

Abstract
Small heat shock proteins (sHSPs) delay protein aggregation in an ATP-independent manner by interacting with client proteins that are in states susceptible to aggregation, including destabilized states related to cellular stress. Up-regulation of sHSPs under stress conditions supports their critical role in cellular viability. Widespread distribution of sHSPs in most organisms implies conservation of function, but it remains unclear whether sHSPs implement common or distinct mechanisms to delay protein aggregation. Comparisons among various studies are confounded by the use of different model client proteins, different assays for both aggregation and sHSP/client interactions, and variable experimental conditions used to mimic cellular stress. To further define sHSP/client interactions and their relevance to sHSP chaperone function, we implemented multiple strategies to characterize sHSP interactions with α-lactalbumin, a model client whose aggregation pathway is well defined. We compared the chaperone activity of human αB-crystallin (HSPB5) with HSPB5 variants that mimic states that arise under conditions of cellular stress or disease. The results show that these closely related sHSPs vary not only in their activity under identical conditions but also in their interactions with clients. Importantly, under nonstress conditions, WT HSPB5 delays client aggregation solely through transient interactions early in the aggregation pathway, whereas HSPB5 mutants that mimic stress-activated conditions can also intervene at later stages of the aggregation pathway to further delay client protein aggregation.
Keywords: small heat shock protein (sHSP), protein aggregation, molecular chaperone, crystallin, stress response, unfolded protein response (UPR), α-lactalbumin, αB-crystallin, client binding, HSPB5, wHsp16.9
Introduction
Small heat shock proteins (sHSPs)3 are a diverse class of ATP-independent molecular chaperones found throughout biology that delay the formation of insoluble protein aggregates through their interactions with destabilized, aggregate-prone proteins (1). Their roles as protein chaperones are implicated in protein aggregation diseases that include Alzheimer's disease, Alexander disease, and tauopathies (2–5). A growing number of inherited disease–associated mutations in human sHSPs further imply critical roles for sHSP function (6–8). Long thought of as “housekeeping” proteins, it is now clear that understanding how sHSPs maintain aggregate-prone proteins (clients) in soluble forms, both under “normal” conditions and during cellular stress, is of great importance for understanding protein homeostasis. Among the ten human sHSPs, HSPB5 (also known as αB-crystallin) is ubiquitously expressed and is the best characterized. Nonetheless, the mechanisms by which it performs as a chaperone remain relatively undefined.
Deciphering the mechanisms of sHSPs is hampered by two fundamental challenges: heterogeneity of HSPB5 oligomeric states and heterogeneity of aggregation states adopted by model clients. The oligomeric distribution of HSPB5 is very sensitive to solution conditions, the presence of posttranslational modifications, and mutations. Likewise, various model clients have been used that differ as to the solution conditions used to promote aggregation (i.e. heat shock, reduction of disulfide bonds, removal of metal ions, etc.). Furthermore, sHSP structure and activity will also be altered by different experimental conditions, complicating the ability to compare different studies and to draw general principles regarding sHSP mechanism.
A key aim to understand the mechanism of sHSP function is to detect HSPB5 interactions with different client species known to exist along an aggregation pathway. To limit the number of experimental variables and facilitate comparisons, we focused on specific variants of HSPB5 (see below) and one client protein whose aggregation pathway has been well described by dynamic light scattering (DLS) studies (9). For this, we chose α-lactalbumin (αLac), a soluble 14-kDa protein whose tertiary structure is stabilized by four disulfide bonds and whose aggregation is initiated by addition of reducing agents (Fig. 1A). Upon addition of a reducing agent, there is an initial concentration-dependent lag phase where species with hydrodynamic radii (RH) consistent with monomeric αLac (approximately 1 nm) are observed (9, 10). Subsequently, in the post-nucleation phase, small aggregates with RH of approximately 102 nm (∼104 αLac molecules) are observed (9). No other intermediate states between monomeric αLac and the small aggregates have been described. Finally, association of the small aggregates leads to formation of large aggregates (103 nm). Numerous studies have shown that the presence of WT HSPB5 results in pronounced lengthening of the lag phase, as detected by DLS.
Figure 1.
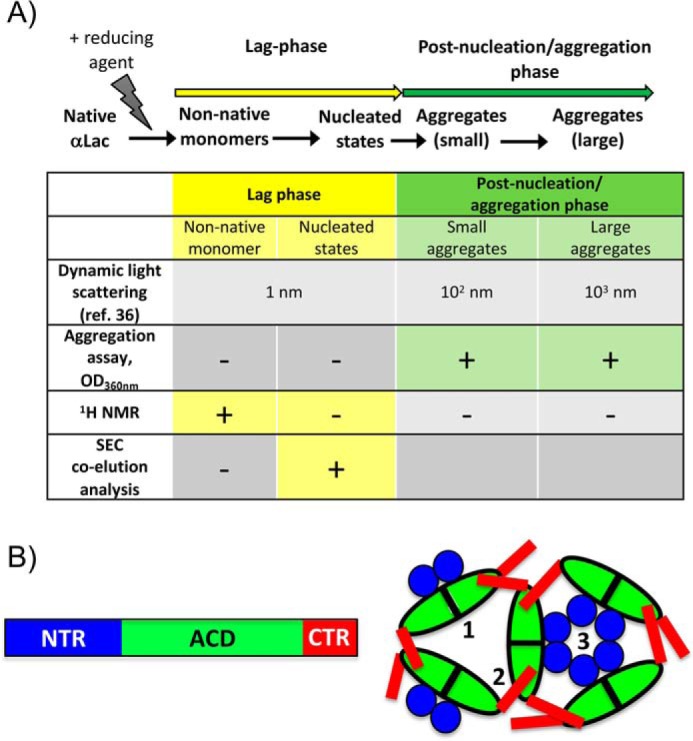
Features of sHSPs and the model client, αLac. A, the aggregation pathway of αLac as defined by previous studies (9, 10). The states of destabilized αLac detected during its aggregation are small nonnative states (1 nm) that self-associate into nucleated states during the lag phase (these are invisible in assays that follow A360 nm) and small aggregates (102 nm) that associate during the post-nucleation/aggregation stage to form large (103 nm) aggregates. The client states detected and/or relevant in the experimental protocols used in our study are summarized. As a point of reference, the size of HSPB5 oligomers ranges from 8–18 nm (23). B, domain architecture of sHSPs and interdomain interactions responsible for oligomerization of HSPB5. Denoted on the right are three types of interactions observed in many (although not all) sHSPs: 1, ACD–ACD interactions; 2, ACD–CTR interactions; 3, NTR interactions. Mutations used in this study disrupt the interactions as follows: 1, ACD/ACD (H104K and R120G); 2, CTR/ACD (GXG); and 3, NTR interactions (D3).
All sHSPs share a domain architecture centered on the structural domain known as the α-crystallin domain (ACD) and flanked by two poorly conserved, mostly disordered N- and C-terminal regions (NTR and CTR, respectively; Fig. 1B). Three types of intersubunit interactions are commonly observed within sHSP oligomers, regardless of their size, heterogeneity, or structure (shown schematically in Fig. 1B): ACD-to-ACD interactions define dimeric units; ACD-to-CTR interactions occur via a three-residue sequence, known as the IXI motif, present in the CTR of most sHSPs; and interactions involving the NTR, which are essential for oligomer formation. Despite these levels of assembly, HSPB5, like many mammalian sHSPs (11), exists in solution as polydisperse distributions of oligomeric species ranging from 10- to 40-mers that are highly sensitive to environmental conditions, posttranslational modifications, and mutations (12, 13). This behavior makes it virtually impossible to trap individual well-defined HSPB5 species to investigate their activities. Nevertheless, progress has been made in defining intersubunit interactions that contribute to the formation, stabilization, and distribution of HSPB5 oligomers. These insights provide avenues to modulate the distribution and prevalent species of HSPB5, yielding a modicum of control and tractability that can be leveraged to better understand sHSP function.
To circumvent the challenges posed by the HSPB5 plasticity, we focus here on five HSPB5 variants (WT and four mutants) that serve as proxies for the protein under differing cellular conditions. Three are mutants thought to mimic states of HSPB5 during different types of cellular stress, and the fourth is a well-known disease mutant. First, stress-induced phosphorylation of three serine residues in the HSPB5 NTR leads to smaller oligomers. The fully phosphorylated state is mimicked by three substitutions, S19D/S45D/S59D, to create the mutant called D3-HSPB5 (14, 15). Second, the H104K-HSPB5 mutant has been shown to mimic HSPB5 under acidosis conditions, where the ACD–ACD dimer interface is destabilized, and, paradoxically, enlarged oligomers are favored (16). The histidine at position 104 is conserved among most human sHSPs (Fig. S1), and pH-dependent, enhanced chaperone activity has been observed in other sHSPs, implying that there may be shared modes of pH-activated chaperone activity (17, 18). Third, the I159G/I161G (GXG-HSPB5) mutant mimics states in which the CTR is released from the ACD–CTR interaction, seen to occur under acidosis conditions and at elevated temperature (19, 20). Fourth, a well-known disease-associated mutation, R120G-HSPB5, substitutes a residue at the ACD–ACD dimer interface (6). Finally, as a first step toward identifying common modes (or a lack thereof) among sHSPs and to put our results into context, we collected analogous data on wheat wHSP16.9, which forms discrete oligomers and well-defined interactions with clients (21, 22). Our results show that HSPB5 variants differ not only in chaperone activity under identical solution conditions but also in their interactions with the client at different stages of protein aggregation. In particular, HSPB5 mutants that mimic states present under stress conditions display the ability to engage client states further along the aggregation pathway, leading to longer delay of the onset of irreversible aggregation.
Results
Aggregation of the model client αLac is initiated by reduction of its disulfide bonds, and formation of αLac species large enough to scatter light is detected as an increase in optical density at 360 nm. Of the species identified in the αLac aggregation pathway, only the small (102 nm) and large (103 nm) aggregates that form in the post-nucleation phase will be detected. HSPB5 oligomers, which range in size from 8 to 18 nm, will also not be detected (23). Under the experimental conditions used, in the absence of sHSP, the signal first appears ∼20 min after addition of a reducing agent (Fig. 2, gray trace). The presence of substoichiometric WT-HSPB5 (molar ratio of αLac to sHSP of 6:1; Fig. 2, open circles) alters the time course of aggregation in two ways: the onset of aggregation is delayed (i.e. a longer lag phase), and the rate of appearance (slope) of aggregates is slowed. In the presence of the disease-associated R120G-HSPB5, onset of observable aggregation occurs earlier than αLac on its own (Fig. 2, blue trace). In contrast, GXG-HSPB5, D3-HSPB5, and H104K-HSPB5 each delay αLac aggregation longer than WT-HSPB5 (Fig. 2, black, red, and green traces, respectively). Thus, these four mutants plus WT-HSPB5 represent a set of highly related HSPB5 variants that exhibit dramatically different chaperone activity under otherwise identical experimental conditions.
Figure 2.

Chaperone activity of HSPB5 and HSPB5 mutants with destabilized αLac. Aggregation of 600 μm αLac, destabilized by addition of DTT, was monitored by the increase in light scattering at 360 nm as a function of time. The presence of WT-HSPB5 or HSPB5 mutants (100 μm) delays the onset of aggregation. Time courses shown are for αLac alone (alac, gray diamonds) and αLac in the presence of WT-HSPB5 (open circles), GXG-HSPB5 (black circles), D3-HSPB5 (red circles), H104K-HSPB5 (green squares), and R120G-HSPB5 (blue circles). The same coloring scheme is used in all figures. Curves shown are the average of duplicates within an experiment and are representative of at least two independent assays (see Fig. S2 and “Experimental procedures” for details).
HSPB5 interactions with early species on the aggregation pathway
The lengthened delay observed in the standard aggregation assay implies that there is a direct interaction between HSPB5 and species of αLac that exist during the lag phase (Fig. 1A). But this assay is blind to what these species might be, as it only reports on species large enough to scatter light. To assess how HSPB5 interacts with earlier states of aggregating αLac, we adapted an NMR approach that allows for direct observation of small protein species (24). NMR signals arising from monomeric states are observable in a standard 1H NMR spectrum, whereas larger intermediates and aggregates will not be detected because of their slow tumbling rates and broad linewidths. Therefore, the progressive loss of NMR signal intensity reports on the disappearance of early states as they proceed along the aggregation pathway. The spectrum of destabilized αLac contains fewer resolved features (i.e. fewer defined peaks and minimal dispersion of peaks in the spectrum) than that of stably folded, disulfide-linked αLac (Fig. 3A, top and bottom spectra, respectively); the top spectrum is consistent with a monomeric protein that is sampling multiple nonnative states.
Figure 3.
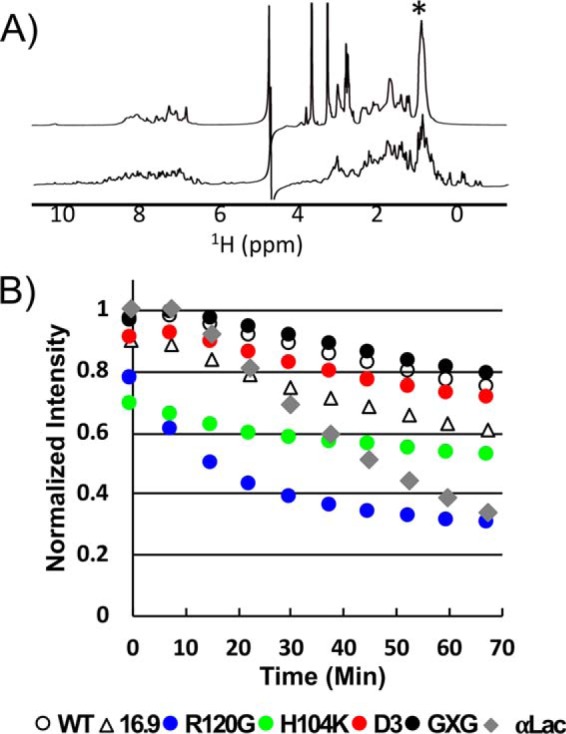
Monitoring early stages of aggregation via 1H NMR. A, 1D 1H spectra of αLac in the presence of 5 mm DTT (top trace) and under nonreducing conditions (bottom trace). The most intense αLac peak in the DTT-destabilized spectrum is at 0.860 ppm, denoted by an asterisk. B, intensity at 0.860 ppm of 400 μm DTT-destabilized αLac in the absence of HSPB5 as a function of time (gray curve) and in the presence of 100 μm HSPB5 (open circles), HSPB5 mutants (colored as in Fig. 2), or wHSP16.9 (open triangles) as a function of time. Intensities were normalized to the value (IαLac alone + IsHSP alone) at 0.860 ppm, which varied depending on the sHSP in each mixture (Fig. S3A). There was a ∼4.5-min delay after addition of DTT/EDTA and prior to the start of acquisition of the first 7.5-min experiment. Thus, the intensity at time 0 min reflects aggregation/binding that occurred in the time period of 4.5–13 min after DTT and EDTA addition. Two independent experiments with R120G- and H104K-HSPB5 were collected (see “Experimental procedures”). Reproducibility of the intensity time courses is demonstrated in Fig. S3.
1D 1H NMR spectra of αLac were collected at 37 °C as a function of time after addition of a reducing agent either in the absence or presence of substoichiometric HSPB5 (molar ratio of αLac to sHSP of 4:1; for greater sensitivity in detecting HSPB5/client interactions, the ratio of αLac to HSPB5 in 1D NMR experiments was changed from 6:1 used in other assays to 4:1; refer to “Experimental procedures” for additional details). The presence of HSPB5 did not alter the αLac spectrum, as the large molecular weight of HSPB5 oligomers virtually eliminate their 1D NMR signals (Fig. S3A). The largest peak in the αLac NMR spectrum (0.860 ppm) contains the resonances of all Leu, Val, and Ile CH3 groups in detectable species. The intensity of this peak represents a useful measure of the fractional population of early states that remain in solution during the experiment (Fig. 3B). Loss of this signal intensity can occur as a result of αLac self-association or as a consequence of binding to HSPB5 oligomers, resulting in high-molecular-weight species. Whether αLac signals are significantly broadened or completely lost depends on the lifetime of the interaction in the high-molecular-weight complex.
In the absence of sHSP, the NMR intensity, and therefore the population of small αLac species, is constant over the first ∼10 min, followed by a rapid decrease (∼70% of the original intensity is gone by 70 min; Fig. 3B, gray curve). The constant signal at early time points is consistent with the previously reported lag in the appearance of αLac aggregates (Fig. 1A) (9). Time courses in the presence of the different HSPB5 variants reveal distinct behaviors (Fig. 3B). All HSPB5 variants, except R120G-HSPB5, maintain more NMR-detectable αLac in solution at the end of the time course. Similar to its behavior observed in the standard aggregation assay (Fig. 2), R120G-HSPB5 accelerates the loss of NMR signal intensity, with the final intensity nearly the same as when no chaperone is present (Fig. 3B). Unexpectedly, although the HSPB5 variants GXG-, D3-, and H104K-HSPB5 are more effective than WT-HSPB5 at extending the lag phase in the standard assay (Fig. 2), they do not maintain NMR-observable αLac species more effectively (Fig. 3B). H104K-HSPB5 is as effective as GXG-HSPB5 in the standard assay but maintains an intermediate amount of NMR-detectable αLac. Thus, the ability to maintain NMR-detectable αLac per se does not predict the relative effectiveness of a chaperone to delay formation of large aggregates, suggesting that the modes of interaction and/or the early species each variant of HSPB5 engages are different.
The behavior of αLac observed by NMR is consistent with the aggregation pathway described by DLS studies, as no loss of NMR signal intensity, which would indicate formation of larger species, is observed during the first 10 min of the aggregation reaction. For αLac alone, this behavior is very consistent (S.D. < 1.5% for the intensity of the methyl signal across five independent experiments; Fig. S3 and Ref. 9). Therefore, a sharp decrease in intensity observed at the early time points in the presence of sHSP must be due to binding of the client to the chaperone. WT- and GXG-HSPB5 show very small but reproducible intensity loss, consistent with transient interactions. Slightly more intensity is lost in the presence of D3-HSPB5, indicating that the phosphomimic form engages early client species with higher affinity and/or a longer lifetime. However, a much larger loss of intensity occurs in the presence of H104K- and R120G-HSPB5, consistent with more avid binding (and/or longer-lived interactions) of early species by these two HSPB5 variants.
During later stages of the aggregation reaction, R120G-HSPB5 continues to promote the formation of larger aggregates, as the intensity at every time point is lower than for αLac alone. As R120G-HPSB5 also enhances the formation of large αLac aggregates in the standard assay (Fig. 2), the NMR result suggests that R120G-HPSB5 binds to early states in the pathway, enhancing αLac self-association and large aggregate formation. These observations imply that R120G-HSPB5 engages early species of αLac in a manner that disfavors maintenance of monomeric species. In contrast, H104K-HSPB5 is effective at lengthening the lag phase in standard assays (Fig. 2) and, after initial interactions with early client states, prevents the formation of large αLac aggregates. Thus, this “activated” form of HSPB5 interacts with early client species in a manner that inhibits their progress along the pathway, presumably by inhibiting their self-association. Together, the observations show that HSPB5 variants do interact with early-lag-phase species but that different HSPB5 species affect the pathway toward αLac aggregation differently.
HSPB5/client interactions at later stages of αLac aggregation
The NMR measurements do not report on sHSP interactions involving higher-molecular-weight client states that are NMR-invisible. To detect sHSP/client interactions that appear later in the lag phase, we looked for co-elution of the client and HSPB5 using size exclusion chromatography (SEC), analyzing 6:1 mixtures of destabilized αLac and HSPB5 that were incubated for 30 min at 42 °C (Fig. 4). As evident from the aggregation assays (Fig. 2), 30 min is well into the lag phase when any of the HSPB5 variants (except R120G) are present, but no post-nucleation aggregate species are detected. Elution profiles of the mixtures (Fig. 4, black) are compared with the profile of each HSPB5 variant (Fig. 4, red) on its own. We previously demonstrated, by SDS-PAGE analysis of SEC fractions from similar experiments, that a shift in elution volume and/or a redistribution of protein absorbance between the two peaks is due to co-elution of the client and sHSP, signifying an interaction (16). Incubation of destabilized αLac with WT-HSPB5 alters neither the elution time nor the peak height of the sHSP oligomer, consistent with a lack of co-elution of αLac (Fig. 4A and Fig. S4). SDS-PAGE of eluted fractions also demonstrated that the distribution of WT-HSPB5 is unaltered during incubation with destabilized αLac (Fig. S4). αLac can be observed throughout the fractions of the mixture, including a faint band in earlier eluting fractions. Because αLac aggregates in the absence of a sHSP, we were unable to acquire an elution profile of destabilized αLac on its own for comparison. From this experiment alone, it is therefore unclear whether there is a weak interaction between WT-HSPB5 and αLac over the time course of this experiment. However, by comparison, interactions with other sHSPs are much more apparent by this method, as described below. Therefore, if there is an interaction with WT-HSPB5, then it must be appreciably weaker and more transient than the interactions with other sHSPs described here.
Figure 4.
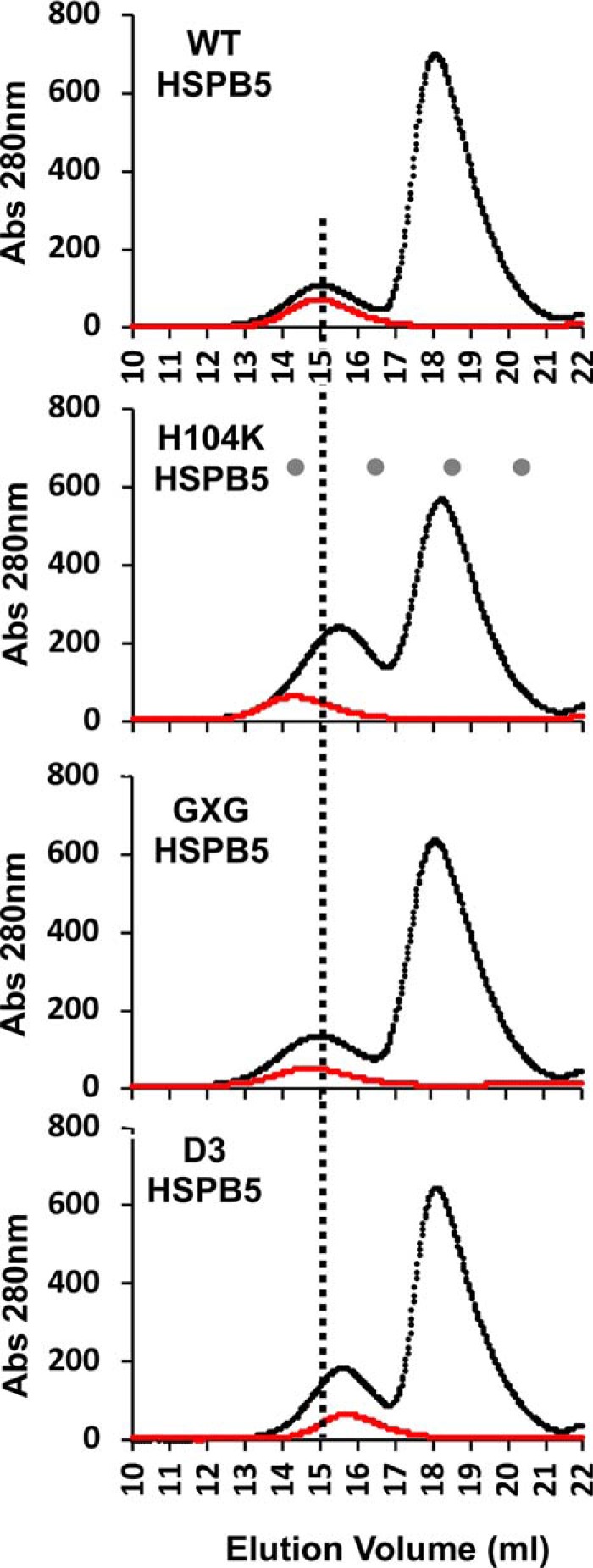
SEC profiles of sHSP/client mixtures. SEC elution profiles of HSPB5 and HSPB5 mutants alone (red curves) and incubated with destabilized αLac for 30 min at 42 °C prior to SEC (black curves). A vertical line shown through all elution profiles denotes the peak elution volume of WT HSPB5 for reference. Elution volumes for molecular mass standards for (from left to right) thyroglobulin (670 kDa, 14.3 ml), bovine γ-globulin (158 kDa, 16.5 ml), equine myoglobin (17 kDa, 18.5 ml) and vitamin B12 (1.35 kDa, 20.4 ml) are noted as gray dots on the H104K chromatogram. SDS-PAGE analyses of fractions collected are shown in Fig. S4.
As described previously, H104K-HSPB5 (16) exists as an enlarged oligomer on its own (elutes earlier than WT-HSPB5). In the mixtures, these oligomers disassemble into smaller oligomeric species that co-elute with a population of αLac that shifts to earlier elution times, indicating the formation of a complex. Isolated GXG-HSPB5 oligomers elute earlier than WT-HSPB5 oligomers but also co-elute with αLac. This is shown by the decreased intensity of the free αLac peak and the increased peak height of the earlier eluting peak. Thus, this variant undergoes long-lived interactions with αLac species, but it does so without dissociating into much smaller oligomers. The phosphomimic mutant D3-HSPB5 elutes later than WT-HSPB5 oligomers, consistent with reports that phosphorylation (or its mimicry) leads to smaller HSPB5 species (14, 25). The increased absorbance under the early-eluting peak in the profile of the mixture indicates co-elution of D3-HSPB5 and client, as confirmed by SDS-PAGE (Fig. S4). Thus, this more dissociated form of HSPB5 engages in long-lived interactions with client species. We could not perform SEC analysis on mixtures including R120G-HSPB5 because of the rapid aggregation observed during its incubation with destabilized αLac, consistent with the accelerated aggregation we observed with R120G in the chaperone assays (Fig. 2). The SEC results reveal a clear difference between WT-HSPB5 and mutants that delay αLac aggregation: the more active holdases form clear, SEC-observable complexes with αLac species that are present during the lag phase whereas WT-HSPB5 does not. The ability to observe complexes between αLac and sHSPs by SEC indicate that the complexes involving forms of αLac that arise later in the lag phase are long-lived, as they remain intact during the chromatography experiment.
The above SEC results suggest a relationship between the ability of specific HSPB5 variants to engage in long-lived interactions with later-lag-phase (prenucleated) species to extend the lag phase. To corroborate these results and their interpretation, we performed light-scattering aggregation assays in which sHSP was introduced at the end of the αLac lag phase rather than having the chaperone present when aggregation is initiated. Therefore, when sHSPs are introduced, samples will contain mixtures of prenucleated, nucleated, and small aggregates of αLac that normally undergo rapid aggregation in the absence of sHSP (i.e. post-lag phase). Each panel in Fig. 5 shows three aggregation curves: in the absence of HSPB5 (black), HSPB5 present at the initiation of aggregation (green), and HSPB5 introduced at the end of lag phase (red). Because it promotes aggregation through interactions with early species of αLac, these experiments were not performed with R120G. Strikingly, when introduced at the end of the lag phase, WT-HSPB5 failed to delay the rapid onset of αLac aggregation, whereas the more active HPSB5 mutants all effectively delay the progression of aggregation. Thus, the ability to delay aggregation of species formed further along an aggregation pathway appears to correlate with enhanced holdase activity, as observed in a standard aggregation assay. Although it fails to delay the onset of aggregation when introduced at the end of the lag phase, WT HSPB5 affects the course of αLac aggregation by producing a reproducible abrupt change in the aggregation curve that signals the end of the very rapid increase in aggregates (Fig. 5, WT black versus red curve). Given our inability to detect an interaction between WT-HSPB5 and any species of αLac other than early-lag-phase forms, the most likely explanation for this effect is that WT-HSPB5 engages monomeric forms still present at the end of lag phase and slows their progression into species that can rapidly form large aggregates.
Figure 5.
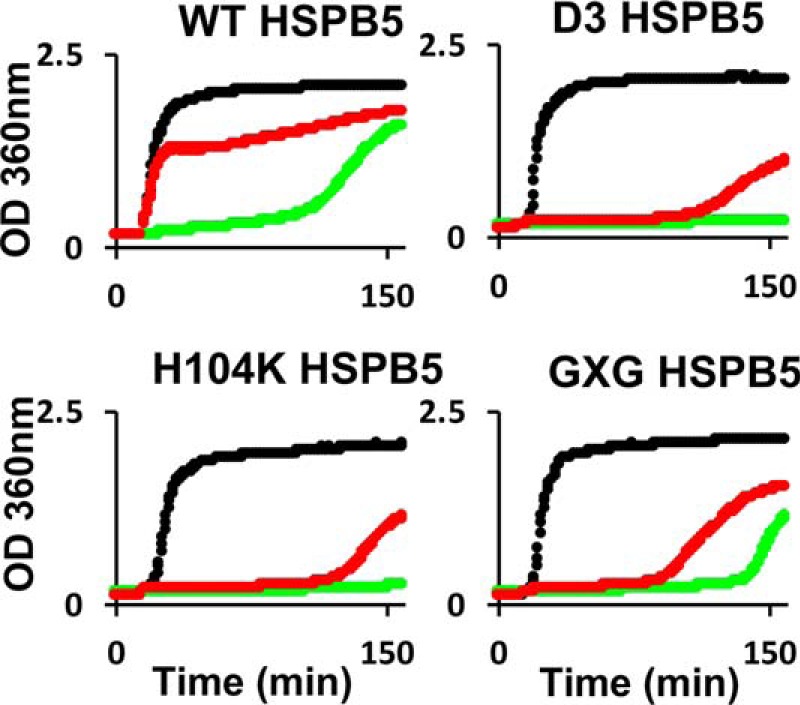
Ability of HSPB5 to chaperone nucleated aggregates. The aggregation of destabilized 600 μm αLac as monitored by light scattering is shown (black curves). When 100 μm HSPB5 or mutant HSPB5 is present at the time of DTT addition, chaperone function is observed (green curves). When addition of the sHSPs was delayed until aggregates of DTT-destabilized αLac were detected (see “Experimental procedures” for details), the activated mutants (H104K, D3, and GXG) effectively delay aggregation (red curves) but HSPB5 does not (red curve, top left).
Altogether, the results indicate that WT-HSPB5 that has not been activated by stress conditions as mimicked by the HSPB5 mutants can intervene in early stages of the αLac aggregation pathway but is ineffective when faced with nucleated forms of αLac that can rapidly form large aggregates. Thus, perturbations in HSPB5, such as those exemplified by the mutations studied here, unmask additional modes of action in which HSPB5 engages with later-stage species to further delay progression of αLac aggregation.
A plant sHSP effectively engages prenucleated client species
As a step toward identifying commonalities in sHSP mechanism and to learn whether HSPB5 behavior reflects general modes of action among sHSPs, we applied the same experimental procedures to a well-characterized sHSP from wheat, wHSP16.9. In aggregation assays, wHSP16.9 produces a similar delay in the onset of αLac aggregation as WT-HSPB5 (Fig. 6A, magenta curve (wHSP16.9) and open circle curves (HSPB5)). However, although WT-HSPB5 and HSPB5 mutants have a minor impact on the final observed OD360 nm in αLac aggregation assays, wHSP16.9 suppresses the large aggregates detected as OD360 nm for a prolonged period of time (Fig. 6A). In 1D NMR assays, a modest loss of αLac NMR intensity is observed at the early time points with wHSP16.9 (Fig. 3B, open triangles), consistent with the plant sHSP engaging early client species transiently. Different from any of the HSPB5 variants studied here, wHSP16.9 co-elutes from SEC earlier with destabilized αLac (Fig. 6B and Fig. S4). This result is consistent with previous reports that wHSP16.9 forms long-lived complexes with client proteins that are larger than the sHSP oligomers themselves (22). Similar to enhanced variants of HSPB5, wHSP16.9 delays formation of large αLac aggregates when introduced at the end of the lag phase of standard aggregation assays (Fig. 6C, red versus green curves). Overall, the comparison indicates that WT wHSP16.9 can engage late-lag-phase client states under the conditions used here without additional perturbations or mutations.
Figure 6.
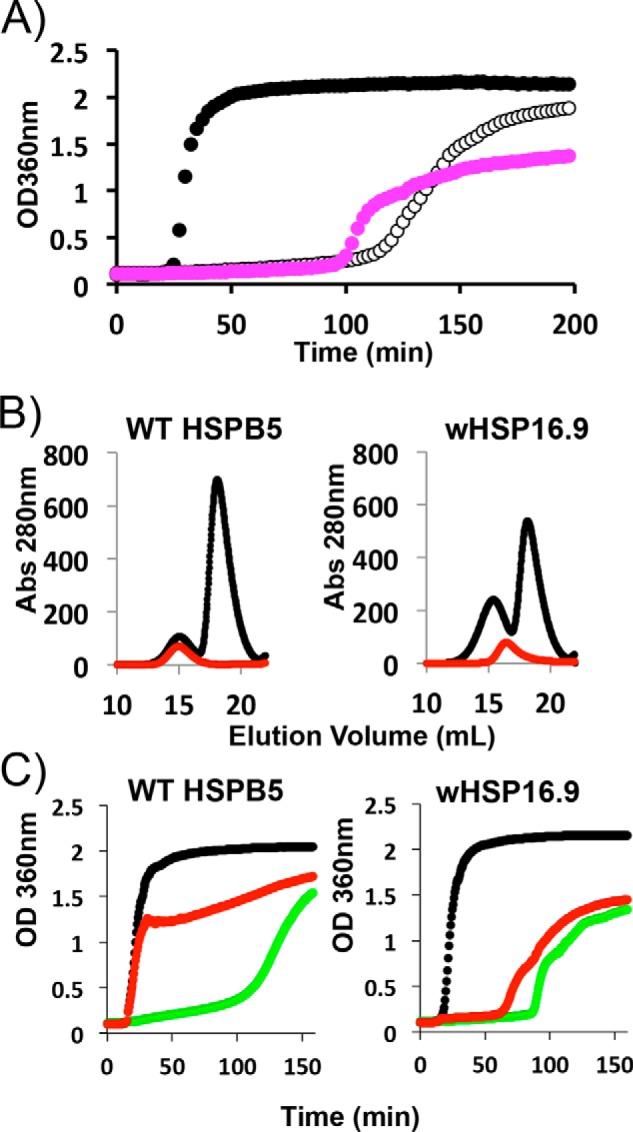
Comparison of human HSPB5 and plant wHSP16.9 properties. A, aggregation of 600 μm DTT-destabilized αLac, as monitored by light scattering at 360 nm as a function of time in the absence of a chaperone (black curve) and in the presence of either 100 μm HSPB5 (open circles) or 100 μm wHSP16.9 (magenta). B, SEC profiles of HSPB5 and wHSP16.9 (red curves) and SEC profiles of HSPB5 and wHSP16.9 incubated with DTT-destabilized αLac for 30 min at 42 °C prior to SEC (black curves). C, aggregation of 600 μm DTT-destabilized αLac, as monitored by light scattering (black curves). When 100 μm HSPB5 or wHSP16.9 was present at the time of DTT addition, chaperone function was observed (green curves). When the addition of the sHSPs was delayed until aggregates of DTT-destabilized αLac were detected (see “Experimental procedures” for details), wHSP16.9 effectively delayed aggregation (red curve, right) but HSPB5 does not (red curve, left).
Discussion
The ability of sHSPs to delay the appearance of large insoluble protein aggregates is believed to be directly responsible for their cytoprotective effects under proteotoxic stress. Despite a growing appreciation of the importance of sHSPs in cellular function and human disease, the molecular mechanisms by which sHSPs perform their critical functions remain undefined. The slow progress toward understanding is in large part due to the peculiar experimental challenges posed both by intrinsic properties of sHSPs that give rise to their exquisite adaptability to changing cellular conditions and by difficulties in following aggregating systems in real time. Additionally, the ability to define general strategies and mechanisms of sHSP chaperone function has been limited by the practice of comparing activity across a variety of conditions, usually dictated by the condition that initiates aggregation for a given model client. It is clear that the structures, dynamics, and, likely, activity of HSPB5 differ under different conditions so that even a comparison of the activity of WT HSPB5 across multiple clients is confounded by the malleability of the chaperone itself. Our study introduces two novel strategies that circumvent the abovementioned challenges and provide readouts for specific species or stages along the αLac aggregation pathway in real time.
Importantly, αLac that is destabilized by reduction of its disulfide bonds aggregates under conditions similar to those normally found in a human cell (pH 7.5, 37 °C), allowing us to characterize client/HSPB5 interactions under nonstress conditions. Our results demonstrate conclusively that HSPB5 inhibits αLac aggregation by prolonging the lag phase of the pathway and that it achieves this effect through weak, transient interactions with αLac species that exist early along the pathway. Because we could detect interactions with other client species in experiments using HSPB5 variants, we can conclude unambiguously that the WT protein under nonstress conditions interacts fleetingly only with small (monomeric) species studied here. We propose that the ability of WT-HSPB5 to engage such species (of αLac) at the earliest stages of aggregation in highly transient interactions is sufficient to support a basal level of chaperone activity under nonstress conditions (see the model in Fig. 7).
Figure 7.
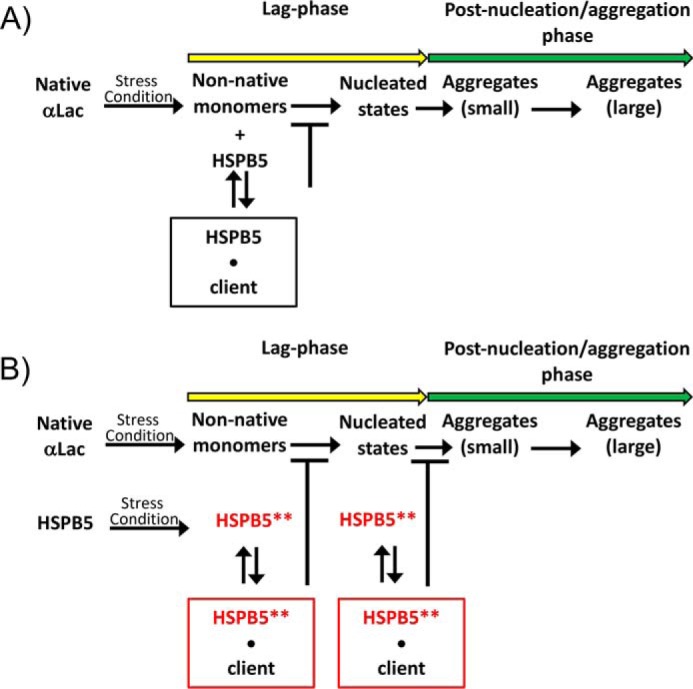
Mechanistic model for HSPB5/client interactions along an aggregation pathway. A, the aggregation pathway of destabilized αLac as defined by DLS (based on Ref. 9). Under nonstress conditions, HSPB5 interacts only with small (monomeric) early-lag-phase species in a highly transient manner. These interactions are sufficient to prolong the lag phase and onset of irreversible aggregates. B, in response to conditions associated with cellular stress (heat, acidosis, phosphorylation), polydisperse HSPB5 undergoes alterations in its oligomeric distribution and/or conformational state(s), signified by HSPB5**. The precise nature of the alterations depends on the perturbation, as both larger and smaller particle distributions are observed across the different HSPB5 mutants. Regardless, HSPB5** can interact with both early- and late-lag-phase species, in modes distinct from WT HSPB5, to further prolong the onset of rapid and irreversible aggregation.
Each of the HSPB5 variants that mimic species or features of species that exist under stress conditions exhibited an enhanced ability to lengthen the amount of time elapsed before aggregates of αLac appeared. Notably, although variants differed in their modes of interactions with early species, a unifying property among the variants with enhanced chaperone activity is their ability to engage client states effectively when introduced at the end of the lag phase, an ability that WT-HSPB5 lacks (Fig. 5). Furthermore, each of the enhanced HSPB5 variants form long-lived complexes with those client states, as judged by co-elution from SEC, although the chaperone–client complexes clearly differ (Fig. 4). Thus, the ability of HSPB5 to engage in long-lived interactions with late- or post-lag phase client states correlates with enhanced ability to delay the onset of aggregation, as viewed in a standard aggregation assay. Both transient and long-lived client complexes have been reported previously for mammalian sHSPs and a variety of model clients, but the ability to rationalize these observations within an aggregation pathway has been missing. Because the “stress-mimicking” variants were all characterized under identical (nonstress) conditions, we can conclude that the differences among them are due to changes in HSPB5 oligomer structure and dynamics associated with stress conditions and not to differences in the client or its aggregation pathway. Thus, the facility of HSPB5 to rapidly alter its structure and oligomer states and distributions in response to cellular environment unmasks cryptic modes of client engagement capable of further lengthening the time to aggregation onset and, importantly, adds functionality by diversifying the number and type of client states with which it interacts.
An important caveat is that the limited ability of nonactivated HSPB5 to engage early client species implies that its “basal” activity will depend on a given client's aggregation pathway. For example, some but not all destabilized mutants of the lens protein γd-crystallin are resistant to chaperoning by WT-HSPB5 (26). Our data suggest that rapidly aggregating clients may be less effectively chaperoned by WT-HSPB5, providing a possible rationale for the range of HSPB5 chaperone activity toward diverse clients (27). Furthermore, each of the enhanced variants investigated here represents extreme cases of stress-related perturbations (complete phosphorylation, pH < 6.8, heat shock), so the degree to which the new modes of interaction are expressed under cellular stress conditions may be on a sliding scale that depends on the severity and nature of the conditions. Nevertheless, the actual HSPB5 activity under such conditions will reflect the properties and interactions defined here.
Our results indicate that HSPB5 chaperone activity is modulated by altering its oligomeric distribution in a number of different ways to generate species with enhanced client binding modes. The plant sHSP studied here offers a counter example, as it displays multiple modes of client binding in its nonstress format. Notably, and as reported previously (28, 29), wHSP16.9 forms large, long-lived complexes containing multiple client molecules to delay aggregation. A noteworthy difference between the two examples here is that HSPB5 forms polydisperse, dynamic oligomeric distributions, and wHSP16.9 forms discrete monodisperse dodecamers. It is tempting to suggest that polydispersity, as observed in HSPB5, is intimately linked to the ability to modulate chaperone activity via the model proposed here, but devising a rigorous test for this hypothesis will be extremely challenging, as it requires the ability to control polydispersity in a given system.
In addition to alterations in oligomeric state and activity in response to changing conditions or posttranslational modification, there is growing appreciation that missense mutations inherited by individuals with a variety of diseases also affect HSPB5 structure and function. Here we assessed the consequence of the best-characterized of the HSPB5 mutations, R120G, associated with cardiomyopathies (6, 30). Similar to two of the enhanced variants (H104K- and GXG-HSPB5), R120G-HSPB5 forms enlarged oligomers. But the disease mutation–carrying oligomers actually promote client aggregation under the nonstress conditions used here, and we found that they engage early-lag-phase client species is a way that promotes their self-association and leads to rapid depletion of monomeric αLac (Fig. 3B). Hence, polydispersity likely comes at a price; although it allows HSPB5 to readily adopt states with enhanced activity, it may also permit the formation of states with enhanced but detrimental client-binding modes. The limited spectrum of diseases associated with the R120G mutation in ubiquitously expressed HSPB5 suggests that specific clients in specific cell types are particularly susceptible to enhanced binding modes. The ability to intervene in such pathological interactions in the future will be aided by a fuller understanding of the structural ramification that gives rise to dysfunction.
The model proposed here (Fig. 7) can be rationalized in a cellular context. HSPB5 is constitutively expressed in a wide array of tissues; at high levels in eye lens, skeletal muscle, and cardiac tissue. The chaperone is therefore available to engage cellular proteins that may become destabilized under normal conditions and/or to perform other, still undefined functions. Under diverse stress conditions, expression of HSPB5 is up-regulated under the control of heat shock transcription factors (31). However, the transcription and translation of new HSPB5 in response to stress is not instantaneous, so the ability of the HSPB5 already present in a cell to immediately adopt additional and more efficacious binding modes can delay the onset of irreversible aggregation while the transcriptional activation of cellular chaperones is underway. Additional layers of complexity (for example, the ability of sHSPs to form hetero-oligomers) and the wide diversity of clients present in a cellular context are challenges for future studies.
Experimental procedures
Protein constructs expression and purification
All protein constructs and methods for their expression and purification have been described previously. Briefly, all proteins were expressed and purified BL21 Escherichia coli. Expression was induced through the addition of isopropyl 1-thio-β-d-galactopyranoside, cultures were grown for 16 h at either 16 °C or 22 °C. wHSP16.9 (UniProt accession number P12810) was purified as described previously (32).
HSPB5 (UniProt accession number P02511) and HSPB5 mutants were purified by a common method, similar to strategies reported previously (33, 34). Cells were resuspended in 20 mm Tris and 200 mm sodium chloride (pH 7.6) and lysed with a French press in the presence of DNase RNase and lysozyme. Lysates were clarified by centrifugation at 38,000 × g for 30 min. 9.46 g of ammonium sulfate was added to 30 ml of clarified lysate to precipitate HSPB5 or mutants. Precipitated HSPB5 or mutants were pelleted by centrifugation at 38,000 × g for 30 min. Precipitated HSPB5 was resuspended in 20 mm Tris (pH 8.0) and applied over a GE G25 column (GE product code 17-0031-01) equilibrated in 20 mm Tris (pH 8.0) to remove excess salts. Buffer-exchanged samples were applied onto a GE DEAE FF column (GE product code 17070901) equilibrated in 20 mm Tris (pH 8.0), and a five-step gradient of 5–25% 20 mm Tris and 1 m sodium chloride (pH 8.0) was applied to elute the protein. Fractions were collected at each step, and the purest fractions were identified by SDS-PAGE and pooled. Pooled fractions were diluted 2-fold with 20 mm Tris (pH 8.0) and were applied onto a GE MonoQ 10/100 GL column equilibrated in 20 mm Tris (pH 8.0). A 0–30% gradient of 20 mm Tris and 1 m sodium chloride was applied, and the purest fractions were identified by SDS-PAGE and pooled. Pooled fractions were concentrated and, as a final step, applied over a SDX200 (GE) gel filtration column equilibrated in 25 mm sodium phosphate and 250 mm sodium chloride (pH 7.5). The purest fractions were identified by SDS-PAGE and dialyzed against 25 mm sodium phosphate and 150 mm sodium chloride (pH 7.5). HSPB5 and mutants were concentrated to ∼1 mm following dialysis.
Chaperone assays by light scattering
Light-scattering chaperone assays were performed in a 96-well plate with 250-μl reactions at 42 °C using a BioTek Synergy HT plate reader. Optical density is a product of light scattering and the occlusion of transmitted light by large visible aggregates. Therefore, optical densities at 360 nm (OD360 nm) are reported as a proxy for light scattering. Use of optical density enables use of a plate reader format, allowing multiple aggregation assays to be monitored simultaneously. This is important because of inherent variability and stochasticity in aggregation. Although trends among different chaperones are highly reproducible, specific aspects of an aggregation time course are best compared within a set of measurements carried out on a single plate. Solutions contained 600 μm bovine α-lactalbumin (Sigma, L6010) in 25 mm sodium phosphate and 150 mm sodium chloride (pH 7.5, PBS 7.5) with and without 100 μm sHSP (subunit concentration). The sHSPs on their own do not contribute to the scattering signal (< 0.1%). Aggregation of αLac was induced by addition of EDTA and DTT to final concentrations of 5 mm. Specifically, 12.5 μl of 100 mm DTT and EDTA were added to 237.5 μl containing α-lactalbumin and sHSP. The addition of DTT and EDTA defines time 0 min. All samples were preincubated for 25 min at 42 °C prior to the addition of DTT and EDTA (also at 42 °C). Data points were collected every 2.5 min. Just prior to every data point collected, the plate was agitated for 5 s.
For assays where aggregates of αLac were allowed to form prior to addition of sHSPs, sHSPs were added to aggregating αLac when ∼5% of the final observed light scattering (based on previous measurement) was detected. 50 μl of 500 μm sHSP at 42 °C were added to wells containing aggregating αLac. PBS 7.5, at 42 °C, was added to αLac on its own and premixed αLac/sHSP wells to control for volume. To facilitate the detection of early aggregates, data points were collected every 38 s. Just prior to every data point collected, the plate was agitated for 5 s.
For all assays, duplicates of each condition were collected in each experiment, and the average curve of the duplicates is presented for clarity. Duplicates from the assay presented in Fig. 2 are shown in Fig. S2. The variability in the H104K curves in this assay is the largest observed across all assays. Additionally, replicates for all assays were collected, and the data presented are consistent across replicates.
SEC of reduced αLac and sHSPs
SEC experiments were performed on an GE Akta Purifier equipped with a 24-ml Superdex 200 preparatory-grade column (GE Life Sciences) and a 100-μl sample loop in PBS 7.5. SEC experiments were performed at room temperature (∼25 °C) with 600 μm αLac and 100 μm sHSP. αLac/sHSP mixtures or sHSP alone were preincubated at 42 °C for 25 min, followed by addition of DTT/EDTA to final concentrations of 5 mm each. Samples were incubated for an additional 30 min at 42 °C after addition of DTT and EDTA and then injected onto the SEC column.
1D NMR spectra of destabilized αLac
Watergated 1D 1H NMR spectra of 400 μm αLac in the presence of 5 mm DTT and 5 mm EDTA were collected over time at 37 °C in the absence and presence of 100 μm sHSP on a Bruker DMX 500-MHz spectrometer equipped with a triple-resonance, triple-axis gradient probe. All samples were in PBS 7.5 containing 10% D2O and preincubated at 37 °C prior to addition of DTT and EDTA.
Data were collected over 7.5 min for each time point and the start of the first spectrum collected was 4.5 min after the addition of DTT and EDTA to allow for instrument calibration. Spectra were processed using Topspin3.0. The progressive loss of intensity because of aggregation was monitored at 0.860 ppm, which provides the strongest signal in the destabilized αLac spectrum (Fig. 3A). The sum of the initial 0.860 ppm intensity of destabilized αLac on its own and the 0.860 ppm intensity sHSP on its own (IαLac alone + IsHSP alone) was used to normalize data collected for each series of experiments (Imixture/(IαLac alone + IsHSP alone)). The retained intensity in the αLac/sHSP mixture, throughout the time series, was reported as the normalized intensity (Imixture/(IαLac alone + IsHSP alone)).
Where appreciable binding was observed (R120G-HSPB5 and H104K-HSPB5), binding was confirmed with additional data collection. For R120G-HSPB5, a replicate was performed using the method described above and the normalized losses in intensity were 22% and 18% at the first time point. To survey potential variability in time, a replicate of H104K was collected, where data collection was shortened to 5 min rather than 7.5 min. In this, the first two data points collected in the 5-min dataset spanned the reported collection window by 2.5 min. Both the 5-min and 10-min time points in this experiment were in agreement with an intensity loss of 25% in the presence of H104K-HSPB5.
1D NMR experiments were collected at a 4:1 αLac:sHSP ratio at 37 °C, which is different from other αLac-related data presented. This ratio allowed detection of αLac/sHSP interactions at early time points without confounding issues with rapid αLac aggregation. Further, maintaining relatively low concentrations of sHSP was important to reduce contributions of sHSP to the spectrum. This was especially important at later time points when the signal from αLac had decayed.
Author contributions
S. P. D. and R. E. K. conceptualization; S. P. D. and R. E. K. data curation; S. P. D. and R. E. K. formal analysis; S. P. D. and R. E. K. investigation; S. P. D. and R. E. K. methodology; S. P. D. writing-original draft; S. P. D. and R. E. K. writing-review and editing; R. E. K. supervision; R. E. K. funding acquisition; R. E. K. project administration.
Supplementary Material
Acknowledgments
We thank Elizabeth Vierling (University of Massachusetts, Amherst) for generously providing plasmids and purification protocols for wHSP16.9. We thank P. Brzovic, H. Baughman, P. DaRosa, K. Reiter, and C. Woods for discussions and critical reading of the manuscript and J. Fields, C. Woods, and S. Witus for contributions to R120G-related NMR experiments.
This work was supported by NEI, National Institutes of Health Grant 5R01 EY017370. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- sHSP
- small heat shock protein
- DLS
- dynamic light scattering
- ACD
- α-crystallin domain
- NTR
- N-terminal region
- CTR
- C-terminal region
- SEC
- size exclusion chromatography
- αLac
- α-lactalbumin
- RH
- hydrodynamic radius/radii.
References
- 1. Horwitz J. (1992) α-Crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. U.S.A. 89, 10449–10453 10.1073/pnas.89.21.10449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Goldstein L. E., Muffat J. A., Cherny R. A., Moir R. D., Ericsson M. H., Huang X., Mavros C., Coccia J. A., Faget K. Y., Fitch K. A., Masters C. L., Tanzi R. E., Chylack L. T. Jr., and Bush A. I. (2003) Cytosolic β-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer's disease. Lancet 361, 1258–1265 10.1016/S0140-6736(03)12981-9 [DOI] [PubMed] [Google Scholar]
- 3. Kato K., Inaguma Y., Ito H., Iida K., Iwamoto I., Kamei K., Ochi N., Ohta H., and Kishikawa M. (2001) Ser-59 is the major phosphorylation site in αB-crystallin accumulated in the brains of patients with Alexander's disease. J. Neurochem. 76, 730–736 [DOI] [PubMed] [Google Scholar]
- 4. Velotta J. B., Kimura N., Chang S. H., Chung J., Itoh S., Rothbard J., Yang P. C., Steinman L., Robbins R. C., and Fischbein M. P. (2011) αB-crystallin improves murine cardiac function and attenuates apoptosis in human endothelial cells exposed to ischemia-reperfusion. Ann. Thorac. Surg. 91, 1907–1913 10.1016/j.athoracsur.2011.02.072 [DOI] [PubMed] [Google Scholar]
- 5. Björkdahl C., Sjögren M. J., Zhou X., Concha H., Avila J., Winblad B., and Pei J. J. (2008) Small heat shock proteins Hsp27 or αB-crystallin and the protein components of neurofibrillary tangles: tau and neurofilaments. J. Neurosci. Res. 86, 1343–1352 10.1002/jnr.21589 [DOI] [PubMed] [Google Scholar]
- 6. Vicart P., Caron A., Guicheney P., Li Z., Prévost M. C., Faure A., Chateau D., Chapon F., Tomé F., Dupret J. M., Paulin D., and Fardeau M. (1998) A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 20, 92–95 10.1038/1765 [DOI] [PubMed] [Google Scholar]
- 7. Laskowska E., Matuszewska E., and Kuczyńska-Winik D. (2010) Small heat shock proteins and protein-misfolding diseases. Curr. Pharm. Biotechnol. 11, 146–157 10.2174/138920110790909669 [DOI] [PubMed] [Google Scholar]
- 8. Nefedova V. V., Muranova L. K., Sudnitsyna M. V., Ryzhavskaya A. S., and Gusev N. B. (2015) Small heat shock proteins and distal hereditary neuropathies. Biochemistry 80, 1734–1747 [DOI] [PubMed] [Google Scholar]
- 9. Bumagina Z. M., Gurvits B. Y., Artemova N. V., Muranov K. O., Yudin I. K., Kurganov B. I. (2010) Mechanism of suppression of dithiothreitol-induced aggregation of bovine α-lactalbumin by α-crystallin. Biophys. Chem. 146, 108–117 10.1016/j.bpc.2009.11.002 [DOI] [PubMed] [Google Scholar]
- 10. Gast K., Zirwer D., Müller-Frohne M., and Damaschun G. (1998) (1998) Compactness of the kinetic molten globule of bovine α-lactalbumin: a dynamic light scattering study. Protein Sci. 7, 2004–2011 10.1002/pro.5560070917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Taylor R. P., and Benjamin I. J. (2005) Small heat shock proteins: a new classification scheme in mammals. J. Mol. Cell Cardiol. 38, 433–444 10.1016/j.yjmcc.2004.12.014 [DOI] [PubMed] [Google Scholar]
- 12. Aquilina J. A., Benesch J. L., Bateman O. A., Slingsby C., and Robinson C. V. (2003) Polydispersity of a mammalian chaperone: mass spectrometry reveals the population of oligomers in αB-crystallin. Proc. Natl. Acad. Sci. U.S.A. 100, 10611–10616 10.1073/pnas.1932958100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baldwin A. J., Lioe H., Robinson C. V., Kay L. E., and Benesch J. L. (2011) αB-crystallin polydispersity is a consequence of unbiased quaternary dynamics. J. Mol. Biol. 413, 297–309 10.1016/j.jmb.2011.07.016 [DOI] [PubMed] [Google Scholar]
- 14. Koteiche H. A., and McHaourab H. S. (2003) Mechanism of chaperone function in small heat-shock proteins: phosphorylation-induced activation of two-mode binding in αB-crystallin. J. Biol. Chem. 278, 10361–10367 10.1074/jbc.M211851200 [DOI] [PubMed] [Google Scholar]
- 15. Ecroyd H., Meehan S., Horwitz J., Aquilina J. A., Benesch J. L., Robinson C. V., Macphee C. E., and Carver J. A. (2007) Mimicking phosphorylation of αB-crystallin affects its chaperone activity. Biochem. J. 401, 129–141 10.1042/BJ20060981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajagopal P., Tse E., Borst A. J., Delbecq S. P., Shi L., Southworth D. R., and Klevit R. E. (May 11, 2015) A conserved histidine modulates HSPB5 structure to trigger chaperone activity in response to stress-related acidosis. eLife 10.7554/eLife.07304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clouser A. F., and Klevit R. E. (2017) pH-dependent structural modulation is conserved in the human small heat shock protein HSBP1. Cell Stress Chaperones 22, 569–575 10.1007/s12192-017-0783-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fleckenstein T., Kastenmüller A., Stein M. L., Peters C., Daake M., Krause M., Weinfurtner D., Haslbeck M., Weinkauf S., Groll M., and Buchner J. (2015) The chaperone activity of the developmental small heat shock protein Sip1 is regulated by pH-dependent conformational changes. Mol. Cell 58, 1067–1078 10.1016/j.molcel.2015.04.019 [DOI] [PubMed] [Google Scholar]
- 19. Pasta S. Y., Raman B., Ramakrishna T., and Rao C. M. (2004) The IXI/V motif in the C-terminal extension of α-crystallins: alternative interactions and oligomeric assemblies. Mol. Vis. 10, 655–662 [PubMed] [Google Scholar]
- 20. Jehle S., Rajagopal P., Bardiaux B., Markovic S., Kühne R., Stout J. R., Higman V. A., Klevit R. E., van Rossum B. J., and Oschkinat H. (2010) (2010) Solid-state NMR and SAXS studies provide a structural basis for the activation of αB-crystallin oligomers. Nat. Struct. Mol. Biol. 17, 1037–1042 10.1038/nsmb.1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Montfort R. L., Basha E., Friedrich K. L., Slingsby C., and Vierling E. (2001) Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 8, 1025–1030 10.1038/nsb722 [DOI] [PubMed] [Google Scholar]
- 22. Basha E., Friedrich K. L., and Vierling E. (2006) The N-terminal arm of small heat shock proteins is important for both chaperone activity and substrate specificity. J. Biol. Chem. 281, 39943–39952 10.1074/jbc.M607677200 [DOI] [PubMed] [Google Scholar]
- 23. Haley D. A., Horwitz J., and Stewart P. L. (1998) The small heat-shock protein, αB-crystallin, has a variable quaternary structure. J. Mol. Biol. 277, 27–35 10.1006/jmbi.1997.1611 [DOI] [PubMed] [Google Scholar]
- 24. Lindner R. A., Kapur A., and Carver J. A. (1997) The interaction of the molecular chaperone, α-crystallin, with molten globule states of bovine α-lactalbumin. J. Biol. Chem. 272, 27722–27729 10.1074/jbc.272.44.27722 [DOI] [PubMed] [Google Scholar]
- 25. Ito H., Kamei K., Iwamoto I., Inaguma Y., Nohara D., and Kato K. (2001) Phosphorylation-induced change of the oligomerization state of αB-crystallin. J. Biol. Chem. 276, 5346–5352 10.1074/jbc.M009004200 [DOI] [PubMed] [Google Scholar]
- 26. Moreau K. L., and King J. A. (2012) (2012) Cataract-causing defect of a mutant γ-crystallin proceeds through an aggregation pathway which bypasses recognition by the α-crystallin chaperone. PLoS ONE 7, e37256 10.1371/journal.pone.0037256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mymrikov E. V., Daake M., Richter B., Haslbeck M., and Buchner J. (2017) The chaperone activity and substrate spectrum of human small heat shock proteins. J. Biol. Chem. 292, 672–684 10.1074/jbc.M116.760413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee G. J., Roseman A. M., Saibil H. R., and Vierling E. (1997) A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 16, 659–671 10.1093/emboj/16.3.659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng G., Basha E., Wysocki V. H., and Vierling E. (2008) Insights into small heat shock protein and substrate structure during chaperone action derived from hydrogen/deuterium exchange and mass spectrometry. J. Biol. Chem. 283, 26634–26642 10.1074/jbc.M802946200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bova M. P., Yaron O., Huang Q., Ding L., Haley D. A., Stewart P. L., and Horwitz J. (1999) Mutation R120G in αB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc. Natl. Acad. Sci. U.S.A. 96, 6137–6142 10.1073/pnas.96.11.6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Thonel A., Le Mouël A., and Mezger V. (2012) Transcriptional regulation of small HSP-HSF1 and beyond. Int. J. Biochem. Cell Biol. 44, 1593–1612 10.1016/j.biocel.2012.06.012 [DOI] [PubMed] [Google Scholar]
- 32. Lee G. J., and Vierling E. (1998) Expression, purification, and molecular chaperone activity of plant recombinant small heat shock proteins. Methods Enzymol. 290, 350–365 10.1016/S0076-6879(98)90031-3 [DOI] [PubMed] [Google Scholar]
- 33. Delbecq S. P., Jehle S., and Klevit R. (2012) Binding determinants of the small heat shock protein, αB-crystallin: recognition of the “IxI” motif. EMBO J. 31, 4587–4594 10.1038/emboj.2012.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delbecq S. P., Rosenbaum J. C., and Klevit R. E. (2015) A mechanism of subunit recruitment in human small heat shock protein oligomers. Biochemistry 54, 4276–4284 10.1021/acs.biochem.5b00490 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.