

Abstract
Obesity is a major risk for patients with chronic metabolic disorders including type 2 diabetes. Sonic hedgehog (Shh) is a morphogen that regulates the pancreas and adipose tissue formation during embryonic development. Peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear receptor superfamily and one of the most important regulators of insulin action. Here, we evaluated the role and mechanism of Shh signaling in obesity-associated insulin resistance and characterized its effect on PPARγ. We showed that Shh expression was up-regulated in subcutaneous fat from obese mice. In differentiated 3T3-L1 and primary cultured adipocytes from rats, recombinant Shh protein and SAG (an agonist of Shh signaling) activated an extracellular signal–regulated kinase (ERK)-dependent noncanonical pathway and induced PPARγ phosphorylation at serine 112, which decreased PPARγ activity. Meanwhile, Shh signaling degraded PPARγ protein via binding of PPARγ to neural precursor cell-expressed developmentally down-regulated protein 4-1 (NEDD4-1). Furthermore, vismodegib, an inhibitor of Shh signaling, attenuated ERK phosphorylation induced by a high fat diet (HFD) and restored PPARγ protein level, thus ameliorating glucose intolerance and insulin resistance in obese mice. Our finding suggests that Shh in subcutaneous fat decreases PPARγ activity and stability via activation of an ERK-dependent noncanonical pathway, resulting in impaired insulin action. Inhibition of Shh may serve as a potential therapeutic approach to treat obesity-related diabetes.
Keywords: sonic hedgehog (SHH), peroxisome proliferator-activated receptor (PPAR), protein degradation, ubiquitylation (ubiquitination), obesity, insulin resistance, type 2 diabetes, NEDD4-1
Introduction
Insulin is a crucial anabolic hormone that stimulates the metabolism of carbohydrates, lipids, and proteins. Adipose tissue receives insulin signals and is responsible for plasma glucose clearance by stimulating its uptake and utilization (1). An inability of tissues to adequately respond to insulin leads to impaired glucose homeostasis and the pathogenesis of type 2 diabetes (2). Obesity resulting from inappropriate food intake is commonly accompanied by metabolic disorders, such as electrolyte imbalance, hyperuricemia, dyslipidemia, and insulin resistance (3). Type 2 diabetes is frequently associated with an accumulation of visceral adipose tissue (4). Nonetheless, the predominant storage of fat in the subcutaneous adipose tissue is indicative of a better metabolic profile (5). The molecular mechanism behind these clinical observations remains unknown. In adipocytes, insulin signaling starts from the binding of insulin to its receptor, leading to the phosphorylation of insulin receptor substrate-1 (IRS-1).2 Activated phosphoinositide 3-kinase (PI3K) triggers the production of specific phosphoinositides, which recruit protein kinase B (Akt) to the plasma membrane (6). Subsequent Akt phosphorylation is responsible for most of the metabolic actions of insulin, such as glucose uptake by inducing the translocation of glucose transporter 4 from intracellular storage to the plasma membrane (7). Thus, Akt phosphorylation normally serves as a downstream marker of insulin. ERK and c-Jun N-terminal kinase (JNK) activations negatively regulate insulin signaling through serine phosphorylation of IRS-1 (8).
Hedgehog (Hh) proteins, including Shh, Indian hedgehog (Ihh), and Desert hedgehog (Dhh), were initially defined as morphogens that regulate the embryo development (9). In mammals, Hh binding to the inhibitory receptor Patched leads to the activation of the cell-surface receptor Smoothened (Smo) and subsequently drives the transcription of Gli target genes (10). Although most Hh-induced biological effects result from the transcriptional program, several noncanonical pathways have been reported, such as mitogen-activated protein kinase ERK (11). Hh signaling is normally quiescent in adults, except during various pathologies, such as cancers (12), immune-demyelinating neuropathy (13), and polycystic kidney disease (14). Shh inhibits adipocyte differentiation by diverting preadipocytes away from adipogenesis (15), which is implicated in the development of white adipose tissue (16). The Hh-Gli2 axis drives lipogenesis in adipocytes, leading to increased obesity in adult mice (17). Moreover, Shh-related ciliopathy is involved in obesity, cognitive impairment, and limb deformities, which has been defined as Bardet-Biedl syndrome in humans (18). Shh regulates epithelial and β-cell expansion during early pancreas morphogenesis (19). Excessively activated Hh signaling impairs β-cell function and insulin secretion, resulting in glucose intolerance in adult mice (20, 21). Furthermore, hepatic Hh signaling contributes to the progression of nonalcoholic fatty liver diseases by promoting liver inflammation (22). Similarly, its signaling in myeloid cells of adipose tissue is suggested to be a trigger of type 2 diabetes (23).
PPARγ is a member of the nuclear receptor superfamily and a master regulator of insulin action. Although specific deletion of PPARγ in muscle, liver, macrophages, or brain alters glucose homeostasis and induces insulin resistance (24–28), adipose tissue is the major site for insulin sensitizing by PPARγ (29). Rosiglitazone, a selective agonist of PPARγ, has been used to treat type 2 diabetes with a glucose lowering effect. Due to side effects including heart failure, rosiglitazone has been withdrawn from the first-line therapy in clinical practice (30). Hence, identifying PPARγ regulators as therapeutic targets would be a novel approach in the treatment of type 2 diabetes.
In this study, we demonstrate that Shh in subcutaneous adipose tissue decreases the activity and stability of PPARγ by activating the ERK-dependent noncanonical pathway. Conversely, vismodegib, an inhibitor of Shh signaling, improves obesity-related insulin resistance by stabilizing PPARγ.
Results
Shh is induced in subcutaneous adipose tissue from obese mice
To determine whether Hh ligands are implicated in obesity, C57Bl/6 mice were first fed with a HFD for 12 weeks. The mice developed pronounced obesity (Fig. S1A) with no difference in food intake (Fig. S1B). Glucose tolerance test (Fig. S1C) and insulin tolerance test (Fig. S1D) revealed impaired insulin sensitivity in these mice. Next, we measured the expressions of Shh, Ihh, and Dhh in subcutaneous, epididymal and brown adipose tissues. ELISA was used to detect the protein level of Shh because of its better sensitivity. Compared with lean mice, both mRNA (Fig. 1A) and protein (Fig. 1B) levels of Shh were significantly increased in subcutaneous fat from obese mice. Although a slight augmentation of Shh mRNA was observed in epididymal and brown adipose tissues, no difference was found at the protein level. No differential expression of Ihh (Fig. 1C) and Dhh (Fig. 1D) was observed in subcutaneous, epididymal, and brown adipose tissues.
Figure 1.

Expression of Hh ligands in adipose tissues of obese mice. Data were obtained from mice fed with a ND or HFD for 12 weeks (n = 6 in each group). Levels of Shh mRNA (A) and protein (B) in subcutaneous (Sub), epididymal (Epi), and brown adipose tissues from lean and obese mice were measured using RT-qPCR and ELISA, respectively. Ihh (C) and Dhh (D) mRNA levels in subcutaneous, epididymal, and brown adipose tissues are shown. The results were normalized to the level of cyclophilin mRNA. *, p < 0.05; NS, no significance.
Shh decreases PPARγ protein level via an ERK-dependent noncanonical pathway
To determine the involvement of Shh signaling in adipocytes, we treated the differentiated 3T3-L1 adipocytes with recombinant Shh protein or SAG, a Smo agonist, for the indicated times or with different doses. Recombinant Shh and SAG activated the mitogen-activated protein kinase pathway with a maximum ERK phosphorylation at 1 h after stimulation (Fig. 2A). ERK phosphorylation induced by recombinant Shh and SAG occurred in a dose-dependent manner (Fig. 2B). In primary rat adipocytes, ERK phosphorylation was also increased by recombinant Shh and SAG (Fig. 2C). These results indicate that Shh signaling activates the ERK pathway in adipocytes.
Figure 2.

PPARγ protein level is decreased via the ERK pathway in adipocytes. A-C, differentiated 3T3-L1 adipocytes were serum-starved and then incubated with Shh (0.5 μg/ml) or SAG (0.5 μmol/liter) for the indicated times (A). Serum-starved 3T3-L1 adipocytes were treated with different concentrations of Shh (0, 0.1, 0.2, 0.5, 1, and 2 μg/ml) or SAG (0, 0.1, 0.2, 0.5, 1, and 2 μmol/liter) for 60 min (B). C, primary rat adipocytes (2 × 105 cells/well) after serum starvation were treated with Shh (0.5 μg/ml) or SAG (0.5 μmol/liter) for 60 min. The level of p-ERK and ERK was analyzed using immunoblotting. D–F, before incubation with Shh (0.5 μg/ml) for 60 min, 3T3-L1 or primary rat adipocytes were transfected with siRNA against Smo (D) or pretreated with vismodegib (100 nmol/liter, E) for 30 min. F, PD98059 (PD, 10 μmol/liter)-pretreated cells were exposed to SAG (0.5 μmol/liter) for 60 min. Levels of p-PPARγ and PPARγ was analyzed by using immunoblotting. G–I, Smo siRNA-transfected (G) or vismodegib-pretreated (H) cells were incubated with Shh (0.5 μg/ml) for 24 h. I, PD98059 (PD, 10 μmol/liter)-pretreated cells were exposed to SAG (0.5 μmol/liter) for 24 h. Levels of PPARγ and β-actin were analyzed using immunoblotting. Immunoblots shown are representative of 3 independent experiments. J, differentiated 3T3-L1 (left) and primary rat adipocytes (right) were pretreated with or without PD98059 (PD, 10 μmol/liter) for 30 min, and then exposed to SAG (0.5 μmol/liter) for 24 h. Cell lysates were analyzed to determine the mRNA level of Gli1. The results were normalized to the level of cyclophilin mRNA. Data were from 3 independent experiments performed in triplicate. *, p < 0.05; NS, no significance.
Because Ser-112 of PPARγ negatively regulates insulin action in adipocytes (31), we examined the effect of recombinant Shh protein on p-PPARγSer-112. Recombinant Shh increased PPARγ phosphorylation at Ser-112 in differentiated 3T3-L1 and primary rat adipocytes. This phosphorylation was attenuated by siRNA against Smo (Fig. 2D) or vismodegib (Fig. 2E), indicating a Smo-dependent mechanism. The efficiency of siRNA against Smo was tested by RT-qPCR (Fig. S2). Moreover, SAG increased p-PPARγSer-112, which was attenuated by PD98059, an ERK inhibitor. This indicates that p-PPARγSer-112 depends on the Shh-activated ERK pathway (Fig. 2F). In addition, Shh decreased the PPARγ protein level 24 h after stimulation. Smo siRNA (Fig. 2G) or vismodegib restored Shh-decreased PPARγ (Fig. 2H). In parallel, SAG-decreased PPARγ was restored by PD98059 (Fig. 2I), demonstrating that ERK phosphorylation by Shh signaling is involved in PPARγ degradation.
To study the cross-talk between the ERK-dependent noncanonical and Gli-dependent canonical pathways we investigated the effect of PD98059 on Gli1 activation in 3T3-L1 and primary rat adipocytes. SAG-induced Gli1 activation was not attenuated by PD98059 (Fig. 2J). These data confirm that ERK phosphorylation and Gli activation are 2 independent pathways. Taken together, these findings suggest that Shh-increased ERK phosphorylation promotes PPARγ phosphorylation at Ser-112 and decreases the protein level of PPARγ.
Shh decreases PPARγ stability via a ubiquitin-dependent mechanism
To explore how Shh signaling decreased the PPARγ protein level, PPARγ-transfected HEK 293 cells were pretreated with cycloheximide and then exposed to SAG. As shown in Fig. 3A, SAG significantly increased PPARγ degradation. Pretreatment with the proteasome inhibitor MG132 significantly prevented the SAG-triggered decrease in PPARγ protein level (Fig. 3B), indicating a role of the proteasome in PPARγ degradation. Depletion of the E3 ubiquitin ligase NEDD4-1 led to PPARγ accumulation (Fig. 3C). Co-immunoprecipitation indicated that the binding of NEDD4-1 to PPARγ was largely augmented by SAG treatment (Fig. 3D). Next, we determined if Shh signaling promoted PPARγ ubiquitination. The level of ubiquitinated PPARγ was markedly higher in SAG-treated cells (Fig. 3E). Collectively, these data indicate that Shh signaling enhances PPARγ degradation via NEDD4-1–dependent ubiquitination.
Figure 3.

Shh signaling decreased PPARγ stability via NEDD4-1–dependent ubiquitination. HEK 293 cells were transfected with PPARγ overexpression plasmid. A, cells were pretreated with cyclohexamide (CHX) (5 μg/ml) for 30 min before exposure to SAG (0.5 μmol/liter), followed by immunoblotting to detect PPARγ and β-actin (left). Quantifications of band intensity normalized to β-actin (right). B, cells were incubated with SAG (0.5 μmol/liter) for 24 h in the presence or absence of MG132 (10 μmol/liter) pretreatment. C, cells were infected with scramble shRNA lentivirus or different lentiviral shRNA constructs against NEDD4-1. PPARγ, NEDD4-1, and β-actin levels were analyzed by immunoblotting. D, immunoblotting (IB) of whole cell lysates (Input) and immunoprecipitates (IP) from PPARγ-overexpressing HEK 293 cells with or without SAG (0.5 μmol/liter) treatment for 24 h. MG132 (10 μmol/liter) was added to the medium 12 h before collecting. E, PPARγ-transfected HEK 293 cells were pretreated with MG132 (10 μmol/liter) and then incubated with or without SAG (0.5 μmol/liter) for 24 h. PPARγ was immunoprecipitated from cell lysates and immunoblotted with an anti-ubiquitin antibody (left). Band intensities were normalized to that of IgG (right). Immunoblots shown are representative of 3 independent experiments. *, p < 0.05.
Vismodegib improves insulin sensitivity in obese mice induced by a HFD
We assessed the potential therapeutic effects of vismodegib on insulin resistance in mice fed a HFD. Vismodegib treatment slightly decreased the body weight of both mice fed with a normal (ND) or HFD. No statistical differences were observed between the two groups (Fig. 4A). Vismodigib did not affect body weight in either ND- or HFD-fed mice after 16 days of treatment (Fig. 4B). Food intake did not differ with vismodegib treatment (Fig. 4C). However, vismodegib significantly improved HFD-induced impairment of glucose tolerance (Fig. 4D) and insulin sensitivity (Fig. 4E). A HFD impaired insulin secretion after glucose stimulation as assessed by the insulin level in the blood. Vismodegib did not affect the glucose-induced insulin level in mice fed with either a ND or HFD (Fig. 4F). Accordingly, the level of p-Akt (Ser-473), a downstream marker of insulin signaling, was increased in ND-fed mice with vismodegib treatment compared with the vehicle group. Importantly, vismodegib restored the HFD-attenuated p-Akt level (Fig. 4G). These results indicate that inhibition of Shh signaling improves HFD-induced insulin resistance.
Figure 4.

Vismodegib improved insulin resistance in obese mice. Mice fed a ND or HFD for 12 weeks (n = 6 in each group) were injected with vismodegib every other day at a dose of 5 mg/kg. A, body weight was measured before every injection. NS: ND V.S. ND+vismo, NS: HFD V.S. HFD+vismo. B, body weights of ND- or HFD-fed mice before and after vismodegib treatment. C, level of average food intake during vismodegib treatment. Glucose tolerance test (D) and insulin tolerance test (E) of ND- or HFD-fed mice with or without vismodegib treatment. *, HFD V.S. HFD+vismo. Areas under the curve (AUC) were determined. F, level of serum insulin in ND- or HFD-fed mice with vismodegib treatment before and after insulin injection (30 min). G, protein levels of p-Akt and Akt in subcutaneous adipose tissue were measured by immunoblotting. Quantifications of band intensity was normalized to β-actin (right). F, tail vein blood was sampled before and 30 min after glucose injection for the measurement of insulin level. *, p < 0.05; NS, no significance.
Vismodegib inhibits ERK activation in subcutaneous adipose tissue of obese mice
Because Gli1 induction is a main reporter of Hh signaling activation, we investigated the effect of vismodegib on Gli1 activation. Expression of Gli1 was up-regulated in adipose tissue of HFD-fed mice. Surprisingly, the induction was not attenuated by vismodegib (Fig. 5A), potentially implying noncanonical pathways. Vismodegib significantly attenuated ERK phosphorylation in HFD-fed mice (Fig. 5B).
Figure 5.

Vismodegib treatment affected the ERK pathway in obese mice. Mice fed a ND or HFD for 12 weeks (n = 6 in each group) were injected with vismodegib every other day at a dose of 5 mg/kg. After sacrifice, the subcutaneous adipose tissues were immediately dissected and underwent quick-freezing in liquid nitrogen. A, level of Gli1 mRNA was measured by RT-qPCR. The result was normalized to the level of cyclophilin. B, protein levels of p-ERK and ERK in subcutaneous adipose tissue were measured by immunoblotting. Quantification of band intensity normalized to β-actin (right). *, p < 0.05; NS, no significance.
Vismodegib restores PPARγ protein level in adipose tissue of obese mice
To confirm the effect of vismodegib on the decreased PPARγ protein level as demonstrated in vitro, we investigated the PPARγ level in subcutaneous fat of mice fed with a ND and HFD. Compared with the vehicle group, the PPARγ protein level did not significantly change in ND mice upon vismodegib treatment. However, vismodegib significantly restored the PPARγ protein level in HFD mice (Fig. 6A). Vismodegib did not affect the PPARγ mRNA level in either ND- or HFD-fed mice (Fig. 6B). Furthermore, it also restored HFD-decreased expression of PPARγ target genes, including adiponectin and CD36 (Fig. 6C).
Figure 6.

Vismodegib restored PPARγ protein levels in subcutaneous fat of obese mice. Mice fed a ND or HFD for 12 weeks (n = 6 in each group) were injected with vismodegib every other day at a dose of 5 mg/kg. After sacrifice, the subcutaneous adipose tissues were immediately dissected and underwent quick-freezing in liquid nitrogen. The protein level of PPARγ in subcutaneous adipose tissue was measured by immunoblotting (left). Quantification of band intensity normalized to β-actin (right). The mRNA levels of PPARγ (B) and PPARγ-targeted genes (C) were measured by RT-qPCR. The results were normalized to the level of cyclophilin. *, p < 0.05; NS, no significance.
Discussion
Obesity is a medical condition associated with the accumulation of excess body fat, triggering metabolic disorders, such as type 2 diabetes (32). Perturbation of fatty acid metabolism is a major factor contributing to whole-body insulin resistance. Fatty acid-induced inflammation and cellular lipid overload inhibit insulin signaling by triggering endoplasmic reticulum stress and oxidative stress (33).
Clinical studies have demonstrated that excess accumulation of visceral adipose tissue is associated with cardio-metabolic risk factors (4). Subcutaneous adipose tissue shows a better metabolic outcome (5). The underlying molecular mechanism remains poorly understood. Our study provides a detailed analyses of the expression of three Hh ligands in epididymal, subcutaneous, and brown fat tissues. Here, we report that Shh expression was increased in subcutaneous fat, whereas it remained unchanged in epididymal and brown fat. This may explain the observation that the PPARγ protein level was decreased mainly in subcutaneous adipose tissue. We did not detect differential expression of Ihh or Dhh in any of the three adipose tissues. In mammals, the 3 Hh genes are located on different chromosomes but have highly conserved sequences at the protein level. Shh shares 91 and 76% identity with Ihh and Dhh, respectively. Ihh has 80% identity with Dhh (34). All three ligands share the same downstream signaling.
Although investigations typically rely on the Gli-dependent canonical pathway, several noncanonical signaling pathways have been reported. For instance, rapid induction of Src family kinases by Shh is implicated in spatial axonal outgrowth (35). Hh signaling triggers Warburg-like metabolic reprogramming via a Smo–Ca2+–AMP-activated protein kinase axis (36). Shh, Ihh, and Dhh drive cytoskeletal rearrangement and endothelial migration via a Smo-Rho A axis (37). Furthermore, Shh activates PI3K to promote mural cell migration and recruitment into neovessels (11). We found that ERK phosphorylation was enhanced by Shh signaling in adipocytes. This noncanonical pathway of ERK activation was mediated by Smo, a member of the G protein–coupled receptor superfamily.
The nuclear receptor PPARγ is a transcription factor and a master regulator of adipogenesis and glucose metabolism (38). In addition to its transcriptional regulation, activity and stability of the PPARγ protein are under precise posttranscriptional modifications, including phosphorylation, ubiquitination, SUMOylation, O-GlcNAcylation, and S-nitrosylation (27, 39–41). Site-specific phosphorylation of PPARγ at Ser-112 by MAPK inhibits PPARγ activity during adipogenesis and insulin action (42), whereas PPARγ agonists cause its phosphorylation at serine 273 by cyclin-dependent kinase 5, leading to its nuclear translocation (31). Our data demonstrated that Shh signaling–induced PPARγ phosphorylation at Ser-112 was responsible for the decreased PPARγ activity in adipocytes. In parallel, the Shh/Smo/ERK axis decreased the PPARγ protein level. Furthermore, the E3 ubiquitin ligases, CUL4B, FBXO9, MuRF2, MuRF3, and Siah2 regulate PPARγ ubiquitination in adipocytes, thus decreasing adipogenesis and lipogenesis (40, 43–46). Other E3 ligases, such as MKRN1 and MDM2, are also involved in PPARγ ubiquitination and proteasome-dependent degradation (47, 48). NEDD4-1, homologous to the E6-AP C terminus-type E3 ubiquitin ligase, plays crucial roles in mediating ubiquitin-dependent trafficking and degradation (49). It has been shown to target PPARγ via binding to a highly conserved proline–proline–X–tyrosine (PPXY) motif (50, 51). We found that the Shh-driven PPARγ degradation was mediated by a ubiquitin-dependent pathway via binding of PPARγ to NEDD4-1 because knockdown of NEDD4-1 preserved the PPARγ protein level. However, the exact link between the Shh-triggered phosphorylation and NEDD4-1–mediated PPARγ ubiquitination warrant further investigation.
One question of interest is whether other nuclear receptors could be regulated by the Hh-triggered and phosphorylation-dependent ubiquitination. In this regard, we looked at the PPARδ protein level, another member of the PPAR subfamily expressed in adipose tissues. Vismodegib did not have a significant effect on the PPARδ protein level in adipose tissues of either ND-fed or HFD mice (Fig. S3). The third member of this subfamily, PPARα, is mainly expressed in the liver, but not in adipose tissue (52). RXRα, the dimerization partner of PPARs, is not affected by a HFD in mice (53). PPARγ degradation by Shh signaling may explain the impaired formation of the PPARγ/RXR heterodimer in insulin resistance. In addition, LXR agonists increase GLUT4 expression, resulting in insulin-mediated glucose uptake into adipose tissue (54). SREBP is also a key transcription factor controlling the synthesis of cholesterol and triglycerol (55). Potential cross-talk between Hh signaling and these metabolic regulators certainly warrants further investigation.
Recently, the Hh signaling pathway was implicated in metabolism and the pathogeneses of several metabolic diseases. Shh signaling switches preadipocytes away from adipogenesis and instead drives them toward osteogenesis. Shh-related ciliopathy is involved in Bardet-Biedl syndrome in humans with severe obesity (18). The Hh-activated Smo–Ca2+–AMP-activated protein kinase axis is involved in Warburg-like metabolic reprogramming (36). Hh signaling promotes the hepatic inflammatory state and controls the progression of nonalcoholic fatty liver diseases (22). Insulin resistance is triggered by Hh signaling in inflammatory cells of obese mice (23). In this study, we provide evidence that Shh-mediated PPARγ ubiquitination via NEDD4-1 is a novel mechanism to illustrate the impaired insulin action in type 2 diabetes.
Vismodegib is the first Hh pathway inhibitor approved in the United States and Europe for the treatment of basal cell carcinoma (56). Recently, Kwon et al. (22) found that intraperitoneal administration of vismodegib decreases hepatic fat accumulation in mice via anti-inflammatory effects. In our study, vismodegib slightly decreased body weight in both ND-fed and HFD mice with statistical significance observed only at a single time point. However, the body weights before and after treatment were not statistically significantly different. Because the sample size was relatively small, we cannot conclude that vismodegib affects body weight. In addition to demonstrating a novel mechanism by which Hh signaling attenuates PPARγ action, our study also provides the first proof of concept that Hh inhibition may represent a new therapeutic strategy to treat metabolic disease featuring with insulin resistance.
Taken together, our data demonstrate that Shh signaling in subcutaneous adipose tissue leads to PPARγ degradation via NEDD4-1–mediated ubiquitination. These results also provide evidence that Shh-induced ERK phosphorylation is a trigger of decreased PPARγ activity and stability (Fig. 7). Improved insulin sensitivity by vismodegib treatment indicates that Shh signaling could be a potential therapeutic target in type 2 diabetes.
Figure 7.
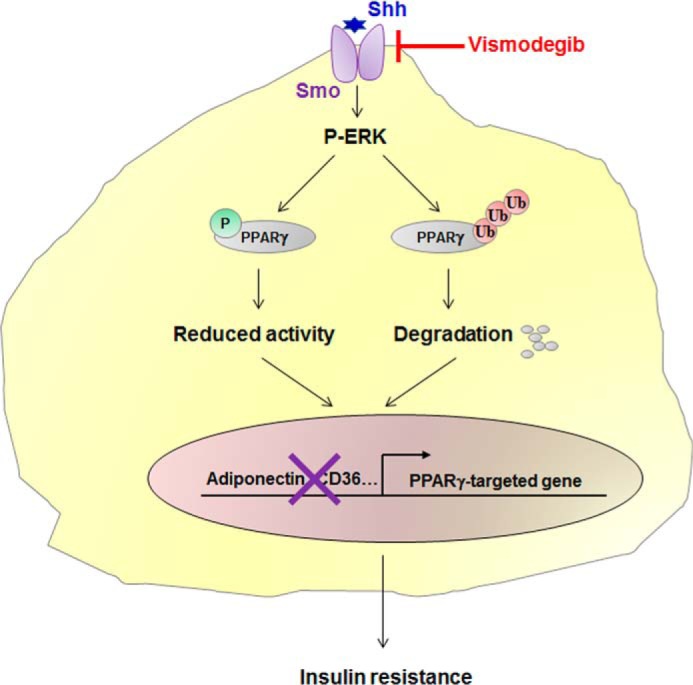
Role of Shh signaling in obesity-induced insulin resistance. Increased Shh expression in subcutaneous fat of obese mice activates p-ERK, which phosphorylates PPARγ at serine 112, leading to an impaired PPARγ activity. On the other hand, Shh-activated ERK phosphorylation promotes NEDD4-1–dependent PPARγ ubiquitination and degradation. Transcription of PPARγ-targeted genes is therefore reduced, leading to an impaired insulin action. Thus, by targeting the Shh receptor Smo, vismodegib instigates HFD–induced insulin resistance in obese mice.
Experimental procedures
Reagents
Murine recombinant Shh protein was from Shenandoah Biotechnology, Warwick, PA (catalog number 200-55), and dissolved in PBS containing 0.2% BSA. Vismodegib, SAG, and PD98059 were from Selleck Chemicals (Houston, TX). Cyclohexamide was from Cayman Chemical (Ann Arbor MI). Antibodies against p-Akt, Akt, p-ERK, ERK, p-JNK, JNK, PPARγ, and NEDD4-1 were from Cell Signaling Biotechnology (Danvers, MA). Rabbit IgG and antibody against β-actin were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture
3T3-L1 preadipocytes and HEK 293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FBS in a humidified 5% CO2 atmosphere at 37 °C. To obtain mature adipocytes, 3T3-L1 cells (1.5 × 105/well) were differentiated in 12-well plates by supplementing with 3-isobutyl-1-methylxanthine, insulin, and dexamethasone.
To obtain primary rat adipocytes, male Sprague-Dawley rats were sacrificed, and the epididymal adipose tissues were then dissected. After incubation with type I collagenase (1 mg/ml) and 1% BSA, the homogenate was filtered through a 50-μm nylon mesh and washed three times in PBS containing 1% BSA. After centrifugation, the adipocytes were re-suspended in Dulbecco's modified Eagle's medium:F-12 medium containing 20% FBS and then incubated in a humidified 5% CO2 atmosphere at 37 °C. Cells were seeded into 12-well plates (2 × 105 cells/well). Differentiated 3T3-L1 or primary rat adipocytes were synchronized with serum starvation (0.5% FBS) for 12 h treatment with vismodegib (100 nmol/liter), PD98059 (10 μmol/liter), recombinant Shh protein (0.5 μg/ml), or SAG (0.5 μmol/liter).
Animal procedures
Male Sprague-Dawley rats and C57BL/6 mice were bred with controlled temperature (25 °C) and a 12-h light and dark cycle. Animal experiments were approved by the institutional ethics review board of Xi'an Jiaotong University (XJTULAC2015-404), and performed in accordance with the NIH guidelines for the care and use of animals. Mice at 8 weeks were fed a HFD (60% fat) or kept on ND (Research Diets, New Brunswick, NJ). Twelve weeks later, mice were treated with intraperitoneal injections of vehicle or vismodegib (5 mg/kg) every other day. Food intake and body weight were measured before every injection and followed every other day.
Glucose tolerance test and insulin tolerance test
For the glucose tolerance test, mice were food-deprived overnight for 14 h with free water supply. Glucose (2 mg/kg) was intraperitoneally injected and blood glucose was determined at 15, 30, 60, and 120 min after the injection. For the insulin tolerance test, mice intraperitoneally received recombinant insulin (0.8 units/kg, Humulin, Eli Lilly and Co., IN). Blood glucose was determined at 0, 15, 30, 60, and 120 min after the injection.
Transfection and lentiviral infection
HEK 293 cells were transfected with a PPARγ expression plasmid using Lipofectamine 2000 (Invitrogen). Experiments were performed 48 h post-transfection. Lentiviruses expressing shRNA–NEDD4-1 were packaged by transfecting 293T cells with pLKO–shRNA constructs. Lentiviral infection was performed using virus-containing media with 4 mg/ml of Polybrene.
RNA extraction, reverse transcriptase-quantitative PCR (RT-qPCR)
Total RNA was extracted by using TRIzol (Invitrogen). RT-qPCR was performed using SYBR Green (Promega, Madison, WI). Primer sequences were described in Table S1. Cyclophilin was used as an internal control.
Immunoblotting, enzyme-linked immunosorbent assay (ELISA), and immunoprecipitation
Proteins were extracted in RIPA buffer supplemented with protease and phosphatase inhibitors. Protein concentrations were measured using the BCA protein assay. Immunoblotting was performed with appropriate primary antibodies and horseradish peroxidase-conjugated secondary antibodies followed by ECL detection. The concentration of Shh protein and serum insulin level was measured using the ELISA kit (Elabsciences, Wuhan, China) according to the manufacturer's instructions.
For immunoprecipitation, cell lysates were incubated with the appropriate antibodies or control IgG at 4 °C overnight followed by incubation with protein A/G-Sepharose beads. Immunoprecipitates were washed with NETN buffer (100 mm NaCl, 1 mm EDTA, 20 mm Tris, pH 8.0, and 0.5% Nonidet P-40). Band intensity was quantified with ImageJ and normalized to loading control.
Statistical analyses
Results are reported as mean ± S.D. We examined the differences between groups using Student's t test (comparison of two groups) or one-way analysis of variance (comparisons more than two groups) test with a Bonferrioni test to correct for multiple comparison testing. For body weight, glucose tolerance test, and insulin tolerance test, we conducted a repeated measures analysis of variance followed by a LSD post hoc test and a Bonferrioni test to correct for multiple comparison testing. p values <0.05 were considered significant.
Author contributions
Q. Y. and N. W. conceptualization; Q. Y. and N. W. resources; Q. Y., J. L., and N. W. data curation; Q. Y. formal analysis; Q. Y. and N. W. supervision; Q. Y., J. L., L. X., and N. W. funding acquisition; Q. Y., J. L., and N. W. validation; Q. Y., J. L., and N. W. investigation; Q. Y. and N. W. visualization; Q. Y., J. L., L. X., and N. W. methodology; Q. Y. and N. W. writing-original draft; Q. Y. and N. W. writing-review and editing.
Supplementary Material
Acknowledgments
We thank MA.Sc Benjamin Peidis for English editing and MA.Sc Chao Zhang for statistical analyses.
This work was supported by National Science Foundation of China Grants 81830015, 81500345, 31430045, 81470373, 81600389, and 81770497 and General Financial Grants 2015M582674 and 2016T90928 from the China Postdoctoral Science Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3 and Table S1.
- IRS-1
- insulin receptor substrate protein-1
- Shh
- Sonic hedgehog
- Hh
- Hedgehog
- PPARγ
- peroxisome proliferator-activated receptor γ
- ERK
- extracellular signal-regulated kinase
- NEDD4-1
- neural precursor cell-expressed developmentally down-regulated protein 4-1
- HFD
- high fat diet
- ND
- normal chow diet
- PI3K
- phosphoinositide 3-kinase
- JNK
- c-Jun N-terminal kinase
- Ihh
- Indian hedgehog
- Dhh
- Desert hedgehog
- Smo
- Smoothened
- SAG
- Smo agonist
- RT-qPCR
- reverse transcriptase-quantitative PCR
- FBS
- fetal bovine serum
- Akt
- protein kinase B.
References
- 1. Tokarz V. L., MacDonald P. E., and Klip A. (2018) The cell biology of systemic insulin function. J. Cell Biol. 217, 2273–2289 10.1083/jcb.201802095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Page M. M., and Johnson J. D. (2018) Mild suppression of hyperinsulinemia to treat obesity and insulin resistance. Trends Endocrinol. Metab. 29, 389–399 10.1016/j.tem.2018.03.018 [DOI] [PubMed] [Google Scholar]
- 3. Koren D., and Taveras E. M. (2018) Association of sleep disturbances with obesity, insulin resistance and the metabolic syndrome. Metabolism 84, 67–75 10.1016/j.metabol.2018.04.001 [DOI] [PubMed] [Google Scholar]
- 4. Tchernof A., and Després J. P. (2013) Pathophysiology of human visceral obesity: an update. Physiol. Rev. 93, 359–404 10.1152/physrev.00033.2011 [DOI] [PubMed] [Google Scholar]
- 5. Abate N., Garg A., Peshock R. M., Stray-Gundersen J., and Grundy S. M. (1995) Relationships of generalized and regional adiposity to insulin sensitivity in men. J. Clin. Investig. 96, 88–98 10.1172/JCI118083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emanuel A. L., Meijer R. I., Muskiet M. H., van Raalte D. H., Eringa E. C., and Serne E. H. (2017) Role of insulin-stimulated adipose tissue perfusion in the development of whole-body insulin resistance. Arterioscler. Thromb. Vasc. Biol. 37, 411–418 10.1161/ATVBAHA.116.308670 [DOI] [PubMed] [Google Scholar]
- 7. Egawa K., Sharma P. M., Nakashima N., Huang Y., Huver E., Boss G. R., and Olefsky J. M. (1999) Membrane-targeted phosphatidylinositol 3-kinase mimics insulin actions and induces a state of cellular insulin resistance. J. Biol. Chem. 274, 14306–14314 10.1074/jbc.274.20.14306 [DOI] [PubMed] [Google Scholar]
- 8. Taniguchi C. M., Emanuelli B., and Kahn C. R. (2006) Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 10.1038/nrn1850,10.1038/nrm1837 [DOI] [PubMed] [Google Scholar]
- 9. Seppala M., Fraser G. J., Birjandi A. A., Xavier G. M., and Cobourne M. T. (2017) Sonic Hedgehog signaling and development of the dentition. J. Dev. Biol. 5, pii e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fernandes-Silva H., Correia-Pinto J., and Moura R. S. (2017) Canonical sonic Hedgehog signaling in early lung development. J. Dev. Biol. 5, pii e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yao Q., Renault M. A., Chapouly C., Vandierdonck S., Belloc I., Jaspard-Vinassa B., Daniel-Lamazière J. M., Laffargue M., Merched A., Desgranges C., and Gadeau A. P. (2014) Sonic hedgehog mediates a novel pathway of PDGF-BB-dependent vessel maturation. Blood 123, 2429–2437 10.1182/blood-2013-06-508689 [DOI] [PubMed] [Google Scholar]
- 12. Bushman W. (2016) Hedgehog signaling in prostate development, regeneration and cancer. J. Dev. Biol. 4, pii E30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chapouly C., Yao Q., Vandierdonck S., Larrieu-Lahargue F., Mariani J. N., Gadeau A. P., and Renault M. A. (2016) Impaired Hedgehog signalling-induced endothelial dysfunction is sufficient to induce neuropathy: implication in diabetes. Cardiovasc. Res. 109, 217–227 10.1093/cvr/cvv263 [DOI] [PubMed] [Google Scholar]
- 14. Quinlan R. J., Tobin J. L., and Beales P. L. (2008) Modeling ciliopathies: primary cilia in development and disease. Curr. Top. Dev. Biol. 84, 249–310 10.1016/S0070-2153(08)00605-4 [DOI] [PubMed] [Google Scholar]
- 15. Suh J. M., Gao X., McKay J., McKay R., Salo Z., and Graff J. M. (2006) Hedgehog signaling plays a conserved role in inhibiting fat formation. Cell Metab. 3, 25–34 10.1016/j.cmet.2005.11.012 [DOI] [PubMed] [Google Scholar]
- 16. Pospisilik J. A., Schramek D., Schnidar H., Cronin S. J., Nehme N. T., Zhang X., Knauf C., Cani P. D., Aumayr K., Todoric J., Bayer M., Haschemi A., Puviindran V., Tar K., Orthofer M., et al. (2010) Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell 140, 148–160 10.1016/j.cell.2009.12.027 [DOI] [PubMed] [Google Scholar]
- 17. Shi Y., and Long F. (2017) Hedgehog signaling via Gli2 prevents obesity induced by high-fat diet in adult mice. eLife 6, e31649 10.7554/eLife.31649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song D. K., Choi J. H., and Kim M. S. (2018) Primary cilia as a signaling platform for control of energy metabolism. Diabetes Metab. J. 42, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas M. K., Rastalsky N., Lee J. H., and Habener J. F. (2000) Hedgehog signaling regulation of insulin production by pancreatic beta-cells. Diabetes 49, 2039–2047 10.2337/diabetes.49.12.2039 [DOI] [PubMed] [Google Scholar]
- 20. Landsman L., Parent A., and Hebrok M. (2011) Elevated Hedgehog/Gli signaling causes beta-cell dedifferentiation in mice. Proc. Natl. Acad. Sci. U.S.A. 108, 17010–17015 10.1073/pnas.1105404108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lau J., and Hebrok M. (2010) Hedgehog signaling in pancreas epithelium regulates embryonic organ formation and adult beta-cell function. Diabetes 59, 1211–1221 10.2337/db09-0914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kwon H., Song K., Han C., Chen W., Wang Y., Dash S., Lim K., and Wu T. (2016) Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 63, 1155–1169 10.1002/hep.28289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Braune J., Weyer U., Matz-Soja M., Hobusch C., Kern M., Kunath A., Klöting N., Kralisch S., Blüher M., Gebhardt R., Zavros Y., Bechmann I., and Gericke M. (2017) Hedgehog signalling in myeloid cells impacts on body weight, adipose tissue inflammation and glucose metabolism. Diabetologia 60, 889–899 10.1007/s00125-017-4223-5 [DOI] [PubMed] [Google Scholar]
- 24. Chao L., Marcus-Samuels B., Mason M. M., Moitra J., Vinson C., Arioglu E., Gavrilova O., and Reitman M. L. (2000) Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J. Clin. Investig. 106, 1221–1228 10.1172/JCI11245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He W., Barak Y., Hevener A., Olson P., Liao D., Le J., Nelson M., Ong E., Olefsky J. M., and Evans R. M. (2003) Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc. Natl. Acad. Sci. U.S.A. 100, 15712–15717 10.1073/pnas.2536828100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hevener A. L., He W., Barak Y., Le J., Bandyopadhyay G., Olson P., Wilkes J., Evans R. M., and Olefsky J. (2003) Muscle-specific Pparg deletion causes insulin resistance. Nat. Med. 9, 1491–1497 10.1038/nm956 [DOI] [PubMed] [Google Scholar]
- 27. Lu M., Sarruf D. A., Talukdar S., Sharma S., Li P., Bandyopadhyay G., Nalbandian S., Fan W., Gayen J. R., Mahata S. K., Webster N. J., Schwartz M. W., and Olefsky J. M. (2011) Brain PPAR-γ promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nat. Med. 17, 618–622 10.1038/nm.2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., Red Eagle A., Vats D., Brombacher F., Ferrante A. W., and Chawla A. (2007) Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 10.1038/nature05894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sugii S., Olson P., Sears D. D., Saberi M., Atkins A. R., Barish G. D., Hong S. H., Castro G. L., Yin Y. Q., Nelson M. C., Hsiao G., Greaves D. R., Downes M., Yu R. T., Olefsky J. M., and Evans R. M. (2009) PPARγ activation in adipocytes is sufficient for systemic insulin sensitization. Proc. Natl. Acad. Sci. U.S.A. 106, 22504–22509 10.1073/pnas.0912487106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Home P. D., Pocock S. J., Beck-Nielsen H., Curtis P. S., Gomis R., Hanefeld M., Jones N. P., Komajda M., McMurray J. J., and RECORD Study Team (2009) Rosiglitazone evaluated for cardiovascular outcomes in oral agent combination therapy for type 2 diabetes (RECORD): a multicentre, randomised, open-label trial. Lancet 373, 2125–2135 10.1016/S0140-6736(09)60953-3 [DOI] [PubMed] [Google Scholar]
- 31. Choi J. H., Banks A. S., Kamenecka T. M., Busby S. A., Chalmers M. J., Kumar N., Kuruvilla D. S., Shin Y., He Y., Bruning J. B., Marciano D. P., Cameron M. D., Laznik D., Jurczak M. J., Schürer S. C., et al. (2011) Antidiabetic actions of a non-agonist PPARγ ligand blocking Cdk5-mediated phosphorylation. Nature 477, 477–481 10.1038/nature10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kabir A., Mousavi S., and Pazouki A. (2019) The Complications of bariatric surgery patients with type 2 diabetes in the world: a systematic review and meta-analysis. Curr. Diabetes Rev. 15, 49–61 10.2174/1573399814666180403164529 [DOI] [PubMed] [Google Scholar]
- 33. Chandrahas V. K., Han J., and Kaufman R. J. (2018) Coordinating organismal metabolism during protein misfolding in the ER through the unfolded protein response. Curr. Top. Microbiol. Immunol. 414, 103–130 [DOI] [PubMed] [Google Scholar]
- 34. Pathi S., Pagan-Westphal S., Baker D. P., Garber E. A., Rayhorn P., Bumcrot D., Tabin C. J., Blake Pepinsky R., and Williams K. P. (2001) Comparative biological responses to human Sonic, Indian, and Desert hedgehog. Mech. Dev. 106, 107–117 10.1016/S0925-4773(01)00427-0 [DOI] [PubMed] [Google Scholar]
- 35. Yam P. T., Langlois S. D., Morin S., and Charron F. (2009) Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron 62, 349–362 10.1016/j.neuron.2009.03.022 [DOI] [PubMed] [Google Scholar]
- 36. Teperino R., Amann S., Bayer M., McGee S. L., Loipetzberger A., Connor T., Jaeger C., Kammerer B., Winter L., Wiche G., Dalgaard K., Selvaraj M., Gaster M., Lee-Young R. S., Febbraio M. A., et al. (2012) Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell 151, 414–426 10.1016/j.cell.2012.09.021 [DOI] [PubMed] [Google Scholar]
- 37. Chinchilla P., Xiao L., Kazanietz M. G., and Riobo N. A. (2010) Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 9, 570–579 10.4161/cc.9.3.10591 [DOI] [PubMed] [Google Scholar]
- 38. Lefterova M. I., Haakonsson A. K., Lazar M. A., and Mandrup S. (2014) PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 25, 293–302 10.1016/j.tem.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ji S., Park S. Y., Roth J., Kim H. S., and Cho J. W. (2012) O-GlcNAc modification of PPARγ reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 417, 1158–1163 10.1016/j.bbrc.2011.12.086 [DOI] [PubMed] [Google Scholar]
- 40. Li P., Song Y., Zan W., Qin L., Han S., Jiang B., Dou H., Shao C., and Gong Y. (2017) Lack of CUL4B in adipocytes promotes PPARγ-mediated adipose tissue expansion and insulin sensitivity. Diabetes 66, 300–313 10.2337/db16-0743 [DOI] [PubMed] [Google Scholar]
- 41. Yin R., Fang L., Li Y., Xue P., Li Y., Guan Y., Chang Y., Chen C., and Wang N. (2015) Pro-inflammatory Macrophages suppress PPARγ activity in adipocytes via S-nitrosylation. Free Radic. Biol. Med. 89, 895–905 10.1016/j.freeradbiomed.2015.10.406 [DOI] [PubMed] [Google Scholar]
- 42. Hu E., Kim J. B., Sarraf P., and Spiegelman B. M. (1996) Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274, 2100–2103 10.1126/science.274.5295.2100 [DOI] [PubMed] [Google Scholar]
- 43. He J., Quintana M. T., Sullivan J., L Parry T., J Grevengoed T., Schisler J. C., Hill J. A., Yates C. C., Mapanga R. F., Essop M. F., Stansfield W. E., Bain J. R., Newgard C. B., Muehlbauer M. J., Han Y., Clarke B. A., and Willis M. S. (2015) MuRF2 regulates PPARγ1 activity to protect against diabetic cardiomyopathy and enhance weight gain induced by a high fat diet. Cardiovasc. Diabetol. 14, 97 10.1186/s12933-015-0252-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kilroy G., Kirk-Ballard H., Carter L. E., and Floyd Z. E. (2012) The ubiquitin ligase Siah2 regulates PPARγ activity in adipocytes. Endocrinology 153, 1206–1218 10.1210/en.2011-1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee K. W., Kwak S. H., Koo Y. D., Cho Y. K., Lee H. M., Jung H. S., Cho Y. M., Park Y. J., Chung S. S., and Park K. S. (2016) F-box only protein 9 is an E3 ubiquitin ligase of PPARγ. Exp. Mol. Med. 48, e234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quintana M. T., He J., Sullivan J., Grevengoed T., Schisler J., Han Y., Hill J. A., Yates C. C., Stansfield W. E., Mapanga R. F., Essop M. F., Muehlbauer M. J., Newgard C. B., Bain J. R., and Willis M. S. (2015) Muscle ring finger-3 protects against diabetic cardiomyopathy induced by a high fat diet. BMC Endocr. Disord. 15, 36 10.1186/s12902-015-0028-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J. H., Park K. W., Lee E. W., Jang W. S., Seo J., Shin S., Hwang K. A., and Song J. (2014) Suppression of PPARγ through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ. 21, 594–603 10.1038/cdd.2013.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu Y., Jin J., Zhang W., Zhang Z., Gao J., Liu Q., Zhou C., Xu Q., Shi H., Hou Y., and Shi J. (2016) EGFR/MDM2 signaling promotes NF-κB activation via PPARγ degradation. Carcinogenesis 37, 215–222 10.1093/carcin/bgv252 [DOI] [PubMed] [Google Scholar]
- 49. Boase N. A., and Kumar S. (2015) NEDD4: the founding member of a family of ubiquitin-protein ligases. Gene 557, 113–122 10.1016/j.gene.2014.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Han L., Wang P., Zhao G., Wang H., Wang M., Chen J., and Tong T. (2013) Upregulation of SIRT1 by 17β-estradiol depends on ubiquitin-proteasome degradation of PPAR-γ mediated by NEDD4–1. Protein Cell 4, 310–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li J. J., Wang R., Lama R., Wang X., Floyd Z. E., Park E. A., and Liao F. F. (2016) Ubiquitin ligase NEDD4 regulates PPARγ stability and adipocyte differentiation in 3T3-L1 cells. Sci. Rep. 6, 38550 10.1038/srep38550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Preidis G. A., Kim K. H., and Moore D. D. (2017) Nutrient-sensing nuclear receptors PPARα and FXR control liver energy balance. J. Clin. Investig. 127, 1193–1201 10.1172/JCI88893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Redonnet A., Groubet R., Noel-Suberville C., Bonilla S., Martinez A., and Higueret P. (2001) Exposure to an obesity-inducing diet early affects the pattern of expression of peroxisome proliferator, retinoic acid, and triiodothyronine nuclear receptors in the rat. Metabolism 50, 1161–1167 10.1053/meta.2001.26759 [DOI] [PubMed] [Google Scholar]
- 54. Faulds M. H., Zhao C., and Dahlman-Wright K. (2010) Molecular biology and functional genomics of liver X receptors (LXR) in relationship to metabolic diseases. Curr. Opin. Pharmacol. 10, 692–697 10.1016/j.coph.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 55. Shao W., and Espenshade P. J. (2012) Expanding roles for SREBP in metabolism. Cell Metab. 16, 414–419 10.1016/j.cmet.2012.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gould S. E., Low J. A., Marsters J. C. Jr, Robarge K., Rubin L. L., de Sauvage F. J., Sutherlin D. P., Wong H., and Yauch R. L. (2014) Discovery and preclinical development of vismodegib. Expert Opin. Drug Discov. 9, 969–984 10.1517/17460441.2014.920816 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.