

Abstract
Introduction:
Ubiquitin-Proteasome System (UPS) has been validated as a novel anticancer drug target in the past 20 years. The UPS contains two distinct steps: ubiquitination of a substrate protein by ubiquitin activating enzyme (E1), ubiquitin conjugating enzyme (E2), and ubiquitin ligase (E3), and substrate degradation by the 26S proteasome complex. The E3 enzyme is the central player in the ubiquitination step, and has a wide range of specific substrates in cancer cells, offering great opportunities for discovery and development of selective drugs.
Areas covered:
This review summarizes the recent advances in small molecule inhibitors of E1s, E2s, and E3s, with a focus on the latest patents (from 2015 to 2018) of E3 inhibitors and modulators.
Expert opinion:
One strategy to overcome limitations of current 20S proteasome inhibitors is to discover inhibitors of the upstream key components of the UPS, such as E3 enzymes. E3s play important roles in cancer development and determine the specificity of substrate ubiquitination, offering novel target opportunities. E3 modulators could be developed by rational design, natural compound or library screening, old drug repurposes, and application of other novel technologies. Further understanding of mechanisms of E3-substrate interaction will be essential for discovering and developing next generation E3 inhibitors as effective anticancer drugs.
Keywords: E3, protein ubiquitination and degradation, ubiquitin-proteasome system, cancer therapy, drug discovery
1. Introduction
1.1. Ubiquitination pathway and E3 ubiquitin ligases
Ubiquitination is an important post-translational process that is involved in the pathophysiological mechanisms of many human diseases, and the ubiquitinated proteins can be recognized and degraded by the 26S proteasome (Figure 1). Ubiquitin (Ub), which plays a central role in this process, contains 76 highly conserved amino acids and is present in all types of cells from yeast to human. Ubiquitination is a multistep enzyme cascade which involves the sequential actions of the ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) (Figure 1). The first step is an ATP-dependent activation of the ubiquitin molecule by an E1 enzyme to form a thioester bond between the Ub carboxyl group and the thiol of the E1 active cysteine residue. Then, the activated ubiquitin is transferred to the cysteine residue of the E2 enzyme, which permits it to be sequentially docked into the E3 ligase. Finally, the ubiquitin moieties attach the specific lysine residue on the protein substrate which is mediated by the E3 ligase [1,2]. The sequential covalent attachment of ubiquitin moieties forms a ubiquitin chain. Ubiquitin contains seven lysine residues (K6, K11, K27, K29, K33, K48, and K63), all of which could be associated with the chain formation. However, K48- and K63-linked mono-ubiquitination and polyubiquitination are the most commonly found and have been investigated extensively [3].
Figure 1. Overview of the UPS.
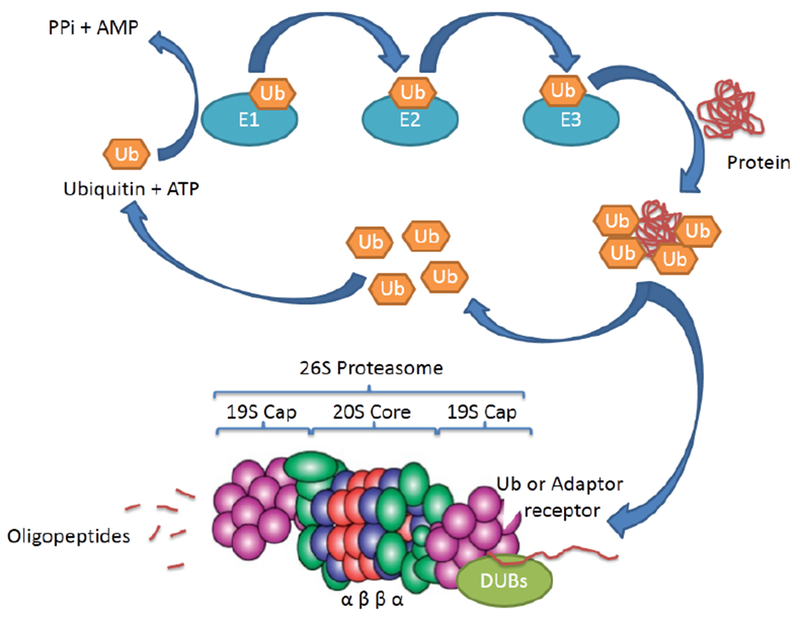
Ubiquitination is a multistep enzyme cascade. The first step is an ATP-dependent activation of ubiquitin (Ub) molecule by an E1 enzyme to form a thioester bond between the Ub carboxyl group and the thiol of the E1 active cysteine residue. Then, the activated ubiquitin is transferred to the cysteine residue of E2 enzymes, which permit to be sequentially docked into E3 ligases. Finally, the ubiquitin moieties attach the specific lysine residue on the protein substrate mediated by the E3 ligases. Deubiquitinating enzymes (DUBs) reverse the ubiquitination process and cleavage of Ub from polypeptides. The ubiquitinated substrate protein is recognized and deubiquitinated by 19S proteasome and subsequently degraded by 20S proteasome. For details, see text.
According to their distinct mechanisms of protein-protein interaction, E3 ligases are currently categorized into three families, (i) Homologous to E6-associated Protein C-terminus (HECT), (ii) Really Interesting New Gene (RING), and (iii) U-box Domain [1,4,5]. HECT E3 ligases can recruit ubiquitin from the E2 enzyme, and directly conjugate ubiquitin moieties to the lysine residue of target proteins, independent of the E2 enzyme. In contrast, RING type and U-box E3 ligases mainly function as scaffolding proteins and recruit the ubiquitin-charged E2 and substrate proteins through docking sites. RING type E3 ligase is characterized as a subunit containing RING domain for recruiting E2 enzyme. Notably, RING E3 ligases are further classified into a single-molecular RING-containing E3 ligases or multi-subunit E3 ligase complexes including various Cullin-containing E3 ligase complexes (e.g. Skp1/Cul1/F-box protein complex or SCF) [2,6,7].
To date, a variety of studies on protein ubiquitination and degradation have confirmed that E1 and many E2 and E3 enzymes are involved in tumorigenesis, which can therefore be considered as specific targets for screening and developing potential anticancer therapeutic agents. However, discovery of small molecule blockers of protein-protein interfaces is challenging. There are up to 1000 E3 ligases encoded by the human genome, and a strategy for inhibiting E3 ligases with small molecules would significantly expand the druggable protein universe. In this review, we first summarize recent emerging findings on the inhibitors of the ubiquitination enzyme cascade (E1, E2, and E3), and then review the latest patents (from 2015 to now) of E3 inhibitors or modulators as well as their applications for cancer treatments.
1.2. E1 enzyme inhibitors
E1 enzyme is indispensable for protein post-translational modification and proteasome-mediated degradation, suggesting that E1 might be a potent pharmacological target for identifying a regulator of the ubiquitin–proteasome pathway. The ATP-competitive molecules, blockers of covalent E1~Ub complex or interaction between E1s and E2s, are essentially available candidates for developing E1 inhibitors [8]. To date, a wide range of natural or synthesized compounds have been identified that are able to target the E1 enzymes in vitro and in vivo [8,9] (Table 1).
Table 1.
E1 and E2 Enzymes Inhibitors
| E1 Enzyme Inhibitors | Effects on cancer cells | Ref. |
|---|---|---|
| Panepophenanthrin | Inhibit the binding of ubiquitin to E1, but has no inhibitory effect on cell growth. [further studies are needed] | [10] |
| Himeic acid A | Inhibitory effect on E1 catalytic activity | [12] |
| Largazole | Induce G1 phase arrest, which is associated with increased expression of p21 and the recruitment of HDAC1 to E2F1 that results in the proteasomal degradation of E2F1 | [13] |
| Hyrtioreticulins A and B | Potent inhibitory ability against the formation of an E1-ubiquitin intermediate | [14] |
| PYR-41 | Irreversible inhibitor of E1, inhibit the activation of NF-ҡB, and preventing the proteasome-mediated degradation of IκBα as well as p53 | [17] |
| JS-K | Reduction of total ubiquitylated proteins, increased levels of p53 expression | [19] |
| NSC624206 | Preventing the ubiquitin-thioester formation step, but has no effect on ubiquitin adenylation in the E1 activation reaction | [18] |
| MLN4924 | Attenuates the function of the various CRLs | [20] |
| 5′-O-sulfamoyl-N(6)-[(1S)-2,3-dihydro-1H-inden-1-yl]-adenosine (Compound I) | Nonselective UAE-inhibitory activity, forming a covalent adduct Ub-I, thereby inhibiting UAE-dependent ATP-PPi exchange activity and E1-E2 transthiolation | [27] |
| MLN7243 | UBA1-selective inhibitor | [28–30] |
| ABPA3 | Dual inhibitor of the Ub and NEDD8 E1s | [31] |
| Deoxyvasicinone-based compounds: - Dipeptide-conjugated deoxyvasicinone derivative - Valine-linked deoxyvasicinone derivative - N-isopropyl-linked deoxyvasicinone derivative |
NAE-inhibitory effects through disrupting the ATP-binding domain [Biological effects and human cancers remain unknown]. | [32,33] |
| Piperacillin 1 | A reversible ATP-competitive inhibitor of NAE; increasing levels of the CRL substrate p27kip1 | [34] |
| M22 (1-benzyl-N-(2,4-dichlorophenethyl) piperidin-4-amine) | Inhibitory activities of NAE against human cancers [further studies are needed] | [35] |
| LP0040 (2H-chromen-2-one derivative with phenyl substituents ) | Inhibitory activities of NAE against human cancers [further studies are needed] | [36] |
| E2 Enzyme Inhibitors | ||
| Peptoid compound (Leucettamol A) | Disrupting the formation of the Ubc13–Uev1A complex, and inactivating of NF-kB and sensitization of tumor cells to chemotherapeutic agents | [40,41] |
| Dimeric sterols (Manadosterols A and B) | Antagonizing the Ubc13–Uev1A interaction, and more potent than Leucettamol A | [42] |
| NSC697923 | Induce cancer cell apoptosis, cell death, initiating cell death by activating JNK pathway | [43] |
| CC0651 | Disrupting the activity of human E2 enzyme Cdc34 | [45] |
| Triazines compounds | Rad6B-inhibitory activities on levels of its conjugation to both ubiquitin and the substrate H2A | [47] |
Panepophenanthrin is extracted from the fermented broth of a mushroom strain Panus rudis Fr. IFO 8994 and identified as the first E1 inhibitor [10] (Table 1). This compound prevented the binding of ubiquitin to E1, but had no inhibitory effect on cell growth [10]. Therefore, its cellular effects had been further studied. The racemic compounds and derivatives of panepophenanthrin were subsequently synthesized, leading to discovery of RKTS-80, −81, and −82 with cell-permeable inhibitory activities against E1 enzyme [11]. Interestingly, natural compounds from marine organisms remain a potential resource for screening and development of anticancer agents. For example, Himeic acid A [12], Largazole [13], and hyrtioreticulins A and B [14] were discovered from a marine fungus Aspergillus sp; the Floridian marine cyanobacterium Symploca sp and the marine sponge Hyrtios reticulatus, respectively. These compounds also exhibit an inhibitory effect on E1 catalytic activity as secondary metabolites (Table 1). Moreover, Largazole, characterized as a putative histone deacetylase (HDAC) inhibitor, essentially impedes the formation of ubiquitin-adenylate complex in vitro [15]. In lung cancer cells, Largazole can induce G1 phrase arrest, which is associated with increased expression of p21 and the recruitment of HDAC1 to E2F1 that results in the proteasomal degradation of E2F1 [16]. Hyrtioreticulins A and B also have potent inhibitory abilities against the formation of an E1-ubiquitin intermediate, with IC50 values of 0.75 and 11 μg/mL, respectively [14].
PYR-41, an irreversible inhibitor of E1, was firstly identified by Yang et al [17], which functionally attenuates the activation of cytokine-mediated nuclear factor-ҡB (NF-ҡB), in part attributing to the blockade of K63-linked ubiquitination of an upstream signaling molecule TRAF6 that results in activation of IҡB kinase (IKK) for the phosphorylation of IҡBα, ultimately preventing the proteasome-mediated degradation of IκBα as well as p53. Hence, PYR-41 may be used as a potential anticancer agonist [17] (Table 1). The E1 inhibitors could also be a useful tool for exploring the mechanisms of ubiquitination [18]. JS-K, a nitric oxide prodrug, can inactivate the formation of ubiquitin~E1 thioester in a dose-dependent manner causing the reduction of total ubiquitylated proteins and increased levels of p53 expression, along with selectively induced apoptosis in the transformed cells retaining wild type p53 [19] (Table 1). NSC624206, a recently reported E1 inhibitor, can prevent in vitro ubiquitination of p27. Mechanistically, NSC624206 specifically impedes the ubiquitin-thioester formation step, but has no effect on ubiquitin adenylation in the E1 activation reaction. Therefore, NSC624206 could be an available agent for the control of excessive ubiquitin-mediated proteolysis in vivo [18] (Table 1).
It should be noted that another conjugation cascade, neddylation, is highly similar to ubiquitylation. One target protein of neddylation is NEDD8 (the neural precursor cell-expressed developmentally downregulated protein 8), whose modification involves the sequential activation of E1 NEDD8-activating enzyme (NAE), E2 NEDD8-conjugating enzyme (Ubc12), and the E3 Cullin-RING ligases (CRLs), ultimately transferring NEDD8 to a target molecule. Being the activator in the first neddylation step, NAE should be a valuable therapeutic target for modulating CRL activity and protein degradation [20–23]. MLN4924 is a selective substrate-assisted inhibitor of NAE, which attenuates the function of the various CRLs to exert its anti-tumor activities in a wide range of cancer types [20,24] (Table 1). Essentially, MLN4924 is an adenosine sulfamate analogue and AMP mimics, which can form the covalent adduct NEDD8-MLN4924 and thereby potently inhibits NAE activity and neddylation of all cullins. This leads to inactivation of CRLs and accumulation of the related substrates such as CDT1, IκBα, and NOXA, further enhancing apoptosis, senescence, and autophagy in cancer cells [21,23]. MLN4924 has exhibited potent anti-tumor activities against a variety of human cancers, including solid (colon, lung, liver, ovarian) and hematological (myeloma, lymphoma) malignancies [21]. In light of this preclinical evidence, several Phase-I clinical trials with MLN4924 are currently ongoing [25,26]. Numerous additional Phase-I and Phase-II studies are undertaken to further investigate its safety and efficacy as a single agent or in combination with other conventional anticancer drugs.
Similar to MLN4924, another adenosine sulfamate analogue, namely 5’-O-sulfamoyl-N(6)-[(1S)-2,3-dihydro-1H-inden-1-yl]-adenosine (Compound I) that has nonselective UAE-inhibitory activity, was identified by Chen et al [27] (Table 1). Mechanistically, Compound I can form a covalent adduct Ub-I, thereby inhibiting UAE-dependent ATP-PPi exchange activity and E1-E2 transthiolation in a dose-dependent manner, along with loss of UAE thioester [27]. Recently, the adenosyl sulfamates MLN7243 [28–30], a UBA1-selective inhibitor, and ABPA3 [31], a dual inhibitor of the Ub and NEDD8 E1s, were reported and tested pre-clinically (Table 1). Therefore, a promising strategy to develop potent and selective E1 inhibitors is through the substrate-assisted inhibition by adenosine sulfamate analogues.
Furthermore, several deoxyvasicinone-based compounds containing inhibitory activities against the function of NAE have been evaluated (Table 1). For example, a dipeptide-conjugated deoxyvasicinone derivative, a valine-linked deoxyvasicinone derivative, and a N-isopropyl-linked deoxyvasicinone derivative were all found to exhibit NAE-inhibitory effects through disrupting the ATP-binding domain [32,33], although the relationship between their biological effects and human cancers remain unknown.
In addition, the FDA-approved drug Piperacillin 1 has been successfully repurposed as a reversible ATP-competitive inhibitor of NAE (Table 1). In both cell-free and cell-based assays, Piperacillin 1 attenuates Ubc12-NEDD8 conjugation mediated by NAE, but exhibits no significant effect on the activity of the related Ub-like enzyme SAE or the E2 enzyme [34]. Although it leads to the increased levels of the CRL substrate p27kip1 in Caco-2 cells, Piperacillin 1 does not exert significant NAE inhibitory activity in HepG2 or THP-1 cell lines. This discovery case demonstrates that a validated library of privileged scaffolds offered by the FDA-approved drugs could be used to develop and screen candidate compounds of NAE inhibitors [34]. The emerging compounds such as M22 (1-benzyl-N-(2,4-dichlorophenethyl) piperidin-4-amine) [35] and LP0040 (2H-chromen-2-one derivative with phenyl substituents) [36] with NAE-inhibitory activities in human cancer cells are also successively identified (Table 1). Future studies should focus on the inhibitory mechanisms of these NAE inhibitors as well as their biological effects in cancer cells.
1.3. E2 enzyme inhibitors
E2 enzymes contain nearly 40 members, which mediate the conjugation of ubiquitin to substrates and therefore control protein stability and interactions in the UPS [37]. Although there is a large amount of structural and biological information available on E2s, only a limited number of E2 inhibitors have been discovered to date.
For example, Ubc13, an E2 enzyme, participates with various E3 ligases in building the K63-linked poly-ubiquitination chains. The K63-linked poly-ubiquitination modification regulates DNA damage repair, mitotic progression, and the NF-κB signaling [38]. Overexpression of the Ubc13 attenuates the abundance of p53. Moreover, knockout of the Ubc13 increases the activity of p53 [39]. Hence, an agent that blocks the formation of the Ubc13-Uev1A complex may function as an anticancer agent. Tsukamoto et al. identified the chiral peptide E2 inhibitor Leucettamol A that was isolated from the marine sponge Leucetta aff. Microrhaphis [40] (Table 1). Leucettamol A disrupts the formation of the Ubc13–Uev1A complex, attenuating the E2 activity [40]. Small molecule antagonists of the Ubc13-Uev1 interaction could inhibit the enzymatic activity of this heterodimer and suppress K63 polyubiquitylation, showing that these antagonists could be specific and potent in nature. This idea was supported by the report by Scheper et al. [41] (Table 1). K63-linked polyubiquitylation of PCNA is inhibited by these peptoid compounds, associated with inactivation of NF-kB and sensitization of tumor cells to chemotherapeutic agents [41]. Recently, two new dimeric sterols, manadosterols A and B derived from the marine sponge Lissodendryx fibrosa, were discovered to contain activities antagonizing the Ubc13–Uev1A interaction, and are more potent than Leucettamol A [42] (Table 1). In addition, Cheng et al. reported a novel UBE2N (Ubc13) inhibitor NSC697923 that was also able to induce cancer cell apoptosis. NSC697923 induced nuclear accumulation of p53 in wild-type p53-containing neuroblastoma (NB) cells, and initiated cell death by activating the c-Jun N-terminal kinase (JNK) pathway in mutant p53-expressing NB cells. Interestingly, NSC697923 impeded cell growth of chemo-resistant LA-N-6 NB cell line, and revealed in vivo antitumor efficacy in NB orthotropic xenografts as well [43] (Table 1), suggesting that NSC697923 may be used as a potential anti-tumor agent for NB patients.
Importantly, the E2 enzyme and ubiquitin exhibit low affinity interactions that mediate its covalent attachment to substrates [44]. Ceccarelli et al. identified a small molecule inhibitor CC0651 that selectively disrupts the activity of human E2 enzyme Cdc34, the specialized E2 for SCF E3 ligases, which forms K48-linked poly-ubiquitination chains on its targets for proteasome-mediated degradation. Proliferation of human cancer cell lines was impeded by CC0651 analogs which also promoted accumulation of the SCFSkp2 substrate p27Kip1 [45] (Table 1). In-depth investigation in cryptic structure has revealed that CC0651 inserts into a cryptic pocket on the Cdc34A surface that is distant from the active site cysteine. CC0651 unexpectedly traps the weak interaction between ubiquitin and the donor site of Cdc34A, thereby impeding catalysis [46]. CC0651 also disrupts the discharge of ubiquitin to acceptor lysine residues, and essentially has no effect on the interaction between Cdc34A and the RING domain subunit of the E3 enzyme [45]. In addition, several newly synthesized triazine compounds exhibit superior Rad6B-inhibitory activities on levels of its conjugation to both ubiquitin and the substrate H2A [47] (Table 1). E2 enzymes are thus susceptible to noncatalytic site inhibition and should be exploited further. A recent study reports a new allosteric pocket on the E2 enzyme Ube2T that plays an important role in the Fanconi anemia DNA repair pathway; by using a biophysical method, the fragment screening, the authors identified several fragments that can bind to the pocket site, causing inhibition of ubiquitin conjugation in vitro [150]. Thus, this finding provides a novel strategy to discover and develop E2 inhibitors.
1.4. E3 ligase inhibitors
Emerging evidence has demonstrated that the target specificity for ubiquitination is essentially determined by E3 ligases. Thus, targeting an E3 ligase should be a novel approach for development of potent anticancer agents with low side effects. Over the past decades, a variety of RING and HECT-type E3 ligases in connection to human cancers have been extensively investigated [48]. In the following sections, we will review the advance on inhibitors of several selected E3 enzymes including F-box and WD repeat domain-containing 7 (FBXW7), β-transducin repeat-containing protein (β-TrCP), S-phase kinase-associated protein 2 (Skp2), mouse double minute 2 homolog (MDM2) and the neuronally expressed developmentally downregulated 4 (NEDD4) (Figure 2) and their potential applications in cancer treatments.
Figure 2. Chemical structure of small molecule E3 inhibitors.
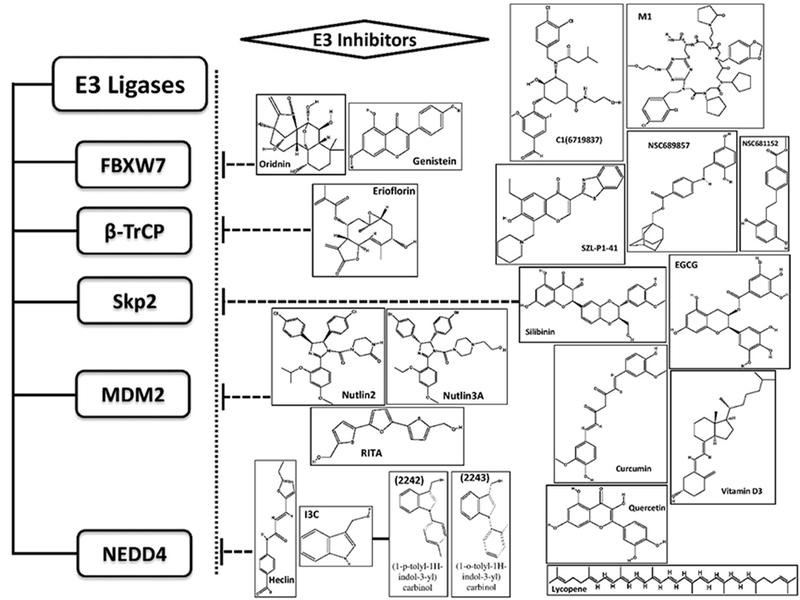
FBXW7 inhibitors Oridonin [61], Genistein [51]; β-TrCP inhibitor Erioflorin [70]; Skp2 inhibitors SZL-P1-41 [97], silibinin [98], Vitamin D3 [99], quercetin [100], lycopene [100], curcumin [100], EGCG [101], C1(6719837) [102], NSC689857 and NSC681152 [103], M1 [104]; MDM2 inhibitors Nutlin2 [108], Nutlin3A[109], RITA [111]; NEDD4 inhibitors Heclin [121], Indole-3-carbinol (I3C) (Compounds 2242 and 2243) [122].
1.4.1. FBXW7
FBXW7 is essentially characterized as a tumor suppressor, which participates in the degradation of multiple key oncoproteins such as c-Myc, c-Jun, cyclin E, mammalian target of rapamycin (mTOR), and other substrates [49]. Recent studies have shown that several regulators including microRNAs (miR-27a, miR-25 and miR-223) [50,51], p53 [52,53], and CCAAT/enhancer-binding protein-d (C/EBP-δ) [54] could govern FBXW7 expression and its substrate degradation in human malignancies. FBXW7 is largely deleted or mutated in a wide range of human cancers [55–58], however the mechanism associated with that is not well addressed [48,59]. Also, FBXW7 can mediate p100 (inhibitor of NF-κB signaling) degradation and function as a pro-survival factor through a GSK3-mediated phosphodegron in multiple myelomas [60]. Thus, targeting F-box protein or its substrate modification could be a promising strategy for screening anticancer agents.
Oridonin is a diterpenoid compound extracted from Chinese medical herb, which exhibits potent anti-leukemia effect [61] (Figure 2). Mechanistically, Oridonin treatment triggers elevation of FBXW7 expression and GSK-3 activity and attenuates the abundance of c-Myc, resulting in induction of cancer cell apoptosis and growth inhibition in the K562 xenograft model [62]. Genistein is an isoflavonoid compound that is isolated from soybean, and has been characterized as an inhibitor of oncogenic progression in various human cancers including pancreatic cancer (PC). Genistein inactivates miR-223 expression and elevates FBXW7 expression (Figure 2), consequently resulting in inhibition of cell growth and induction of apoptosis in PC cells [51]. In addition, treatment of PC cells with Genistein enhances the accumulation of miR-34a, resulting in the downregulation of Notch-1 signal pathway, which is involved in induction of cell apoptosis, inhibition of cell growth, and suppression of cell invasion [63]. SCF-I2, a biplanar dicarboxylic acid compound, has been shown to distort its substrate binding pocket and prevents recognition of phosphodegron on substrates by SCFCdc4 [64]. Hence, SCF-I2 represents the first example of a WD40 domain inhibitor. Interestingly, the inhibitors (e.g. imatinib [65]) targeting F-box proteins have been proposed to sensitize cancer stem cells to chemotherapies. However, the long-term effect of FBXW7 inhibition still needs to be assessed.
1.4.2. β-TrCP
β-TrCP (β-TrCP1 or β-TrCP2) exhibits a critical role in targeting specific substrates for ubiquitination and degradation. The diversity of β-TrCP function is essentially determined by a wide range of specific substrates of β-TrCP, such as Emi1, Cdc25A, Wee1A, cyclin D1, BTG, REST, PLK4, CEP68, Snail, the extracellular matrix protein fibronectin, Twist, Mcl-1, BimEL, Pdcd4, and Pro-caspase-3 that have been characterized to play a crucial role in cell cycle, apoptosis and migration (reviewed in [66]). In vitro cell culture and in vivo mouse model investigations have been conducted to confirm the tumorigenic activity of β-TrCP to promote tumor cell growth and development. For example, elevated accumulation of β-TrCP has been widely observed in a variety of cancers, including gastric [67], prostate [68], and breast [69]. Thus, targeting β-TrCP represents a novel strategy for cancer treatments in a context dependent manner. Recently, Erioflorin (Figure 2), extracted from Eriophyllum lanatum, has been identified as a stabilizer to enhance the abundance of Pdcd4, a well-characterized β-TRCP substrate, via impeding its proteasomal degradation. Erioflorin can specifically perturb the interaction between β-TrCP1 and its substrates, including Pdcd4, consequently causing a decrease in ubiquitination and degradation of Pdcd4, while having no effect on degradation of targets of other E3 ligases, such as p21 and HIF-1α [70]. This is the first reported compound that can specifically attenuate the activity of β-TrCP E3 ligase, although its effects on other β-TrCP substrates remain largely unknown. Due to the fact that β-TrCP targets specific substrates and exerts tumor suppressing function under some conditions, it is therefore difficult to develop an anticancer drug targeting β-TrCP in a tissue-specific manner [71,72]. Therefore, further investigation is required to delineate the exact role of β-TrCP in tumorigenesis or tumor suppression with the help of animal models in different tissue contexts.
1.4.3. Skp2
Skp2, a well characterized oncoprotein, regulates cancer development and progression via targeting its substrates (including p27, p21, p57, TOB1, RASSF1, FOXO1, and RBL2) for degradation [73–79]. For example, upregulation of Skp2 expression, which subsequently targets p27 for degradation, leads to enhanced cell proliferation in prostate epithelial cells [80,81]. Moreover, the data from experiments using Skp2 knockdown mice has further confirmed the oncogenic function of Skp2 in T-cell lineage, B-cell lineage, bone marrow, liver, breast, prostate, and skin [82–88]. Consistently, overexpression of Skp2 largely occurs in a variety of human cancers such as lymphomas [89,90], pancreatic cancer [91], breast carcinomas [92,93], and prostate cancer [94,95]. Altogether, targeting Skp2 represents a feasible approach to screen and develop novel E3 inhibitors for cancer therapies.
It has been reported that two small molecules, namely compound A (SMIP0004) and compound 25 (SZL-P1-41) (Figure 2), were able to attenuate Skp2 activities [96,97]. Compound A prevents incorporation of Skp2 into the SCF ligase, resulting in cell growth inhibition, apoptosis induction, and G1/S cell cycle arrest in multiple myeloma cells [97]. Compound 25 suppresses Skp2 E3 ligase activity, leading to attenuation of cell survival and Akt-mediated glycolysis as well as activation of p53-dependent senescence. Strikingly, compound 25 restricts cancer stem cell traits and cancer progression in animal trials [96]. Further research is needed to guide future chemical optimization of compound 25 derivatives in order to develop potential anticancer agents [96]. Interestingly, several natural agents including curcumin, quercetin, lycopene, silibinin, epigallocatechin-3-gallate (EGCG), and Vitamin D3 have also been found to downregulate expression of Skp2 in human cancers [98–101] (Figure 2). In addition, targeting the Skp2-Cks1’s p27-binding interface is largely used to screen selective inhibitors in recent studies. Wu et al. used this approach and obtained four compounds [namely, C1 (6719837), C2 (6544607), C16 (6744881). and C20 (A067/0031209) ] that were able to stabilize the expression of p27 in different cancer cell lines and induce cell cycle arrest in G1 or G2/M phases [102]. Ungermannova et al. reported other two compounds (NSC689857 and NSC681152) that perturb the Skp2-Cks1 interaction, thereby preventing p27Kip1 ubiquitination [103] (Figure 2). Recent studies also show the compound M1 disrupts Skp2/p300 interaction by promoting p300-mediated p53 acetylation and stabilization, ultimately inducing p53-mediated apoptosis in cancer cells without affecting Skp2 proteolytic activity [104] (Figure 2). Further biological studies and structure modifications could further improve the potency of this Skp2 inhibitor for cancer therapies. In addition, Skp2 nonproteolytic function in tumorigenesis should also be considered in the development of its chemical probes and inhibitors.
1.4.4. MDM2
MDM2 is a RING type E3 ligase, which plays a vital role in modulating the stability of the tumor suppressor protein p53 that regulates cell stress and limits cancer development. As an oncoprotein, MDM2 expression is upregulated in various tumors that retain wild type p53, thereby promoting cancer development [105,106]. Thus, targeting MDM2 in these cancer cells is a promising strategy for anticancer drug development [107].
Nutlins (Figure 2), a family of cis-imidazoline derivatives, are identified by high-throughput screening, cause the inactivation of MDM2 and therefore mediate p53 ubiquitination [108]. Nutlin2 binds to MDM2 directly in its p53-binding pocket, causing inhibition and therefore initiating p53-dependent signal transduction pathway in cancer cells. This leads to cell-cycle arrest, apoptosis, and cellular senescence of human tumor xenografts in nude mice with no significant toxic effect on healthy tissues [108]. Nutlin 3A/R7112 has entered clinical trials for solid tumors and leukemia because of its favorable pharmacological properties and toxicity in the preclinical studies [109]. Further development of its derivatives attempts to improve the pharmacological properties of the inhibitors whilst retaining potency and cellular selectivity for wild-type p53- over mutant p53-containing cell lines [110]. RITA (NSC652287) (Figure 2) is another MDM2 small molecule inhibitor that inhibits cell growth selectively in p53-positive cancer lines [111]. Mechanistically, RITA binds to the N-terminus of p53, thereby inducing a conformational change to prevent the formation of the α-helix, which is required for the recognition of p53 by MDM2 [111]. Furthermore, RITA does not attenuate the transcriptional activity of p53, instead that it impedes the interaction of p53 with other regulatory proteins, such as p300, which can catalyze p53 polyubiquitination with MDM2 [112]. Like Nutlins, RITA activates apoptosis in human tumor cells with little effect on healthy cells, and exhibits the growth-suppressing effect in tumor xenograft model mouse as well [111]. In light of this similarity, it will be interesting to elucidate whether RITA and Nutlins can synergistically enhance apoptosis in cancer cells [113]. In addition, several MDM2 inhibitors (e.g. HLI98, 5-deazaflavin compounds) that can promote p53 stabilization and activation have been studied in various cancer cells [114,115]. Further research is required to determine whether these molecules are useful agents for cancer treatments. Currently, some MDM2 small molecule inhibitors, including RG7112, RG7388, MI-77301/ SAR405838, AMG 232, MK-8242, DS3032b, HDM20, CGM097, RO5353, and RO2468, have entered clinical trials for potential cancer treatments [116–118].
1.4.5. NEDD4
NEDD4 is categorized into HECT-type E3 ligases, which ubiquitinate protein substrates to control virtually biochemical processes and are dysregulated in various diseases and cancers [119]. Like other E3 ligases, NEDD4 has received a continuing attention about its oncogenic role in human cancer progression [119]. However, it is difficult to develop specific agents for selectively modulating HECT E3 ligases due to the rotations of the N and C lobes of the HECT domains accompanying catalysis, a shallow active site, and dynamic regulation of HECT E3 activity [4,120]. Thus, little is known about small molecule enzymatic inhibitors of HECT domain-containing ubiquitin ligases. To meet this challenge, some novel methods (e.g. microphage display-based ubiquitin variants and Alpha screening technology) have been introduced into the screening of E3 ligase inhibitors and have shown interesting findings. Zhang et al. systematically developed ubiquitin variants (UbVs) that inhibit or activate HECT E3s. UbV inhibitors hijack the E2-binding site while UbV activators saturate the N-lobe exosite as shown by structural analysis of 6 HECT UbV complexes [120]. With the help of the establishment of a systematic UbVs-based library, there is an anticipation to address the complete interaction mechanisms for modulating enzyme activity across the human HECT family, thereby developing modulators for targeting a wide range of key signaling proteins.
In addition, Mund et al. isolated bicyclic peptides (bicycles) and a small molecule, heclin, by phage display and Alpha screening technology, respectively. Bicyclic peptides can obstruct the E2 binding sites, thereby exhibit in vitro inhibitory activity to the E3 ligase, via specific binding to the HECT domains of Smurf2, NEDD4, Mule/ Huwe1, and WWP. Heclin (Figure 2) inhibits several HECT ligases in vitro and in tissue culture cells. Furthermore, heclin has no inhibitory effect on E2 binding, but perturbs its conformation, thereby inducing in vitro oxidation of the active site CYS [121]. Further research on heclin may extend our knowledge of its multiple biological functions (e.g., blocking the modification of substrates targeted by HECT ligases). Quirit et al. reported a small molecule, indole-3-carbinol (I3C) (Figure 2), derived from cruciferous vegetables, which provides a new chemical scaffold with NEDD4-1 inhibitory activity. Further, its synthetic derivative, 1-benzyl-I3C, and the two indole-3-carbinol analogues with added methyl groups, Compounds 2242 and 2243, could insert into the catalytic site of NEDD4-1 and exhibit their NEDD4-1 inhibitory activities, associated with inhibition of melanoma cell proliferation. Thus, I3C analogues may be potential candidate anticancer agents for treatments of human cancers [122].
2. Patents on ubiquitin ligase system (from 2015 to 2018)
2.1. Category A: Modulating methods and inhibitors of E3
2.1.1. N-acetyl-L-phenylalanylamide (US9725708B2)
E6AP is a member of HECT family E3 ligases. HECT domain contains Lys48-linked polyubiquitin degradation signals that are recognized by the 26S proteasome [123]. The inventors provided methods for modulating the E6AP ligase activities by increasing or decreasing E6AP ligase oligomerization. Furthermore, their study showed that non-competitive small molecule inhibitors are useful in reducing ligase oligomer formation (Table 2).
Table 2.
Summary of reviewed patents on ubiquitin ligase
| Patent ID | Invention | Application | Invention type | Ref. |
|---|---|---|---|---|
| Category A: Modulating methods and inhibitors of ligase E3 | ||||
| US9725708B2 | New strategies for regulating E6AP ligase function using oligomerization modulatory agents | modulating E6AP-related pathologies |
Modulating methods | [126] |
| US2016/0068490A1 | Sulfonamidoquinoline compounds | Increasing the tumor suppressor p27 levels | Ubiquitnation inhibitors | [128] |
| US2016/0194644 A1 | E3 ligase RNF185 inhibitors | Identifying E3 ligase RNF185 inhibitors | Ligase inhibitors and screening method. | [129] |
| US2017/0275341A1 | Ubiquitin variant |
Modulating HECT E3 ligases | Modulating methods | [132] |
| US2017/0321205A1 | Ubiquitin variant |
Modulating SCF E3 ligases | Modulating methods | [133] |
| US2015/0374678A1 | Methods of inducing a CRBN conformation change to alter the substrate specificity by CMAs | Modulating biological activities such as immune-mediated effect, tumoricidal effect, apoptosis, proliferation, toxicity, or substrate degradation | Modulating methods | [134] |
| EP2954896A1 | Small molecule inhibitors | Therapeutic agents for activation of autophagy and the treatment of depression, and neurodegenerative diseases | Ubiquitnation inhibitors | [135] |
| US9464311B2 | A screening method, that specifically and covalently modifying the active site cysteine thiol residue of the HECT and RBR family of E3 ligase | Screening for small molecule modulators of ubiquitin E3 ligases that contain an active site cysteine | Method for identifying modulators | [137] |
| Category B: Assay methods of ligase E3 activity | ||||
| EP2564193B1 | Different stages of the ubiquitination cascade | In vitro assaying ubiquitination | Assay | [138] |
| US9587265B2 | Ubiquitin Chain Analysis | The newly characterized ovarian tumor protease (OTU) family DUBs and developing a method for determining the linkage specificity in the polyubiquitin chain | Assay | [139] |
| US2016/0076074A1 | The developed method for assaying the ubiquitination reaction, the test components of which consists of an E3 ligase, a ubiquitin C-terminal thioester fluorophore, and a candidate molecule for E3 ligase modulator | The novel fluorescent probe “UbFlu” has been developed to design high-throughput screening assays to search for modulators of E3 ligases with catalytic cysteine | Assay | [140] |
| Category C: Novel ubiquitination system and ligase | ||||
| US2015/0105446A1 | A novel activating enzyme Uba6 and methods for identifying ubiquitin inhibitors and also methods for reducing ubiquitination via the Uba6 pathway | Treatment of inflammatory disorders, cellular proliferative disorders, and neurodegenerative disorders | Novel Activation and Transfer Cascade for Ubiquitin | [141] |
| US2017/0283852A1 | A novel ATP-independent ubiquitination system | Designing ATP-independent ubiquitination process modulators | Identifying ubiquitination mechanism | [142] |
| US 9255129B2 | Modified polynucleotides encoding SIAH1 ubiquitin protein ligase1 | Modified mRNA sequence can be used as therapeutics with benefits beyond just evading, avoiding, or diminishing the immune response | Modified-mRNA encoding E3 enzyme | [143] |
2.1.1.1. Mechanism of action
E6AP ligase oligomer is a catalytically functional form of the enzyme. Both activation and deficiency of E6AP function are implicated in various human diseases [124]. The inventors contemplated that the fully functional form of E6AP is a trimer. Small molecule mimetics are sufficient to dissociate the trimer and, consequently, E6AP-catelyzed chain assembly is reduced, while Cys820-ubiquitin thioester formation is not influenced. Small molecule mimetics can be used to modulate E6AP ligase activity by reducing or increasing ligase oligomer formation [126]. It was further found that N-acetyl-L-phenylalanylamide, a non-competitive inhibitor of ligase activity, could be used to reduce ligase oligomer formation by connecting a ubiquitin-protein E3 ligase with it. It was reported that the E6 viral protein improves E6AP ligase activity by promoting oligomerization of the E6AP ligase with the ability to dimerize through its N-terminal Zn2+ binding domain [125]. The E6AP ligase oligomer formation can be increased by E6 polypeptide or a peptide fragment from a human papilloma virus [126].
2.1.1.2. Application
Mutations in E6AP are associated with pathological conditions in human or animal subjects. According to the embodiments, methods and therapies have been developed to regulate E6AP ligase activities by blocking the catalytic actions of overactive E6AP ligase or increasing E6AP ligase activity that are pathologically deficient. Angelman syndrome is associated with the loss-of-function mutation of E6AP as a consequence of the deletion, imprinting defects, or mutation of the UBE3A gene. E6-induced oligomerization rescues Angelman syndrome loss-of-function mutations by contributing to subunit assembly and stabilization. This invention provided new strategies for regulating E6AP ligase function using oligomerization modulatory agents as therapeutic agents for modulating E6AP-related pathologies [126].
2.1.2. Sulfonamidoquinoline compounds (US2016/0068490A1)
This invention is about certain sulfonamidoquinoline compounds that inhibit ubiquitination, as well as their pharmaceutical compositions and methods of use. Compounds having the structural formula shown in Figure 3 and pharmaceutically acceptable salts, prodrugs, and N-oxides can be used to treat diseases by inhibiting SCFSkp2/Cks1 protein-protein interaction (PPI) and blocking the degradation of tumor suppressor p27 [128] (Table 2).
Figure 3. Patented E3 Ligases Inhibitors.
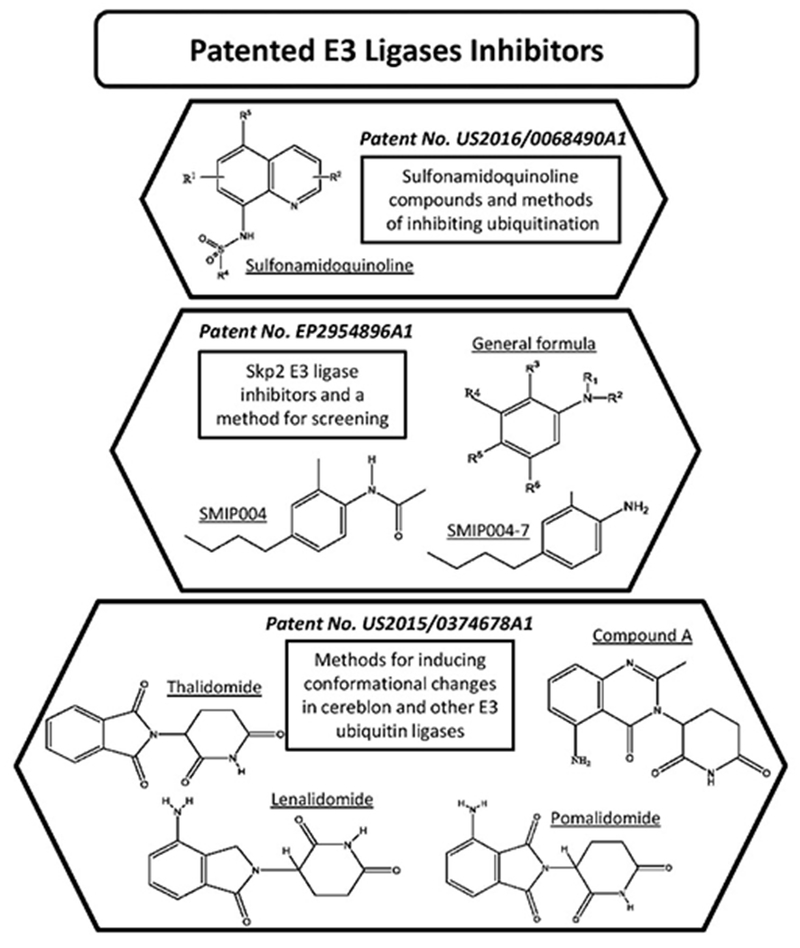
2.1.2.1. Mechanism of action
The SCF family of E3 ligases is a well characterized family via PPI. SCF ligase complex interacts with p27 (a negative regulator of cell cycle progression), an adaptor protein, Cks1, and the substrate recognition component, Skp2. Elevated levels of Cks1 are associated with the low levels of p27 in a number of cancers [127]. Small molecule inhibitors of SCF ligases, which have an ability to decrease activity of an enzyme or receptor or disrupt binding, are more efficient at inhibiting PPI’s between Skp1-Skp2 and Skp2-Cks1-p27. The inventor demonstrated that sulfonamidoquinoline compounds modulate activity of SCFSkp2/Cks1 PPI and inhibit ubiquitination, and, consequently, increase levels of p27 [128].
2.1.2.2. Application
Compounds can be used to modulate SCFSkp2/Cks1 PPI in an animal or human cell by interacting with the proteins. Compounds can also be used to treat or prevent diseases, such as cancers in a subject, by increasing the tumor suppressor p27 levels. It can be achieved by administering an effective amount of a compound or pharmaceutical composition that increases the p27 levels to the subject. The therapeutic dosages of compounds vary according to the usage of the compounds, the administering method, the health condition of the patient, and the judgment of the physician. Combination therapies of these compounds with chemotherapeutic drugs may produce higher antitumor effects [128].
2.1.3. E3 RNF185 inhibitors (US2016/0194644A1)
A mutation in the Cystic Fibrosis Transmembrane conductance regulator (CFTR) gene, which encodes a cyclic-AMP dependent chloride channel at the apical membrane of epithelial cells, is associated with Cystic Fibrosis. The invention identified a component of the endoplasmic reticulum associated with degradation (ERAD) machinery as a novel target and a new E3 ligase RNF185, which targets the CFTR and CFTR△F508. A combination of RNF185 and RNF5 inhibitors can be used for treating cystic fibrosis and chronic obstructive pulmonary disease (COPD) (Table 2).
2.1.3.1. Mechanism of action
About 70% of CFTR mutation involves the deletion of phenylalanine at position 508. E3 ligase RNF185, which specifically targets both CFTR and CFTR△F508 to proteasome degradation, is a homolog of RNF5. Using the E3 ligase RNF185 inhibitor in combination with the E3 ligase RNF5 inhibitor provides a synergistic effect on the inhibition of the degradation of CFTR△F508 and leads to dramatic stabilization of CFTR△F508. The inventors discovered that such stabilization is due to the function of RNF5 and RNF185 in CFTR co-translational quality control and their reinforced function in regulating CFTR post-translational turnover rate. Due to the loss of CFTR function and the elevation of RNF5 levels in COPD patients, the inventors proposed using a combination of RNF185 and RNF5 inhibitors to hinder CFTR degradation and increase the amount of CFTR reaching the plasma membrane [129].
2.1.3.2. Application
The invention can be used to screen or identify E3 ligase RNF185 inhibitors, which are useful for the treatment of cystic fibrosis and COPD by determining whether or not a candidate compound inhibits the activity of an E3 ligase RNF185. E3 ligase RNF185 inhibitors can be used in the manufacture of a medicament for the treatment of cystic fibrosis. The pharmaceutical composition may be comprised of an E3 ligase RNF185 inhibitor and an E3 ligase RNF5 inhibitor, which are combinations of siRNA or shRNA. The invention also provides a method for treatment of cystic fibrosis associated with CFTR△F508 by administering a pharmaceutically effective dose of E3 ligase RNF185 inhibitor [129].
2.1.4. Ubiquitin variant modulators of HECT E3 ligases (US 2017/0275341 A1)
HECT E3 ligases are implicated in cancer, hypertension, neurological disorders, and other disease [130,131]. The invention relates to UbVs that specifically target HECT E3 ligases, methods of identifying and characterizing ubiquitin variants, and the use of these variants to modulate the activity of HECT E3 ligases (Table 2).
2.1.4.1. Mechanism of action
The UbVs are screened from the libraries of Ub variants and generated by randomizing the entire sequence of human ubiquitin or particular regions. The UbVs are identified by their binding affinity with one or more HECT E3 ligases or fragments of one or more HECT. The invention includes 70 specific examples of UbVs’ sequences, which have one or more mutations in region 1 (amino acid 2-14), region 2 (42-49), or region 3 (62-78) of Ub containing two C-terminal glycine residues added to the wild type Ub sequence (SEQ ID NO:1). The inventor suggested that if the conserved amino acid of the UbVs is replaced and/or deleted, the resulting molecule may have the same or similar function as the original molecule. The UbVs can be made using standard recombinant methods. The recombinant expression vectors may contain a selectable marker gene that facilitates the selection of transformed host cells. In one example of the invention, a UbV binds to the active site of a HECT E3 ligase with greater affinity than Ub, resulting in competitive inhibition. Thus, HECT E3 ligase activity can be modulated by administration of a UbV or a nucleic acid molecule encoding a UbV to a subject [132].
2.1.4.2. Application
The UbVs can be used to design peptide mimetics. Peptide mimetics contain synthetic structures that are designed to obtain desired functional features. UbVs can also be used to identify additional agents that can modulate the HECT E3 ligase activity. An agent that affects the binding affinity can be considered as a candidate for use in therapeutic protocols.
The UbVs can be used in the prevention or treatment of cancer, hypertension, autoimmune diseases, and neurological disorders. Additionally, diseases or conditions that can be treated include wound healing, transplantation, and organ culture. Furthermore, UbV treatment can be used therapeutically in the context of microbial infection because HECT-like E3 ligases have been identified as virulence factors in certain pathogenic bacteria [132].
2.1.5. Ubiquitin variant modulators of SCF E3 ligases (US 2017/0321205 A1)
The multi-subunit SCF enzyme complexes are composed of invariant Rbx1, Cul1, and Skp1 subunits and a variable F-box protein that binds substrates and dictates specificity. The invention provides ubiquitin variants that specifically bind to SCF E3 ligases and use of these variants to modulate the activity of SCF E3 ligases (Table 2).
2.1.5.1. Mechanism of action
The invention provides methods for obtaining UbVs that bind to a Cul1–Rbx1–Skp1–F boxSkp2 SCF complex to modulate the activity of SCF E3 ligases. The invention provides the sequence of 35 specific examples of UbVs. UbVs can be subjected to further mutagenesis in order to identify additional UbVs that have desirable features. The activity of SCF E3 ligase can be modulated in a cell with a UbV polypeptide or a nucleic acid molecule encoding a UbV polypeptide. The UbVs can block or decrease the activity of SCF E3 ligase by preventing the Cul1 binding to the surface of Skp1 and F-box protein and disrupting Cul1–Rbx1–Skp1–F boxSkp2 SCF complex formation [133].
2.1.5.2. Application
The UbVs can be used to design peptide mimetics, which may serve as peptide inhibitor analogs. The invention can be used in the prevention or treatment of cancer, sleep and metabolic disorders, immune disorders, hepatitis C virus-related conditions, and muscle atrophy. UbVs can also be used to identify SCF E3 ligase inhibitors by detecting whether the candidate agent impacts the binding of the UbV to the SCF E3 ligase. Candidate agents can be screened from peptides, nucleic acid molecules, natural products, and small organic or inorganic molecules [133].
2.1.6. Cereblon modifying agents (CMAs) (US 2015/0374678A1)
The invention describes drug screening methods, computational methods, and therapeutic methods by the elucidation of the interaction among cereblon (CRBN), CRBN-associate protein and CMAs (Table 2).
2.1.6.1. Mechanism of action
CRBN has been identified as a therapeutic drug target for the treatment of various diseases. It is bound to DDB1-CUL4-ROC1 E3 ubiquitin ligase complex. With the binding of CMAs, changes within the CMA-binding pocket or other alterations in the properties of the CRBN surface result in different E3 ubiquitin ligase surfaces being exposed. Therefore, CRBN recruits different substrates to the E3 ubiquitin ligase and leads to differential downstream biological or therapeutic effects. CMAs can be screened by the comparison of the CRBN conformational change or alteration on the adjacent region of the protein. The conformational change or alteration can be assessed by X-ray crystallography or by comparing the difference in atomic coordinates after the binding of CMAs [134].
2.1.6.2. Application
This invention provides methods of inducing a CRBN conformation change to alter the substrate specificity by CMAs. Lenalidomide, compound A, or any other immunomodulatory compounds (Figure 3) induce the conformational change of CRBN and can be used to modulate biological activities such as immune-mediated effect, tumoricidal effect, apoptosis, proliferation, toxicity, or substrate degradation. Compounds provided in the invention can be used to treat or alleviate CRBN-mediated cancer in a patient. It can also be used in combination with other therapeutic agents useful in the treatment or prevention of any cancer [134].
2.1.7. Skp2 E3 Ligase inhibitors (EP2954896 A1)
The invention relates to small molecule Skp2 inhibitors that were used in the therapeutic activation of autophagy, and especially for use as an antidepressant. Methods of determining if a subject suffering from depression responds to antidepressant and methods of screening novel compounds that activate autophagy are also disclosed in this patent (Table 2).
2.1.7.1. Mechanism of action
E3 enzyme Skp2 is a ubiquitin ligase that determines the protein stability of the autophagy initiator and executor Beclin1. The inventor found that inhibition of Skp2 elicits antidepressant-like effects in mice and shows the comparable effects on synaptic neurotransmission in brain slices using the antidepressant Paroxetine. N-(4-butyl-2-methylphenyl) acetamide (SMIP004), and analogues (Figures 3) thereof, were identified as cancer cell apoptosis selective inducers [136]. They can also be used as pharmaceutically active agents for the treatment of depression [135].
2.1.7.2. Application
Screening methods of the invention can be used to find therapeutic agents for activation of autophagy and the treatment of depression, infectious disease, and myopathy or neurodegenerative diseases. SMIP004 and SMIP004-7 (4-butyl-2-methylaniline) (Figure 3) are the most suitable compounds for use as antidepressants [135].
2.1.8. Method for identifying modulators of ubiquitin ligases (US9464311B2)
The invention provides a screening method, which exploits mechanism-based activity probes that specifically and covalently modify the active site cysteine thiol residue of the HECT and RBR family of E3 ligase (Table 2).
2.1.8.1. Mechanism of action
The HECT and RBR families of E3 ligase have an active cysteine that forms an intermediate thioester bond with Ub. The present invention uses an activity probe to identify a modulator of ubiquitin ligase activity. The activity probe is comprised of an E2 peptide, a reactive chemical moiety, a ubiquitin peptide, and a label. The E2 peptide forms an E2-E3 interaction domain; the chemical moiety which is selected from the group consisting of acrylates, vinyl sulfonyls, acyloxymethyl ketones, beta-lactones, cyanamides, and epoxysuccinates, which act with the active site cysteine of a specific ubiquitin ligase. The ubiquitin peptide originating from the C-terminus of Ub contains an E3-ubiquitin interaction domain. The label may be fluorescent, enzymatic, or radioactive. The decrease in probe labels on the ligase is indicative of an inhibitor, while, on the contrary, an increase in probe labels is indicative of an activator of the ligase [137].
2.1.8.2. Application
The activity probes of this invention can be used to screen for small molecule modulators of ubiquitin E3 ligases that contain an active site cysteine. They can also be used in identification of functional E3 ligases that are activated in tumors. Labelled activity probes and mass spectrometry can be used to identify E3 ligases with enhanced enzyme activity in tumor samples. This approach is efficient in accurately measuring enzyme activity instead of identifying causative genes by transcriptional profiling of human cancers [137].
2.2. Category B: Assay methods of ligase E3 activity
2.2.1. Ubiquitination Assay (EP2564193B1)
The inventors found that the different stages of the ubiquitination cascade can be investigated directly from a single assay through specifically and differentially labelling the various components of the ubiquitination cascade with different tags. In addition, it is possible to sequentially determine activity at each stage of the cascade without changing the assay composition (Table 2).
2.2.1.1. Mechanism of action
The invention conducts a system for assaying ubiquitination in a sample. The assay sample is comprised of ubiquitin, a specific indicated enzyme (i.e. E1, E2 or E3), and a substrate protein. The sample is exposed to a labelled binding partner that is specific to the ubiquitin and the amount of labelled ubiquitin bound to any one of the components E1, E2, E3, and a substrate protein in the sample is measured. The labelled binding proteins of the invention may be polyclonal or monoclonal antibodies. The antibodies contain an Fv region which has specific antigen activity. All of the components (E1, E2, and E3), and a substrate protein in the sample are comprised of a specific immobilization tag, which vary among components and facilitate its immobilization onto a solid surface. These tags include HIS, GST, HA, c-Myc, and FLAG. The ubiquitin tag is recognized by the labelled binding partner to produce a detectable signal [138].
2.2.1.2. Application
To date, it is still difficult to provide an effective in vitro ubiquitination assay due to the specific modulation of each member of E3 ligases by a variety of different proteins. The inventors have overcome these difficulties and provided an optimized method for assaying ubiquitination. The high-throughput screening for modulators of ubiquitination has become available and possible [138].
2.2.2. Ubiquitin chain analysis (US9587265B2)
Analysis of naturally occurring ubiquitination is important to assess the likely function of the ubiquitination. However, the architecture of heterotypic ubiquitin chains is difficult to assess by the current technology. Thus, there is need for developing a method to determine linkage-specificity of polyubiquitin chains. The invention describes a procedure to determine ubiquitin polymers using linkage-specific deubiquitinating enzymes (DUBs) (Table 2).
2.2.2.1. Mechanism of action
The invention provides the newly characterized ovarian tumor protease (OTU) family DUBs and develops a method for determining the linkage specificity in the polyubiquitin chain. OTUD1, OTUD4, and OTUD3 are used to cleave K63, K48, and K6 and/or K11 linkages in a ubiquitin polymer, respectively. Also, the novel OTULIN DUB is used to determine Met-1 linked polyubiquitin chains [139].
2.2.2.2. Application
The diverse applications of methods have been described in the invention. For example, the DUBs described in the invention can be used to analyze the nature of the polyubiquitin chains occurring on proteins which have been ubiquitinated in vivo or in vitro. In another example, ubiquitinated proteins in a cell can be isolated on a ubiquitin affinity resin and analyzed for the presence of a particular ubiquitin linkage by exposing them to DUBs and monitoring release of the protein from the matrix [139].
2.2.3. Probes and assays for measuring E3 ligase activity (US20160076074)
This invention is a technology relating to the biological process of protein ubiquitination and is also related to compositions and methods for studying protein ubiquitination and developing therapeutics to modulate protein ubiquitination (Table 2).
2.2.3.1. Mechanism of action
The invention highlights that C-terminal ubiquitin thioesters undergo a direct trans-thiolation reaction with the catalytic cysteine residues of the model HECT E3 ubiquitin ligase Rsp5, thus forming catalytically active Rsp5~Ub thioesters. The developed reaction will work in the absence of the E1 and E2 enzymes and ATP, thereby recapitulating the enzymatic mechanism of native ubiquitination reaction. Additionally, small molecule thiols can mimic the E2 enzyme, and efficiently trigger E3 ligase autoubiquitination. In the present invention, the novel fluorescent probe “UbFlu” has been developed to design high-throughput screening assays to search for modulators of E3 ligases with catalytic cysteine such as HECT E3s, RBR E3s, and NEL E3s [140].
2.2.3.2. Application
This invention provides a potent method for screening of E3 ligase modulators. The test composition of the method consists of an E3 ligase, a ubiquitin C-terminal thioester fluorophore, and a candidate molecule for E3 ligase modulator [140].
2.3. Category C: Novel ubiquitination system and ligase
2.3.1. Novel Activation and Transfer Cascade for Ubiquitin (US20150105446)
This invention provides a novel activating enzyme Uba6. Also, methods for identifying ubiquitin inhibitors and methods for reducing ubiquitination via the Uba6 pathway are provided in the invention (Table 2).
2.3.1.1. Mechanism of action
The enzymatic properties of Uba6 are different from the classical ubiquitin activating enzyme Ube1. Uba6 promotes the activation of ubiquitination and the formation of a Uba6-S∼ubiquitin thioester. Ube1 and Uba6 have distinct substrate specificities because of the difference in the C-terminal ubiquitin-like domain. Small molecular compounds that inhibit thiol esterification of UbA6 or inhibit transfer of Ub to E2s can be used to inhibit the activity of Uba6. The activity of Uba6 can also be inhibited using RNAi. siRNAs, which are complementary to the biologically active portion of Uba6 mRNA, can be used to reduce Uba6 activity. The active portion of the Uba6, such as a ThiF domain, a catalytic cysteine domain, an adenylate domain, or a ubiquitin-like domain, can be used to screen small molecular inhibitors or siRNAs [141].
2.3.1.2. Application
Inhibition of Uba6 activity influences the resultant downstream biological effects of ubiquitin-conjugation. It also affects the integrity of cell division, cell signaling, and several aspects of cellular physiology, which are potential mechanisms involved in some diseases. Uba6 is therefore a potentially therapeutic target. Modulatory agents of Uba6 or Ube1 can be used for the treatment of inflammatory disorders, cellular proliferative disorders, and neurodegenerative disorders [141].
2.3.2. Novel ubiquitination system and the uses thereof (US 2017/0283852 A1)
This invention disclosed a novel ATP-independent ubiquitination system in which ubiquitin is activated by ADP-ribosylation and is mediated by the SidE family effector of Legionella pneumophila (Table 2).
2.3.2.1. Mechanism of action
The inventor demonstrates a method in which a single enzyme can be used to carry out ubiquitination. The ATP-independent ubiquitination system comprised of β-nicotinamide adenine dinucleotide (β-NAD), a ubiquitin or a variant of the ubiquitin, a substrate, and a ubiquitin activating protein that transfers an ADP from β-NAD to the ubiquitin to form an ADP-ribosylated ubiquitin. The ubiquitin activating protein was expressed by the group which encoded a putative mono-ADP-ribosyltransferase motif (R-S-ExE). R-S-ExE are members of the SidE family effector proteins of L.Pneumophila and ubiquitinate multiple Rab small GTPases. Ubiquitin was activated by changing to an ADP-ribosylated form without the need of E1 and E2 enzymes. The ADP-ribosylated ubiquitin can then be conjugated to the substrate through a ribose-phosphate link. Mutations of SidE family effectors result in the loss of ubiquitination activating activity [142].
2.3.2.2. Application
Preventing β-NAD-dependent ADP-ribosylation of ubiquitin may lead to inhibition of the ATP-independent ubiquitination process. The invention can be used to design ATP-independent ubiquitination process modulators such as antibodies to ADP-ribosylated ubiquitin or antibodies to the activating proteins identified in this disclosure. The methods of this invention contribute to identifying additional substrates of ATP-independent ubiquitination [142].
2.3.3. Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase1 (US 9255129B2)
The invention deals with compositions, methods, processes, kits, and devices for the design, preparation, manufacture, and/or formulation of oncology-related polynucleotides (encoding SIAH E3 ubiquitin protein ligase 1). The inventors discovered that certain modified mRNA sequences can be used as therapeutics with benefits beyond just evading, avoiding, or diminishing the immune response (Table 2).
2.3.3.1. Mechanism of action
The present invention provides a modified and purified polynucleotide comprising a first region of linked nucleosides. The said first region encodes the oncology-related polypeptide of interest that maintains a modular organization and contains two flanking regions, which are located at the 5’ and 3’ terminus of said first region, respectively. The 5’ terminus contains a sequence of linked nucleosides selected from the group consisting of the native 5’ untranslated region (UTR) of any of the reference sequences. Also, the 3’ terminus contains a sequence of linked nucleosides selected from the group consisting of the native 3’ UTR of any of the reference sequences and a 3’ tailing sequence. The modified polynucleotides in the present invention may comprise at least two modifications and a translatable region. The modification may be on at least one nucleoside and/or the backbone of said nucleotides, on both a nucleoside and a backbone linkage or on a sugar of the nucleoside. The modification may include replacing at least one phosphodiester linkage with a phosphorothioate linkage. The oncology-related polynucleotides, oncology-related primary constructs, or oncology-related mRNA may alter and modulate a biological or physiological process or compound such as, but not limited to, the cell cycle, the DNA damage response (e.g. DNA damage repair), apoptosis, senescence, tumor metastasis, tumorigenesis, and a wide range of key signaling pathways (e.g. the phosphatidyl inositol 3 (PI3) kinase/Akt cellular signaling pathway, receptor tyrosine kinase signaling, G protein signaling, Ras signaling) in cancerous, precancerous, and other cells [143].
2.3.3.2. Application
The inventors have explored the uses of modified polynucleotides in the fields of antibodies, viruses, veterinary applications, and in diverse in vivo settings to enhance one or more of the stability and/or clearance in tissues, receptor uptake and/or kinetics, cellular access by the compositions, engagement with translational machinery, mRNA half-life, translation efficiency, immune evasion, protein production capacity, secretion efficiency (when applicable), accessibility to circulation, protein half-life and/or modulation of a cell’s status, function and/or activity [143].
3. Conclusions
In the first step of the UPS, E1, E2, and E3 participate in the enzymatic cascade reaction of ubiquitination, which is a critically regulatory process widely found in human diseases including cancer. E3 ligases have various disease-specific substrates, which offers an opportunity to develop E3 inhibitors with improved potency, specificity, selectivity, and toxicity. Candidates of E3 modulators have been extensively investigated for treatment of cancers in the past decades. As described, a wide range of natural and synthesized compounds (e.g. PROTACs or molecular glues) have been characterized to exhibit inhibitory and modulatory activities toward the interaction between E3 and substrates. However, the growing in-depth research on target modulators remains a challenge due to the complexity of protein-protein interaction between E3s and their target substrates. With the applications of novel technologies including high-throughput screening, protein-engineered technology, bioinformatics, identification of protein crystal structure, and using small molecules such as PROTACs/ molecular glues in this area, it is hopefully possible to develop more specific E3 modulators as potential anticancer drugs in the near future.
4. Expert opinion:
Over the past two decades, a notable progress has been made in drug discovery by targeting the 20S proteasome in the UPS. One of the examples is the 20S proteasome inhibitor bortezomib, which was approved by the U.S. FDA in 2003 and currently has become a stable for the treatment of multiple myeloma and mantle cell lymphoma. Despite the capabilities of 20S proteasome inhibitors to selectively kill cancer cells, there is an increasing consideration on their potential adverse effects. Clinical trials have shown that 20S proteasome inhibitors may potentially result in the disorder of protein degradation, which is mediated by proteasomes in healthy cells. Additionally, there is emerging clinical evidence of development of tumors resistant to 20S proteasome inhibitors [114]. Thus, one of the key issues to reduce some of the limitations associated with 20S proteasome inhibitors is to discover inhibitors of the upstream key components of the ubiquitination cascade in the UPS.
Along this line, E3 ubiquitin ligases have received particular attention because they play an essential role in the determination of the specificity of substrate ubiquitination. E3 ligases are important regulators of many cellular processes including cell growth, cell death, transformation, and tumor development and hence serve as the key molecules in the UPS for modification and degradation of target proteins. Targeting E3 ligases and understanding the detailed mechanisms of interaction between E3 and target proteins could lead to a new route for screening and developing novel E3 inhibitors or modulators as potential anticancer agents and a strategy to overcome the shortcomings of current 20S proteasome inhibitors, e.g., toxicity and resistance.
One strategy to target an E3 ubiquitin ligase is to block its PPIs. PPIs have defined specificity which play critical roles in determining their biological functions and thus have been considered as promising targets for therapeutic intervention. Great bioavailability and bio-accessibility of oral administration are the advantageous properties of small molecule modulators of PPIs. However, it remains as a challenge to get specific binding affinity from the wide range of binding interfaces when using small molecules to target PPIs. This has questioned whether E3 ligases could be used as druggable targets. However, the recent progress is strong in the favor of targeting E3 ligases, especially against PPIs, with small molecules. Examples of successfully targeted E3s include IAP (inhibitor of apoptosis), MDM2, VHL/HIF-1 α [144, 151], and CRBN [134,152].
One of the most effective strategies for development of specific small molecule inhibitors is structure-based drug design. The main obstacle for this approach is availability of structural data as crystallization and assays of large multi-subunit protein complexes may be rather difficult. Additionally, large size of the full complexes and their multi-subunit composition pose a challenge for NMR spectroscopic studies. Some of these interfaces are already being targeted for potential therapeutic intervention. Crystal structures of subunits, components, and full-size complexes, alone or with bound small molecules and substrate peptides, should provide increasing opportunities to the rational structure-based design of chemical probes and potential small-molecule therapeutics [144]. The successful examples include newly characterized PROTACs [151], CMAs [134] and bifunctional molecule dBET1 [152].
Apart from rational design, natural products could be another source for the development of E3 modulators. Several natural small molecules have been found to inhibit the cascade enzymes in the UPS. For example, plant-derived active compounds, such as Ginsenoside Rg1 isolated from Chinese herbal medicines [145], turn out to be a critical compound library for inhibitors or modulators of the ubiquitination enzymes and candidate anticancer agents. The lead structures derived from natural compounds may offer various novel scaffolding classes of modulators with specific pharmacological properties, which can be used functionally in the modification and synthesis of E3 modulatory compounds.
Another useful approach to target cascade enzymes in the ubiquitination process is drug repurposing [146]. The repurposing achieved by high-throughput screening system could potentially hit a compound with a desired biological or clinical effect. This can be then validated for its new indications as E3 modulators. The candidate compounds could have dramatically less unpredictable toxicity to healthy cells and less development cost than de novo discovered drugs. Essentially, a database of approved drugs might provide a validated library of privileged scaffolding compounds, on the basis of available pharmacokinetic and toxicological evaluation in human subjects.
Novel technologies including proteome analysis (e.g. chemical proteomics and tandem mass spectrometry [147], AlphaScreen assay [121]), protein engineered methods (e.g. phage display-based UbVs [120,121,132], yeast two hybrid screen [148]), gene manipulation (e.g. RNAi [141,149]) and PROTACs/ molecular glues [134, 151–152] have been applied in the identification and development of E3 modulators. High-throughput properties or high sensitivity and selectivity are some of the advantages these technologies have. However, potential limitations of E3 modulators identified by novel technologies must be considered. For example, the subtle structure differentiation of UbVs binding surfaces will result in different stabilities and specificities to target cells, although Ub interacts with its bound partners via a similar interface. Although siRNAs are able to regulate activities of E3s in target cells, their efficacies are largely dependent on the implementation of the systematic delivery approaches, which require further optimization. We expect that the wider availability of powerful assays for the detection of enzymatic activities in the UPS will speed up the discovery of more natural product inhibitors of ubiquitination. This will improve our understanding of the biological functions of UPS and Ub-like modifiers. In addition, it is necessary to further investigate the molecular mechanisms and targets of some previously characterized compounds, such as MLN4924 [23,26], to uncover more of their potentials.
Importantly, synergistic effects are found when E3 modulators are used in combination with chemotherapeutic drugs in: inducing tumor cell death, overcoming drug resistance, and enhancing therapeutic efficacy. As a result, MDM2 inhibitors Nutlins, used in combination with other drugs for cancer therapeutic treatments, have entered clinical trials [118]. Further investigations on roles and mechanisms of action of E3s in the initiation and progression of human tumorigenesis will help overcome limitations of current E3 modulators and help discover and develop new generation small molecule E3 inhibitors as potent, selective anticancer drugs.
Article highlights.
Protein ubiquitination is a crucial post-translational process that is involved in the development and progression of human cancers.
E3 ligase is the central player in ubiquitination process and in the ubiquitin-proteasome system, and has a wide range of specific substrate proteins, suggesting it as a novel anticancer target.
Discovery of E3 inhibitors has challenges in selectivity, given the complexity of E3 substrate proteins.
Understanding the mechanism of action of E3 is critical for developing E3 inhibitors or modulators.
Availability of novel technologies (e.g. phage display-based ubiquitin variant, AlphaScreen assay, using small molecules such as PROTACs/ molecular glues, etc.) makes it feasible to screen and discover potent, specific E3 inhibitors.
Acknowledgements
We thank Hassan Cheaito and Rania Fardous for critical reading of the manuscript. This work was partially supported by Guangdong Province Key Laboratory for Green Processing of Natural Products and Product Safety Fund (No: 201605; to Xin Li), National Cancer Institute grant R21CA184788 (to QP Dou) and NIH grant P30 CA022453 (to the Karmanos Cancer Institute at Wayne State University) as well as Karmanos internal funds.
Funding
This work is supported by Guangdong Province Key Laboratory for Green Processing of Natural Products and Product Safety Fund (No: 201605; To: Dr. Xin Li), and by the National Natural Science Foundation of China (Grant No. 81772492/H1615), as well as by a National Cancer Institute grant R21CA184788 (to QP Dou) and a National Institutes of Health grant P30 CA022453 (to the Karmanos Cancer Institute at Wayne State University).
Abbreviations
- CFTR
the cystic fibrosis transmembrane conductance regulator
- CMAs
cereblon modifying agents
- COPD
chronic obstructive pulmonary disease
- CRBN
cereblon
- CRLs
the Cullin-RING ligases
- Cys
cysteine
- DUBs
deubiquinating enzymes
- E1
ubiquitin activating enzyme
- E2
ubiquitin conjugating enzyme
- E3
ubiquitin ligase
- EGCG
epigallocatechin-3-gallate
- ERAD
the endoplasmic reticulum associated degradation
- FBXW7
F-box and WD repeat domain-containing 7
- HDAC
histone deacetylase
- HECT
homologous to E6-associated protein C-terminus
- IAP
inhibitor of apoptosis protein
- JNK
c-Jun N-terminal kinase
- K
lysine residue
- MDM2
mouse double minute 2 homolog
- mTOR
mammalian target of rapamycin
- NAE
NEDD8-activating enzyme
- NB
neuroblastoma
- NEDD4
the neuronally expressed developmentally downregulated 4
- NEDD8
the neural precursor cell-expressed developmentally downregulated protein 8
- NF-ҡB
nuclear factor-ҡB
- NMR
nuclear magnetic resonance
- OTU
ovarian tumor protease
- PC
pancreatic cancer
- PCNA
proliferating cell nuclear antigen
- PI3
phosphatidyl inositol 3
- PPIs
protein-protein interactions
- RBR
RING between RING
- RING
really interesting new gene
- RNAi
RNA interference
- SCF
Skp1/Cul1/F-box protein complex
- siRNA
small/short interfering RNA
- Skp2
S-phase kinase-associated protein 2
- TNF-α
tumor necrosis factor α
- TRAF6
TNF receptor associated factor 6
- Ub
Ubiquitin
- UbVs
ubiquitin variants
- UPS
the ubiquitin-proteasome system
- UTR
untranslated region
- β-NAD
β-nicotinamide ademine dinucleotide
- β-TrCP
β-transducin repeat-containing protein
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Bibliography:
Paper of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem 2012; 81: 203–229 [DOI] [PubMed] [Google Scholar]
- 2.Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta 2014; 1843: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swatek KN, Komander D. Ubiquitin modifications. Cell Res 2016; 26(4): 399–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 2008; 14(1):10–21 [DOI] [PubMed] [Google Scholar]
- 5.Lipkowitz S, Weissman AM. RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer 2011; 11: 629–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deshaies RJ, Joazeiro CAP. RING Domain E3 Ubiquitin Ligases. Ann Rev Biochem 2009; 78: 399–434 [DOI] [PubMed] [Google Scholar]
- 7.Landré V, Rotblat B, Sonia Melino S. Screening for E3-Ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget 2014; 18(5): 7988–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang W, Sidhu SS. Development of inhibitors in the ubiquitination cascade. FEBS Lett 2014; 588: 356–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res 2016; 26: 484–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekizawa R, Ikeno S, Nakamura H, et al. Panepophenanthrin, from a mushroom strain, a novel inhibitor of the ubiquitin activating enzyme. J Nat Prod 2002; 65: 1491–1493 [DOI] [PubMed] [Google Scholar]
- 11.Moses JE, Commeiras L, Baldwin JE, Adlington RM. Total synthesis of panepophenanthrin. Org Lett 2003; 5: 2987–2988 [DOI] [PubMed] [Google Scholar]
- 12.Tsukamoto S, Hirota H, Imachi M, et al. Himeic acid A: a new ubiquitin-activating enzyme inhibitor isolated from a marine-derived fungus, Aspergillus sp. Bioorg Med Chem Lett 2005; 15: 191–194 [DOI] [PubMed] [Google Scholar]
- 13.Taori K, Paul VJ, Luesch H. Structure and activity of largazole, a potent antiproliferative agent from the Floridian marine cyanobacterium Symploca sp. J Am Chem Soc 2008; 130: 1806–1807 [DOI] [PubMed] [Google Scholar]
- 14.Yamanokuchi R, Imada K, Miyazaki M, et al. Hyrtioreticulins A–E, indole alkaloids inhibiting the ubiquitin-activating enzyme, from the marine sponge Hyrtios reticulatus. Bioorg Med Chem 2012; 20: 4437–4442 [DOI] [PubMed] [Google Scholar]
- 15.Ungermannova D, Parker SJ, Nasveschuk CG, et al. Largazole and its derivatives selectively inhibit ubiquitin activating enzyme (e1). PLoS One 2012; 7: e29208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu LC, Wen ZS, Qiu YT, et al. Largazole arrests cell cycle at G1 phase and triggers proteasomal degradation of E2F1 in lung cancer cells. ACS Med Chem Lett 2013; 4: 921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Kitagaki J, Dai RM, et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res 2007; 67: 9472–9481 [DOI] [PubMed] [Google Scholar]; • This paper described an E1 inhibitor PYR-41 that blocked K63-linked ubiquitination of TRAF6 and prevented the proteasome-based degradation of IκBα and p53, which is currently used as a tool for exploring ubiquitylation.
- 18.Ungermannova D, Parker SJ, Nasveschuk CG, et al. Identification and mechanistic studies of a novel ubiquitin E1 inhibitor. J Biomol Screen 2012; 17(4): 421–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitagaki J, Yang Y, Saavedra JE, et al. Nitric oxide prodrug JS-K inhibits ubiquitin E1 and kills tumor cells retaining wild type p53. Oncogene 2009; 28(4): 619–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009; 458: 732–736 [DOI] [PubMed] [Google Scholar]; •• This paper first reported that MLN4924, an adenosine sulfamate analogue, potently inhibits NAE activity against a wide range of human cancers. Phase I clinical trials with MLN4924 are currently ongoing.
- 21.Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs 2012; 21: 1563–1573 [DOI] [PubMed] [Google Scholar]
- 22.Watson IR, Irwin MS, Ohh M. NEDD8 pathways in cancer, sine quibus non. Cancer Cell 2011; 19: 168–176 [DOI] [PubMed] [Google Scholar]
- 23.Oladghaffari M, Islamian JP, Baradaran B, Monfared AS. MLN4924 therapy as a novel approach in cancer treatment modalities. J Chemother 2016; 28(2): 74–82 [DOI] [PubMed] [Google Scholar]
- 24.Soucy TA, Dick LR, Smith PG, et al. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer 2010; 1: 708–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shah JJ, Jakubowiak AJ, O’Connor OA, et al. Phase I study of the novel investigational NEDD8-activating enzyme inhibitor Pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res 2016; 22(1): 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong KM, Micel LN, Selby HM, et al. Targeting the protein ubiquitination machinery in melanoma by the NEDD8-activating enzyme inhibitor Pevonedistat (MLN4924). Invest New Drugs 2017; 35(1): 11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen JJ, Tsu CA, Gavin JM, et al. Mechanistic studies of substrate-assisted inhibition of ubiquitin activating enzyme by adenosine sulfamate analogues. J Biol Chem 2011; 286: 40867–40877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traore T, Huck JH, Shi JS, et al. 255 Pre-clinical in vivo characterization of MLN7243, an investigational ubiquitin activating enzyme inhibitor, in solid tumor models. Eur J Cancer 2014; 50: 8524054023 [Google Scholar]
- 29.Hyer ML, Milhollen MA, Ciavarri J, et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 2018; 24(2): 186–193 [DOI] [PubMed] [Google Scholar]
- 30.McHugh A, Fernandes K, South AP, et al. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: the identification of mechanisms of differential sensitivity. Oncotarget 2018; 9 (29): 20265–20281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.An H, Statsyuk AV. An inhibitor of ubiquitin conjugation and aggresome formation. Chem Sci 2015; 6(9): 5235–5245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong HJ, Ma VP, Cheng Z, et al. Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening. Biochimie 2012; 94: 2457–2460 [DOI] [PubMed] [Google Scholar]
- 33.Zhong HJ, Leung KH, Lin S, et al. Discovery of deoxyvasicinone derivatives as inhibitors of NEDD8-activating enzyme. Methods 2015; 71: 71–76 [DOI] [PubMed] [Google Scholar]
- 34.Zhong HJ, Liu LJ, Chan DSH, et al. Structure-based repurposing of FDA-approved drugs as inhibitors of NEDD8-activating enzyme. Biochimie 2014; 102: 211–215 [DOI] [PubMed] [Google Scholar]; • This paper provides a strategy to establish a validated library of privileged scaffolds offered by the FDA-approved drugs for screening and development of E1 inhibitors.
- 35.Lu P, Liu X, Yuan X, et al. Discovery of a novel NEDD8 activating enzyme inhibitor with piperidin-4-amine scaffold by structure-based virtual screening. ACS Chem Biol 2016; 11: 1901–1907 [DOI] [PubMed] [Google Scholar]
- 36.Lu P, Guo Y, Zhu L, et al. A novel NAE/UAE dual inhibitor LP0040 blocks neddylation and ubiquitination leading to growth inhibition and apoptosis of cancer cells. Eur J Med Chem 2018; 154: 294–304 [DOI] [PubMed] [Google Scholar]
- 37.Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res 2016; 26: 423–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodge CD, Spyracopoulos L, Mark Glover JN. Ubc13: the Lys63 ubiquitin chain building machine. Oncotarget 2017; 39(7): 64471–64504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laine A, Topisirovic I, Zhai D, et al. Regulation of p53 localization and activity by Ubc13. Mol Cell Biol 2006; 26(23): 8901–8913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsukamoto S, Takeuchi T, Rotinsulu H, et al. Leucettamol A: A new inhibitor of Ubc13-Uev1A interaction isolated from a marine sponge, Leucetta aff. microrhaphis. Bioorg Med Chem Lett 2008; 18: 6319–6320 [DOI] [PubMed] [Google Scholar]; • This paper reported that Leucettamol A isolated from the marine sponge can disrupt the formation of the UBC13–UEV1A complex to attenuate the E2 activity.
- 41.Scheper J, Guerra-Rebollo M, Sanclimens G, et al. Protein-protein interaction antagonists as novel inhibitors of non-canonical polyubiquitylation. PLoS One 2010; 5(6): e11403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ushiyama S, Umaoka H, Kato H, et al. Manadosterols A and B, Sulfonated Sterol Dimers Inhibiting the Ubc13–Uev1A Interaction, Isolated from the Marine Sponge Lissodendryx fibrosa. J Nat Prod 2012; 75 (8):1495–1499 [DOI] [PubMed] [Google Scholar]
- 43.Cheng J, Fan YH, Xu X, et al. A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis 2014; 5(2): e1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol 2009; 10: 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ceccarelli DF, Tang X, Pelletier B. An allosteric inhibitor of the Human Cdc34 ubiquitin-conjugating enzyme. Cell 2011; 145: 1075–1087 [DOI] [PubMed] [Google Scholar]
- 46.Huang H, Ceccarelli DF, Orlicky S, et al. E2 enzyme inhibition by stabilization of a low-affinity interface with ubiquitin. Nat Chem Biol 2014; 10(2): 156–163 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper reported that CC0651 unexpectedly traps the weak interaction between ubiquitin and the donor site of Cdc34A and thereby impedes catalysis, suggesting that E2 enzymes are susceptible to noncatalytic site inhibition and should be exploited as a novel drug target in the UPS.
- 47.Kothayer H, Spencer SM, Tripathi K, et al. Synthesis and in vitro anticancer evaluation of some 4,6-diamino-1,3,5-triazine-2-carbohydrazides as Rad6 ubiquitin conjugating enzyme inhibitors. Bioorg Med Chem Lett 2016; 26(8): 2030–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Y, Mallampalli RK. Small molecule therapeutics targeting F-box proteins in cancer. Semin Cancer Biol 2016; 36: 105–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akhoondi S, Sun D, von der Lehr N, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007; 67:9006–9012 [DOI] [PubMed] [Google Scholar]
- 50.Spruck C miR-27a regulation of SCF(Fbw7) in cell division control and cancer. Cell Cycle 2011; 10: 3232–3233 [DOI] [PubMed] [Google Scholar]
- 51.Ma J, Cheng L, Liu H, et al. Genistein down-regulates miR-223 expression in pancreatic cancer cells. Curr Drug Targets 2013; 14: 1150–1156 [DOI] [PubMed] [Google Scholar]
- 52.Kimura T, Gotoh M, Nakamura Y, Arakawa H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci 2003; 94: 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mao JH, Perez-Losada J, Wu D, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004; 432: 775–779 [DOI] [PubMed] [Google Scholar]
- 54.Balamurugan K, Wang JM, Tsai HH, et al. The tumour suppressor C/EBPdelta inhibits FBXW7 expression and promotes mammary tumour metastasis. EMBO J 2010; 29: 4106–4117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c- Myc protein degradation. Proc Natl Acad Sci U S A 2004; 101: 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.von der Lehr N, Johansson S, Wu S, et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 2003; 11: 1189–1200 [DOI] [PubMed] [Google Scholar]
- 57.Kim SY, Herbst A, Tworkowski KA, et al. Skp2 regulates Myc protein stability and activity. Mol Cell 2003; 11: 1177–1188 [DOI] [PubMed] [Google Scholar]
- 58.Yada M, Hatakeyama S, Kamura T, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004; 23: 2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gregory MA, Hann SR. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt’s lymphoma cells. Mol Cell Biol 2000; 20: 2423–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Busino L, Millman SE, Scotto L, et al. Fbxw7α- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol 2012; 14: 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou GB, Chen SJ, Wang ZY, Chen Z. Back to the future of oridonin: again, compound from medicinal herb shows potent antileukemia efficacies in vitro and in vivo. Cell Res 2007; 17: 274–276 [DOI] [PubMed] [Google Scholar]
- 62.Huang HL, Weng HY, Wang LQ, et al. Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol Cancer Ther 2012; 11: 1155–1165 [DOI] [PubMed] [Google Scholar]
- 63.Xia J, Duan Q, Ahmad A, et al. Genistein inhibits cell growth and induces apoptosis through up-regulation of miR-34a in pancreatic cancer cells. Curr Drug Targets 2012; 13: 1750–1756 [DOI] [PubMed] [Google Scholar]; • This paper described that the isoflavonoid compound Genistein derived from soybean can inhibit cell growth and induce apoptosis through up-regulation of miR-34a in pancreatic cancer cells.
- 64.Orlicky S, Tang X, Neduva V, et al. An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat Biotechnol 2010; 28: 733–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reavie L, Buckley SM, Loizou E, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell 2013; 23: 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng N, Zhou Q, Wang Z, Wei W. Recent advances in SCF ubiquitin ligase complex: Clinical implications. Biochim Biophys Acta 2016; 1866: 12–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim CJ, Song JH, Cho YG, et al. Somatic mutations of the beta-TrCP gene in gastric cancer. APMIS 2007; 115: 127–133 [DOI] [PubMed] [Google Scholar]
- 68.Gerstein AV, Almeida TA, Zhao G, et al. APC/CTNNB1 (beta-catenin) pathway alterations in human prostate cancers. Genes Chromosom Cancer 2002; 34: 9–16 [DOI] [PubMed] [Google Scholar]
- 69.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318: 1108–1113 [DOI] [PubMed] [Google Scholar]
- 70.Blees JS, Bokesch HR, Rübsamen D, et al. Erioflorin stabilizes the tumor suppressor Pdcd4 by inhibiting its interaction with the E3-ligase beta-TrCP1. PLoS One 2012; 7: e46567. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper delineated the molecular mechanism of Erioflorin targeting β-TrCP E3 ligase.
- 71.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer 2008; 8: 438–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmid T, Jansen AP, Baker AR, et al. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res 2008; 68: 1254–1260 [DOI] [PubMed] [Google Scholar]
- 73.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1: 193–199 [DOI] [PubMed] [Google Scholar]
- 74.Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci U S A 1998; 95: 11324–11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kamura T, Hara T, Kotoshiba S, et al. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proc Natl Acad Sci U S A 2003; 100: 10231–10236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hiramatsu Y, Kitagawa K, Suzuki T, et al. Degradation of Tob1 mediated by SCFSkp2-dependent ubiquitination. Cancer Res 2006; 66: 8477–8483 [DOI] [PubMed] [Google Scholar]
- 77.Song MS, Song SJ, Kim SJ, et al. Skp2 regulates the antiproliferative function of the tumor suppressor RASSF1A via ubiquitin mediated degradation at the G1-S transition. Oncogene 2008; 27: 3176–3185 [DOI] [PubMed] [Google Scholar]
- 78.Wang H, Cui J, Bauzon F, Zhu L. A comparison between Skp2 and FOXO1 for their cytoplasmic localization by Akt1. Cell Cycle 2010; 9: 1021–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bhattacharya S, Garriga J, Calbo J, et al. SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene 2003; 22: 2443–2451 [DOI] [PubMed] [Google Scholar]
- 80.Waltregny D, Leav I, Signoretti S, et al. Androgen-driven prostate epithelial cell proliferation and differentiation in vivo involve the regulation of p27. Mol Endocrinol 2001; 15: 765–782 [DOI] [PubMed] [Google Scholar]
- 81.Lu L, Schulz H, Wolf DA. The F-box protein SKP2 mediates androgen control of p27 stability in LNCaP human prostate cancer cells. BMC Cell Biol 2002; 3: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin HK, Chen Z, Wang G, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 2010; 464: 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nakayama K, Nagahama H, Minamishima YA, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 2000; 19: 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kratzat S, Nikolova V, Miething C, et al. Cks1 is required for tumor cell proliferation but not sufficient to induce hematopoietic malignancies. PLoS One 2012; 7: e37433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Agarwal A, Bumm TG, Corbin AS, et al. Absence of SKP2 expression attenuates BCR-ABL-induced myeloproliferative disease. Blood 2008; 112: 1960–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakayama K, Nagahama H, Minamishima YA, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6: 661–672 [DOI] [PubMed] [Google Scholar]
- 87.Minamishima YA, Nakayama K, Nakayama K. Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in Skp2-deficient mice. Cancer Res 2002; 62: 995–999 [PubMed] [Google Scholar]
- 88.Sistrunk C, Kim SH, Wang X, et al. Skp2 deficiency inhibits chemical skin tumorigenesis independent of p27(Kip1) accumulation. Am J Pathol 2013; 182: 1854–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kullmann MK, Grubbauer C, Goetsch K, et al. The p27-Skp2 axis mediates glucocorticoid-induced cell cycle arrest in T-lymphoma cells. Cell Cycle 2013; 12: 2625–2635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lim MS, Adamson A, Lin Z, et al. Expression of Skp2, a p27(Kip1) ubiquitin ligase, in malignant lymphoma: correlation with p27(Kip1) and proliferation index. Blood 2002; 10: 2950–2956 [DOI] [PubMed] [Google Scholar]
- 91.Schüler S, Diersch S, Hamacher R, et al. SKP2 confers resistance of pancreatic cancer cells towards TRAIL-induced apoptosis. Int J Oncol 2011; 38: 219–225 [PubMed] [Google Scholar]
- 92.Hulit J, Lee RJ, Li Z, et al. p27Kip1 repression of ErbB2-inducedmammary tumor growth in transgenicmice involves Skp2 andWnt/beta-catenin signaling. Cancer Res 2006; 66: 8529–8541 [DOI] [PubMed] [Google Scholar]
- 93.Fujita T, Liu W, Doihara H, et al. Dissection of the APCCdh1-Skp2 cascade in breast cancer. Clin Cancer Res 2008; 14: 1966–1975 [DOI] [PubMed] [Google Scholar]
- 94.Wei S, Chu PC, Chuang HC, et al. Targeting the oncogenic E3 ligase Skp2 in prostate and breast cancer cells with a novel energy restriction-mimetic agent. PLoS One 2012; 7: e47298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhao H, Bauzon F, Fu H, et al. Skp2 deletion unmasks a p27 safeguard that blocks tumorigenesis in the absence of pRb and p53 tumor suppressors. Cancer Cell 2013; 24: 645–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan CH, Morrow JK, Li CF, et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013; 154: 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen Q, Xie W, Kuhn DJ, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008; 111: 4690–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roy S, Kaur M, Agarwal C, et al. p21 and p27 induction by silibinin is essential for its cell cycle arrest effect in prostate carcinoma cells. Mol Cancer Ther 2007; 6: 2696–2707 [DOI] [PubMed] [Google Scholar]
- 99.Yang ES, Burnstein KL. Vitamin D inhibits G1 to S progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J Biol Chem 2003; 278: 46862–46868 [DOI] [PubMed] [Google Scholar]
- 100.Huang HC, Lin CL, Lin JK. 1,2,3,4,6-penta-O-galloyl-beta-D-glucose, quercetin, curcumin and lycopene induce cell-cycle arrest in MDA-MB-231 and BT474 cells through downregulation of Skp2 protein. J Agric Food Chem 2011; 59: 6765–6775 [DOI] [PubMed] [Google Scholar]
- 101.Huang HC, Way TD, Lin CL, Lin JK. EGCG stabilizes p27kip1 in E2-stimulated MCF-7 cells through down-regulation of the Skp2 protein. Endocrinology 2008; 149: 5972–5983 [DOI] [PubMed] [Google Scholar]
- 102.Wu L, Grigoryan AV, Li Y, et al. Specific small molecule inhibitors of Skp2-mediated p27 degradation. Chem Biol 2012; 19: 1515–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ungermannova D, Lee J, Zhang G, et al. High-throughput screening AlphaScreen assay for identification of small-molecule inhibitors of ubiquitin E3 ligase SCF Skp2-Cks1. J Biomol Screen 2013;18(8): 910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]; • This paper described a successful example of AlphaScreen assay for identification of small-molecule inhibitors of ubiquitin E3 ligase SCF Skp2-Cks1.
- 104.Oh M, Lee JH, Moon H, et al. A chemical inhibitor of the Skp2/p300 interaction that promotes p53 mediated apoptosis. Angew Chem Int Ed 2016; 55: 602–606 [DOI] [PubMed] [Google Scholar]
- 105.Momand J, Jung D, Wilczynski S Niland J. The MDM2 gene amplification database. Nucleic Acids Res 1998; 26: 3453–3459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reifenberger G, Liu L, Ichimura K, et al. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 1993; 53: 2736–2739 [PubMed] [Google Scholar]
- 107.Hock AK, Vousden KH. The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta 2014; 1843 (1): 137–149 [DOI] [PubMed] [Google Scholar]
- 108.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303: 844–848 [DOI] [PubMed] [Google Scholar]; •• This paper reported that Nutlins, a family of cis-imidazoline derivatives, inactivate MDM2 and mediate p53 ubiquitination; these compounds have entered clinical trials for solid tumors and leukemia.
- 109.Cohen P, Tcherpakov M. Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 2010; 143: 686–693 [DOI] [PubMed] [Google Scholar]
- 110.Vu B, Wovkulich P, Pizzolato G et al. Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett 2013; 4(5): 466–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10: 1321–1328 [DOI] [PubMed] [Google Scholar]
- 112.Grossman SR, Deato ME, Brignone C, et al. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003; 300: 342–344 [DOI] [PubMed] [Google Scholar]
- 113.Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin–proteasome system. Nat Rev Drug Discov 2006; 5: 596–613 [DOI] [PubMed] [Google Scholar]
- 114.Yang Y, Ludwig RL, Jensen JP, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005; 7: 547–559 [DOI] [PubMed] [Google Scholar]
- 115.Roxburgh P, Hock AK, Dickens MP, et al. Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis 2012; 33: 791–798 [DOI] [PubMed] [Google Scholar]
- 116.Zhang Z, Chu XJ, Liu JJ, et al. Discovery of Potent and Orally Active p53-MDM2 Inhibitors RO5353 and RO2468 for Potential Clinical Development. ACS Med Chem Lett 2014; 5: 124–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang B, Golding BT, Hardcastle IR. Small-molecule MDM2-p53 inhibitors: recent advances. Future Med Chem 2015; 7(5): 631–645 [DOI] [PubMed] [Google Scholar]; • This paper provided an overview of small-molecule MDM2-p53 inhibitors.
- 118.Tisato V, Voltan R, Gonelli A, et al. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncolog 2017; 10(1): 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Uchida C, Kitagawa M. RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr Cancer Drug Tar 2016; 16(2): 157–174 [DOI] [PubMed] [Google Scholar]
- 120.Zhang W, Wu K, Sartori MA. System-wide modulation of HECT E3 ligases with selective ubiquitin variant probes. Mol Cell 2016; 62: 121–136 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper described a systematic phage display-based ubiquitin variant strategy to screen E3 modulators.
- 121.Mund T, Lewis MJ, Maslen S, Pelham HR. Peptide and small molecule inhibitors of HECT-type ubiquitin ligases. Proc Natl Acad Sci U S A 2014; 47(111): 16736–16741 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper reported discovery of bicyclic peptides(bicycles) by phage display and heclin by Alpha screening technology, both targeting the HECT-type E3 enzyme.
- 122.Quirit JG, Lavrenov SN, Poindexter K, et al. Indole-3-carbinol (I3C) analogues are potent small molecule inhibitors of NEDD4–1 ubiquitin ligase activity that disrupt proliferation of human melanoma cells. Biochem Pharmacol 2017; 127: 13–27 [DOI] [PubMed] [Google Scholar]
- 123.Wang M, Pickart CM. Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J 2005; 24(24): 4324–4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Beaudenon S, Huibregtse JM. HPV E6, E6AP and cervical cancer. BMC Biochem 2008; 9 (1): 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ronchi VP, Klein JM, Haas AL. E6AP/UBE3A ubiquitin ligase harbors two E2-ubiquitin binding sites. J Biol Chem 2013; 288: 10349–10360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haas AL, Ronchi VP. Methods of modulating ubiquitin ligase activity. US9725708 (granted patents)(2017)
- 127.Spruck C, Strohmaier H, Watson M, et al. A CDK-Independent Function of Mammalian Cks1: Targeting of SCFSkp2 to the CDK Inhibitor p27Kip1. Mol Cell 2001; 7(3): 639–650 [DOI] [PubMed] [Google Scholar]
- 128.Carroll D, Sran A, Singh R, et al. Ubiquitination inhibitors. US20160068490 (applications)(2016)
- 129.Delaunay-Moisan A, Toledano M. Ligase E3 RNF185 inhibitors and uses thereof. US 20160194644(applications)(2016)
- 130.Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 2009; 10(6): 398–409 [DOI] [PubMed] [Google Scholar]
- 131.Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta 2014; 1843(1): 61–74 [DOI] [PubMed] [Google Scholar]
- 132.Zhang W, Sidhu S. Ubiquitin variant modulators of HECT E3 ligases and their uses. US 20170275341(applications)(2017)
- 133.Gorelik M, Sidhu S. Ubiquitin variant modulators of SCF E3 ligases and their uses. US 20170321205(applications) (2017)
- 134.Chamberlain P, Cathers EB, Lopez-girona A. Compositions and methods for inducing conformational changes in cereblon other E3 ubiquitin ligases. US20150374678 (applications)(2015)
- 135.Nils G, Theo R. Small molecule inhibitors of the E3 ligase Skp2 in depression and other disease. EP2954896 (2015)
- 136.Rico-Bautista E, Zhu W, Kitada S, et al. Small molecule-induced mitochondrial disruption directs prostate cancer inhibition via upr signaling. Oncotarget 2013; 4: 1212–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Riley B, Johnston J, Morgans D. Method for identifying modulators of ubiquitin ligases. US9464311 (granted patents)(2016)
- 138.Clark JP. Ubiquitination Assay. EP2564193(2016)
- 139.Mevissen TET, Hospenthal MK, Komander D. Ubiquitin chain analysis. US9587265 (granted patents)(2017)
- 140.Statsyuk AV, Park S. Probes and assays for measuring E3 ligase activity. US20160076074 (applications) (2016)
- 141.Harper JW, Jin J. Novel activation and transfer cascade for ubiquitin.US20150105446 (applications) (2015)
- 142.Luo ZQ, Qiu J, Das C, Sheedlo M. Novel ubiquitination system and the uses thereof. US 20170283852 (applications) (2017)
- 143.Chakraaaaborty T, de Fougerolles A. Modified polynucleotides encoding siah E3 ubiquitin protein ligase1. US 9255129B2 (granted patents)(2016)
- 144.Bulatov E, Ciulli A. Targeting Cullin–RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem J 2015; 467: 365–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chang TL, Huang YH, Ou YD. The role of ginsenosides in inhibiting ubiquitin activating enzyme (E1) activity. J Funct Foods 2014; 7: 462–470 [Google Scholar]
- 146.Patel K, Ahmed ZS, Huang X, et al. Discovering proteasomal deubiquitinating enzyme inhibitors for cancer therapy: lessons from rational design, nature and old drug reposition. Future Med Chem; 2018. (Epub ahead of print, doi: 10.4155/fmc-2018-0091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Stein ML, Groll M. Applied techniques for mining natural proteasome inhibitors. Biochim Biophys Acta 2014; 1843: 26–38 [DOI] [PubMed] [Google Scholar]
- 148.Wong JH, Alfatah M, Sin MF, et al. A yeast two-hybrid system for the screening and characterization of small-molecule inhibitors of protein-protein interactions identifies a novel putative Mdm2-binding site in p53. BMC Biol 2017; 15(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen X, Mangala LS, Rodriguez-Aguayo C, et al. RNA interference-based therapy and its delivery systems. Cancer Metat Rev 2018; 37:107–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Morreale FE, Bortoluzzi A, Chaugule VK, et al. Allosteric targeting of the Fanconi anemia ubiquitin-conjugating enzyme Ube2T by fragment screening. J Med Chem 2017; 60: 4093–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study identified a new allosteric pocket on Ube2T through a fragment screening using biophysical methods.
- 151.Buckley DL, Raina K, Darricarrerre N, et al. HaloPROTACS: Use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem. Biol 2015; 10(8): 1831–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This paper reports the design of a novel class of PROTACs that incorporate small molecule VHL ligands to successfully degrade HaloTag7 fusion proteins.
- 152.Winter GE, Buckley DL, Paulk J, et al. Phthalimide conjugation asastrategy for in vivo target protein degradation. Science 2015; 348(6241): 1376–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study provides a chemical strategy that promotes ligand-dependent target protein degradation using as an example transcriptional coactivator BRD4. As a result, a bifunctional molecule dBET1 was identified that induced highly selective cereblon-dependent BET protein degradation in vitro and in vivo and delayed leukemia progression in mice.