

SETD2 is commonly mutated in clear cell renal cell carcinoma (ccRCC), and exploring the underlying mechanism is urgent for developing targeted therapies for ccRCC. Here, we systematically analyzed the alterations in histone epigenetic marks (H3K36me3 and H4K16ac), mRNA transcriptome and chromatin-bound proteins evoked by SETD2 depletion. This work provided a robust foundation for understanding SETD2's role in ccRCC.
Keywords: Cancer Biology*, Proteogenomics, Cell biology*, Oncogenes*, RNA SEQ, PLK1-CDK1 Pathway, SETD2
Graphical Abstract

Highlights
Integrated analysis of histone epigenetic marks, mRNA transcriptome and chromatin-bound proteome in SETD2 KO cells.
A crosstalk between H3K36me3 and H4K16ac.
Changes of metabolic pathways and RNA polymerase II-mediated transcription initiation by SETD2 depletion.
Mechanistic insights into SETD2-mediated cell cycle-related PLK1-CDK1/CCNB1 regulatory pathway.
Abstract
Clear cell renal cell carcinoma (ccRCC) is the most common type of RCC in humans. SET domain-containing 2 (SETD2), a lysine methyltransferase for histone and other proteins, has been found to be frequently lost in ccRCC. However, the mechanisms through which deficiency in SETD2 contributes to ccRCC development remain largely unknown. Here, we used a human embryonic kidney epithelial cell line with the SETD2 gene being knocked out using CRISPR/Cas9 technology. Using ChIP-seq analysis, we showed that SETD2 loss leads to diminished occupancy of histone H3K36me3 and H4K16ac on actively transcribed genes. Transcriptome sequencing of the knockout cells revealed diminished expression of genes involved in metabolic pathways and elevated expression of genes involved in regulation of RNA polymerase II-mediated transcription. Quantitative proteomic analysis of chromatin-associated proteins showed that genetic ablation of SETD2 leads to elevated chromatin occupancy of proteins involved in chromatin remodeling and RNA polymerase II transcription regulation, and diminished chromatin binding of proteins involved in translation elongation and RNA splicing. Interestingly, we found that SETD2 depletion attenuates cell proliferation, and this can be rescued by knockdown of CDK1. Taken together, we illustrate multiple SETD2-regulated cellular pathways that suppress cancer development and uncover mechanisms underlying aberrant cell cycle regulation in SETD2-depleted cells.
Kidney cancer is a chemotherapy-resistant disease, and it comprises numerous different cancers with distinct histopathological features and genetic alterations (1–3). Clear cell renal cell carcinoma (ccRCC)1 accounts for the majority (∼92%) of kidney cancer cases (3, 4). Typically, ccRCC cells are originated from malignant epithelial cells with clear cell morphology, which is accompanied with abnormal lipid and glycogen accumulation, suggesting a role of dysregulated metabolic pathways in ccRCC development (1).
Large-scale genomic sequencing studies on ccRCC revealed many frequently mutated genes, such as von Hippel-Lindau tumor suppressor (VHL), polybromo 1 (PBRM1), SET domain-containing 2 (SETD2), and BRCA1-associated protein 1 (BAP1) (3). In this vein, VHL encodes an E3 ubiquitin ligase regulating the levels of hypoxia inducible factors (HIF) in cells, and it is involved in cellular response pathways toward metabolic stress and nutrient stimulation (5). PBRM1, SETD2, and BAP1 proteins are involved in regulating histone epigenetic marks, indicating the importance of epigenetic reprogramming in ccRCC development (3, 6, 7).
SETD2 is the major histone methyltransferase for the trimethylation of lysine 36 in histone H3 (H3K36me3) in mammalian cells (8, 9). Mutation in SETD2 confers a loss of DNA methylation in non-promoter regions, suggesting its role in maintaining DNA epigenetic marks (3). SETD2 also plays roles in DNA damage repair, where the SETD2-mediated H3K36me3 promotes the chromatin localization of protein factors involved in mismatch repair and homologous recombination repair (10–12). Thus, SETD2-deficient cells exhibit increased mutation frequency and microsatellite instability as well as heightened sensitivity toward DNA double strand break-inducing agents (10–12). Additionally, SETD2 interacts with phosphorylated C-terminal domain (CTD) of RNA polymerase II to promote transcription elongation (13). However, the downstream cellular pathways modulated by SETD2 remain largely unknown.
In the present study, we sought to understand the underlying biological processes/pathways regulated by SETD2. Toward this end, we performed an integrated analysis of two histone epigenetic marks (H3K36me3 and H4K16ac) by ChIP-seq, the mRNA transcriptome by RNA-seq and the chromatin-bound proteins by SILAC (stable isotope labeling by amino acids in cell culture)-based quantitative proteomic workflow. Our results reveal that deficiency in SETD2 leads to the activation of multiple cellular pathways that are conducive for cancer development.
EXPERIMENTAL PROCEDURES
Cell Lines
HEK293T (293T) cells were purchased from ATCC (Manassas, VA). SETD2 KO cells were previously generated by CRISPR/Cas9-mediated genome editing in 293T background (11). UOK121 and UOK143 cells were generously provided by Professor Guo-Min Li (10). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and 100 unit/ml penicillin/streptomycin at 37 °C in a humidified incubator with 5% CO2.
5-Bromouridine (BrU) Incorporation Assay
BrU incorporation experiment was conducted following the previously described protocols (14). Briefly, cultured cells were collected and washed for 3 times with ice-cold PBS. The pellet was lysed in a buffer containing 10 mm Tris-HCl (pH 7.4), 10 mm NaCl, 3 mm MgCl2 and 0.5% (v/v) NP-40. After washing, the nuclear pellet was resuspended in nuclei storage buffer containing 50 mm Tris-HCl (pH 8.3), 0.1 mm EDTA, 5 mm MgCl2 and 40% (v/v) glycerol. The semi-in vivo transcription was subsequently performed in a transcription reaction buffer, which contained 10 mm Tris-HCl (pH 8.3), 2.5 mm MgCl2, 150 mm KCl, 2 mm DTT and RNase inhibitor, along with 1 mm dNTP and 1 mm BrU. After incubation at 30 °C for 1 h, the nuclear RNA was extracted with Omega Total RNA Kit I (Omega, Norcross, GA). About 200 ng total RNA was digested with nuclease P1 and alkaline phosphatase, as previously described (15). For the measurement of BrU incorporation, the digestion mixture was subjected to LC-MS/MS analysis on an LTQ XL linear ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA).
ChIP Sequencing
ChIP experiments were conducted as previously described (11). Approximately 2 × 106 cells were washed with ice-cold PBS and resuspended in a douncing buffer containing 10 mm Tris-HCl (pH 7.5), 4 mm MgCl2, 1 mm CaCl2, and 1× protease inhibitor mixture. After homogenization, micrococcal nuclease (Worthington Biochemicals, Lakewood, NJ) was used to fragment the chromatin, and the reaction was terminated by using 0.5 m EDTA. A hypotonic buffer (0.2 mm EDTA, pH 8.0, 0.1 mm benzamidine, 0.1 mm phenylmethylsulfonyl fluoride, 1.5 mm dithiothreitol, 1× protease inhibitor mixture) was then added to the mixture and incubated on ice for 1 h. ChIP experiments were performed with 1 μg of anti-H3K36me3 (Abcam, Cambridge, UK, ab9050) or anti-H4K16ac (Millipore, Burlington, MA, 07–329) antibody. DNA was subsequently retrieved from the immunoprecipitation mixture with 100 mm NaHCO3 and 1% SDS at 68 °C for 2 h and purified using QIAquick PCR Purification Kit (Qiagen, Venlo, Netherlands).
A sequencing library was prepared from the retrieved DNA with TruSeq ChIP Sample Preparation Kit (Illumina, San Diego, CA) following the manufacturer's instructions. Briefly, the DNA was end repaired with End Repair Mix, and 3′ ends were adenylated with A-Tailing Mix. The RNA Adaptor Indices were then ligated to DNA fragment. After purification with AMPure XP Beads (Beckman Coulter, Brea, CA) and size selection on a 2% agarose gel, the product was amplified by PCR using the PCR Master Mix with PCR Primer Mixture for 18 cycles and purified with AMPure XP Beads. The purified DNA libraries were subsequently quantified using Agilent 2100 Bioanalyzer and multiplexed for sequencing. Sequencing was performed on an Illumina HiSeq4000 at the DNA Technologies Core at UC Davis.
RNA Sequencing
For RNA sequencing, the RNA was extracted with Omega Total RNA Kit I (Omega) and quantified using NanoDrop One Spectrophotometer (Thermo Fisher Scientific). Generally, the RNA library preparation was conducted following the instructions for NEBNext® Ultra™ II RNA Library Prep Kit for Illumina. Total RNA was pretreated using the NEBNext rRNA Depletion Kit to remove the rRNA. After DNase I digestion and cleanup, the rRNA-depleted RNA was fragmentized in NEBNext First Strand Synthesis Reaction Buffer by heating at 94 °C for 15 min on a thermal cycler. The first strand cDNA was synthesized using First Strand synthesis enzyme Mix (NEB, Ipswich, MA) with random primers. The second strand cDNA was then synthesized using NEBNext Second Strand Synthesis Enzyme Mix (NEB). The DNA sample was purified with AMPure XP Beads. The sequencing library was subsequently prepared as described above. The DNA was ligated with NEBNext Multiplex Oligos for Illumina (NEB) and multiplexed for sequencing. Sequencing was again performed on an Illumina HiSeq4000 by the DNA Technologies Core at UC Davis.
SILAC-based Quantitative Profiling of Chromatin-binding Proteins
Cells were cultured in SILAC DMEM medium (Thermo Fisher Scientific) containing 10% dialyzed FBS (Invitrogen) and 1% penicillin and streptomycin (Invitrogen), supplemented with unlabeled l-arginine and l-lysine (Sigma, St. Louis, MO) (light medium), or 13C6-l-arginine and 13C6,15N2-l-lysine (Cambridge Isotope Laboratories) (heavy medium). 293T and SETD2 KO cells were cultured in light or heavy medium. After complete labeling in heavy medium for more than 10 cell doublings, an equal number of light and heavy labeled cells were harvested and washed with PBS.
Chromatin fractionation was performed following the previously described procedures (11). The cells were lysed using a cytoplasmic lysis buffer (10 mm Tris-HCl, pH 8.0, 0.34 m sucrose, 3 mm CaCl2, 2 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, 0.5% NP-40, protease inhibitor mixture) for 30 min on ice. The intact nuclei were subsequently collected by centrifugation at 5000 rpm for 2 min. The nuclear pellets were then homogenized in a nuclear lysis buffer (20 mm HEPES, pH 7.9, 1.5 mm MgCl2, 1 mm EDTA, 150 mm KCl, 0.1% NP-40, 1 mm DTT, 10% glycerol, protease inhibitor mixture). After centrifugation at 14,000 rpm for 30 min, the chromatin-enriched pellet fraction was incubated in a chromatin isolation buffer, which contained 20 mm HEPES (pH 7.9), 1.5 mm MgCl2, 150 mm KCl, 10% glycerol, protease inhibitor mixture and 0.15 unit/μl benzonase (Sigma), on ice for 2 h with gentle resuspension in every 10 min. After centrifugation at 5000 rpm for 2 min, the supernatant was collected as the chromatin fraction.
The chromatin fractions from equal amounts of light- and heavy-labeled cells were mixed together and loaded onto a 10% SDS-PAGE gel. After electrophoretic separation, gel slices containing the proteins were cut into small pieces, and the proteins were digested, following a previously published protocol (16). After digestion, the peptide mixtures were desalted using OMIX C18 Tips (Agilent, Santa Clara, CA) and subjected to LC-MS/MS analysis on a Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer coupled with an EASY-nLC 1200 UPLC (Thermo Fisher Scientific).
Raw data acquired from the Q Exactive Plus mass spectrometer were processed in parallel with MaxQuant Version 1.5.0.3 for protein identification and quantification. MaxQuant multiplicity was set to 2, and 13C6-l-arginine and 13C6,15N2-l-lysine were selected as heavy amino acids. Methionine oxidation and cysteine carboamidomethylation were set as variable and fixed modifications, respectively. The maximum number of missed cleavages for trypsin was set to two per peptide. The tolerances in mass accuracy were 20 ppm for both MS and MS/MS. The maximum false discovery rates (FDRs) were 0.01 at both the peptide and protein levels. Raw MS data were searched against the IPI human proteome database (with 87061 protein entries, version: 3.68) to which contaminants and reverse sequences were added. Raw output results were analyzed, and known contaminant proteins were removed from analysis.
Experimental Design and Statistical Rationale
For quantitative profiling of chromatin-binding proteins, four biological replicates were processed to obtain statistically robust results. The 293T and SETD2 KO cells were employed as control and experimental cell lines, respectively. To minimize experimental error or bias, SILAC was used to achieve accurate protein quantification. For quantitative analysis, at least one unique and razor peptide was considered for the identification and quantification of the proteins. In addition, unmodified peptides and peptides with methionine oxidation, or asparagine and glutamine deamination were employed for quantification. The H/L normalized ratios of biological replicates were employed to calculate the average ratios of identified proteins. The ratios were normalized by finely adjusted normalization factors to minimize the variability between samples, based on the assumption that most proteins do not or only minimally change between samples. All RNA-seq and other quantitative assays were performed with at least three biological replicates, and Student's t test was used to verify the significance of the results.
Real-time Quantitative PCR (RT-qPCR)
For RT-qPCR, total RNA was extracted using Omega Total RNA Kit I (Omega) and quantified. Reverse transcription was performed using M-MLV Reverse Transcriptase (Promega) for first strand cDNA synthesis. RT-qPCR was performed using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) on the CFX96 RT-qPCR detection system (Bio-Rad). Primers used in RT-qPCR are listed in supplemental Table S1.
Cell Proliferation and Clonogenic Survival Assay
For proliferation assay, cells were plated in 6-well plates in triplicate in DMEM supplemented with 10% FBS. After 6 h, the cells were transfected with plasmids expressing shRNA, siRNA or treated with 100 nm GSK461364 for 72 h. For cell counting, the cells were detached by trypsin-EDTA (Life Technologies), and the number of cells was counted with a hemocytometer using a microscope.
For clonogenic survival assay, the cells were plated in 6-well plates in triplicate. After 6 h, the cells were transfected with plasmids expressing shRNA, siRNA, or treated with the indicated concentrations of GSK461364. After treatment for 2 days, the medium was exchanged with fresh DMEM supplemented with 10% FBS. Cell colonies grown for 10–14 days were then fixed using 6% (v/v) glutaraldehyde and stained with 0.5% (w/v) crystal violet. Colonies containing at least 50 cells were counted under a microscope.
Chromatin Fractionation and Western Blotting
Chromatin fractionation was performed as described above. The protein concentrations in the soluble and chromatin fractions were determined by Bradford assay (Bio-Rad). For Western blot analysis, protein samples were separated on a 12% SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). After blocking with blotting-grade blocker (Bio-Rad), the membrane was incubated in a solution containing primary antibody and 5% BSA for 2 h, and then incubated in a 5% blotting-grade blocker containing the HRP-conjugated secondary antibody. The immunoblots were detected using ECL Western blotting detection reagent (Amersham Biosciences, Little Chalfont, UK). Primary antibodies used in this study included H3K36me3 (Abcam, ab9050; 1:5000), histone H3 (Cell Signaling Technology, Danvers, MA, 9715S; 1:10000), H4K16ac (Millipore, 07–329; 1:10000), histone H4 (Cell Signaling Technology, 13919S; 1:5000), β-actin (Cell Signaling Technology, 4967S; 1:5000), CDK1 (Cell Signaling Technology, 77055S; 1:5000), phosphor-CDK1 (Cell Signaling Technology, 9111S; 1:5000), WEE1 (Santa Cruz Biotechnology, Dallas, TX, sc-5285; 1:3000), CDC25C (Santa Cruz Biotechnology, sc-13138; 1:3000) and PLK1 (Santa Cruz Biotechnology, sc-17783; 1:2000).
Statistical Analysis
All data in this study are presented as the mean ± standard deviation (S.D.), unless otherwise indicated. Dot plots were generated in Origin 9 (OriginLab, Northampton, MA) (Fig. 4B). For all tests, p values of < 0.05 were considered statistically significant. All statistical analyses were performed using Origin 9 or Microsoft Office Excel (Microsoft, Redmond, WA). The Gene Ontology (GO) analysis was performed using the DAVID bioinformatics resources (17). Kaplan-Meier survival curves for the Cancer Genome Atlas (TCGA) samples were generated using the OncoLnc online tool (18). For the analysis, up and bottom 33% percentile slices were used.
Fig. 4.

A comparison of chromatin-binding protein profile between 293T and SETD2 KO cells obtained from SILAC-based quantitative proteomic experiments. A, A scheme depicting the procedures for the SILAC-based quantitative profiling of chromatin-binding proteins. B, GO analysis of upregulated proteins in chromatin fraction of SETD2 KO cells. C, GO analysis of downregulated proteins in chromatin fraction of SETD2 KO cells. D, Fold changes of quantified proteins in different pathways. GO:0045944, positive regulation of transcription from RNA polymerase II promoter; GO:0000122, negative regulation of transcription from RNA polymerase II promoter; GO:0006414, translational elongation; GO:0008380, RNA splicing.
ChIP-seq Analysis
The sequencing reads of ChIP-seq were aligned to human hg19 reference genome using the Bowtie 1.1.2 (19). Peak calling were generated using the Model-based Analysis of ChIP-Seq (MACS) (20). The UCSC Genome Browser was used to visualize the mapping results (21), and the deepTools2 was used for visualization of the analysis results (22).
RNA-seq Analysis
For RNA-seq analysis, the sequencing results were first aligned to human hg19 reference genome using the TopHat Gapped-read mapper for RNA-seq data (23). The differentially expressed transcripts were subsequently generated using the Cuffdiff tools following the developers' instructions (24).
RESULTS
SETD2 is Frequently Mutated in ccRCC and Correlated With Patient Survival
Recent large-scale sequencing studies revealed SETD2 as one of the most frequently mutated genes in cancer with an alteration frequency of 4.4% in 10,945 patients with advanced cancers (Fig. 1A) (3, 6, 25, 26). Especially high frequency of mutations were observed in ccRCC, where 21.4% (96 of 448) and 16.0% (67 of 418) of cases had mutated SETD2 gene in two cohort studies (Fig. 1A) (3). Kaplan-Meier survival analysis also supported the correlation between SETD2 deficiency and poorer prognosis in ccRCC patients, where patients with higher levels of SETD2 expression manifest significantly better prognosis (Fig. 1B), suggesting that SETD2 is a potent tumor suppressor for ccRCC.
Fig. 1.
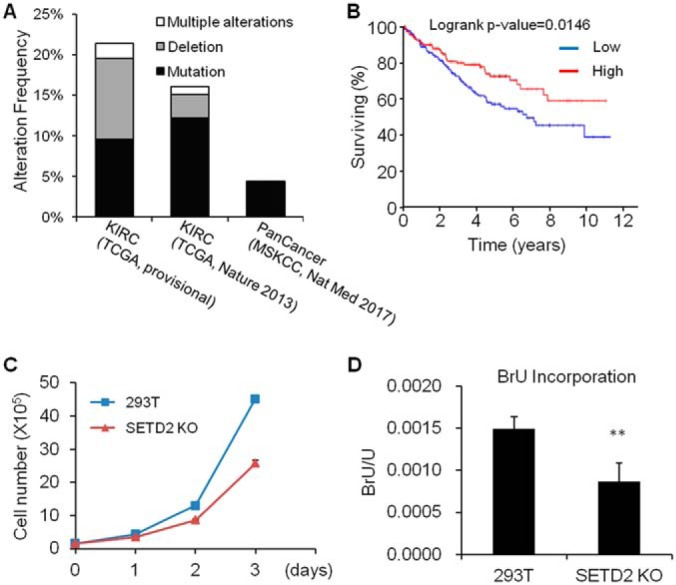
SETD2 is a tumor suppressor in clear cell renal cell carcinoma (ccRCC), and knockout of SETD2 affects cell proliferation and RNA transcription. A, SETD2 is frequently mutated in different cancers, especially in ccRCC. The mutation frequency of SETD2 is about 4.4% in all cancers (481 of 10945 cases), whereas 21.4% (96 of 448) and 16% (67 of 418) of ccRCC cases had mutated SETD2 gene (TCGA, Provisional and TCGA, Nature 2013, respectively). B, Higher level of SETD2 expression is correlated with better survival in ccRCC patients (n = 172 each in Low and High groups). C, Knockout of SETD2 in 293T cells results in diminished proliferation. D, Knockout of SETD2 confers a reduced rate of RNA transcription in SETD2 KO cells, as manifested by decreased 5-bromouridine (BrU) incorporation into RNA.
Knockout of SETD2 Leads to Decreased Proliferation and RNA Transcription
Despite the importance of mutant SETD2 in cancer development, the mechanisms through which SETD2 suppresses tumor development remain elusive. To better understand these mechanisms, we first generated SETD2 knockout (SETD2 KO) cells by using the CRISPR/Cas9 technology (11). We found that SETD2 KO (clone 5) cells display diminished proliferation rate (Fig. 1C). Because the SETD2-mediated H3K36me3 is commonly associated with transcription (27, 28), we measured the transcription efficiency based on 5-bromouridine (BrU) incorporation (14). Our result reveals a markedly decreased rate of BrU incorporation in SETD2 KO cells, supporting that SETD2 depletion lead to diminished transcription efficiency (Fig. 1D).
SETD2 KO Cells Display Drastically Reduced H3K36me3, which is Correlated with Diminished Acetylation of Lysine 16 in Histone H4 (H4K16ac)
We next performed ChIP-seq experiment to analyze the genome-wide occupancy of H3K36me3 in HEK293T (293T) and SETD2 KO cells (Fig. 2A and 2C). Consistent with the previous findings (29, 30), our results show that the H3K36me3 level increases gradually after the transcription start site (TSS) and peaks at around the transcription end site (TES) in 293T cells (Fig. 2A and 2C). The H3K36me3 levels are, however, markedly attenuated in SETD2 KO cells (Fig. 2A and 2C). H4K16ac is a histone epigenetic mark for transcriptionally active chromatin (31, 32). Considering our previous finding that deletion of SETD2 compromises the DNA damage-induced H4K16ac (11), we analyzed the genome-wide occupancy of H4K16ac in 293T and SETD2 KO cells (Fig. 2B and 2D). Our results reveal that the genome-wide occupancy of H4K16ac is decreased in SETD2 KO cells, with the distribution of changes mirroring that of H3K36me3. In this vein, we observe a decrease in H4K16ac across the region from 3 kb upstream of the TSS to 3 kb downstream of the TES, except for the region surrounding the TSS (Fig. 2B and 2D). In addition, an analysis of the loci with the levels of H3K36me3 and H4K16ac at around the TES being diminished by at least 1.5-fold in SETD2 KO cells shows that, among the 2042 sites with decreased levels of H4K16ac, 1624 sites (∼80%) are also with reduced levels of H3K36me3 (Fig. 2E–2F and supplemental Fig. S1). Hence, the above results demonstrate, for the first time, that the SETD2-medidated H3K36me3 promotes H4K16ac in cells without exposure to DNA damaging agents.
Fig. 2.
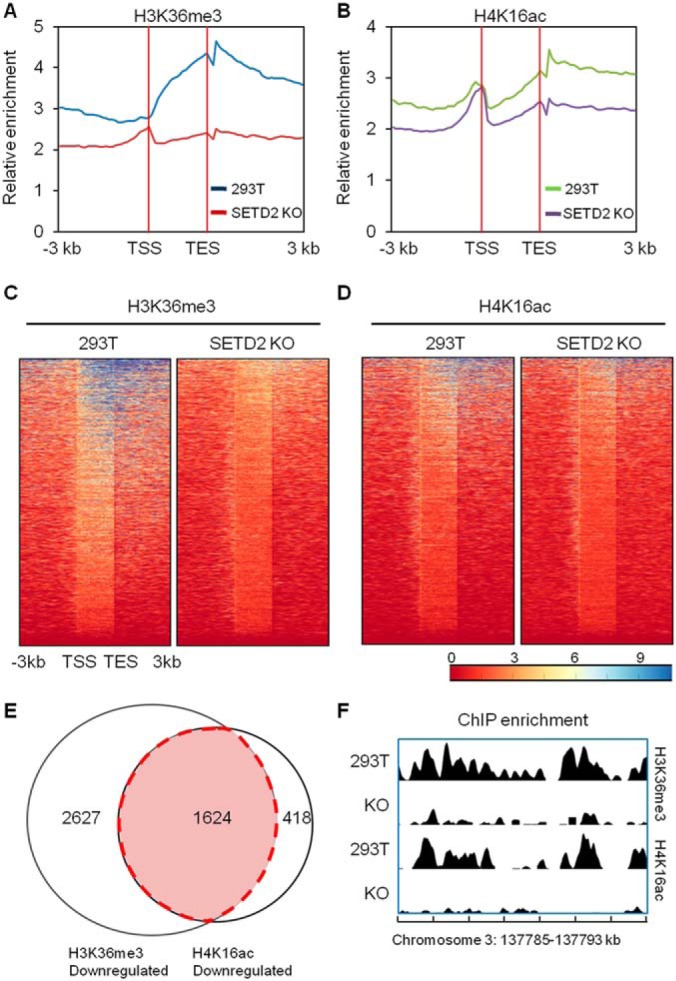
A comparison of genome-wide occupancy of H3K36me3 and H4K16ac in 293T and SETD2 KO cells by ChIP-seq revealed a strong correlation between the two histone epigenetic marks. A, Average distribution of H3K36me3 around the transcription start site (TSS) and transcription end site (TES). B, Average distribution of H4K16ac around the TSS and TES. C–D, The density of ChIP-seq reads of H3K36me3 (C) and H4K16ac (D) around the TSS and TES in 293T cells and SETD2 KO cells. Regions are ranked according to H3K36me3 density. E, A Venn diagram depicting the overlap of downregulated H3K36me3 and H4K16ac sites in SETD2 KO cells. Nearly 80% (1624/2042) of sites with diminished H4K16ac display decreased levels of H3K36me3. F, A comparison of the density of H3K36me3 and H4K16ac ChIP-seq reads across the indicated genomic region in 293T and SETD2 KO cells.
RNA-seq Showed Altered Expression of Genes in Metabolic Pathways and RNA Polymerase II Transcription Regulation in SETD2 KO Cells
Because of the importance of H3K36me3 and H4K16ac in transcriptional regulation, we next examined the effects of SETD2 deficiency on transcription. RNA-seq analyses of 293T and SETD2 KO cells show that 68 and 167 genes were significantly (q < 0.05) up- and downregulated, respectively, in SETD2 KO cells relative to 293T cells (Fig. 3A). We found that deletion of SETD2 results in the suppression of multiple metabolic pathways, including creatine (involved in energy metabolism), glycosaminoglycan (involved in kidney agenesis, tumor progression, and fibrosis processes), fucose (involved in carbohydrate metabolism, signaling pathway by the Notch receptor family) metabolic processes, etc. (Fig. 3C–3D). To corroborate these findings, we measured the creatine content in SETD2 KO cells, and the results show that indeed the creatine level in SETD2 KO cells is significantly lower than that in 293T cells (Fig. 3F and supplemental Fig. S2). The dysregulation of metabolic pathways suggests a profound and unexpected effect of SETD2 on cancer development. In this context, it is worth noting that kidney cancer is considered a metabolic disease, where alterations in several cellular metabolic pathways involved in oxygen, iron, energy and nutrient sensing were noted (1–3).
Fig. 3.

A comparison of mRNA expression levels in 293T and SETD2 KO cells obtained by RNA-Seq analysis. The results showed a positive correlation between mRNA expression levels and H3K36me3/H4K16ac levels. In SETD2 KO cells, RNA polymerase II transcription-related genes were upregulated and a large number of genes involved in metabolic pathways were downregulated. A, A scatter plot comparing the log2(FPKM+1) values for RNA-seq data in 293T cells and SETD2 KO cells. A total of 580 and 651 transcripts were significantly (p < 0.05) up- and downregulated, respectively, in SETD2 KO cells. B, Cumulative distribution of the log2(fold changes) of RNA expression in SETD2 KO cells for genes with high H3K36me3 ratio or low H3K36me3 ratio between 293T and SETD2 KO cells. C, Gene Ontology (GO) analysis of upregulated genes in SETD2 KO cells. (D) GO analysis of downregulated genes in SETD2 KO cells. E, The interaction network of upregulated RNA polymerase II transcription-related proteins in SETD2 KO cells. Lines indicate the interaction between proteins. The size and the color of each node are proportional to its interaction protein number. F, Relative levels of creatine in 293T cells and SETD2 KO cells. ***, p < 0.001.
GO analysis of the upregulated genes reveals the pathways associated with positive or negative regulation of transcription from RNA polymerase II promoter, indicating the heightened regulation of transcription initiation by RNA polymerase II (Fig. 3C–3D). This finding is consistent with the observation that deletion of SETD2 results in elevated occupancy of H3K36me3 and H4K16ac near the TSS of genes in SETD2 KO cells (Fig. 2).
A number of oncogenes are known to be involved in transcriptional regulation. When over-expressed, these oncogenes can lead to uncontrolled gene transcription and ultimately result in cancer development (33). An interaction network analysis shows that many RNA polymerase II transcription-related genes that are upregulated in SETD2 KO cells are associated with cancer. These include JUN, PLK1, GLI2, RCN3, IRS2, GAS1, FOXF1 and MAFA (Fig. 3E), and we validated the altered expression of some of these genes by real-time quantitative PCR (RT-qPCR) analysis (supplemental Fig. S3). It is worth noting that we obtain consistent results in gene expression alterations for another clone of SETD2 KO cells, i.e. SETD2 KO-clone 20 (supplemental Fig. S4). Collectively, these data suggest that deficiency in SETD2 results in aberrant cell metabolism and RNA polymerase II-mediated transcription initiation, both of which are hallmarks of cancer (34).
The mRNA Expression Levels Are Highly Correlated with H3K36me3 and H4K16ac Levels in the Corresponding Genomic Regions
Owing to their effects on chromatin structure, epigenetic modifications of histone proteins are strongly linked with transcription (35). To elucidate whether gene transcription is correlated with the occupancy of H3K36me3 and H4K16ac, we conducted statistical analysis of the ChIP-seq and RNA-seq data (Fig. 3B and supplemental Fig. S5). Our results show that those genes with higher levels of H3K36me3 and H4K16ac exhibit elevated levels of mRNA expression (Fig. 3B). In addition, by comparing the upregulated genes with downregulated genes, average levels of H3K36me3 and H4K16ac occupancy between TSS and TES are positively correlated with gene expression (supplemental Fig. S5).
Quantitative Profiling of Chromatin-binding Proteins Revealed Alterations in Chromatin Remodeling and Transcription Regulation in SETD2 KO Cells
Histone post-translational modifications serve as docking sites for recruiting proteins to chromatin, where the chromatin-binding proteins function in numerous biological processes, including DNA replication, transcription and cell cycle regulation (36). Aberrant transcription regulation and cell cycle control are frequently accompanied with cancer development. Considering the marked decreases of H3K36me3 and H4K16ac in SETD2 KO cells, we reasoned that the loss of SETD2 may also result in substantial changes in the profile of chromatin-binding proteins. Hence, we performed a SILAC-based quantitative profiling of the chromatin-binding proteome in 293T cells and the isogenic SETD2 KO cells (Fig. 4A and supplemental Fig. S6–S10).
We were able to identify ∼2220 proteins in the chromatin fraction, among which 153 and 203 are decreased and increased by at least 1.5-fold, respectively, in SETD2 KO cells relative to the isogenic 293T cells. Gene Ontology (GO) analysis shows that many proteins involved in translation elongation, RNA splicing and RNA processing exhibit diminished chromatin localization, indicating that the decrease in H3K36me3 affects various aspects of mRNA-related processes (Fig. 4C and 4D). By contrast, proteins involved in chromosome organization, cell cycle regulation and DNA metabolic process exhibit increased chromatin localization. These results support that deficiency in SETD2 led to pronounced reprogramming of the chromatin-binding proteome (Fig. 4B and 4D). We also analyzed the enrichment of proteins involved in regulation of transcription by RNA polymerase II. The results show that these proteins exhibit elevated chromatin localization in the SETD2-deficient background, which is in keeping with the increased mRNA expression of the corresponding genes (Fig. 4D).
We also conducted an interaction network analysis on proteins in the chromatin fraction that are upregulated by at least 2-fold in SETD2 KO cells. Our results illustrate that several proteins encoded by cancer-related genes display heightened chromatin occupancy in SETD2 KO cells, such as SMARCA1/2, CDK1, CDK2, CCNB1 and KIF2C (supplemental Fig. S12).
Knockdown of CDK1 Rescued the Decreased Proliferation of SETD2 KO Cells
We next performed an interaction network analysis of the commonly altered targets based on RNA-seq data and chromatin proteomic data. Protein interaction network analysis of these targets reveals the upregulation of the PLK1-CDK1/CCNB1 pathway in SETD2 KO cells (Fig. 5A). In addition, we assessed whether the elevated expression and chromatin localization of CDK1 influence the proliferation rate of SETD2 KO cells (Fig. 5B and supplemental Fig. S13). It turns out that the diminished proliferation of SETD2 KO cells is fully rescued upon the siRNA-mediated knockdown of CDK1 (Fig. 5B and supplemental Fig. S14), underscoring the crucial role of CDK1 in impeding cell cycle progression in SETD2 KO cells. Knockdown of CCNB1 could suppress the proliferation of both 293T and SETD2 KO cells. We then investigated the expression of some oncogenes after knockdown of CDK1 in SETD2 KO cells (supplemental Fig. S14E). Indeed, the expression level of JUN, GLI2, IRS2 and GAS1 were rescued by knockdown of CDK1, suggesting that they are regulated by CDK1 (supplemental Fig. S14E). The level of creatine was, however, not rescued by knockdown of CDK1, indicating that the creatine level is regulated by distinct mechanisms (supplemental Fig. S14). In addition, suppression of PLK1 with shRNA or with a small-molecule inhibitor (GSK461364) could diminish significantly the proliferation of 293T and SETD2 KO cells. PLK1 could modulate CDK1 activation by regulating the phosphorylation of the latter protein (37, 38). These results suggest that PLK1 and CCNB1 cooperate with CDK1 in regulating cell cycle progression (Fig. 5B and supplemental Fig. S13).
Fig. 5.
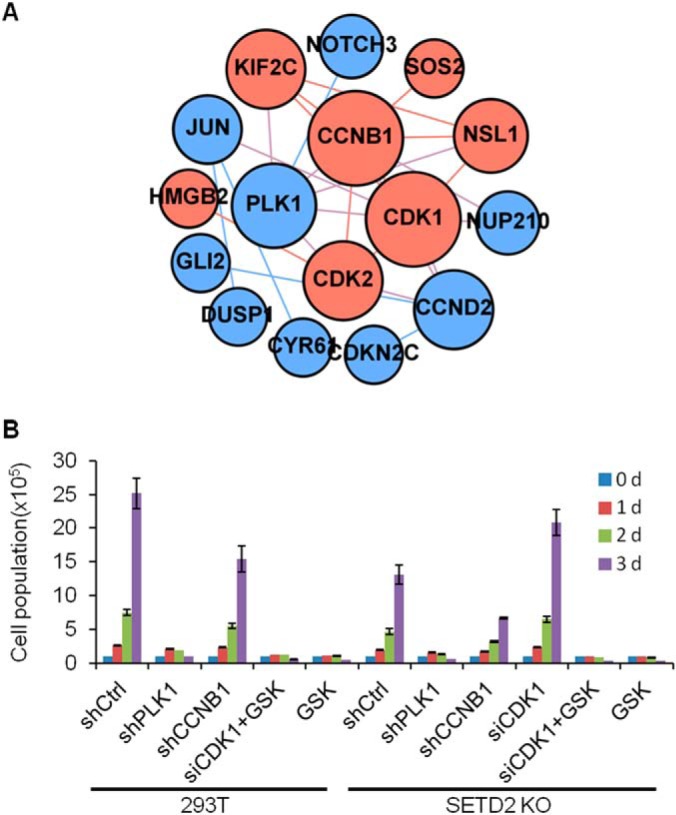
Cell proliferation rate measurement of 293T and the isogenic SETD2 KO cells after shRNA- or siRNA-mediated knockdown of PLK1, CCNB1 or CDK1, or treatment with a PLK1 inhibitor (GSK461364). A, Interaction network of targets from RNA-seq and quantitative proteomic analysis of chromatin-binding proteins in PLK1-CDK1 pathway. Blue, potential tumor targets from RNA-seq; Red, potential tumor targets based on results about chromatin-binding proteins. The size of the nodes is proportional to the interaction numbers of each nodes. B, Proliferation of 293T and SETD2 KO cells after knockdown of PLK1, CCNB1, CDK1 or treatment with a PLK1 inhibitor (GSK461364, GSK).
PLK1 Modulates the Chromatin Localization and Activation of CDK1 in SETD2 KO Cells
CDK1-CCNB1 complex is a key player in cell cycle control, and multiple mechanisms are associated with its activation. Along this line, CDK1 is phosphorylated and dephosphorylated by WEE1/MYT1 and CDC25C, respectively, during cell cycle progression (37). Phosphorylation of CDK1 led to its inactivation and suppression of mitotic entry. PLK1 could regulate the activity of CCNB1, WEE1 and CDC25C by direct phosphorylation and consequently control CDK1 activation and entry into mitosis (37, 38). To examine whether CDK1 activation is modulated by PLK1 in SETD2-deficient background, we next assessed the chromatin localization and phosphorylation of CDK1 by Western blot analysis (Fig. 6). First, we observed heightened chromatin localization of CDK1 in SETD2 KO cells relative to 293T cells, which is in line with the data obtained from SILAC-based quantitative proteomic experiment (Fig. 6A and supplemental Fig. S12). Considering the different sensitivities of 293T and SETD2 KO cells toward PLK1 inhibition, we compared the chromatin localization of CDK1 in these cells after PLK1 knockdown (Fig. 6B–6C). The results show that knockdown of PLK1 could significantly increase the chromatin occupancy of CDK1 in SETD2 KO cells, but not in 293T cells (Fig. 6B–6C). The same finding is made with the use of the PLK1 inhibitor, GSK461364 (Fig. 6D–6E).
Fig. 6.

Chromatin localization of CDK1 after knockdown or inhibition of PLK1. A, Chromatin localization of CDK1 in 293T and SETD2 KO cells. B–E, Chromatin localization of CDK1 after knockdown of PLK1 (B–C) or treatment with a PLK1 inhibitor, GSK461364 (D–E) in 293T and SETD2 KO cells. G–H, Protein levels of WEE1 and CDC25C after knockdown of PLK1 (G) or treatment with GSK461364 (H) in 293T and SETD2 KO cells. CF, chromatin fraction; SF, soluble fraction.
We next assessed whether the CDK1 activation was modulated by PLK1 inhibition. Our results show that treatment with GSK461364 lead to a higher level of inactive, phosphorylated form of CDK1 on chromatin in SETD2 KO cells relative to 293T cells (Fig. 6D–6F), indicating that PLK1 plays a more important role in CDK1 activation in SETD2-deficient background. In addition, Western blotting results reveal heightened levels of WEE1, but not CDC25C, in SETD2 KO cells relative to 293T cells (Fig. 6G–6H), suggesting that the over-inactivation of CDK1 in SETD2 KO cells following PLK1 inhibition arises from elevated level of WEE1. Taken together, our results support the notion that the inactive, phosphorylated CDK1 on chromatin acts as a guardian for cell cycle progression, and CDK1 can be activated by PLK1 (Fig. 6).
We next assessed whether the alteration in the PLK1-CDK1 pathway is a common feature for SETD2-deficient ccRCC cells. To this end, we measured the PLK1 expression levels in patient-derived UOK121 (SETD2-proficient) and UOK143 (SETD2-deficient) ccRCC cells. It turns out that PLK1 is expressed at a higher level in UOK143 than UOK121 cells (supplemental Fig. S15). We also analyzed the publicly available RNA-seq data for ccRCC patients retrieved from the TCGA database (3). The results show that, in patients carrying a mutated SETD2 gene, the expression level of PLK1 is also significantly increased (supplemental Fig. S15), supporting that PLK1 is commonly upregulated in SETD2-deficient ccRCC patients.
In summary, our results demonstrate that SETD2 assumes crucial roles in ccRCC through regulating multiple cellular pathways. In SETD2-deficient cells, decreased levels of H3K36me3 and H4K16ac lead to reduced transcription, which results in the downregulation of numerous genes involved in cellular metabolism. To compensate for repressed cellular metabolism, cells over-express many genes in regulating RNA polymerase II-mediated transcription initiation, and these include several tumor-related genes. In SETD2-deficient cells, potential tumor target PLK1 is upregulated, which stimulates the chromatin localization of its downstream target CDK1, thereby resulting in decreased proliferation (Fig. 6).
DISCUSSION
SETD2 is known to be frequently mutated in ccRCC; however, there were limited analyses about the genetic and proteomic alterations elicited by deficiency in SETD2 (1, 3, 39, 40). Here we embarked on a systematic, multi-pronged approach to elucidate the perturbations in genome-wide occupancies of two relevant histone epigenetic marks (H3K36me3 and H4K16ac), mRNA transcriptome and chromatin-bound proteins evoked by SETD2 depletion. Our analysis revealed the dark side of SETD2's effects on ccRCC and provided important knowledge about the cellular pathways that are altered by deficiency in SETD2.
The Crosstalk Between H3K36me3 and H4K16ac
Histone post-translational modifications (PTMs) assume critical roles in regulating gene expression by altering chromatin structure and/or modulating the recruitment of chromatin-binding proteins (41–45). SETD2-mediated H3K36me3 is an epigenetic mark associated with transcription elongation, where it gradually increases after the transcription start site and peaks at around the transcription end site (Fig. 2A) (27, 32). H4K16ac is another active chromatin mark, which localizes mainly at the transcription start sites (Fig. 2B) (31, 46). In Arabidopsis, H3K36me3 recruits MRG1/2 and HAM1/2 to acetylate histone H4 and regulates FLOWING LOCUS T (FT) gene expression (47). Recently, we found that H3K36me3 directly promotes H4K16ac at DNA double strand break (DSB) sites, thereby facilitating DNA DSB repair (11). By employing ChIP-seq analysis, we demonstrate here, for the first time, that the SETD2-mediated H3K36me3 stimulates H4K16ac in cells without genotoxic agent exposure (Fig. 2A–2F), and that the expression levels of genes are positively correlated with the occupancy of these two histone epigenetic marks (Fig. 3B and supplemental Fig. S5). These results suggest that these two histone epigenetic marks may cooperate with each other to promote active transcription.
Dysregulation of Metabolic Pathways and RNA Polymerase II-mediated Transcription Initiation by SETD2
Dysregulation of cellular metabolism is a hallmark of cancer. The changes in metabolism result in alterations in metabolites in cancer cells, which provide the microenvironment and satisfy chemical demands for cancer cell growth and metastasis (1, 2, 34). ccRCC is a disease associated with several metabolic alterations including energy, nutrient and oxygen sensing (1–3). VHL, a gene frequently mutated in ccRCC (5), encodes an E3 ligase for HIF1α and HIF2α ubiquitination, through which it regulates metabolisms in oxygen and energy sensing (48). SETD2 is another commonly mutated gene in ccRCC (Fig. 1A) (3). In this context, the SETD2-catalyzed H3K36me3 is required for recruiting DNA repair machinery to damage site, thereby activating DNA DSB repair and mismatch repair (10–12, 49). Thus, deficiency in SETD2 increases the genomic mutation rate in cells (10). Recently, it was reported that WEE1 inhibition selectively kills H3K36me3-deficient cancer cells owing to the low dNTP content in cells, indicating metabolic alterations in SETD2-deficient cells (50). Here we found that deficiency in SETD2 led to the downregulation of many metabolic pathways, such as creatine, glycosaminoglycan, and carbohydrate metabolic processes. Moreover, our LC-MS/MS quantification results reveal a significant decline in creatine level in SETD2 KO cells (Fig. 3F). These results together support that metabolic alterations may constitute a very important mechanism in the development of ccRCC in SETD2-deficient individuals (Fig. 3).
Aberrant regulation of gene expression is another hallmark of cancer cells, and many oncogenes regulate the transcription of genes involved in the proliferation, apoptosis, invasion and metastasis of cancer cells (34). Our results reveal that genetic deletion of SETD2 leads to the overexpression of many cancer-related genes, including GAS1, GLI2, FOXO1, FOXO3, IRS2, JUN, PLK1, RCN3, SMAD1, SMAD3, TOP2A, and VEGFA (Fig. 3, 4). Likewise, genetic ablation of SETD2 lead to heightened chromatin binding of proteins encoded by cancer-related genes, such as SMARCA1/2, CDK1, CDK2, CCNB1 and KIF2C (Fig. 4 and supplemental Fig. S11). Thus, our results support a role of SETD2 in regulating cancer-associated genes.
Cell Cycle-related PLK1-CDK1/CCNB1 Regulatory Pathway in SETD2 KO Cells
ccRCC is the major type of kidney cancer that is resistant to chemotherapy and difficult to cure (1, 2). Most therapies for ccRCC target downstream genes in VHL-deficient ccRCC, but the responses are not optimistic, and most patients eventually progress (51). Thus, novel therapeutic targets for ccRCC are urgently needed. By analyzing transcriptome and chromatin-binding proteome, we reveal the aberrant activation of the PLK1-CDK1/CCNB1 regulatory pathway in SETD2 KO cells (Fig. 5 and 6). CDK1 is a key determinant of cell cycle progression, and it interacts directly with its partner CCNB1 to fulfill their functions in regulating entry or exit of mitosis (52, 53). We find that SETD2 KO cells exhibit diminished proliferation, which could be rescued by genetic depletion of CDK1 (Fig. 5). These results demonstrate that the aberrant cell cycle progression, arising from CDK1 dysregulation, might contribute to tumor development in SETD2-deficient cancers.
Together, our results support that SETD2 deficiency promotes cancer development through at least four distinct mechanisms: (1) repression of DNA repair and perturbation of genome stability; (2) alteration of cellular metabolism; (3) aberrant activation of cancer-associated genes; and (4) dysregulation of cell cycle progression. Hence, this study reveals the molecular underpinnings of SETD2's role in tumor suppression.
Data Availability
The ChIP sequencing and mRNA sequencing data have been deposited in the NCBI GEO database with accession number GSE107391 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107391). The proteomic data in this paper have been deposited into PeptideAtlas (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View? with the identifier PASS012793).
Supplementary Material
Acknowledgments
We thank Prof. Guo-Min Li for providing cell lines.
Footnotes
* This work was supported by the National Institutes of Health (R01 CA210072). M. H. was supported in part by an NRSA T32 Institutional Research Training Grant (ES018827).
 This article contains supplemental Tables and Figures. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables and Figures. The authors declare that they have no conflicts of interest with the contents of this article.
1 The abbreviations used are:
- ccRCC
- clear cellrenal cell carcinoma
- HIF
- hypoxia-inducible factor
- SETD2
- SET domain-containing 2
- CTD
- C-terminal domain.
REFERENCES
- 1. Hakimi A. A., Reznik E., Lee C. H., Creighton C. J., Brannon A. R., Luna A., Aksoy B. A., Liu E. M., Shen R., Lee W., Chen Y., Stirdivant S. M., Russo P., Chen Y. B., Tickoo S. K., Reuter V. E., Cheng E. H., Sander C., and Hsieh J. J. (2016) An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell 29, 104–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Linehan W. M., Srinivasan R., and Schmidt L. S. (2010) The genetic basis of kidney cancer: a metabolic disease. Nat. Rev. Urol. 7, 277–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. The Cancer Genome Atlas Research Network. (2013) Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 499, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chow W. H., Dong L. M., and Devesa S. S. (2010) Epidemiology and risk factors for kidney cancer. Nat. Rev. Urol. 7, 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guo G., Gui Y., Gao S., Tang A., Hu X., Huang Y., Jia W., Li Z., He M., Sun L., Song P., Sun X., Zhao X., Yang S., Liang C., Wan S., Zhou F., Chen C., Zhu J., Li X., Jian M., Zhou L., Ye R., Huang P., Chen J., Jiang T., Liu X., Wang Y., Zou J., Jiang Z., Wu R., Wu S., Fan F., Zhang Z., Liu L., Yang R., Wu H., Yin W., Liu Y., Peng H., Jiang B., Feng Q., Li C., Xie J., Lu J., Kristiansen K., Li Y., Zhang X., Li S., Wang J., Yang H., and Cai Z. (2011) Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat. Genet. 44, 17–19 [DOI] [PubMed] [Google Scholar]
- 6. Varela I., Tarpey P., Raine K., Huang D., Ong C. K., Stephens P., Davies H., Jones D., Lin M. L., Teague J., Bignell G., Butler A., Cho J., Dalgliesh G. L., Galappaththige D., Greenman C., Hardy C., Jia M., Latimer C., Lau K. W., Marshall J., McLaren S., Menzies A., Mudie L., Stebbings L., Largaespada D. A., Wessels L. F., Richard S., Kahnoski R. J., Anema J., Tuveson D. A., Perez-Mancera P. A., Mustonen V., Fischer A., Adams D. J., Rust A., Chan-on W., Subimerb C., Dykema K., Furge K., Campbell P. J., Teh B. T., Stratton M. R., and Futreal P. A. (2011) Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469, 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li J., Duns G., Westers H., Sijmons R., van den Berg A., and Kok K. (2016) SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget 7, 50719–50734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edmunds J. W., Mahadevan L. C., and Clayton A. L. (2008) Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 27, 406–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park I. Y., Powell R. T., Tripathi D. N., Dere R., Ho T. H., Blasius T. L., Chiang Y. C., Davis I. J., Fahey C. C., Hacker K. E., Verhey K. J., Bedford M. T., Jonasch E., Rathmell W. K., and Walker C. L. (2016) Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li F., Mao G., Tong D., Huang J., Gu L., Yang W., and Li G. M. (2013) The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 153, 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li L., and Wang Y. (2017) Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 292, 11951–11959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pfister S. X., Ahrabi S., Zalmas L. P., Sarkar S., Aymard F., Bachrati C. Z., Helleday T., Legube G., La Thangue N. B., Porter A. C., and Humphrey T. C. (2014) SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 7, 2006–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li M., Phatnani H. P., Guan Z., Sage H., Greenleaf A. L., and Zhou P. (2005) Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc. Natl. Acad. Sci. U.S.A. 102, 17636–17641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roberts T. C., Hart J. R., Kaikkonen M. U., Weinberg M. S., Vogt P. K., and Morris K. V. (2015) Quantification of nascent transcription by bromouridine immunocapture nuclear run-on RT-qPCR. Nat. Protoc. 10, 1198–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fu L., Guerrero C. R., Zhong N., Amato N. J., Liu Y., Liu S., Cai Q., Ji D., Jin S. G., Niedernhofer L. J., Pfeifer G. P., Xu G. L., and Wang Y. (2014) Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bing T., Shangguan D., and Wang Y. (2015) Facile discovery of cell-surface protein targets of cancer cell aptamers. Mol. Cell. Proteomics 14, 2692–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 18. Anaya J. (2016) OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. Peer J. Computer Sci. 2, e67 [Google Scholar]
- 19. Langmead B., Trapnell C., Pop M., and Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., and Liu X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., and Haussler D. (2002) The human genome browser at UCSC. Genome Res. 12, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramirez F., Ryan D. P., Gruning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dundar F., and Manke T. (2016) deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., and Salzberg S. L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., and Pachter L. (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zehir A., Benayed R., Shah R. H., Syed A., Middha S., Kim H. R., Srinivasan P., Gao J., Chakravarty D., Devlin S. M., Hellmann M. D., Barron D. A., Schram A. M., Hameed M., Dogan S., Ross D. S., Hechtman J. F., DeLair D. F., Yao J., Mandelker D. L., Cheng D. T., Chandramohan R., Mohanty A. S., Ptashkin R. N., Jayakumaran G., Prasad M., Syed M. H., Rema A. B., Liu Z. Y., Nafa K., Borsu L., Sadowska J., Casanova J., Bacares R., Kiecka I. J., Razumova A., Son J. B., Stewart L., Baldi T., Mullaney K. A., Al-Ahmadie H., Vakiani E., Abeshouse A. A., Penson A. V., Jonsson P., Camacho N., Chang M. T., Won H. H., Gross B. E., Kundra R., Heins Z. J., Chen H. W., Phillips S., Zhang H., Wang J., Ochoa A., Wills J., Eubank M., Thomas S. B., Gardos S. M., Reales D. N., Galle J., Durany R., Cambria R., Abida W., Cercek A., Feldman D. R., Gounder M. M., Hakimi A. A., Harding J. J., Iyer G., Janjigian Y. Y., Jordan E. J., Kelly C. M., Lowery M. A., Morris L. G. T., Omuro A. M., Raj N., Razavi P., Shoushtari A. N., Shukla N., Soumerai T. E., Varghese A. M., Yaeger R., Coleman J., Bochner B., Riely G. J., Saltz L. B., Scher H. I., Sabbatini P. J., Robson M. E., Klimstra D. S., Taylor B. S., Baselga J., Schultz N., Hyman D. M., Arcila M. E., Solit D. B., Ladanyi M., and Berger M. F. (2017) Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Campbell J. D., Alexandrov A., Kim J., Wala J., Berger A. H., Pedamallu C. S., Shukla S. A., Guo G., Brooks A. N., Murray B. A., Imielinski M., Hu X., Ling S., Akbani R., Rosenberg M., Cibulskis C., Ramachandran A., Collisson E. A., Kwiatkowski D. J., Lawrence M. S., Weinstein J. N., Verhaak R. G., Wu C. J., Hammerman P. S., Cherniack A. D., Getz G., Artyomov M. N., Schreiber R., Govindan R., and Meyerson M. (2016) Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 48, 607–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wagner E. J., and Carpenter P. B. (2012) Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sims R. J. 3rd, and Reinberg D. (2009) Processing the H3K36me3 signature. Nat. Genet. 41, 270–271 [DOI] [PubMed] [Google Scholar]
- 29. Wen H., Li Y., Xi Y., Jiang S., Stratton S., Peng D., Tanaka K., Ren Y., Xia Z., Wu J., Li B., Barton M. C., Li W., Li H., and Shi X. (2014) ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang C. I., Alekseyenko A. A., LeRoy G., Elia A. E., Gorchakov A. A., Britton L. M., Elledge S. J., Kharchenko P. V., Garcia B. A., and Kuroda M. I. (2013) Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat. Struct. Mol. Biol. 20, 202–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taylor G. C., Eskeland R., Hekimoglu-Balkan B., Pradeepa M. M., and Bickmore W. A. (2013) H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 23, 2053–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang Z., Zang C., Rosenfeld J. A., Schones D. E., Barski A., Cuddapah S., Cui K., Roh T. Y., Peng W., Zhang M. Q., and Zhao K. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aerts S., and Cools J. (2013) Cancer: Mutations close in on gene regulation. Nature 499, 35–36 [DOI] [PubMed] [Google Scholar]
- 34. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 35. Berger S. L. (2002) Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 12, 142–148 [DOI] [PubMed] [Google Scholar]
- 36. Kustatscher G., Wills K. L., Furlan C., and Rappsilber J. (2014) Chromatin enrichment for proteomics. Nat. Protoc. 9, 2090–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zitouni S., Nabais C., Jana S. C., Guerrero A., and Bettencourt-Dias M. (2014) Polo-like kinases: structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 15, 433–452 [DOI] [PubMed] [Google Scholar]
- 38. Petronczki M., Lenart P., and Peters J. M. (2008) Polo on the rise-from mitotic entry to cytokinesis with Plk1. Dev Cell 14, 646–659 [DOI] [PubMed] [Google Scholar]
- 39. Su X., Zhang J., Mouawad R., Comperat E., Roupret M., Allanic F., Parra J., Bitker M. O., Thompson E. J., Gowrishankar B., Houldsworth J., Weinstein J. N., Tost J., Broom B. M., Khayat D., Spano J. P., Tannir N. M., and Malouf G. G. (2017) NSD1 Inactivation and SETD2 mutation drive a convergence toward loss of function of H3K36 writers in clear cell renal cell carcinomas. Cancer Res. 77, 4835–4845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu L., Guo R., Zhang X., Liang Y., Kong F., Wang J., and Xu Z. (2017) Loss of SETD2, but not H3K36me3, correlates with aggressive clinicopathological features of clear cell renal cell carcinoma patients. Biosci. Trends 11, 214–220 [DOI] [PubMed] [Google Scholar]
- 41. Giannini G., Cabri W., Fattorusso C., and Rodriquez M. (2012) Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med. Chem. 4, 1439–1460 [DOI] [PubMed] [Google Scholar]
- 42. Dokmanovic M., Clarke C., and Marks P. A. (2007) Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. 5, 981–989 [DOI] [PubMed] [Google Scholar]
- 43. Greer E. L., and Shi Y. (2012) Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bannister A. J., and Kouzarides T. (2011) Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castillo J., Lopez-Rodas G., and Franco L. (2017) Histone post-translational modifications and nucleosome organisation in transcriptional regulation: some open questions. Adv. Exp. Med. Biol. 966, 65–92 [DOI] [PubMed] [Google Scholar]
- 46. Verdin E., and Ott M. (2015) 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 16, 258–264 [DOI] [PubMed] [Google Scholar]
- 47. Xu Y., Gan E. S., Zhou J., Wee W. Y., Zhang X., and Ito T. (2014) Arabidopsis MRG domain proteins bridge two histone modifications to elevate expression of flowering genes. Nucleic Acids Res. 42, 10960–10974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kaelin W. G., Jr. (2004) The von Hippel-Lindau tumor suppressor gene and kidney cancer. Clin. Cancer Res. 10:6290S–6295S [DOI] [PubMed] [Google Scholar]
- 49. Carvalho S., Vitor A. C., Sridhara S. C., Martins F. B., Raposo A. C., Desterro J. M., Ferreira J., and de Almeida S. F. (2014) SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife 3, e02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pfister S. X., Markkanen E., Jiang Y., Sarkar S., Woodcock M., Orlando G., Mavrommati I., Pai C. C., Zalmas L. P., Drobnitzky N., Dianov G. L., Verrill C., Macaulay V. M., Ying S., La Thangue N. B., D'Angiolella V., Ryan A. J., and Humphrey T. C. (2015) Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell 28, 557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rini B. I., and Atkins M. B. (2009) Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 10, 992–1000 [DOI] [PubMed] [Google Scholar]
- 52. Malumbres M., and Barbacid M. (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 [DOI] [PubMed] [Google Scholar]
- 53. Asghar U., Witkiewicz A. K., Turner N. C., and Knudsen E. S. (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ChIP sequencing and mRNA sequencing data have been deposited in the NCBI GEO database with accession number GSE107391 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE107391). The proteomic data in this paper have been deposited into PeptideAtlas (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View? with the identifier PASS012793).