

Abstract
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infants and children. Recessive inactivating mutations in the ABCC8 and KCNJ11 genes account for approximately 50% of all CHI cases. Hyperinsulinaemic hypoglycaemia in infancy and diabetes in later life have been reported in patients with HNF1A, HNF4A and ABCC8 mutations. Herein, we present a child who was diagnosed with CHI at birth, then developed diabetes mellitus at the age of nine years due to a novel homozygous missense, p.L171F (c.511C>T) mutation in exon 4 of ABCC8. The parents and one sibling were heterozygous carriers, whilst a younger sibling who had transient neonatal hypoglycemia was homozygous for the mutation. The mother and (maternal) uncle, who was also heterozygous for the mutation, developed diabetes within their third decade of life. The preliminary results of sulphonylurea (SU) treatment was suggestive of SU responsiveness. Patients with homozygous ABCC8 mutations can present with CHI in the newborn period, the hyperinsulinism can show variability in terms of clinical severity and age at presentation and can cause diabetes later in life. Patients with homozygous ABCC8 mutations who are managed medically should be followed long-term as they may be at increased risk of developing diabetes after many years.
Keywords: Congenital hyperinsulinism, MODY, ABCC8 mutation, children
What is already known on this topic?
Homozygous ABCC8 mutations cause severe, persistent, diffuse hyperinsulinemic hypoglycaemia (HH) which is usually diazoxide unresponsive and requires surgical therapy. In medically managed patients with congenital hyperinsulinism, disease symptoms become milder over time. HH in the neonatal period, and subsequent diabetes, have been reported in heterozygous mutations of HNF4A and HNF1A as well as heterozygous ABCC8 mutations.
What this study adds?
We describe the first homozygous ABCC8 mutation with hyperinsulinemic hypoglycaemia (HH) in the neonatal period and its evolution to complete insulin deficient, sulphonylurea responsive diabetes mellitus. Findings from this present work, which show a broad clinical spectrum from asymptomatic to mild symptomatic hypoglycemia and severe hypoglycemia as well as insulin deficient diabetes mellitus in family members with identical mutation confirm the phenotypical variations in ABCC8 mutations. This present case report emphasizes the need for long-term follow up of patients with HH in the neonatal period due to ABCC8 mutations, particularly in those who have received medical therapy for risk of developing diabetes in later life.
Introduction
Adenosine triphosphate (ATP)-sensitive potassium (KATP) channels play an essential role in the regulation of insulin secretion from the pancreatic beta-cell; the key mechanism maintaining the blood glucose level in a narrow range of 3.5-5.5 mmol/L (1,2,3). KATP channels are open at low glucose levels (1). Increased metabolism, resulting in an increased ATP/adenosine diphosphate ratio, leads to closure of the KATP channel, depolarisation of the beta cell membrane and subsequent calcium influx through voltage-gated calcium channels. This in turn leads to insulin secretion via the exocytosis of secretory granules (2,3). Dysfunction of the KATP channel can cause either congenital hyperinsulinism (CHI) or diabetes (neonatal or adult onset) (1,4,5,6,7,8,9). CHI occurs when KATP channels are absent on the cell membrane or when they remain closed despite low glucose levels. In contrast, diabetes occurs if KATP channels remain open despite high blood glucose concentrations and increased metabolism in the beta cell (1,4). Recessive inactivating mutations of the KATP channel genes (ABCC8 and KCNJ11) are the most common cause of severe, diazoxide unresponsive, diffuse CHI which usually requires pancreatectomy (1,10). Patients with dominant mutations of KATP channel genes, ABCC8 and KCNJ11, may cause variable phenotype ranging from asymptomatic macrosomia, mild diazoxide responsive CHI to severe persistent hyperinsulinaemic hypoglycaemia (HH) as well as diabetes mellitus in later life (7,8,9,11).
CHI within the neonatal period and evolution to diabetes later in life has been reported in individuals with heterozygous inactivating mutations in the hepatocyte nuclear factor 4A and 1A genes (HNF4A and HNF1A) (12,13,14) and dominant mutations in ABCC8 genes in a very limited number of cases (1,7,11,13,15,16,17,18,19,20,21).
To the best of our knowledge, CHI due to homozygous ABCC8 mutations and evolution to complete insulin deficient-diabetes later in life has not been reported. Herein, we present a patient with a novel, homozygous ABCC8 mutation who was diagnosed with CHI in the neonatal period and developed diabetes at the age of nine years.
Case Report
A nine year-old Turkish boy (VI.2 in Figure 1) presented with abdominal pain and fever. He was diagnosed with perforated appendicitis and was referred to the endocrine clinic for coexisting hyperglycaemia (blood glucose level was 27.75 mmol/L). A detailed family history revealed the presence of diabetes in multiple members of the maternal family (see details on the pedigree and footnotes). Specifically, the patient’s mother was on insulin therapy for diabetes mellitus which had been diagnosed during the first trimester of pregnancy, when she was 24 years of age. A maternal uncle was also affected. There was also a history of neonatal hypoglycaemia of varying duration and severity affecting two of the patient’s siblings.
Figure 1.
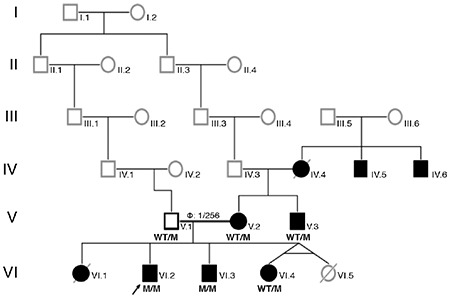
Pedigree of the family. The members developed either hypoglycaemia, diabetes or both are indicated as affected and shown with black-filled boxes. IV.4: Insulin dependent diabetes since 35 years-old, developed diabetic nephropathy (chronic renal failure) (reportedly), IV.5: Had insulin dependent diabetes and diabetic nephropathy (reportedly), IV.6: Diabetes and bilateral visual loss was reported, V.1: Father, 41 years old, apparently healthy with normal glucose and HbA1c (5.6%) levels, V.2: Mother 37 years old, developed insulin dependent diabetes during pregnancy and has been on insulin treatment since 24 years old, changing the treatment to SU therapy is in progress (see the section of the case report concerning sulphonylurea treatment), V.3: 40 years old, had insulin dependent diabetes mellitus since 32 years-old, VI.1: Born at term, macrosomic birth weight (4750 gram; 2.8 SDS), hypoglycaemia was detected during the neonatal period, died at 1-month during hospitalization with unknown etiology, VI.2: Index patient, VI.3: 9.5 years-old, born at term, birth weight was 3750 gram (0.4 SDS), had transient hypoglycemia during the neonatal period, latest blood glucose and HbA1c levels were normal, VI.4: 6.5 years old, born at seven months of gestation from a twin pregnancy, birth weight was 1250 gram (1.05 SD), HbA1c is normal
M: mutated, WT: wild type, SDS: standard deviation score, SU: sulphonylurea, HbA1c: haemoglobin A1c
The patient was the first living child of the family and was born with a birth weight of 3750 grams (+6.6 SD) via caesarian section at a gestation age of 29 weeks. Parents were distantly related. He presented with a hypoglycaemic episode at postnatal day one (blood glucose was 1.33 mmol/L and simultaneous insulin level was 22.7 µIU/mL, C-peptide 5.42 ng/mL (0.9-7.1). A diagnosis of HH was considered and diazoxide was commenced. The patient developed pulmonary edema, which was considered likely to be a complication of treatment with diazoxide. Diazoxide was subsequently stopped and octreotide therapy was introduced. Hypoglycaemia remitted at the age of three months and the child remained free of hypoglycaemic episodes until nine years of age, when he was admitted to our hospital.
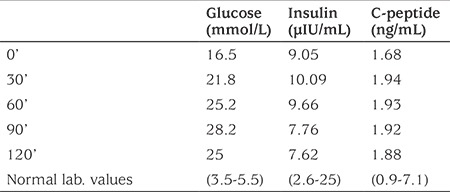
On admisson, the child was lethargic and had pale and grayish colour skin. His height was 140 cm [0.7 standard deviation score (SDS)], weight was 35 kg (0.8 SDS), and body mass index (BMI) 17.8 (0.7 SDS). Respiratory rate was 20 breaths/minute, heart rate was 72 beats/minute and blood pressure was 90/60 mmHg. There was abdominal distention, rigidity and rebound tenderness on physical examination. The patient underwent emergency appendectomy. During the post-operative period hyperglycaemia persisted and subcutaneous insulin therapy was introduced. Laboratory investigations revealed a blood glucose of 13.2 mmol/L with a simultaneous insulin of 8.82 µIU/mL (2.6-25), C-peptide: 1.28 ng/mL (0.9-7.1). Glycosylated haemoglobin A1c (HbA1c) was 9.1% (76 mmol/L). Islet cell, anti-insulin and anti-glutamic acid decarboxylase antibodies were negative. Over the following two months the insulin requirement gradually decreased until insulin treatment could be completely withdrawn. During the follow-up, HbA1c remained within the range of the high normal limits (6.2% to 6.4%) with dietary intervention and lifestyle changes. At the age of 11.5 years HbA1c was shown to be again significantly raised at 9.6% (81 mmol/L). At this time his weight was 46 kg (0.9 SDS), height was 152 cm (0.8 SDS) and BMI was 19.9 (0.9 SDS). The patient and family refused recommencement of insulin therapy. Subsequently, HbA1c increased to 11.4% (101 mmol/l) at the age of 12 years when an oral glucose tolerance test suggested insulin deficient-diabetes mellitus (Table 1).
Table 1. Oral glucose tolerance test results of index case at age 12 years.
Genetic Testing
Genomic DNA was extracted from peripheral leukocytes using standard procedures and the coding regions and intron/exon boundaries of the ABCC8, KCNJ11, HNF4A and HADH genes were amplified by polymerase chain reaction (primers available on request). Amplicons were sequenced using the BigDye Terminator Cycle Sequencing Kit V.3.1 (Applied Biosystems, Warrington, UK) according to manufacturer’s instruction and reactions were analysed on an ABI 3730 Capillary sequencer (Applied Biosystems, Warrington, UK). Sequences were compared with the reference sequences (NM_001287174.1, NM_000525.3, NM_175914.4 and NM_005327.4) using Mutation Surveyor v5.0.1 software (SoftGenetics, State College, Pennsylvania, USA). The variant was classified using the American College of Medical Genetic and Genomics/Association for Molecular Pathology guidelines (22).
A written informed consent was obtained from the patients and/or their legal guardians.
Results
The index patient (VI.2, see Figure 1) was found to be homozygous for a novel missense c.511C>T (p.L171F) variant in exon 4 of ABCC8 (Figure 2). The p.L171F variant affects a highly conserved amino acid and in silico analysis predicted the variant to be disease-causing (Alamut Visual V2.10 Software, Rouen, France). Mutation testing showed that the variant co-segregated with diabetes and hypoglycemia within the family, with an incomplete penetrance of heterozygous carriers (Figure 1).
Figure 2.
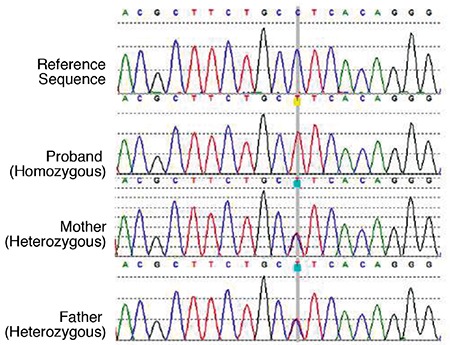
Electropherograms of the reference, index case and parents for c.511C>T (p.L171F) mutation
Treatment and Follow-up
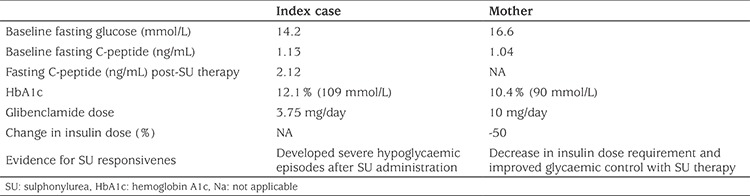
Following detection of the ABCC8 mutation, a trial of sulphonylurea (SU) treatment was commenced in the index case and his mother, who had been on insulin therapy for 13 years in an outpatient setting (Table 2). The mother’s daily insulin dose requirement was reduced by approximately 50% from the baseline at the first week of the SU therapy with improved blood glucose measurements. The index case also responded to SU therapy and even developed one hypoglycaemic episode following SU therapy. Although the SU doses were adjusted accordingly, the family avoided giving the glibenclamide regularly due to the severe hypoglycaemic episodes which had not been observed while he was on insulin therapy or during fasting.
Table 2. Sulphonlyurea treatment trial results in the index case and his heterozygous carrier mother with diabetes mellitus.
Discussion
Herein, we present a patient with a novel homozygous ABCC8 mutation who was diagnosed with HH in the neonatal period and diabetes at the age of nine years. Hyperglycemia was first recognized during acute appendicitis which suggested stress-induced hyperglycemia. However, the patient had persistent hyperglycemia which required insulin therapy, a history of HH and relatives with autoantibody-negative diabetes. These findings were suggestive of monogenic diabetes, which was confirmed by molecular genetics analysis.
Homozygous KATP channel gene mutations are the most common cause of severe, diazoxide unresponsive HH which often requires pancreatectomy (3,13). However, clinical heterogeneity is observed in patients with dominant ABCC8 mutations (9). Kapoor et al (23) reported a marked clinical heterogenity in siblings with identical mutations in ABCC8, ranging from asymptomatic hypoglycaemia to macrosomia, transient HH or severe HH and development of diabetes mellitus later in life. Besides, a heterogeneous nature is observed regarding severity and response to medical treatment and age of onset of symptoms (23).
Variations in the severity of HH and clinical course have also been reported in a mother and her daughter with a heterozygous E1506K mutation in ABCC8 (20). In this report the child had severe symptoms and hypoglycaemic convulsions at age three months while the mother had subtle symptoms of hypoglycaemia followed by gestational diabetes which persisted after delivery.
Similarly, a marked clinical heterogenity was also observed in this present family. While one of the homozygous siblings (VI.2, index case) had prolonged HH and required medical therapy, the other sibling with an identical homozygous mutation (VI.3) suffered from transient hypoglycemia in the first week of life which remitted within three months. Diabetes was observed in the heterozygous mother and a maternal uncle, but the father (V.1) who was also heterozygous for the ABCC8 mutation, had normal fasting plasma glucose and HbA1c levels. Unfortunately, the family refused performance of oral glucose tolerance test in the individuals who carried the mutation, but had not yet developed fasting hyperglycaemia or elevated HbA1c. We could also not perform genetic analysis in other relatives who also had diabetes with renal and ocular complications. We, therefore, were not able to confirm whether the diabetes in these additional family members was due to the ABCC8 mutation.
Indeed, the clinical course for patients with ABCC8 mutations is also substantially variable (1,7,13,15,16,17,18,19,20). Clinical features include hypoglycaemia within the neonatal period which remits over time, coexistence of hypoglycaemia and post-fed hyperglycaemic episodes, impaired fasting glucose or impaired glucose tolerance in response to a glucose load and in a few cases the development of diabetes (1,7,11,13,15,16,17,18,19,20). Regarding the type of mutation reported in ABCC8 which caused neonatal HH and diabetes later in life, only a few cases were reported to have a homozygous mutation (18), whilst the majority had heterozygous or compound heterozygous mutations (7,11,15,17,19,20).
Biallelic (either homozygous or compound heterozygous) ABCC8 mutations usually cause severe, diazoxide unresponsive HH which often requires surgical management (3). Therefore, the number of medically managed cases, particularly those with long-term follow-up, is very small. This limits our understanding of the underlying mechanisms and experience in the management of cases who develop hyperglycemia later in life. The data from the reported cases and experimental studies suggest that the key mechanisms are dysregulated insulin secretion, impaired first phase insulin secretion, delayed insulin response and b-cell apoptosis mediated via enhanced b-cell depolarisation, resulting in increased calcium ion entry into the cell (7,8,9,13,15,16,17,18,19,20,24,25,26).
Taking into account the previously reported cases, our patient is the only case with a homozygous ABCC8 mutation who presented with CHI (confirmed by clinical and biochemical evidence and mutation analysis) within the neonatal period which evolved into complete insulin deficient diabetes later in life. Therefore, this family provides novel insights into the clinical heterogeneity of CHI and later onset diabetes in patients with homozygous ABCC8 mutations.
Neonatal diabetes due to a dominant activating mutation of a KATP channel gene (KCNJ11 or ABCC8) is usually sulphonylurea responsive (27,28). We also performed a trial of SU therapy in the index case and his heterozygous mother who had insulin dependent diabetes. Preliminary results suggested a favorable SU responsiveness. Since SU drugs work by binding to the SUR1 subunit of the KATP channel, a positive response to SU therapy suggested that the presence of a homozygous mutation may not completely abolish the channel function.
In conclusion, we present the novel missense c.511C>T (p.L171F) ABCC8 mutation causing neonatal HH and SU-responsive diabetes mellitus later in life. There are, however, some limitations in interpreting the phenotype-genotype relationships observed in this family. Firstly, we were not able to analyse the mutation status of other family members with diabetes mellitus. Secondly, although clinical evidences and bioinformatic tools confirmed the pathogenicity of the novel mutation, functional analyses have not been undertaken to assess the role of the variant in vitro. These results highlight the need for the long-term follow up of a larger series of CHI patients with homozygous ABCC8 mutations who have been managed medically. In addition further evaluation of these variants, including functional analysis, to better understand the underlying molecular mechanism and phenotype-genotype relationships should be performed.
Footnotes
Ethics
Informed Consent: A written informed consent was obtained from the patients and/or their legal guardians.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Concept: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Sian Ellard, Sarah E. Flanagan, Khalid Hussain, Design: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Sian Ellard, Sarah E. Flanagan, Khalid Hussain, Data Collection or Processing: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Sian Ellard, Sarah E. Flanagan, Analysis or Interpretation: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Sian Ellard, Sarah E. Flanagan, Khalid Hussain, Literature Search: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Writing: Emregül Işık, Hüseyin Demirbilek, Jayne A. Houghton, Sian Ellard, Sarah E. Flanagan, Khalid Hussain.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Bennett K, James C, Hussain K. Pancreatic beta-cell KATP channels: Hypoglycaemia and hyperglycaemia. Rev Endocr Metab Disord. 2010;11:157–163. doi: 10.1007/s11154-010-9144-2. [DOI] [PubMed] [Google Scholar]
- 2.Demirbilek H, Rahman SA, Buyukyilmaz GG, Hussain K. Diagnosis and treatment of hyperinsulinaemic hypoglycaemia and its implications for paediatric endocrinology. Int J Pediatr Endocrinol. 2017;2017:9. doi: 10.1186/s13633-017-0048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah P, Rahman SA, Demirbilek H, Guemes M, Hussain K. Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol. 2017;5:729–742. doi: 10.1016/S2213-8587(16)30323-0. [DOI] [PubMed] [Google Scholar]
- 4.Clark R, Proks P. ATP-sensitive potassium channels in health and disease. Adv Exp Med Biol. 2010;654:165–192. doi: 10.1007/978-90-481-3271-3_8. [DOI] [PubMed] [Google Scholar]
- 5.Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, Hattersley AT, Ellard S. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. 2012;55:123–127. doi: 10.1007/s00125-011-2319-x. [DOI] [PubMed] [Google Scholar]
- 6.Hartemann-Heurtier A, Simon A, Bellanne-Chantelot C, Reynaud R, Cave H, Polak M, Vaxillaire M, Grimaldi A. Mutations in the ABCC8 gene can cause autoantibody-negative insulin-dependent diabetes. Diabetes Metab. 2009;35:233–235. doi: 10.1016/j.diabet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Huopio H, Otonkoski T, Vauhkonen I, Reimann F, Ashcroft FM, Laakso M. A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet. 2003;361:301–307. doi: 10.1016/S0140-6736(03)12325-2. [DOI] [PubMed] [Google Scholar]
- 8.Huopio H, Reimann F, Ashfield R, Komulainen J, Lenko HL, Rahier J, Vauhkonen I, Kere J, Laakso M, Ashcroft F, Otonkoski T. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J Clin Invest. 2000;106:897–906. doi: 10.1172/JCI9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, Shield JP, Tinker A, Ellard S, Hussain K. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011;54:2575–2583. doi: 10.1007/s00125-011-2207-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K. Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism. Eur J Endocrinol. 2013;168:557–564. doi: 10.1530/EJE-12-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ocal G, Flanagan SE, Hacihamdioglu B, Berberoglu M, Siklar Z, Ellard S, Savas Erdeve S, Okulu E, Akin IM, Atasay B, Arsan S, Yagmurlu A. Clinical characteristics of recessive and dominant congenital hyperinsulinism due to mutation(s) in the ABCC8/KCNJ11 genes encoding the ATP-sensitive potasium channel in the pancreatic beta cell. J Pediatr Endocrinol Metab. 2011;24:1019–1023. doi: 10.1515/jpem.2011.347. [DOI] [PubMed] [Google Scholar]
- 12.Pearson ER, Boj SF, Steele AM, Barrett T, Stals K, Shield JP, Ellard S, Ferrer J, Hattersley AT. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med. 2007;4:118. doi: 10.1371/journal.pmed.0040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rahman SA, Nessa A, Hussain K. Molecular mechanisms of congenital hyperinsulinism. J Mol Endocrinol. 2015;54:119–129. doi: 10.1530/JME-15-0016. [DOI] [PubMed] [Google Scholar]
- 14.Stanescu DE, Hughes N, Kaplan B, Stanley CA, De Leon DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. 2012;97:2026–2030. doi: 10.1210/jc.2012-1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S, Perry A, Abu- Libdeh A, Glaser B, Shyng SL, Zangen DH. Novel De Novo Mutation in Sulfonylurea Receptor 1 Presenting as Hyperinsulinism in Infancy Followed by Overt Diabetes in Early Adolescence. Diabetes. 2008;57:1935–1940. doi: 10.2337/db08-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bin-Abbas BS, Al-Ashwal AA. Diabetes in a nonpancreatectomized child with nesidioblastosis. Diabetes Care. 2004;27:626–627. doi: 10.2337/diacare.27.2.626. [DOI] [PubMed] [Google Scholar]
- 17.Gussinyer M, Clemente M, Cebrian R, Yeste D, Albisu M, Carrascosa A. Glucose intolerance and diabetes are observed in the long-term followup of nonpancreatectomized patients with persistent hyperinsulinemic hypoglycemia of infancy due to mutations in the ABCC8 gene. Diabetes Care. 2008;31:1257–1259. doi: 10.2337/dc07-2059. [DOI] [PubMed] [Google Scholar]
- 18.Harel S, Cohen AS, Hussain K, Flanagan SE, Schlade-Bartusiak K, Patel M, Courtade J, Li JB, Van Karnebeek C, Kurata H, Ellard S, Chanoine JP, Gibson WT. Alternating hypoglycemia and hyperglycemia in a toddler with a homozygous p.R1419H ABCC8 mutation: an unusual clinical picture. J Pediatr Endocrinol Metab. 2015;28:345–351. doi: 10.1515/jpem-2014-0265. [DOI] [PubMed] [Google Scholar]
- 19.Saito-Hakoda A, Yorifuji T, Kanno J, Kure S, Fujiwara I. Nateglinide is Effective for Diabetes Mellitus with Reactive Hypoglycemia in a Child with a Compound Heterozygous ABCC8 Mutation. Clin Pediatr Endocrinol. 2012;21:45–52. doi: 10.1297/cpe.21.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vieira TC, Bergamin CS, Gurgel LC, Moises RS. Hyperinsulinemic hypoglycemia evolving to gestational diabetes and diabetes mellitus in a family carrying the inactivating ABCC8 E1506K mutation. Pediatr Diabetes. 2010;11:505–508. doi: 10.1111/j.1399-5448.2009.00626.x. [DOI] [PubMed] [Google Scholar]
- 21.Haghverdizadeh P, Sadat Haerian M, Haghverdizadeh P, Sadat Haerian B. ABCC8 genetic variants and risk of diabetes mellitus. Gene. 2014;545:198–204. doi: 10.1016/j.gene.2014.04.040. [DOI] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapoor RR, Flanagan SE, Ellard S, Hussain K. Congenital hyperinsulinism: marked clinical heterogeneity in siblings with identical mutations in the ABCC8 gene. Clin Endocrinol (Oxf) 2012;76:312–313. doi: 10.1111/j.1365-2265.2011.04203.x. [DOI] [PubMed] [Google Scholar]
- 24.Miki T, Tashiro F, Iwanaga T, Nagashima K, Yoshitomi H, Aihara H, Nitta Y, Gonoi T, Inagaki N, Miyazaki J, Seino S. Abnormalities of pancreatic islets by targeted expression of a dominant-negative KATP channel. Proc Natl Acad Sci U S A. 1997;94:11969–11973. doi: 10.1073/pnas.94.22.11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols CG, Koster JC, Remedi MS. Beta-cell hyperexcitability: from hyperinsulinism to diabetes. Diabetes Obes Metab. 2007;(9 Suppl 2):81–88. doi: 10.1111/j.1463-1326.2007.00778.x. [DOI] [PubMed] [Google Scholar]
- 26.Remedi MS, Rocheleau JV, Tong A, Patton BL, McDaniel ML, Piston DW, Koster JC, Nichols CG. Hyperinsulinism in mice with heterozygous loss of K(ATP) channels. Diabetologia. 2006;49:2368–2378. doi: 10.1007/s00125-006-0367-4. [DOI] [PubMed] [Google Scholar]
- 27.Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Sovik O, Polak M, Hattersley AT; Neonatal Diabetes International Collaborative Group. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6. 2 mutations. N Engl J Med. 2006;355:467–477. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 28.Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT; Neonatal Diabetes International Collaborative Group. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31:204–209. doi: 10.2337/dc07-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]