

Abstract
Inactivating autosomal recessive mutations in fibroblast growth factor 23 (FGF23), klotho (KL) and polypeptide N-acetylgalactosaminotransferase 3 (GALNT3) genes lead to a rare disorder, hyperphosphatemic familial tumoral calcinosis (HFTC). Patients with HFTC present with hyperphosphatemia and tumor like soft tissue calcifications. Although 78% of patients develop their first symptoms between the ages of 2-13 years, diagnosis is usually delayed until adulthood. Some individuals with the same genetic defect develop a condition named hyperphosphatemic hyperostosis syndrome. Herein we report two siblings suffering from periarticular, warm, hard and tender subcutaneous masses. Subcutaneous calcifications were present on X-ray and biopsy results were consistent with calcinosis in both patients. Laboratory results showed marked hyperphosphatemia and elevated renal tubular phosphate reabsorption rates, normal renal function tests and normal serum 25-hydroxyvitamin D levels. Thus, we suspected HFTC and performed next generation sequencing for the GALNT3 gene, reported as the most frequent cause. A novel homozygote P85Rfs*6 (c.254_255delCT) mutation in GALNT3 was identified in both siblings. Our report adds two new patients to the literature about this rare genetic disease and suggests that small deletions in the GALNT3 gene may be related with HFTC phenotype.
Keywords: GALNT3, hyperphosphatemia, tumoral calcinosis
What is already known on this topic?
Mutations in the FGF23, KL and GALNT3 genes cause hyperphosphatemic familial tumoral calcinosis (HFTC), which is a rare disorder. Patients with HFTC commonly present with hyperphosphatemia and tumor-like soft tissue calcifications. The main management strategy for HFTC is pain control and phosphate depletion.
What this study adds?
We describe two siblings with hyperphosphatemic familial tumoral calcinosis due to a novel homozygote GALNT3 mutation and add to the scarce literature. We wish to emphasize that physicians should also consider this rare condition in the differential diagnosis of calcinosis.
Introduction
Hyperphosphatemic familial tumoral calcinosis (HFTC) is a very rare disorder of phosphate homeostasis resulting from decreased fibroblast growth factor 23 (FGF23) synthesis or activity (1). FGF23 gene encodes this protein which inhibits the sodium phosphate cotransporter in proximal renal tubules and 25-hydroxyvitamin D 1-α-hydroxylase expression, by its co-receptor klotho (KL). The polypeptide N-acetylgalactosaminotransferase 3 (GALNT3) gene codes the enzyme known variously as UDP-N-acetyl-alpha-D galactosamine or polypeptide N-acetylgalactosaminyltransferase-3 (GalNAc-T3), which protects intact FGF23 from catabolism and inactivation by posttranslational glycosylation (2). Inactivating autosomal recessive mutations in FGF23, KL or the GALNT3 genes lead to increased renal tubular phosphate reabsorption and, usually, elevated 1,25-dihydroxyvitamin D3 (1,25-OH2D3), promoting gastrointestinal absorption of calcium and phosphorus (1,3,4).
Patients with HFTC usually present with hyperphosphatemia and tumor-like soft tissue calcifications. Although 78% of patients develop their first symptoms between two and 13 years of age, diagnosis is usually delayed until adulthood. Some individuals with the same genetic defect develop hyperphosphatemic hyperostosis syndrome (HHS), a condition which was formerly described as a distinct entity (5). Here we report childhood onset HFTC in two siblings with a novel homozygote GALNT3 mutation.
Case Reports
Case 1
A previously healthy 10 year-old female patient presented with complaints of pain and swelling in her left elbow. Due to the limitation of movement of the elbow, surgery was performed in another medical center at the age of eight years. Excisional biopsy revealed well-circumscribed subcutaneous tissue including widespread dystrophic calcification and multinuclear giant cells. She was referred to us upon recurrence of bilateral calcinosis in her elbows and in her right upper thigh.
The patient was the offspring of a first-degree cousin marriage. Her past medical history revealed no myositis, skin lesions or renal disease. Physical examination revealed calcinous masses of approximately 3 cm-6 cm diameters in the left elbow, the right elbow and in the right upper thigh (Figure 1). The masses were warm, hard and tender. Laboratory results showed marked hyperphosphatemia, normal serum creatinine, 25-hydroxyvitamin D and parathormone levels and an elevated ratio of tubular maximum reabsorption of phosphorus/glomerular filtration rate (TmP/GFR), consistent with HFTC (Table 1). Direct radiographs demonstrated radio-opaque soft tissue masses around the elbows bilaterally and right upper femur diaphysis (Figure 2). Bone mineral density Z-score was 0.7. Dental and ophthalmological examination showed no involvement. Milimetric calcified plaques were present inside the right lower eyelid. A novel homozygote P85Rfs*6 (c.254_255delCT) mutation in exon 1 of the GALNT3 gene was detected by next generation sequencing (NGS). In silico analyses was performed with Mutation Taster, which confirmed that the mutation led to frameshift and a premature stop codon. Both parents were heterozygous carriers for the same mutation.
Figure 1.
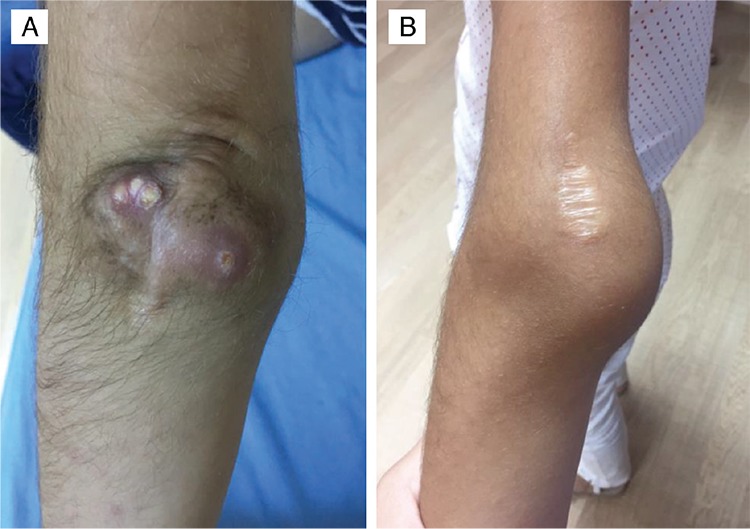
A) Calcinosis in the left elbow of Case 1. B) Subcutaneous mass around the left elbow of Case 2
Table 1. Clinical characteristics, laboratory and genetic results of the two patients with hypertrophic familial tumoral calcinosis.
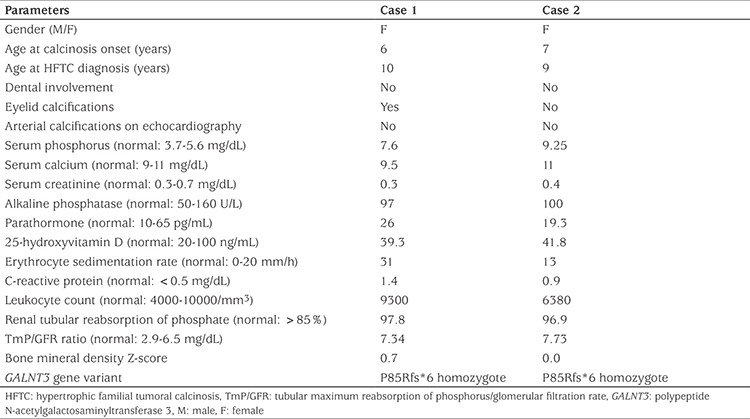
Figure 2.
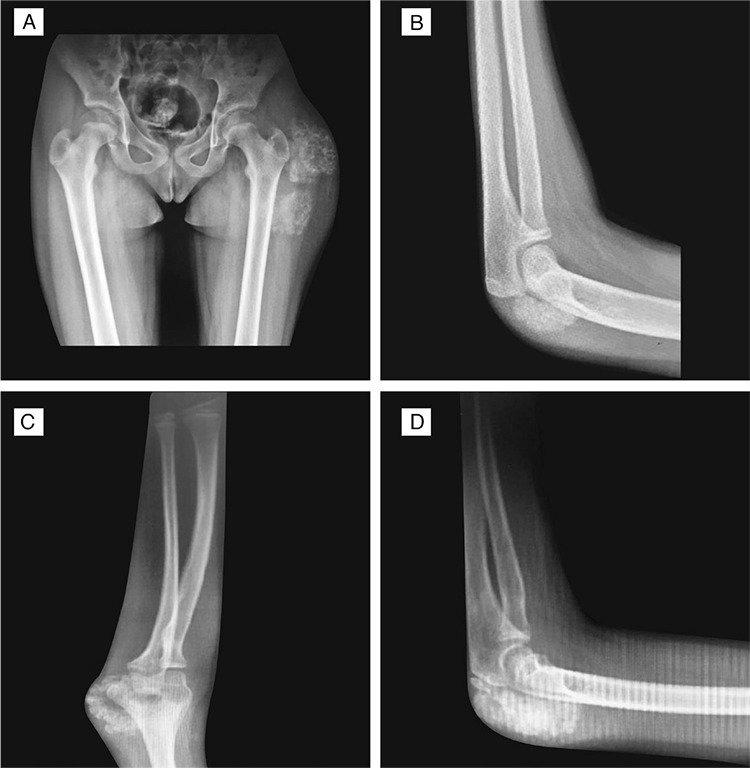
Radiographic findings in our two patients. Two giant radio-opaque soft tissue masses around the left femur neck of Case 1 in anteroposterior view (A). Lateral view of the left elbow of Case 1, revealing subcutanous calcifications (B). Anteroposterior view of upper extremities shows a radio-opaque mass around the elbow joint of Case 2 (C). A subcutaneous calcified mass behind the olecranon is identified in a lateral view of the left elbow (D)
Case 2
This nine year-old female patient was simultaneously referred to our department with her older sister, Case 1. She had developed similar but milder complaints over the preceding two years, including swelling of the left elbow which required surgery due to joint contracture and bilateral recurrence in her elbows thereafter. Direct radiographs demonstrated radio-opaque soft tissue masses around both elbows (Figure 2). Dental and ophthalmological examination showed no involvement. Hyperphosphatemia, elevated TmP/GFR ratio, family history, biopsy result and presence of the same homozygote P85Rfs*6 (c.254_255delCT) mutation in GALNT3 gene confirmed the diagnosis of HFTC.
Figure 3 shows the pedigree and NGS results of our patients.
Figure 3.
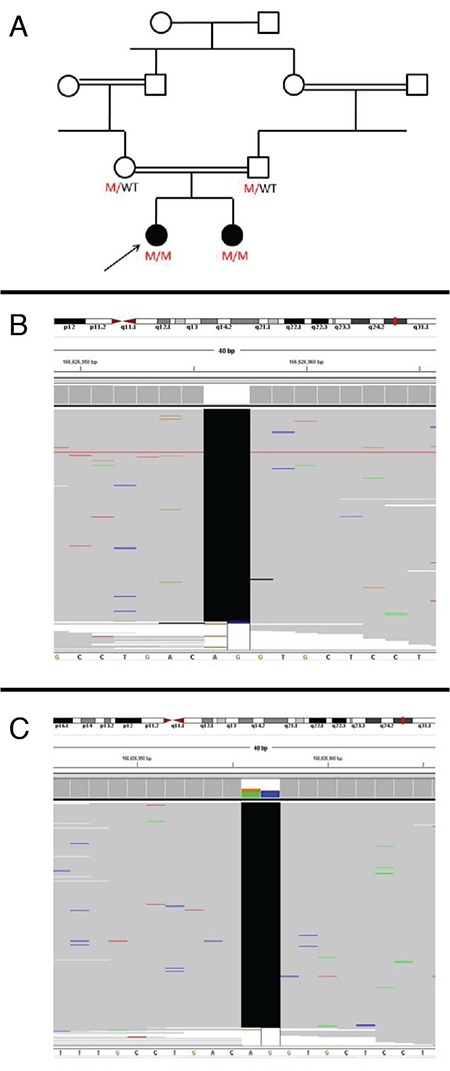
Genetic pedigree of the hyperphosphatemic familial tumoral calcinosis patients is presented in A. Next generation sequence view of the variant identified in the two cases are shown in B (Case 1) and C (Case 2)
A written informed consent for this report was obtained from the parents of the patients.
Discussion
Tumoral calcinosis (TC) is a condition in which calcium crystals accumulate in soft tissues, particularly in periarticular regions. HFTC is the autosomal recessive inherited form of TC with hyperphosphatemia and normal renal function. Differential diagnosis includes chronic renal failure, hypervitaminosis D, primary hyperparathyroidism or connective tissue diseases with particular emphasis on dermatomyositis and scleroderma. HFTC is very rare, approximately 75 cases have been genetically described worldwide to the best of our knowledge and almost all of the information is based on case reports (5,6,7,8,9,10). Homozygote mutations in the GALNT3, FGF23 and KL genes were found in patients with the HHS phenotype. HHS is characterized by painful diaphyseal hyperostosis and may overlap with the TC phenotype in some cases. One study speculated that nonsense and missense GALNT3 mutations are associated with TC and HHS phenotypes, respectively (11). Indeed, the majority of reported GALNT3 mutations are missense or nonsense, and only five distinct small deletions were identified in HFTC patients, according to The Human Gene Mutation Database. Small deletions were reported to cause only the TC phenotype, as was also true for our patients (8,12,13).
Besides subcutaneous calcifications, patients often present with dental abnormalities and occasionally anemia, low-grade fever, regional lymphadenopathy, splenomegaly, amyloidosis, chronic recurrent osteomyelitis and eyelid calcifications (14,15,16). Vascular calcifications may rarely occur and can cause significant morbidity (17). Some HFTC patients develop hyperphosphatemia several years after the onset of dental abnormalities and calcinosis (11). Eyelid calcification was present in one of our patients. However, other clinical traits had not yet developed at presentation.
The management of HFTC mainly targets pain control and phosphate depletion. Surgery is not recommended, due to recurrences, until the calcinosis causes restricted joint movement. A phosphate restricted diet and phosphate binders are the mainstays of the medical treatment (18). A calcium-free phosphate binder, Sevelamer alters the intestinal absorption of phosphorus. Although Sevelamer and dietary phosphate restriction is reported to lead to complete or partial recovery of the calcinosis, recurrences have been reported due possibly to self-discontinuation or ineffectiveness of the drug in a significant proportion of patients. Other agents, including acetazolamide, probenocid and topical sodium thiosulfate, have been reported to be beneficial with variable outcomes (8,19,20,21).
The limitation of our study was the unavailability of serum 1,25-OH2D3 level determinations in both our patients. However, the patients had other clinical and laboratory results consistent with HFTC diagnosis. We believe that elevated serum 1,25-OH2D3 levels are only supportive in the diagnosis of HFTC.
In conclusion, we report two siblings with a novel homozygote GALNT3 mutation representing an HFTC phenotype. HFTC is a rare cause of tenderness and pain around the joints in children and should be kept in mind in the differential diagnosis of arthritis. Our report adds two new patients to the information on a rare genetic disease and we wish to highlight the need for attention to this rare disorder. We speculate that small deletions in GALNT3 gene may be related with HFTC phenotype. However, this speculation can be confirmed only with genotype-phenotype correlation studies including long-term outcomes of more patients in the future.
Footnotes
Ethics
Informed Consent: A written informed consent for this report was obtained from the parents of the patients.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Rabia Miray Kışla Ekinci, Sibel Balcı, Fatih Gürbüz, Concept: Rabia Miray Kışla Ekinci, Mustafa Yılmaz, Bilgin Yüksel, Design: Mustafa Yılmaz, Bilgin Yüksel, Data Collection or Processing: Atıl Bişgin, Fatih Gürbüz, Analysis or Interpretation: Sibel Balcı, Mehmet Taştan, Atıl Bişgin, Literature Search: Mehmet Taştan, Rabia Miray Kışla Ekinci, Writing: Rabia Miray Kışla Ekinci, Mustafa Yılmaz.
Financial Disclosure: The authors declared that this study received no financial support.
References
- 1.Benet-Pages A, Orlik P, Strom TM, Lorenz-Depiereux B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 2.Kato K, Jeanneau C, Tarp MA, Benet-Pagès A, Lorenz-Depiereux B, Bennett EP, Mandel U, Strom TM, Clausen H. Polypeptide GalNActransferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J Biol Chem. 2006;281:18370–18377. doi: 10.1074/jbc.M602469200. [DOI] [PubMed] [Google Scholar]
- 3.Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–581. doi: 10.1038/ng1358. [DOI] [PubMed] [Google Scholar]
- 4.Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Musculoskelet Neuronal Interact. 2007;7:318–319. [PubMed] [Google Scholar]
- 5.Rafaelsen S, Johansson S, Ræder H, Bjerknes R. Long-term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet. 2014;15:98. doi: 10.1186/s12863-014-0098-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keskar VS, Imel EA, Kulkarni M, Mane S, Jamale TE, Econs MJ, Hase NK. The Case | Ectopic calcifications in a child. Kidney Int. 2015;87:1079–1081. doi: 10.1038/ki.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah A, Miller CJ, Nast CC, Adams MD, Truitt B, Tayek JA, Tong L, Mehtani P, Monteon F, Sedor JR, Clinkenbeard EL, White K, Mehrotra R, LaPage J, Dickson P, Adler SG, Iyengar SK. Severe vascular calcification and tumoral calcinosis in a family with hyperphosphatemia: a fibroblast growth factor 23 mutation identified by exome sequencing. Nephrol Dial Transplant. 2014;29:2235–2243. doi: 10.1093/ndt/gfu324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramnitz MS, Gourh P, Goldbach-Mansky R, Wodajo F, Ichikawa S, Econs MJ, White KE, Molinolo A, Chen MY, Heller T, Del Rivero J, Seo-Mayer P, Arabshahi B, Jackson MB, Hatab S, McCarthy E, Guthrie LC, Brillante BA, Gafni RI, Collins MT. Phenotypic and Genotypic Characterization and Treatment of a Cohort With Familial Tumoral Calcinosis/Hyperostosis-Hyperphosphatemia Syndrome. J Bone Miner Res. 2016;31:1845–1854. doi: 10.1002/jbmr.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shawar SM, Ramadan AR, Ali BR, Alghamdi MA, John A, Hudaib FM. FGF23-S129F mutant bypasses ER/Golgi to the circulation of hyperphosphatemic familial tumoral calcinosis patients. Bone. 2016;93:187–195. doi: 10.1016/j.bone.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Masi L, Beltrami G, Ottanelli S, Franceschelli F, Gozzini A, Zonefrati R, Galli G, Ciuffi S, Mavilia C, Giusti F, Marcucci G, Cioppi F, Colli E, Fossi C, Franchi A, Casentini C, Capanna R, Brandi ML. Human Preosteoblastic Cell Culture from a Patient with Severe Tumoral Calcinosis-Hyperphosphatemia Due to a New GALNT3 Gene Mutation: Study of In Vitro Mineralization. Calcif Tissue Int. 2015;96:438–452. doi: 10.1007/s00223-015-9974-8. [DOI] [PubMed] [Google Scholar]
- 11.Dumitrescu CE, Kelly MH, Khosravi A, Hart TC, Brahim J, White KE, Farrow EG, Nathan MH, Murphey MD, Collins MT. A case of familial tumoral calcinosis/hyperostosis-hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporos Int. 2009;20:1273–1278. doi: 10.1007/s00198-008-0775-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garringer HJ, Fisher C, Larsson TE, Davis SI, Koller DL, Cullen MJ, Draman MS, Conlon N, Jain A, Fedarko NS, Dasgupta B, White KE. The role of mutant UDP-N-acetyl-alpha-D-galactosaminepolypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006;91:4037–4042. doi: 10.1210/jc.2006-0305. [DOI] [PubMed] [Google Scholar]
- 13.Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V, Quartier P, Cormier- Daire V, Dechaux M, Malandrinou FCh, Singhellakis PN, Le Merrer M, Econs MJ. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010;152:896–903. doi: 10.1002/ajmg.a.33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman M, Koerber F, Topaz O, Bergman R, Sprecher E, Schoenau E. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118:261–266. doi: 10.1007/s00439-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 15.Demellawy DE, Chang N, de Nanassy J, Nasr A. GALNT3 gene mutationassociated chronic recurrent multifocal osteomyelitis and familial hyperphosphatemic familial tumoral calcinosis. Scand J Rheumatol. 2015;44:170–172. doi: 10.3109/03009742.2014.958100. [DOI] [PubMed] [Google Scholar]
- 16.Ichikawa S, Imel EA, Sorenson AH, Severe R, Knudson P, Harris GJ, Shaker JL, Econs MJ. Tumoral Calcinosis Presenting with Eyelid Calcifications due to Novel Missense Mutations in the Glycosyl Transferase Domain of the GALNT3 Gene. J Clin Endocrinol Metab. 2006;91:4472–4475. doi: 10.1210/jc.2006-1247. [DOI] [PubMed] [Google Scholar]
- 17.Chefetz I, Heller R, Galli-Tsinopoulou A, Richard G, Wollnik B, Indelman M, Koerber F, Topaz O, Bergman R, Sprecher E, Schoenau E. A novel homozygous missense mutation in FGF23 causes familial tumoral calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118:261–266. doi: 10.1007/s00439-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 18.Claramunt-Taberner D, Bertholet-Thomas A, Carlier MC, Dijoud F, Chotel F, Silve C, Bacchetta J. Hyperphosphatemic tumoral calcinosis caused by FGF23 compound heterozygous mutations: what are the therapeutic options for a better control of phosphatemia? Pediatr Nephrol. 2018;33:1263–1267. doi: 10.1007/s00467-018-3945-z. [DOI] [PubMed] [Google Scholar]
- 19.Finer G, Price HE, Shore RM, White KE, Langman CB. Hyperphosphatemic familial tumoral calcinosis: Response to acetazolamide and postulated mechanisms. Am J Med Genet Part A. 2014;164:1545–1549. doi: 10.1002/ajmg.a.36476. [DOI] [PubMed] [Google Scholar]
- 20.Balachandran K, Kamalanathan S, Sahoo JP, Das AK, Halanaik D. Differential response of idiopathic sporadic tumoral calcinosis to bisphosphonates. Indian J Endocr Metab. 2014;18:521–525. doi: 10.4103/2230-8210.137511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jost J, Bahans C, Courbebaisse M, Tran TA, Linglart A, Benistan K, Lienhardt A, Mutar H, Pfender E, Ratsimbazafy V, Guigonis V. Topical Sodium Thiosulfate: A Treatment for Calcifications in Hyperphosphatemic Familial Tumoral Calcinosis? J Clin Endocrinol Metab. 2016;101:2810–2815. doi: 10.1210/jc.2016-1087. [DOI] [PubMed] [Google Scholar]