Staining and antigen retrieval
A tissue section microarray of 50 paired primary and metastatic breast tumors was obtained from U.S. BioMax (Br10010e) (Table S1) as formalin-fixed paraffin-embedded samples. Sections were deparaffinated (Citrasolv, Thermo Fisher Scientific), antigen retrieval was performed (R-buffer A, Retriever 2100 pressure cooker, Electron Microscopy Sciences), and tissue sections were blocked in 10% (v/v) donkey serum with 5% (w/v) BSA in PBS for at least 1 h before overnight incubation with the relevant primary antibodies.
Imaging and analysis
Cells were imaged on a Keyence BZ-X710 microscope (Keyence, Itasca, IL) using the relevant filter cubes for bright field (no filter), DAPI (blue filter), and dAKAP1 staining (red filter). All images were acquired with the same magnification (100×, oil immersion), exposure time, and illumination intensity. Images were quantified and processed using ImageJ software.
Quantification of mitochondrial tumor enrichment
A quality review of each pair of tissue samples was completed by blindly scoring representative images with a quality score of 0 (poor), 1 (intermediate), or 2 (excellent). All pairs containing at least one score of 0 were disregarded for quantification (5 of 50 pairs were discarded). Tumor cells and tumor stroma were identified by nuclear staining and tissue morphology. The fluorescence intensity of dAKAP1 staining was measured as the average of 3 equivalent rectangles in regions of tumor cells and tumor stromal cells. Values were normalized to DAPI staining to account for differences in cell density. A tumor enrichment ratio was calculated as a ratio of background-corrected, DAPI-normalized dAKAP1 fluorescence intensity in tumor cells to that of stromal cells. Ratios ≥ 2.5 were considered “strong” staining, 2.5 > ratios > 1.5 were “intermediate,” and ratios ≤ 1.5 were scored as “weak” staining.
Live-cell imaging
Mitochondrial membrane potential of cell lines
Cells were grown in medium containing 10% FBS in a 5% CO2 incubator at 37 °C. On the day of imaging, the medium was aspirated and replaced with medium containing 40 nm TMRM (T668, Thermo Fisher Scientific) and 50 nm MitoTracker Green FM (M7514, Invitrogen), and the mixture was incubated for 45 min at 37 °C. The cells were rinsed twice in PBS and further incubated in medium containing NucBlue Hoescht 33342 stain (R37605, Invitrogen, 1 drop/ml) for 15 min at 37 °C. The medium was aspirated and replaced with FluoroBrite DMEM + 10% FBS.
Cells were imaged on a GE Deltavision OMX SR microscope (GE Life Healthcare Sciences). After loading (as above), cells were placed in a humidified chamber with 5% CO2 at 37 °C and imaged using a 60× oil immersion objective (Olympus, Shinjuku, Tokyo, Japan). Nuclei were excited with a 405-nm laser at 15% T for 100 ms and captured at 435 nm. MitoTracker Green FM was excited with a 488-nm laser at 30% T for 150 ms and captured at 528 nm. TMRM was excited with a 568-nm laser at 5% T for 50 ms and captured at 609 nm. All images were acquired with the same settings. Images were quantified and processed using ImageJ software. For the ΔΨm microscopy analyses presented in Figs. 4A, S2A, and S2G, the ratios were calculated by dividing TMRM fluorescence by MitoTracker Green FM fluorescence in individual cells.
Mitochondrial membrane potential of dAKAP1-depleted MCF7
To determine ΔΨm in dAKAP1-depleted MCF7 cells (Fig. S2, A–C), siRNA knockdown was achieved in MCF7 cells with either 50 nm AKAP1 targeting siRNA or 50 nm nontargeting siRNA and transfected with Lipofectamine 2000, as described above. The transfection medium was replaced with DMEM+10% FBS after 24 h to achieve a total of 48 h of serum starvation prior to assay. Cells were loaded with 40 nm TMRM for 45 min as described above.
For microscopy analysis (Fig. S2B) of ΔΨm, images were acquired every 30 s for 15 min. At time = 5 min, 25 μm FCCP was added to the medium. Membrane potentials were determined by measuring fluorescent intensity in three rectangles/cell and averaged. Data were normalized to average depolarized membrane potential at 15 min. Images were acquired using a 63× oil immersion objective with a DMI8 automated inverted microscope (Leica), an EL6000 component (light source, filter wheel, and ultra-fast shutter, Leica), and a CoolSnap HQ camera (Photometrics).
For microplate analysis (Fig. S2C) of ΔΨm, cells were plated in six replicates. Cells were treated with siRNA and loaded with 40 nm TMRM, as described above. For depolarized control, cells were treated with FCCP for 5 min after loading with TMRM. The medium was aspirated and replaced with whole FluoroBrite medium. The fluorescence intensity of each well was measured with 530 nm excitation and 590 nm emission. Wells containing only medium were used to determine background fluorescence. Quantification of ΔΨm reflects background-subtracted fluorescence intensity normalized to FCCP-depolarized wells.
Mitochondrial morphology
MCF7 cells were plated onto 35-mm poly-l-lysine (Sigma)–coated No.1.5 glass-bottom dishes (P35G-1.5-10-C, MatTek, Ashland, MA) and treated as described above for siRNA transfection with starvation. On the day of imaging, the medium was aspirated and replaced with medium containing 50 nm MitoTracker Green FM (M7514, Invitrogen), and the mixture was incubated for 45 min. Cells were rinsed twice in PBS and incubated in medium containing NucBlue Hoescht 33342 stain (R37605, Invitrogen, 1 drop/ml) for 15 min at 37 °C. The medium was aspirated and replaced with serum-free FluoroBrite DMEM with 1 g/liter glucose. Cells were imaged on a GE Deltavision OMX SR microscope. After loading (as described above), cells were placed in a humidified chamber with 5% CO2 at 37 °C and imaged using a 60× oil immersion objective (Olympus). Nuclei were excited with a 405-nm laser at 15% T for 100 ms and captured at 435 nm. MitoTracker Green FM was excited with a 488-nm laser at 30% T for 150 ms and captured at 528 nm. All images were acquired with the same settings. Images were quantified and processed using ImageJ software. The mitochondrial morphology quantification shown in Fig. 5, F and G, was done using the “Mitochondrial Morphology” ImageJ plugin (68).
Mitochondrial photoactivation
Cells were co-transfected with siRNAs (see above) and plasmids encoding mito-paGFP, mito-dsRed, and murine dAKAP1–SNAP (rescue cells) using Lipofectamine 2000. Cells were incubated in transfection medium for 24 h at 37 °C. The transfection medium was removed and replaced with serum-free DMEM with 1 g/liter glucose, and the mixture was incubated for an additional 48 h. On the day of imaging, cells were removed from the medium, loaded with 30 nm SNAP-Cell® 647-SiR (New England Biolabs, Ipswich, MA), and incubated at 37 °C for 30 min to label dAKAP1–SNAP. After incubation, the cells were rinsed three times in PBS followed by incubation in serum-free medium for an additional 30 min. The medium was replaced with warmed serum-free medium and moved to a 5% CO2 microscope chamber for imaging at 37 °C.
Cells were imaged on a GE Deltavision OMX SR microscope and imaged using a 60× oil immersion objective (Olympus). Z-stacks were acquired every 0.5 μm over 2 μm. Quantification was done on sum intensity Z-projections, whereas representative images are displayed as maximum intensity Z-projections. Mitochondrial paGFP was photoactivated with a 405-nm laser at 1% T for 2 ms in a 1-μm diameter spot. The activated mito-paGFP fluorescent signal was excited at 488 nm and captured at 528 nm. Mitochondrial dsRed fluorescence was excited at 568 nm and captured at 609 nm. For dAKAP1–SNAP rescue cells, SNAP cell 647-SiR was excited at 640 nm at 20% T for 30 ms and captured at 683 nm. Images were captured every 2 min, starting 2 min before activation (time = −2 min) for 30 min. Time 0 min corresponds to the time point immediately following initial photoactivation. All images were acquired with the same settings.
Quantification of photoactivated mitochondria diffusion
To quantify the diffusion of fluorescence intensity outside of the activated region over time, a polygon was drawn using ImageJ software to contain the entire cell, carefully excluding only the photoactivated region at t = 0 min, and integrated fluorescence intensity was measured. The excluded area was defined by connecting the outermost edges of the photoactivated mitochondrial region and varied dependent on the elongation and interconnection of the mitochondrial network. The diffusion of photoactivated fluorophore outside of the activated region was then calculated by subtracting background so that t = 0 min was equal to 0 for each time point and expressed as a percentage of the initial (t = 0 min) integrated fluorescence intensity within the activated region.
Rate of diffusion and mitochondrial connectivity
The rate of diffusion was calculated as the difference between the integrated fluorescent intensity outside of the activated region (quantification described above) at the final time point (t = 28 min) from that at the time of activation (t = 0 min) divided by time (28 min). Mitochondrial connectivity was determined as the area of activated region determined by binary mask (Isodata ImageJ algorithm) of image at time of activation (t = 0). Images were quantified and processed using ImageJ software.
Migration into wounds
Relevant cell lines were seeded into 4-chamber, glass bottom, tissue culture–treated dishes (Grenier Bio-One, Monroe, NC) to achieve a confluent monolayer at the time of the assay. Cells were loaded with 50 nm MitoTracker Green FM for 1 h in a 37 °C, 5% CO2 incubator. Cells were rinsed once in PBS and incubated in CO2-buffered FluoroBrite (Gibco) medium + 10% FBS for 30 min. Monolayers were gently scratched with a P10 pipette tip to induce a wound-healing response and then immediately rinsed twice in PBS. Cells were returned to CO2-buffered FluoroBrite medium + 10% FBS in a humidified 37 °C, 5% CO2 imaging chamber on a Keyence BZ-X710 using a 20× magnification objective. Cells were imaged every 10 min for 18 h.
Quantification of mitochondrial migration and MLI
Velocity of migration was quantified by analyzing the migration of individual cells into the wound region. Distance traveled at the leading edge was determined for a minimum of 15 cells over the span of 1 h (6 images) under each condition, over the course of three independent experiments.
The MLI of individual cells migrating into the wound region was calculated as described (58), with a slight modification to avoid the photosensitivity observed when nuclei were imaged. This modification involved setting the cell centroid as the centroid of the line drawn from the trailing edge to the leading edge (Fig. 7, J–L, black dots) rather than the nucleus. The MLI was calculated as the ratio of integrated density anterior of the centroid over that of the total line (thus MLI ≤ 1). The MLI of each migrating cell was measured repeatedly in a sequence of five time steps and averaged. Indexes less than 0.35 were scored as “posterior” localization, greater than 0.65 were scored as “anterior” localization, and MLI numbers between these values were considered generally mislocalized, as described previously (58).
Patient-derived samples
Breast cancer tumor samples (Fig. 2I, patients A–C) were obtained from Northwest BioSpecimens following the guidelines of “nonhuman research” study procedures from the Human Subjects Division at the University of Washington. The remaining samples were obtained through commercial sources such that patient privacy was protected by the manufacturer (tumor lysate, OriGene, Fig. 2I, patient D; tissue slice microarray, U.S. BioMax, Fig. 1, A–H).
Statistical analysis
All data are presented as the mean ± S.E. unless otherwise indicated. Sample size (n) indicates the number of independent experiments represented in amalgamated data; total cell numbers used in these experiments are indicated. Statistical significance was typically determined using unpaired, two-tailed Student's t test. When variances were determined to be significantly different, Welch's correction was applied. Significance between >2 samples (Figs. 3C and 4B) was calculated using one-way ANOVA with Tukey's test for multiple comparisons. In all cases, significance was determined as p ≤ 0.05. Significance as indicated in figures: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; and ****, p ≤ 0.0001. All statistical analyses were performed in GraphPad Prism7 software.
Data and materials availability
All data generated that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
S. A., F. D. S., and J. D. S. conceptualization; S. A., L. G., M. G., S.-E. O., F. D. S., and J. D. S. resources; S. A. and F. D. S. data curation; S. A. and F. D. S. formal analysis; S. A., L. G., M. G., S.-E. O., and F. D. S. methodology; S. A. and J. D. S. writing-original draft; S. A., L. G., L. K. L., F. D. S., and J. D. S. writing-review and editing; L. K. L. visualization; F. D. S. and J. D. S. supervision; J. D. S. funding acquisition; J. D. S. project administration.
Supplementary Material
Acknowledgments
We thank Suzanne Hoppins for providing plasmids, protocols, and editorial assistance. We also thank members of the Judit Villén laboratory for graciously providing the MCF7 cells. Finally, we thank Paula Bucko for generating the dAKAP1–SNAP construct.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2, Table S1, and Movies S1–S6.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- EMT
- epithelial-to-mesenchymal transition
- AKAP
- A-kinase–anchoring protein
- PKA
- protein kinase A
- CCLE
- Cancer Cell Line Encyclopedia
- TMRM
- tetramethylrhodamine
- mito-paGFP
- mitochondrially targeted photoactivatable GFP
- MLI
- mitochondrial localization index
- OCR
- oxygen consumption rate
- ECAR
- extracellular acidification rate
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- FCCP
- carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- RIPA
- radioimmune precipitation assay (buffer)
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- DAPI
- 4′,6-diamidino-2-phenylindole
- ANOVA
- analysis of variance.
References
- 1. Harbeck N., and Gnant M. (2017) Breast cancer. Lancet 389, 1134–1150 10.1016/S0140-6736(16)31891-8 [DOI] [PubMed] [Google Scholar]
- 2. Noone A. M., Howlader N., Krapcho M., Miller D., Brest A., Yu M., Ruhl J., Tatalovich Z., Mariotto A., Lewis D. R., Chen H. S., Feuer E. J., and Cronin K. A. (eds) (2018) SEER Cancer Statistics Review, 1975–2015, National Cancer Institute, Bethesda, MD: https://seer.cancer.gov/csr/1975_2015/, based on November 2017 SEER data submission, posted to the SEER Website, April 2018 [Google Scholar]
- 3. Quail D. F., and Joyce J. A. (2013) Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalluri R., and Weinberg R. A. (2009) The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thiery J. P., and Sleeman J. P. (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 7, 131–142 10.1038/nrm1835 [DOI] [PubMed] [Google Scholar]
- 6. Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Brooks M., Reinhard F., Zhang C. C., Shipitsin M., Campbell L. L., Polyak K., Brisken C., Yang J., and Weinberg R. A. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 10.1016/j.cell.2008.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McAllister S. S., and Weinberg R. A. (2014) The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 16, 717–727 10.1038/ncb3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cavalli L. R., Varella-Garcia M., and Liang B. C. (1997) Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ. 8, 1189–1198 [PubMed] [Google Scholar]
- 9. Vander Heiden M. G., and DeBerardinis R. J. (2017) Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 10.1016/j.cell.2016.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Viale A., Corti D., and Draetta G. F. (2015) Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 75, 3685–3686 10.1158/1538-7445.AM2015-3685 [DOI] [PubMed] [Google Scholar]
- 11. Zeisberg M., and Neilson E. G. (2009) Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 119, 1429–1437 10.1172/JCI36183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gujral T. S., Chan M., Peshkin L., Sorger P. K., Kirschner M. W., and MacBeath G. (2014) A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 159, 844–856 10.1016/j.cell.2014.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langeberg L. K., and Scott J. D. (2015) Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 16, 232–244 10.1038/nrm3966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smith F. D., Esseltine J. L., Nygren P. J., Veesler D., Byrne D. P., Vonderach M., Strashnov I., Eyers C. E., Eyers P. A., Langeberg L. K., and Scott J. D. (2017) Local protein kinase A action proceeds through intact holoenzymes. Science 356, 1288–1293 10.1126/science.aaj1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kjällquist U., Erlandsson R., Tobin N. P., Alkodsi A., Ullah I., Stålhammar G., Karlsson E., Hatschek T., Hartman J., Linnarsson S., and Bergh J. (2018) Exome sequencing of primary breast cancers with paired metastatic lesions reveals metastasis-enriched mutations in the A-kinase anchoring protein family (AKAPs). BMC Cancer 18, 174 10.1186/s12885-018-4021-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith F. D., Reichow S. L., Esseltine J. L., Shi D., Langeberg L. K., Scott J. D., and Gonen T. (2013) Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. Elife 2, e01319 10.7554/eLife.01319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith F. D., Langeberg L. K., Cellurale C., Pawson T., Morrison D. K., Davis R. J., and Scott J. D. (2010) AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12, 1242–1249 10.1038/ncb2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pattabiraman D. R., Bierie B., Kober K. I., Thiru P., Krall J. A., Zill C., Reinhardt F., Tam W. L., and Weinberg R. A. (2016) Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351, aad3680 10.1126/science.aad3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rinaldi L., Sepe M., Delle Donne R., Conte K., Arcella A., Borzacchiello D., Amente S., De Vita F., Porpora M., Garbi C., Oliva M. A., Procaccini C., Faicchia D., Matarese G., Zito Marino F., et al. (2017) Mitochondrial AKAP1 supports mTOR pathway and tumor growth. Cell Death Dis. 8, e2842 10.1038/cddis.2017.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sotgia F., Whitaker-Menezes D., Martinez-Outschoorn U. E., Salem A. F., Tsirigos A., Lamb R., Sneddon S., Hulit J., Howell A., and Lisanti M. P. (2012) Mitochondria “fuel” breast cancer metabolism: Fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle 11, 4390–4401 10.4161/cc.22777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang L. J., Durick K., Weiner J. A., Chun J., and Taylor S. S. (1997) Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J. Biol. Chem. 272, 8057–8064 10.1074/jbc.272.12.8057 [DOI] [PubMed] [Google Scholar]
- 22. Lin R. Y., Moss S. B., and Rubin C. S. (1995) Characterization of S-AKAP84, a novel developmentally regulated A-kinase anchor protein of male germ cells. J. Biol. Chem. 270, 27804–27811 10.1074/jbc.270.46.27804 [DOI] [PubMed] [Google Scholar]
- 23. Smith F. D., Omar M. H., Nygren P. J., Soughayer J., Hoshi N., Lau H. T., Snyder C. G., Branon T. C., Ghosh D., Langeberg L. K., Ting A. Y., Santana L. F., Ong S. E., Navedo M. F., and Scott J. D. (2018) Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. U.S.A. 115, E11465–E11474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H., Scimia M. C., Wilkinson D., Trelles R. D., Wood M. R., Bowtell D., Dillin A., Mercola M., and Ronai Z. A. (2011) Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44, 532–544 10.1016/j.molcel.2011.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merrill R. A., Dagda R. K., Dickey A. S., Cribbs J. T., Green S. H., Usachev Y. M., and Strack S. (2011) Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 9, e1000612 10.1371/journal.pbio.1000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chiarugi P., and Cirri P. (2016) Metabolic exchanges within tumor microenvironment. Cancer Lett. 380, 272–280 10.1016/j.canlet.2015.10.027 [DOI] [PubMed] [Google Scholar]
- 27. Casey T., Bond J., Tighe S., Hunter T., Lintault L., Patel O., Eneman J., Crocker A., White J., Tessitore J., Stanley M., Harlow S., Weaver D., Muss H., and Plaut K. (2009) Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res. Treat. 114, 47–62 10.1007/s10549-008-9982-8 [DOI] [PubMed] [Google Scholar]
- 28. Sciacovelli M., and Frezza C. (2017) Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 284, 3132–3144 10.1111/febs.14090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parker J. S., Mullins M., Cheang M. C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z., Quackenbush J. F., Stijleman I. J., Palazzo J., Marron J. S., Nobel A. B., Mardis E., Nielsen T. O., Ellis M. J., Perou C. M., and Bernard P. S. (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 10.1200/JCO.2008.18.1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mertins P., Mani D. R., Ruggles K. V., Gillette M. A., Clauser K. R., Wang P., Wang X., Qiao J. W., Cao S., Petralia F., Kawaler E., Mundt F., Krug K., Tu Z., Lei J. T., et al. (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 10.1038/nature18003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: The next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 34. Lawrence R. T., Perez E. M., Hernández D., Miller C. P., Haas K. M., Irie H. Y., Lee S. I., Blau C. A., and Villén J. (2015) The proteomic landscape of triple-negative breast cancer. Cell Rep. 11, 630–644 10.1016/j.celrep.2015.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pelicano H., Zhang W., Liu J., Hammoudi N., Dai J., Xu R. H., Pusztai L., and Huang P. (2014) Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 16, 434 10.1186/s13058-014-0434-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neve R. M., Chin K., Fridlyand J., Yeh J., Baehner F. L., Fevr T., Clark L., Bayani N., Coppe J. P., Tong F., Speed T., Spellman P. T., DeVries S., Lapuk A., Wang N. J., et al. (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527 10.1016/j.ccr.2006.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang L. J., Wang L., Ma Y., Durick K., Perkins G., Deerinck T. J., Ellisman M. H., and Taylor S. S. (1999) NH2-Terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (D-AKAP1) to either mitochondria or endoplasmic reticulum. J. Cell Biol. 145, 951–959 10.1083/jcb.145.5.951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ma Y., and Taylor S. S. (2008) A molecular switch for targeting between endoplasmic reticulum (ER) and mitochondria: Conversion of a mitochondria-targeting element into an ER-targeting signal in DAKAP1. J. Biol. Chem. 283, 11743–11751 10.1074/jbc.M710494200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merrill R. A., and Strack S. (2014) Mitochondria: A-kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int. J. Biochem. Cell Biol. 48, 92–96 10.1016/j.biocel.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Livigni A., Scorziello A., Agnese S., Adornetto A., Carlucci A., Garbi C., Castaldo I., Annunziato L., Avvedimento E. V., and Feliciello A. (2006) Mitochondrial AKAP121 links cAMP and src signaling to oxidative metabolism. Mol. Biol. Cell 17, 263–271 10.1091/mbc.e05-09-0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saris N. E., and Carafoli E. (2005) A historical review of cellular calcium handling, with emphasis on mitochondria. Biochemistry (Mosc.) 70, 187–194 [DOI] [PubMed] [Google Scholar]
- 42. Hoppins S. (2014) The regulation of mitochondrial dynamics. Curr. Opin. Cell Biol. 29, 46–52 10.1016/j.ceb.2014.03.005 [DOI] [PubMed] [Google Scholar]
- 43. Liu X., Weaver D., Shirihai O., and Hajnóczky G. (2009) Mitochondrial “kiss-and-run”: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 28, 3074–3089 10.1038/emboj.2009.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Caino M. C., Seo J. H., Aguinaldo A., Wait E., Bryant K. G., Kossenkov A. V., Hayden J. E., Vaira V., Morotti A., Ferrero S., Bosari S., Gabrilovich D. I., Languino L. R., Cohen A. R., and Altieri D. C. (2016) A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 7, 13730 10.1038/ncomms13730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Caino M. C., Seo J. H., Wang Y., Rivadeneira D. B., Gabrilovich D. I., Kim E. T., Weeraratna A. T., Languino L. R., and Altieri D. C. (2017) Syntaphilin controls a mitochondrial rheostat for proliferation-motility decisions in cancer. J. Clin. Invest. 127, 3755–3769 10.1172/JCI93172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Campello S., Lacalle R. A., Bettella M., Mañes S., Scorrano L., and Viola A. (2006) Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J. Exp. Med. 203, 2879–2886 10.1084/jem.20061877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cunniff B., McKenzie A. J., Heintz N. H., and Howe A. K. (2016) AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol. Biol. Cell 27, 2662–2674 10.1091/mbc.e16-05-0286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao J., Zhang J., Yu M., Xie Y., Huang Y., Wolff D. W., Abel P. W., and Tu Y. (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824 10.1038/onc.2012.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu K., Chen G., Li X., Wu X., Chang Z., Xu J., Zhu Y., Yin P., Liang X., and Dong L. (2017) MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 7, 41718 10.1038/srep41718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chang C. R., and Blackstone C. (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587 10.1074/jbc.C700083200 [DOI] [PubMed] [Google Scholar]
- 51. Cribbs J. T., and Strack S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 10.1038/sj.embor.7401062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smirnova E., Griparic L., Shurland D. L., and van der Bliek A. M. (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256 10.1091/mbc.12.8.2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gomes L. C., Di Benedetto G., and Scorrano L. (2011) During autophagy, mitochondria elongate are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 10.1038/ncb2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li J., Huang Q., Long X., Guo X., Sun X., Jin X., Li Z., Ren T., Yuan P., Huang X., Zhang H., and Xing J. (2017) Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene 36, 4901–4912 10.1038/onc.2017.98 [DOI] [PubMed] [Google Scholar]
- 55. Rambold A. S., Kostelecky B., Elia N., and Lippincott-Schwartz J. (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. U.S.A. 108, 10190–10195 10.1073/pnas.1107402108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Flippo K. H., Gnanasekaran A., Perkins G. A., Ajmal A., Merrill R. A., Dickey A. S., Taylor S. S., McKnight G. S., Chauhan A. K., Usachev Y. M., and Strack S. (2018) AKAP1 protects from cerebral ischemic stroke by inhibiting Drp1-dependent mitochondrial fission. J. Neurosci. 38, 8233–8242 10.1523/JNEUROSCI.0649-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McKenzie A. J., Campbell S. L., and Howe A. K. (2011) Protein kinase A activity and anchoring are required for ovarian cancer cell migration and invasion. PLoS ONE 6, e26552 10.1371/journal.pone.0026552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Desai S. P., Bhatia S. N., Toner M., and Irimia D. (2013) Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys. J. 104, 2077–2088 10.1016/j.bpj.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prat A., Parker J. S., Karginova O., Fan C., Livasy C., Herschkowitz J. I., He X., and Perou C. M. (2010) Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 12, R68 10.1186/bcr2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Carlucci A., Adornetto A., Scorziello A., Viggiano D., Foca M., Cuomo O., Annunziato L., Gottesman M., and Feliciello A. (2008) Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 27, 1073–1084 10.1038/emboj.2008.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DeBerardinis R. J., and Chandel N. S. (2016) Fundamentals of cancer metabolism. Sci. Adv. 2, e1600200 10.1126/sciadv.1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johnson L. V., Walsh M. L., Bockus B. J., and Chen L. B. (1981) Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 88, 526–535 10.1083/jcb.88.3.526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Aggarwal-Howarth S., and Scott J. D. (2017) Pseudoscaffolds and anchoring proteins: The difference is in the details. Biochem. Soc. Trans. 45, 371–379 10.1042/BST20160329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alto N. M., Soderling J., and Scott J. D. (2002) Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J. Cell Biol. 158, 659–668 10.1083/jcb.200204081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kovanich D., van der Heyden M. A., Aye T. T., van Veen T. A., Heck A. J., and Scholten A. (2010) Sphingosine kinase interacting protein is an A-kinase anchoring protein specific for type I cAMP-dependent protein kinase. Chembiochem 11, 963–971 10.1002/cbic.201000058 [DOI] [PubMed] [Google Scholar]
- 66. Means C. K., Lygren B., Langeberg L. K., Jain A., Dixon R. E., Vega A. L., Gold M. G., Petrosyan S., Taylor S. S., Murphy A. N., Ha T., Santana L. F., Tasken K., and Scott J. D. (2011) An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, E1227–1235 10.1073/pnas.1107182108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pidoux G., Witczak O., Jarnæss E., Myrvold L., Urlaub H., Stokka A. J., Küntziger T., and Taskén K. (2011) Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. 30, 4371–4386 10.1038/emboj.2011.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dagda R. K., Cherra S. J. 3rd, Kulich S. M., Tandon A., Park D., and Chu C. T. (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 284, 13843–13855 10.1074/jbc.M808515200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pryde K. R., Smith H. L., Chau K. Y., and Schapira A. H. (2016) PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J. Cell Biol. 213, 163–171 10.1083/jcb.201509003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cardone L., Carlucci A., Affaitati A., Livigni A., DeCristofaro T., Garbi C., Varrone S., Ullrich A., Gottesman M. E., Avvedimento E. V., and Feliciello A. (2004) Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol. Cell. Biol. 24, 4613–4626 10.1128/MCB.24.11.4613-4626.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Glaven J. A., Whitehead I. P., Nomanbhoy T., Kay R., and Cerione R. A. (1996) Lfc and Lsc oncoproteins represent two new guanine nucleotide exchange factors for the Rho GTP-binding protein. J. Biol. Chem. 271, 27374–27381 10.1074/jbc.271.44.27374 [DOI] [PubMed] [Google Scholar]
- 72. Meiri D., Greeve M. A., Brunet A., Finan D., Wells C. D., LaRose J., and Rottapel R. (2009) Modulation of Rho guanine exchange factor Lfc activity by protein kinase A-mediated phosphorylation. Mol. Cell. Biol. 29, 5963–5973 10.1128/MCB.01268-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Steen R. L., Martins S. B., Tasken K., and Collas P. (2000) Recruitment of protein phosphatase 1 to the nuclear envelope by A-kinase anchoring protein AKAP149 is a prerequisite for nuclear lamina assembly. J. Cell Biol. 150, 1251–1261 10.1083/jcb.150.6.1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.