

Comprehensive analysis of MHC-II immunopeptidomes from cells lacking DO (human HLA-DO or murine H2-O) shows that DO controls the diversity of the MHC-II peptide repertoire. DO expression leads to presentation of a broader distribution of peptides, with many low-abundance epitopes presented only in the presence of DO. We suggest that regulated expression of DO allows the immune system to maintain tolerance to a wide variety of self-antigens and allows for an immune response focused on immunodominant antigens for pathogen recognition.
Keywords: Immunology*, Peptidomics, Absolute quantification, Immunoaffinity, Label-free quantification, Mass Spectrometry
Graphical Abstract

Highlights
MHC-II-bound peptide repertoires from DO-sufficient and DO-deficient cells.
Fewer unique peptides and core epitopes were presented in the absence of DO.
Immunopeptidome differences appeared to result from reduced DM editing.
DO-dependent self-epitopes elicited CD4 T cell responses in mice.
Abstract
Presentation of antigenic peptides on MHC-II molecules is essential for tolerance to self and for initiation of immune responses against foreign antigens. DO (HLA-DO in humans, H2-O in mice) is a nonclassical MHC-II protein that has been implicated in control of autoimmunity and regulation of neutralizing antibody responses to viruses. These effects likely are related to a role of DO in selecting MHC-II epitopes, but previous studies examining the effect of DO on presentation of selected CD4 T cell epitopes have been contradictory. To understand how DO modulates MHC-II antigen presentation, we characterized the full spectrum of peptides presented by MHC-II molecules expressed by DO-sufficient and DO-deficient antigen-presenting cells in vivo and in vitro using quantitative mass spectrometry approaches. We found that DO controlled the diversity of the presented peptide repertoire, with a subset of peptides presented only when DO was expressed. Antigen-presenting cells express another nonclassical MHC-II protein, DM, which acts as a peptide editor by preferentially catalyzing the exchange of less stable MHC-II peptide complexes, and which is inhibited when bound to DO. Peptides presented uniquely in the presence of DO were sensitive to DM-mediated exchange, suggesting that decreased DM editing was responsible for the increased diversity. DO-deficient mice mounted CD4 T cell responses against wild-type antigen-presenting cells, but not vice versa, indicating that DO-dependent alterations in the MHC-II peptidome could be recognized by circulating T cells. These data suggest that cell-specific and regulated expression of HLA-DO serves to fine-tune MHC-II peptidomes, in order to enhance self-tolerance to a wide spectrum of epitopes while allowing focused presentation of immunodominant epitopes during an immune response.
Antigen presentation by MHC-II molecules is required for development of CD4 T cells and regulation of CD4-mediated cellular and humoral immune responses. The nonclassical MHC-II molecule DO (human HLA-DO or murine H2-O) has been implicated in modulating immune responses to both self and foreign antigens. Mouse strains with inactive H2-Ob genes have increased neutralizing antibody responses to some persistent viruses (1), possibly related to increased ability of DO-deficient B cells to enter the germinal center (2), and HLA-DO variants in humans have been associated with resistance to HBV and HCV (1). DO knockout (DO-KO) mice also exhibit increased titers of autoantibodies, together somewhat paradoxically with decreased antibody responses to immunized protein antigens (3). DO expression is regulated differently from the coordinate regulation of other proteins involved in MHC-II antigen processing (4), with expression restricted to medullary thymic epithelial cells, immature dendritic cells (DCs), and mature B cells (5–8). DO expression is also downregulated following entry of B cells into the germinal center and following maturation of DCs (8–13), coinciding with the onset of inflammation. This pattern of expression suggests a potential role for DO in maintenance of T cell tolerance. Ectopic overexpression of DO in dendritic cells has been shown to prevent diabetes in NOD mice, consistent with this idea (14), and CD4 T cells from DO-KO mice show differential TCRBV usage, also indicating a potential role for DO in regulating T cell selection in the thymus (3, 15).
Presumably, these effects of DO result from modulation of MHC-II antigen processing. DO is posited to regulate peptide loading onto classical MHC-II proteins through interaction with the MHC-II peptide-exchange factor DM (16), which is required for efficient MHC-II peptide exchange (17, 18) and which has been shown to control CD4 epitope selection (19–22). DO has been shown to act as a tight-binding competitor of DM (23–25). The inhibition of DM by DO is pH-dependent, potentially restricting efficient antigen presentation to low pH late endosomal compartments (26–29). Several studies have shown that DO-KO murine and human cells have alterations in MHC-II antigen presentation (8, 9, 15, 28, 30–32). However, in these studies, the effect of DO seems to vary with the epitope studied, with presentation of peptides increased, decreased, or unchanged by expression of DO (15, 28, 30, 31, 33). This variation is difficult to reconcile with the biochemical effect of DO as a competitive inhibitor of DM, and the fundamental question of how DO affects the overall spectrum of MHC-II bound peptides has remained unanswered.
To definitively determine the role of DO in modulating the MHC-II peptidome, we used CRISPR/Cas9 gene editing to target DO in a human lymphoblastoid cell line and characterized MHC-II bound peptides from DO-deficient and DO-sufficient cells by quantitative mass spectrometry. We found that DO regulates the diversity of MHC-II antigen expression, by increasing the number of different peptides presented without changing the overall MHC-II expression level. Many low abundance peptides were presented only in the presence of DO. We confirmed these observations using a mouse model of DO deletion and determined through immunization experiments that the immune system is sensitive to these DO-dependent alterations in the peptide repertoire. This work defines a role for DO in mediating qualitative and quantitative changes in the MHC-II peptidome and provides a mechanistic basis for the biological consequences of DO expression.
EXPERIMENTAL PROCEDURES
Generation of DO-knockout (DO-KO)1 and WT clones
Single-guide RNAs (sgRNAs) designed to target exon 1 of HLA-DOβ (sgRNA-1: 5′ gACTAGCAGAGCCACCACCCA 3′ and sgRNA-2: 5′ GCTAGTGAATCTGACCCGAC 3′) using the CRISPR Design Tool (http://tools.genome-engineering.org) (34) were cloned into the pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid, which was a gift from Feng Zhang (35) (Plasmid #42230, Addgene, Cambridge, MA), and which was modified by insertion of a GFP sequence. The px330-GFP vector was then transfected into the human lymphoblastoid HLA-DR1 homozygous LG-2 cell line (36, 37) using the Amaxa Nucleofector Kit V (Lonza, Walkersville, MD) according to the manufacturer's instructions, with protocol Y-001 and the Nucleofector II system (Lonza). Transfected cells were sorted for GFP expression on a FACSAria (Becton Dickinson, Franklin Lakes, NJ) and then grown at 37 °C, 5% CO2 in RPMI supplemented with 10% fetal bovine serum, 100 IU/ml/100 μg/ml penicillin/streptomycin (Corning, Corning, NY), and 2 mm Glutamax (Thermo Fisher, Waltham, MA) for 1 week, followed by limiting dilution to isolate single-cell clones. PCR with primers specific for HLA-DOβ was performed using DNA isolated from expanded clonal populations, and amplicons were then gel-purified and cloned into the pCR2.1 vector using the TOPO-TA cloning kit (Thermo Fisher Scientific, Waltham, MA). DNA isolated from mini-preps of bacterial clones was sequenced to determine whether indels were present in exon 1 of HLA-DOβ. Clones were cultured in the medium described above.
Western Blotting
Total protein was isolated from a negative control T cell line (SUP-T1, [ATCC, Manassas, VA]), from the parental LG-2 line, and from the WT, DO-KO-1, and DO-KO-2 clones by cell lysis in cold RIPA buffer (50 mm Tris-HCl, 150 mm NaCl, 1% [v/v] Triton X-100, 1% [w/v] sodium deoxycholate, 0.1% [w/v] SDS, pH 7.4) containing protease inhibitor (Roche, Indianapolis, IN). Protein was quantified using a bicinchoninic acid protein assay (Thermo Fisher Scientific), and 40 μg of each lysate was loaded onto a Novex 12% Tris-Glycine gel (Thermo Fisher Scientific) and then transferred to a PVDF membrane. Membranes were blocked overnight with 10% nonfat dry milk, probed for HLA-DOβ (DOB.L1, Santa Cruz Biotechnology, Santa Cruz, CA), and then reprobed with anti-GAPDH (Millipore, Burlington, MA) to confirm equal protein loading.
Flow Cytometric Analysis
DO-KO and WT clones were blocked with 10 μg/ml human IgG (Sigma Aldrich, St. Louis, MO), and then stained for surface expression of HLA-DR (Thermo Fisher Scientific). Co-staining of viable cells was performed using the Live/Dead Fixable Dead Cell Stain Kit (Thermo Fisher Scientific) for all antibodies. For HLA-DM staining, cells were permeabilized using the BD Cytofix/Cytoperm kit according to the manufacturer's instructions (BD Biosciences, San Jose, CA) and then similarly blocked using human IgG. Permeabilized cells were incubated with PE-conjugated MaPDM.1 (Santa Cruz Biotechnology) to evaluate DM expression. Isotype controls were used for all antibodies. To assess presentation of the CLIP and HLA-A2(104–117) (A2) epitopes, LG-2 clones were incubated with the CerCLIP (FITC-conjugated, BD Biosciences) or UL-5A1 (38, 39) (followed by incubation with FITC-conjugated anti-mouse IgG F(ab′)2 [Thermo Fisher Scientific]) antibodies together with HLA-DR to calculate ratios of expression for mean fluorescence intensity (MFI) of epitopes to HLA-DR to account for any small differences in DR expression. Antibodies used in additional experiments (details below) were anti-mouse CD4 (RM4–5), CD8α (53–6.7), CD25 (PC61), CD69 (H1.2F3), and I-Ab (AF6–120.1) (BD Biosciences), as well as anti-mouse CD11b (M1/70) and CD43 (S11) (BioLegend, Dedham, MA) and CD45R/B220 (RA3–6B2) (Thermo Fisher Scientific). Prior to staining, mouse cells were blocked with 50 μg/ml anti-mouse CD16/CD32 (2.4G2, BioXCell, West Lebanon, NH). Cells were acquired on an LSR II flow cytometer (Becton Dickinson) and analyzed using FlowJo version 9.8.5 software (Tree Star, Ashland, OR).
RNAseq
RNA isolation, library preparation, and sequencing were performed at the Broad Institute (Cambridge, MA). Briefly, RNA was isolated using a Trizol-based method followed by purification using silica spin columns. Quantification of RNA was then performed using the Quant-iT RiboGreen RNA Assay Kit (Thermo Fisher Scientific), and RNA quality was measured as RNA Quality Score via Caliper GX (PerkinElmer, Waltham, MA). 200 ng of total RNA was used for library preparation, which was performed with an automated variation of the Illumina TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, CA) according to the manufacturer's instructions, with indexed adapters designed by the Broad Institute. Pooled libraries were normalized to 2 nm and then denatured with 0.1 N NaOH. Flowcell cluster amplification and sequencing were performed according to the manufacturer's instructions (Illumina) using either the HiSeq 2000 or HiSeq 2500 sequencing platform, with a 101bp paired-end read. The Broad Picard Pipeline was used for data de-multiplexing and data aggregation.
For analysis of sequencing data, filtering of rRNA and low-quality reads was first performed. Transcripts were then quantified by RSEM v1.2.7 (40) with Bowtie 2 (41) using the hg19(GRCh37) assembly. RefSeq annotations were downloaded on 2/5/17 from the UCSC genome browser (42, 43). Functional annotation clustering was performed using DAVID 6.8 (44, 45). A correlation analysis was also performed using RNAseq and mass spectrometric data (below) to evaluate whether clonal or CRISPR off-target effects could account for presentation of fewer peptides in DO-KO. The Pearson correlation coefficients, when comparing intensities of peptides found with greater intensity in WT or found only in WT versus the fold-change expression of corresponding genes downregulated in DO-KO, were −0.012 and −0.102 for DO-KO-1 and DO-KO-2 compared with WT, indicating that reduced peptide numbers in the absence of DO were not because of changes in gene expression.
Experimental Design and Statistical Rationale
WT, DO-KO-1 and DO-KO-2 human LG-2 clones were used to determine the qualitative and quantitative differences in peptide diversity modulated by HLA-DO. LG-2 cells express the MHC-II proteins HLA-DR1 (DRA1*01:01,DRB1*01:01), HLA-DQ5 (DQA1*01:01,DQB1*05:01), and HLA-DP4 (DQA1*01:03,DQB1*04:01) at an approximate 100:17:2 ratio (37); only HLA-DR1 was analyzed in the present study. HLA-DR1-bound peptides were isolated from ∼108 cells using immunoaffinity purification and were further identified using LC-MS/MS. For DO-KO-1, five biological replicates from independent cell cultures with paired WT replicates were analyzed. For DO-KO-2, three biological replicates from independent cell cultures with paired WT replicates were analyzed. To evaluate the role of the murine HLA-DO ortholog H2-O, we used splenic B cells from littermate H2-O−/− and WT C57BL/6 mice. These mice express I-Ab as their only MHC-II protein. Three biological replicates of paired WT and DO-KO samples from ∼10 mice/replicate were used, totaling 3 × 108 cells/replicate. For both human and mouse samples, pairs of WT and DO-KO samples were processed in parallel, with each biological replicate tested in three technical replicates. Only peptides identified with 1% FDR were considered. For systematic comparison across the peptides between different samples using their MS1 intensities for volcano plot analysis, we applied a multiple comparison correction using the original Benjamini-Hochberg method to calculate p values. The Benjamini, Krieger and Yekutieli method was used for comparison of differential source protein localization analysis in WT and DO-KO samples using GO terms. We used a paired parametric t test to calculate p values for differences in numbers of peptides, cores and diversity measures between WT and DO-KO. Also, paired nonparametric t-tests, unpaired nonparametric Mann-Whitney t-tests, and specific t-tests were used for different analyses, which are indicated in the figure legends of each plot. Prism (version 7.03, GraphPad, San Diego, CA) was used for statistical analysis and graphing data. R version 3.3.2 was used for histogram and kernel density plots.
Isolation of HLA-DR1-bound Peptides
Membrane solubilized fractions isolated from ∼108 cells of each WT and DO-KO LG-2 clone were used for elution experiments. Five independent samples each for WT and DO-KO from separate cell cultures were analyzed, with five sets of WT and DO-KO pairs processed in parallel. Cells were suspended in ice-cold hypotonic buffer (10 mm Tris-HCl, pH 8.0, containing protease inhibitors). Repeated (4–5) freeze-thaw cycles were used for cell disruption. Cellular debris was removed by centrifuging the lysate at 4000 × g for 5 min at 4 °C. The supernatant was collected and further centrifuged at 100,000 × g for 1 h at 4 °C to pellet the membrane fraction. The membrane pellet was solubilized in ice-cold 50 mm Tris-HCl, 150 mm NaCl, pH 8.0, containing protease inhibitors and 5% β-octylglucoside, and then mixed slowly overnight at 4 °C. Supernatant containing the solubilized membrane was recovered by centrifuging the lysate at 100,000 × g for 1 h at 4 °C. 2.5 μg of DR1-GAG or DR1-HA complex was added to the membrane fraction as controls. An immunoaffinity column of protein A agarose-LB3.1 antibody, prepared as previously described (37), was used for isolation of DR1-bound peptide complexes. The column was equilibrated with buffer (50 mm Tris-HCl, 150 mm NaCl, pH 8.0, containing protease inhibitors) for 2 h. The membrane fraction was first equilibrated with protein A agarose beads for 1 h at 4 °C and then allowed to mix slowly to prevent nonspecific binding of proteins to beads. The precleared supernatant was incubated with LB3.1 antibody conjugated to the protein A agarose affinity column for 1 h at 4 °C and allowed to mix slowly. The column was washed with several buffers in succession as follows: (1) 50 mm Tris-HCl, 150 mm NaCl, pH 8.0, containing protease inhibitors and 5% β-octylglucoside (5 times the bead volume); (2) 50 mm Tris-HCl, 150 mm NaCl, pH 8.0, containing protease inhibitors and 1% β-octylglucoside (10 times the bead volume); (3) 50 mm Tris-HCl, 150 mm NaCl, pH 8.0, containing protease inhibitors (30 times the bead volume); (4) 50 mm Tris-HCl, 300 mm NaCl, pH 8.0, containing protease inhibitors (10 times the bead volume); (5) 1× PBS (30 times the bead volume); and (6) HPLC water (100 times the bead volume). Bound complexes were acid-eluted, and MHC-peptide concentration from the membrane fraction was measured by ELISA. Peptides were further separated using a Vydac C4 macrospin column (Hichrom, Berkshire, UK). First, the mixture of DR1 and peptides were bound to the column, and after subsequent washes with 0.1% TFA, the peptides were eluted using 30% acetonitrile in 0.1% TFA. Eluted peptides were lyophilized using a SpeedVac and were resuspended in 25 μl of 5% acetonitrile and 0.1% TFA. This fraction was further divided into 3 different portions that were considered as technical replicates of the same sample. 2 pmols of ADH digest was added, and a total of 3/25 μl was injected, so that the amount of ADH per injection was 240 fmols. Each fraction was analyzed using a Q Exactive™ Hybrid Quadrupole-Orbitrap™ Mass Spectrometer (Thermo Fisher Scientific). 3 samples from clone DO-KO-2, 3 additional WT clone samples, and a single sample of the parental LG-2 line were analyzed similarly.
Liquid Chromatography–Mass Spectrometry (MS)
For LC/MS/MS analysis, peptide extracts were reconstituted in 25 μl 5% acetonitrile containing 0.1% (v/v) trifluoroacetic acid and separated on a nanoACQUITY (Waters Corporation, Milford, MA) UPLC with technical triplicate injections. In brief, a 3.0 μl injection was loaded in 5% acetonitrile containing 0.1% formic acid at 4.0 μl/min for 4.0 min onto a 100 μm I.D. fused-silica precolumn packed with 2 cm of 5 μm (200Å) Magic C18AQ (Bruker-Michrom, Auburn, CA) and eluted using a gradient at 300 nL/min onto a 75 μm I.D. analytical column packed with 25 cm of 3 μm (100Å) Magic C18AQ particles to a gravity-pulled tip. The solvents were A) water (0.1% formic acid); and B) acetonitrile (0.1% formic acid). A linear gradient was developed from 5% solvent A to 35% solvent B in 90 min. Ions were introduced by positive electrospray ionization via liquid junction into a Q Exactive hybrid mass spectrometer (Thermo Fisher Scientific). Mass spectra were acquired over m/z 300–1750 at 70,000 resolution (m/z-200), and data-dependent acquisition selected the top 10 most abundant precursor ions in each scan for tandem mass spectrometry by HCD fragmentation using an isolation width of 1.6 Da, collision energy of 27, and a resolution of 17,500.
Peptide Identification
Raw data files were peak processed with Proteome Discoverer (version 2.1, Thermo Fisher Scientific) prior to database searching with Mascot Server (version 2.5, Matrix Science, Boston, MA) against the combined database of UniProt_Human, UniProt_Bovine and UniProt_EBV databases, with 115,105 entries downloaded on 8/5/16. (The LG-2 cell line carries the Epstein-Barr virus genome and was cultured in medium containing fetal bovine serum.) Search parameters included “no enzyme” specificity to detect peptides generated by cleavage after any residue. The variable modifications of oxidized methionine and pyroglutamic acid for N-terminal glutamine were considered. The mass tolerances were 10 ppm for the precursor and 0.05Da for the fragments. Search results were then loaded into the Scaffold Viewer (Proteome Software, Inc., Portland, OR) for peptide/protein validation and label-free quantitation. Scaffold assigns probabilities using PeptideProphet or the LDFR algorithm for peptide identification and the ProteinProphet algorithm for protein identification, allowing the peptide and protein identification to be scored on the level of probability. An FDR of 1% was adjusted for reliable identification of peptides. Spectra files for all samples are shown in supplemental Tables S7–S9. Peptide lists were filtered to remove contaminants such as keratins and IgG-derived peptides. Core epitopes were identified for the HLA-DRB1*0101 allele using the NetMHCIIpan3.0 server; the top scoring 9-residue sequence within each sequenced peptide was used as the core epitope (46). Peptides with a length of less than 9 amino acids were excluded from the core epitope analysis. A similar analysis using a different prediction algorithm, P9 (47), identified essentially the same cores (∼92% identical).
Label-free Quantitation
Label-free relative quantitation of all peptides eluted from WT and DO-KO LG-2 cells was performed using precursor intensity analysis in Scaffold, Scaffold Q+/Q+S (48–50). Scaffold uses the precursor intensity information from the Thermo Proteome Discoverer. The software normalizes total precursor intensity values across the samples and calculates fold change or log2 normalized intensity across the samples while considering different statistical parameters like t test, ANOVA and coefficient of variance. The log2 normalized intensity values were converted to intensities for subsequent analyses. Triplicate technical replicates were run for each sample. Only peptides that were observed in at least two of three technical replicates in a sample were used for intensity analysis, with missing values imputed as the minimum intensity observed in that sample, and a single average value used to represent the three technical replicates. For analysis of core epitope intensities, the intensity values for all peptides sharing the same core epitope were summed within each technical replicate, using an approach like PLAtEAU (51), except that NetMHCIIpan rather than overlap analysis was used to identify core epitopes. Missing values were imputed, and technical replicates were averaged for core epitopes as described above for peptides. For calculation of diversity statistics, rank abundance plots, density histograms, and volcano plots, only core epitopes present in all the biological replicates were considered. To determine fractional intensities in the rank abundance plot, the intensity for each core was divided by the total intensity for all core epitopes present in that sample. To determine average fractional intensities in the density plot, an average of all biological replicates was calculated. The datasets used for intensity analysis are shown in supplemental Tables S10–S12. For correlation analysis, SIEVE software (48, 52) was used, with frame parameters adjusted to m/z range of 300–1700, frame time width of 1.5 min, m/z width of 10ppm, and retention time from 10–80 min. The intensity analysis shown for pairs of biological replicates in supplemental Fig. S1A–S1B included 10,000 frames.
Diversity Calculations
Shannon's diversity index (H) and Simpson's diversity index (D) were calculated to analyze the diversity of WT and DO-KO peptidomes. These indices consider not only the number of species (peptides) but also how evenly peptide abundances are distributed in the entire sample. Diversity calculations were performed only for peptides identified in all biological samples as described above. Shannon's entropy (H) was calculated as:
| (1) |
where R is the number of peptides and pi is the proportion of the total ion intensity represented by peptide i. The higher the entropy value, the more diverse the sample. Simpson's diversity index was calculated as:
| (2) |
Simpson's reciprocal diversity index was calculated as 1/D, with higher values representing more diverse samples. The Chao2 index (for replicated incidence data) for peptides was calculated as:
| (3) |
where Sobs is the number of observed species, Q1 is the number of singletons (species occurring once), and Q2 is the number of doubletons (species occurring twice).
Absolute Quantification Using Stable Isotope-labeled Peptides
Nine peptides were selected for intensity validation by referencing a volcano plot analysis of differences in intensities of the WT and DO-KO-1 versus their significance values adjusted using Benjamini-Hochberg's correction: one peptide that was observed with higher core epitope intensity in WT versus DO-KO-1 samples (WT>DO-KO), two peptides that were observed at higher core epitope intensity in DO-KO-1 versus WT samples, (WT<DO-KO), and six peptides that were observed with relatively equal core epitope intensities in the two samples (WT≈DO-KO) (Fig. 5D, supplemental Table S13). We selected core epitopes that were observed in each of the five biological replicates, and for differentially expressed core epitopes, that showed a statistically significant difference of 2-fold or greater after adjustment for multiple comparisons. For each core epitope, we selected the most abundant peptide containing that epitope for synthesis and absolute quantification studies. Peptides with 13C and 15N labels incorporated at specific residues were synthesized by 21st Century Biochemicals (Marlborough, MA) and spiked into new samples of WT and DO-KO-1 as internal controls to quantify the chemically-identical unlabeled (light) peptides present in these samples. The purity and quantification of these peptides were confirmed using amino acid analysis. DR1-bound peptides were eluted from WT or DO-KO-1 LG-2 cells as described in the previous section, and a mixture of isotope-labeled peptides at 60 fmols/injection was spiked into the sample. The data were analyzed as 3 technical replicates. Quantitation of the selected peptides was performed using Skyline software (V3.7, University of Washington, Seattle, WA) by generating extracted ion chromatograms of the MS1 signals for the M, M+1, and M+2 isotopes of each precursor. For most peptides, both +3 and +4 charge state ions were observed; intensities of these were summed for each peptide. Summed areas were then compared with the corresponding heavy peptide areas to determine absolute amount of peptide.
Fig. 5.
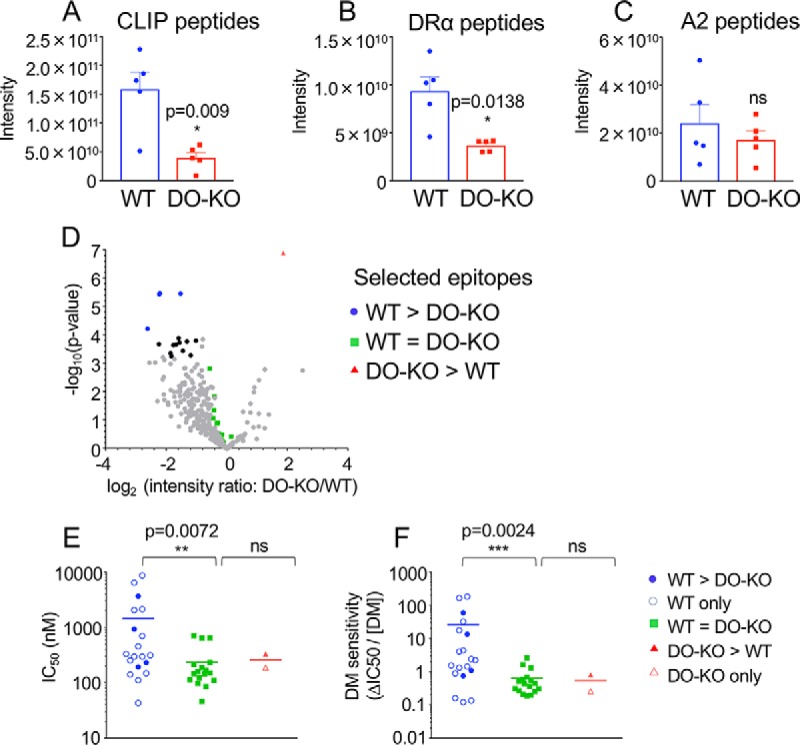
DO expression increases presentation of low affinity and DM-sensitive peptides. A–C, Relative abundance levels for peptides with known DM sensitivity were analyzed in 5 WT and DO-KO-1 samples: DM-sensitive CLIP peptides (58) with core epitope MRMATPLLM (A), DM-sensitive DRα peptides (56) with core epitope FASFEAQGA (B), and DM-resistant A2 peptides (38) with the core epitope WRFLRGYHQ (C). Mean ± S.D. (n = 5) shown. Paired parametric t test used to calculate p values. D, Volcano plot for cores identified in each of all 5 biological samples for WT and DO-KO-1 samples. Cores with intensity ratio differences >2-fold and p values <5.75E-04 (Benjamini-Hochberg-adjusted) are shown as black dots. Cores showing significant differences and selected for binding affinity studies, DM sensitivity studies and absolute quantification studies are shown in blue for WT>DO-KO, red for DO-KO>WT, and green for WT≈DO-KO. E, Binding affinity was characterized for sets of peptides observed in DO-KO only (1 peptide), WT only (15 peptides), or for peptides with intensities of WT>DO-KO (4 peptides), WT<DO-KO (1 peptide), or WT = DO-KO (17 peptides). F, The DM sensitivities for the same sets of peptides were assessed. Mean ± S.D. from 3 independent experiments shown for all peptides in each group; unpaired nonparametric Mann-Whitney test used to calculate p values.
Soluble Recombinant HLA-DR1 and HLA-DM
Soluble extracellular domains of recombinant HLA-DR1 (DR1) (DRA*0101/DRB1*0101) and DM (DMA*0101/DMB*0101) for binding affinity and DM sensitivity measurements were expressed in Drosophila S2 cells and purified by immunoaffinity chromatography followed by Superdex200 (GE Healthcare, Chicago, IL) size exclusion chromatography as previously described (18, 53).
Binding Affinity and DM Sensitivity Measurements
For three abundant self-peptides, DM sensitivity has been previously characterized: CLIP, the invariant chain chaperone fragment efficiently removed by DM (18, 54), DRα52–68, the human ortholog of the YAe epitope (55) known to be highly sensitive to DM exchange (56), and the transplantation alloepitope A2104–117 (39) previously demonstrated to be highly resistant to DM-mediated exchange (38). For these peptides, we summed the intensities of all peptides sharing the respective core epitopes and compared summed intensities between replicate samples of WT and DO-KO-1 cells. For a broader analysis, we selected 38 peptides comprising the 9 peptides used for stable isotope quantitation, 2 additional peptides with greater core epitope intensity in WT peptidomes than in DO-KO-1 as indicated by volcano plot analysis (WT>DO-KO), 11 with average core epitope intensity similar in WT and DO-KO-1 samples as indicated by volcano plot analysis (WT≈DO-KO), 15 core epitopes identified exclusively in WT, and one core epitope identified exclusively in DO-KO-1 (Fig. 5D and supplemental Table S14). For each core epitope, we selected the most abundant peptide containing that epitope for synthesis and binding analysis. A fluorescence polarization (FP) assay was used to measure the IC50 of each selected peptide, using N-terminally-acetylated influenza hemagglutinin HA306–318 (Ac-PRFVKQNTLRLAT) labeled with Alexa Fluor 488 tetrafluorophenyl ester (Invitrogen, Carlsbad, CA) via the primary amine at K5 as probe peptide as previously described (57). The DR1 concentration used was selected by titrating DR1 against fixed labeled peptide concentration (25 nm) and choosing the concentration of DR1 that showed ∼50% maximum binding. For calculating IC50 values, 100 nm DR1 was incubated with 25 nm Alexa488-labeled HA306–318 probe peptide, in combination with a serial dilution of test peptides, beginning at 10 μm followed by 2-fold dilutions. The reaction mixture was incubated at 37 °C. The capacity of each test peptide to compete for binding of probe peptide was measured by FP after 72 h at 37 °C. FP values were converted to fraction bound by calculating [(FP_sample - FP_free)/(FP_no_comp - FP_free)], where FP_sample represents the FP value in the presence of test peptide; FP_free represents the value for free Alexa488-conjugated HA306–318; and FP_no_comp represents values in the absence of competitor peptide. We plotted fraction bound versus concentration of test peptide and fit the curve to the equation y = 1/(1 + [pep]/IC50), where [pep] is the concentration of test peptide, y is the fraction of probe peptide bound at that concentration of test peptide, and IC50 is the 50% inhibitory concentration of the test peptide. To measure DM sensitivity, an IC50,DM was obtained by including 500 nm DM in the binding competition assay, and ΔIC50 was calculated as (IC50,DM - IC50) as described (58). DM sensitivity was calculated as ΔIC50/[DM], where [DM] is the concentration of DM.
Whole-cell Proteomics
For in-gel digestion and LC-MS/MS analysis, total protein was isolated as above for Western blotting. 50 μg of whole-cell lysate was run on an SDS-PAGE system to separate proteins from lower molecular weight contaminants, and the entire protein region of the gel was then excised and subjected to in-gel trypsin digestion after reduction with DTT and alkylation with IAA. Peptides eluted from the gel were lyophilized and resuspended in 100 μl of 5% acetonitrile (0.1% [v/v] TFA) with 1 pmol ADH digest. An injection of 1.5 μl was loaded by a Waters nanoACQUITY UPLC in 5% acetonitrile (0.1% formic acid) at 4.0 μl/min for 4.0 min onto a 100 μm I.D. fused-silica precolumn packed with 2 cm of 5 μm (200Å) Magic C18AQ (Bruker-Michrom). Peptides were eluted at 300 nL/min from a 75 μm I.D. gravity-pulled analytical column packed with 25 cm of 3 μm (100Å) Magic C18AQ particles using a linear gradient from 5–35% of mobile phase B (acetonitrile + 0.1% formic acid) in mobile phase A (water + 0.1% formic acid) for 120 min. Ions were introduced by positive electrospray ionization via liquid junction at 1.5kV into a Thermo Scientific Q Exactive hybrid mass spectrometer. Mass spectra were acquired over m/z 300–1750 at 70,000 resolution (m/z 200) with an AGC target of 1e6, and data-dependent acquisition selected the top 10 most abundant precursor ions for tandem mass spectrometry by HCD fragmentation using an isolation width of 1.6 Da, max fill time of 110ms, and AGC target of 1e5. Peptides were fragmented by a normalized collisional energy of 27, and fragment spectra acquired at a resolution of 17,500 (m/z 200). Raw data files were peak-processed with Proteome Discoverer (version 1.4, Thermo Scientific) followed by identification using Mascot Server (version 2.5, Matrix Science) against an Epstein-Barr virus (Swiss-Prot), Human (Swiss-Prot), Bovine (UniProt) FASTA file (downloaded 8/2016). Search parameters included Trypsin/P specificity, up to 2 missed cleavages, a fixed modification of carbamidomethyl cysteine, and variable modifications of oxidized methionine, pyroglutamic acid for Q, and N-terminal acetylation. Assignments were made using a 10-ppm mass tolerance for the precursor and 0.05 Da mass tolerance for the fragments. All nonfiltered search results were processed by Scaffold (version 4.4.4, Proteome Software, Inc.) utilizing the Trans-Proteomic Pipeline (Institute for Systems Biology) with a 0.96% false-discovery rate.
Mice
H2-O-deficient mice were provided by Dr. Xinjian Chen at the University of Utah School of Medicine, following backcrossing to C57BL/6 mice for 10 generations. H2-O−/− mice were bred at the University of Massachusetts Medical School with C57BL/6 mice obtained from Jackson Laboratory (Bar Harbor, ME), and mice heterozygous for H2-O−/− were bred to obtain H2-O−/− and WT littermate controls for B cell isolation and immunization experiments. Mice were cared for and used in accordance with institutional guidelines.
Isolation of B Cells from H2-O-deficient and WT Mice
Spleens were isolated from H2-O-deficient and WT littermate mice, dissociated into single-cell suspensions, and splenic B cells were evaluated for I-Ab expression by first gating on the B220+CD43−CD11b− population and performing flow cytometric analysis as above. To isolate mature B cells from the splenocyte population, CD43- and CD11b-expressing cells were depleted using biotinylated anti-mouse CD43 and CD11b (BioLegend) in conjunction with the EasySep Mouse Streptavidin RapidSpheres Isolation Kit (Stem Cell Technologies, Cambridge, MA) according to the manufacturer's instructions. Purity post-isolation was determined by FACS to be >90% for each sample, with an average purity of 94 ± 2.6%.
Isolation and Characterization of I-Ab-bound Peptides
Mouse B cells (∼3 × 108) were solubilized in 50 mm Tris-HCl, 150 mm NaCl, pH 8.0, containing protease inhibitors and 5% β-octylglucoside and were processed as for LG-2 membrane fractions described above, except that whole cell lysates instead of solubilized membrane fractions were used, before loading on an affinity column of I-Ab-specific mAb M5114 coupled to CNBr-activated Sepharose, with elution and analysis as described above for isolation of HLA-DR1-bound peptides. Peptide sequences were identified as described above except that the UniProt Mouse database which was downloaded on 10/7/16 with 57,984 entries.
Mouse Immunization
6–8-week-old H2-O-deficient and WT littermate mice were immunized i.p. with 4 × 107 irradiated (3000 rads) age- and sex-matched splenocytes from WT or H2-O-deficient mice. Spleens from recipient mice were harvested 15h later, and single-cell suspensions were prepared. Following red blood cell lysis, splenocytes were subjected to flow cytometric analysis as above, using anti-mouse CD4, CD8, CD25, and CD69 antibodies, by first gating on the CD4+CD8− population and then assessing expression of CD25 and CD69. Similar CD4 T cell activation results were observed at 3 days after immunization.
RESULTS
Generation and Validation of DO-KO and WT Clones
To study the effect of DO on the self-peptide repertoire, we used CRISPR/Cas9 gene editing to delete HLA-DO from the HLA-DR-expressing lymphoblastoid cell line LG-2. Following transfection of sgRNAs, cellular clones were isolated, expanded, and sequenced to evaluate DNA modifications at target sites in HLA-DOβ exon 1 (Fig. 1A). A clone (DO-KO-1) targeted by sgRNA-1 was shown to harbor 1-nt and 7-nt deletions, whereas a clone targeted by sgRNA-2 (DO-KO-2) showed 1-nt and 4-nt deletions. An additional clone (WT)—subjected to the identical transfection process with sgRNA-2 but without any modifications in the HLA-DOβ locus on either chromosome—was selected as a control. Deletion of DOβ was confirmed by Western blotting in DO-KO cells (Fig. 1B). HLA-DM and HLA-DR expression was determined by FACS analysis to be consistent among all clones (Fig. 1C–1E). To examine presentation of specific epitopes previously demonstrated to be DM-sensitive (CLIP) or DM-resistant (A2[104–117]), we used antibodies specific for their DR-bound forms (CerCLIP and UL-5A1, respectively). Expression of CerCLIP was decreased ∼2-fold in DO-KO clones compared with WT, whereas expression of UL-5A1 was unchanged (Fig. 1F–1G), consistent with the expected DO inhibition of DM editing activity.
Fig. 1.
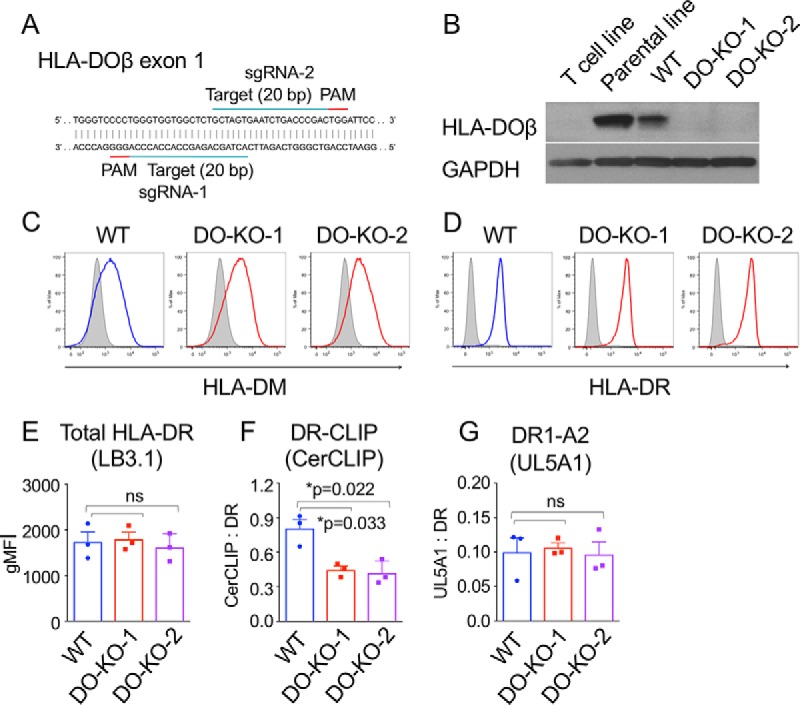
Generation of DO-KO and WT clones. A, sgRNAs and target sequences for HLA-DO knockout. B, Western blotting for HLA-DOβ performed with lysates from indicated cell lines. C, D, Intracellular HLA-DM (C) and surface HLA-DR (D) expression are similar among WT and DO-KO clones by FACS analysis. Isotype controls, gray. E, F, G, Surface expression of peptide-MHC complexes, using mAbs LB3.1 (total HLA-DR), CerCLIP (DR-CLIP(79)), and UL5A1 (DR1-A2104–117). Mean ± S.D. (n = 3) shown. Paired parametric t test used to calculate p values. gMFI, geometric mean intensity.
Reduced HLA-DR Immunopeptidome Presented by DO-KO as Compared with WT Cells
To evaluate the influence of HLA-DO on the full spectrum of peptides presented by MHC-II, we characterized the immunopeptidomes presented by HLA-DR1 from DO-KO and WT cells. We purified HLA-DR1 from DO-KO-1 and WT clones by immunoaffinity, released peptides by acid treatment, and characterized the resultant peptide pools by HPLC/MS/MS using a high-sensitivity mass spectrometer with search parameters set to provide a conservative false discovery rate. Similarly to previous comparative immunopeptidome reports (59–61), we observed ∼70% overlap between peptides identified in replicate WT or DO-KO samples (supplemental Fig. S1A–S1B). Integrated parent ion peak areas were highly reproducible sample-to-sample (supplemental Fig. S1C–S1D), whereas overlap of WT and DO-KO samples was comparatively lower (supplemental Fig. S1A-S1D). A total of 6116 distinct peptide sequences were identified in 5 samples of WT cells (Fig. 2A, supplemental Table S1), similar to the numbers of peptides identified in recent high-density immunopeptidome studies for other human and mouse MHC proteins (61). A smaller number of peptides, 5207, was identified in samples from DO-KO-1 cells processed in parallel (Fig. 2A, supplemental Table S1). A broad distribution of peptide lengths centered around 15–16 residues was observed similarly for both WT and DO-KO-1 (Fig. 2A). As is typical for MHC-II peptidomes, many peptides were present as nested sets surrounding a common core epitope. This is illustrated in Fig. 2B for 11 peptides from the human transferrin receptor protein, found in both WT and DO-KO, which share the FLYQDSNWA core that binds to HLA-DR1 (Fig. 2B, supplemental Table S4). We used the MHC-II binding prediction resource NetMHCIIpan3.1 (62) to predict 9-residue HLA-DR1-binding cores for each of the eluted peptides (supplemental Table S4). Sequence preferences within these cores were essentially identical for WT and DO-KO-1 (Fig. 2C), as was the average number of nested peptides per core and their distribution (Fig. 2D). Thus, many features of the HLA-DR1 peptidome were not significantly altered as a result of DO.
Fig. 2.

DO expression results in presentation of a greater number of peptides. A, Similar length distribution of eluted peptides. B, Core epitope illustrated for nested set of peptides from human transferrin receptor. C, Similar sequence motifs within aligned core sequences for WT and DO-KO-1 cells. D, Similar trimming of peptides for WT versus DO-KO-1. E, Species richness. The number of unique peptides eluted from WT was significantly greater than for DO-KO-1 in each of 5 independent experiments. F, G, H, Diversity indices. Chao2 index (F), Shannon's entropy (G) and Simpson's reciprocal diversity index (H) are greater for WT than for DO-KO-1, calculated for peptides observed in each of five biological replicates of both WT and DO-KO-1. Paired parametric t test used to calculate p values.
The most apparent difference between WT and DO-KO peptidomes was that in the absence of DO, fewer different peptide sequences were presented. For each of 5 replicate sets of WT and DO-KO-1 cultures processed in parallel, or 3 replicate sets for WT and DO-KO-2, fewer peptides were isolated from DO-KO than from WT (Fig. 2E, supplemental Fig. S2A, supplemental Tables S1–S2). In biodiversity analysis, the number of different species (“richness”) is a primary criterion of diversity, but other diversity measures are available that differentially weight the contribution of rare versus abundant species (63). The Chao2 index provides an extrapolated estimate of the total richness including rare species missed by undersampling (64). The Shannon diversity index considers the relative abundance of different species, with even distributions assigned higher diversity values than skewed distributions. Simpson's entropy also considers relative abundance, preferentially weighting more abundant species (63). By all these measures, diversity was significantly larger for the set of peptides eluted from WT as compared with DO-KO-1 (Fig. 2F–2H) or DO-KO-2 (supplemental Fig. S2B–S2D).
Validation of Immunopeptidome Differences
We investigated several potential explanations for the reduced number of peptides observed for DO-KO cells. We used whole-cell quantitative proteomics to determine whether deletion of DO had any effect on overall protein levels; no significant skewing was observed (supplemental Fig. S3). We used RNASeq to evaluate potential alterations because of CRISPR/Cas9 off-targeting effects. Both the DO-KO clones and the WT clone exhibited some changes in gene expression compared with the parental LG-2 line, but there was no correlation of these differences with peptides differentially present in WT or DO-KO, and clustering of functional annotations for differentially expressed genes did not indicate any systematic effect on antigen presentation pathways. The reduced number of peptides in DO-KO potentially could be explained by elution of peptides from fewer MHC molecules, but the amounts of HLA-DR1 purified from WT and DO-KO cells were not different (supplemental Fig. S4A), consistent with cell surface expression levels (Fig. 1). Peptides from recombinant peptide-MHC complexes spiked into cell lysates before immunoaffinity isolation as internal controls were recovered similarly for WT and DO-KO samples (supplemental Fig. 4E), indicating that differential purification did not contribute significantly to the observed differences. For 3 pairs of samples, we removed a fraction of the eluted peptide mixture and measured the total amount of peptide by quantitative amino acid analysis. No significant differences in the total amount of peptidic material were observed between the samples (supplemental Fig. S4B), indicating that the elution efficiency did not differ between WT and DO-KO. In addition, no significant differences in the distribution of amino acid residues was observed (supplemental Fig. S4C), suggesting that the observed differences between WT and DO-KO were not because of factors such as overall hydrophobicity, which can limit the ability to volatilize and enter the spectrometer for analysis. Sample-dependent factors that could suppress ionization did not contribute to differential detection of peptides in WT and DO-KO samples, as control peptides from yeast alcohol dehydrogenase spiked into each sample were detected with similar efficiency (supplemental Fig. S4D). Thus, the reduced number of peptides identified for DO-KO as compared with WT samples represents an actual difference in the cellular peptide abundances and is not a consequence of experimental factors.
Fewer Epitopes Presented in the Absence of DO
As each peptide in a nested set represents a different version of the same epitope, we asked whether DO deletion reduced the number of distinct epitopes presented as it did the number of individual peptides. We combined records for each peptide sharing the same core epitope, thus counting each set of related epitopes only once (supplemental Table S4). DO-KO-1 and DO-KO-2 cells presented fewer core epitopes then did WT cells, with each DO-KO replicate having fewer cores than WT samples processed in parallel (Fig. 3A, supplemental Fig. S2E, supplemental Tables S4–S5).
Fig. 3.
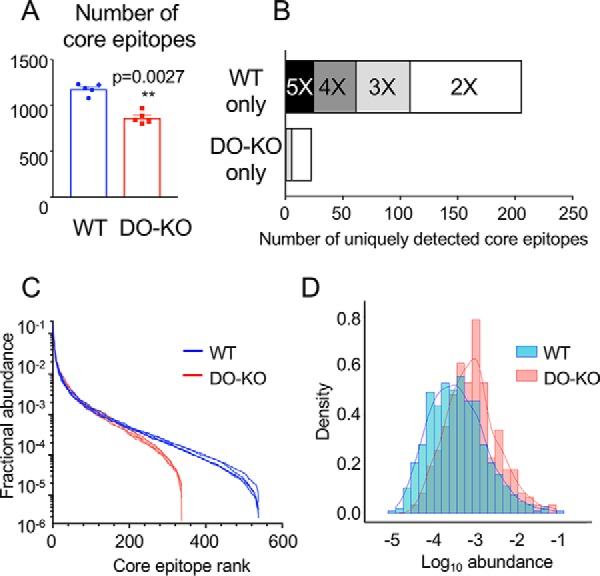
DO expression results in presentation of greater numbers of and a broader distribution of core epitopes. A, The number of unique core epitopes observed in WT was significantly greater than for DO-KO-1 cells in each of 5 independent experiments. Mean ± S.D. (n = 5) shown. Paired parametric t test used to calculate p values. B, More core epitopes are identified uniquely in WT as compared with DO-KO-1 samples. Bar shading indicates number of replicate samples for which the core epitope was identified. For example, the black bar labeled “5X” indicates epitopes identified in each of 5 WT samples and none of the 5 DO-KO-1 samples, the dark gray bar labeled “4X” indicates samples identified in 4/5 WT samples and no DO-KO-1 samples, etc. C, Rank abundance plot. Fractional intensity of core epitopes from WT (blue) or DO-KO-1 (red) in each biological sample is represented as an individual line. D, Histogram of fractional intensities of core epitopes, overlaid with a kernel density plot.
To help understand the greater diversity of epitopes presented by WT as compared with DO-KO cells, we looked for core sequences present in both peptidomes, or unique to only WT or DO-KO. Of 704 distinct core epitopes detected in each of the 5 WT replicates tested, 25 core sequences were not detected in any of the DO-KO-1 samples. By contrast, only one core epitope was present in each of the DO-KO-1 samples but was not detected in any of WT samples (Fig. 3B). This same pattern held if we relaxed the identification criteria and considered peptides present in fewer replicates. For example, 37 additional core epitopes were present in 4 of the 5 WT replicate samples but were absent from any DO-KO-1 sample, whereas no additional core epitopes were detected in 4 of 5 DO-KO-1 samples and were absent from any WT sample. A similar pattern was observed for the second DO-deficient clone DO-KO-2, although in this case the test is less stringent because only three replicates samples were analyzed (supplemental Fig. S2F). Overall, the WT peptidome appears to contain many peptides that are absent from or present at much lower frequency in the DO-KO peptidome.
One method to visualize diversity is by a rank abundance plot, in which the relative abundance of each species is plotted on a logarithmic scale against the species rank (i.e. 1 for the most abundant species, 2 for the next most abundant, etc.). This analysis shows a steeper profile for DO-KO-1 as compared with WT, with the WT curve showing a more even distribution and a long tail, indicating that many more low-abundance peptides are present in WT as compared with DO-KO-1 (Fig. 3C). Because the total molar amount of MHC (and peptide) present in WT and DO-KO samples was identical (supplemental Fig. S4A), this would imply that the average fractional abundance of peptides in the DO-KO-1 samples was higher than for WT, as the same total amount of peptide was represented by fewer different sequences. This can be seen in the density plot (histogram) of abundances of identified core epitopes, which shows a slight shift to higher abundances for the DO-KO peptides (Fig. 3D). Similar shifts in rank abundance and density plots were observed for DO-KO-2 relative to WT samples processed in parallel (supplemental Fig. S2G–S2H). Thus, one component of the increased diversity of WT as compared with DO-KO peptidomes is an increased representation of lower-abundance species.
Validation of Intensity Differences
Although individual peptides are detected in the mass spectrometer with different efficiencies depending on their charge, hydrophobicity and other factors, parent ion intensities as used above in diversity and rank abundance analyses provide reliable quantitation when averaged over many ions (65). To validate the quantitation of individual peptides, we used a stable isotope-labeling approach, in which synthetic peptides carrying 13C and/or 15N labels were introduced into eluted peptide samples before analysis and used as internal standards (supplemental Table S13). Using volcano plot analysis (Fig. 5D), we selected 9 peptides: 1 with parent ion intensities greater in WT than in DO-KO samples, 2 with intensities greater in DO-KO than in WT samples, and 6 with intensities approximately equal between the samples. We used the most abundant peptide containing each core epitope for analysis. The peptides sampled a range of masses, charges, hydrophobicities, and observed intensities. For most of the peptides, both +3 and +4 ions were observed; these were summed for the quantitation (supplemental Table S13). A good correlation was observed between the observed intensities and the calculated amounts of peptides present (Fig. 4A), validating the peak integration and sample normalization procedures. Three replicates of one peptide fell off the line defined by the other peptides, presumably because of sequence-specific factors. The calculated amount of each peptide present in the WT and DO-KO eluates was used to determine an abundance ratio, which varied 20-fold between peptides, and which clearly distinguished peptides in the WT>DO-KO, WT≈DO-KO, and DO-KO>WT sets (Fig. 4B).
Fig. 4.
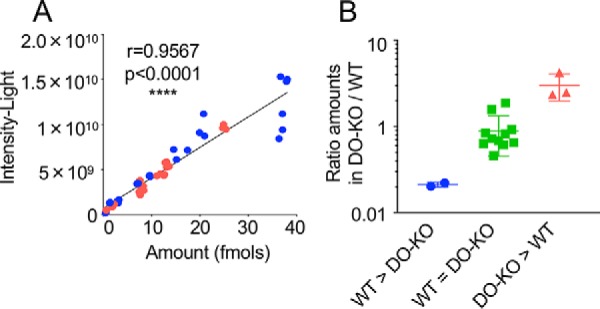
Stable isotope-labeled peptide analysis validates MS1 intensity analysis. A, Observed intensity versus amount measured using 13C- and 15N-labeled internal standards, for 9 selected peptides (supplemental Table S13), each assayed in 3 replicate injections of a WT (blue) or DO-KO-1 (red) sample. Pearson correlation coefficient indicated. B, Observed DO-KO-1/WT abundance ratio for peptides selected from groups with intensity greater in WT than DO-KO (blue), approximately equal intensity in WT and DO-KO (green), or greater in DO-KO than WT (red). Mean ± S.D. shown (n = 3–10).
Analysis of Epitope Source Protein Intracellular Localization
Because DO has been suggested to control the intracellular location of antigen loading through its effects on DM (26, 27, 66, 67), we evaluated the GO-annotated cellular compartments (68, 69) for source proteins from which the eluted peptides were derived. These were not appreciably different between WT and DO-KO, suggesting that the location of antigen loading was not substantially altered by the presence of DO (supplemental Fig. S5A). We repeated this analysis for core epitopes identified uniquely in WT or DO-KO samples (identified as in Fig. 3B.) For core epitopes unique to WT, the distribution of source proteins was similar to that which was observed for all peptides (supplemental Fig. S5B). Comparison with DO-KO is hampered by the paucity of peptides found uniquely in these samples, but an increased representation of peptides derived from extracellular sources (secreted proteins and medium components) is apparent (supplemental Fig. S5B), providing one possible explanation for some of the peptidome differences observed in the presence of DO. DO-KO mice previously have been observed to have increased capacity as compared with WT to present peptides derived from exogenous soluble protein antigens (28). However, extracellular proteins comprised only a small part of the overall peptidome, and we observed DO-dependent differences in peptide presentation for epitopes derived from many intracellular sources, both intracellular and extracellular. Moreover, many epitopes were presented preferentially in WT as compared to DO-KO. Thus, we sought other explanations for the effect of DO on MHC-II peptidome diversity.
DO Expression Allows for Presentation of a Population of DM-sensitive Peptide Antigens
Given the role of DM in epitope selection (22, 70, 71) and the function of DO as an inhibitor of DM (23–25), we sought to determine whether DO-dependent differences in the MHC-II peptidome were related to sensitivity to DM-mediated exchange. We first examined the relative abundance of three epitopes for which DM sensitivity has been previously characterized, summing the intensities of individual peptides that contain the respective core epitopes. CLIP and DRα peptides were observed at lower abundance in the absence of DO (Fig. 5A–5B, supplemental Fig. S2I–S2J), whereas A2 peptides were not significantly different (Fig. 5C, supplemental Fig. S2K), as expected from their known DM sensitivities, and consistent with surface expression data by FACS (Fig. 1F–1G). To extend this analysis to additional peptides, we characterized the binding affinity and DM sensitivity of 38 additional peptides, grouped into sets according to their representation in WT and DO-KO-1 peptidomes (Fig. 5D–5F, supplemental Table S14). The DO-KO>WT and DO-KO-only peptides fell within the range of the WT≈DO-KO peptide set, whereas the WT>DO-KO and WT-only peptides included several species with lower binding affinity and higher DM sensitivity. These results support the idea that the increased complexity of the WT as compared with the DO-KO peptidome is due at least in part to increased representation of DM-sensitive epitopes that are lost when DO modulation of DM editing activity is absent.
DO Control of Peptide Diversity Evaluated in a Mouse Model
Given the DO-dependent peptide differences observed in human B cells, we sought to evaluate whether such differences would be similarly observed in a mouse model of DO deficiency. We made use of H2-O−/− mice, which previously have been shown to exhibit autoimmune and immunodeficient phenotypes (3, 28). As was observed for human B cells, H2-O deletion did not affect surface expression of MHC-II on mouse B cells (Fig. 6A), consistent with previous reports (3, 28). We immunoaffinity-purified I-Ab from WT and H2-O−/− splenic B cells, eluted bound peptides, and characterized peptidomes by mass spectrometry. As was observed for human B cells, a broad length distribution was observed for peptides eluted from both WT and H2-O−/− mouse B cells (Fig. 6B). As observed for human B cells, DO deletion in mice caused reductions in the number of unique peptides and the number of core epitopes presented by MHC-II, with reduced peptide diversity as measured by several indices, and skewed rank abundance and density plots (Fig. 6C–6I, supplemental Tables S3 and S6). To test whether the immune system is sensitive to DO-dependent peptide differences, we performed a cross-immunization experiment. H2-O-deficient and WT mice were immunized with irradiated splenocytes from WT and H2-O-deficient mice, with syngeneic splenocyte immunizations serving as controls. CD4 T cells of H2-O−/− recipient mice were activated when immunized with WT splenocytes, but CD4 T cells of WT recipients immunized with H2-O−/− splenocytes were not significantly activated, nor were CD4 T cells of syngeneic control recipient mice (Fig. 6J–6K). This unidirectional pattern, in which T cells were selectively activated only after having developed in the H2-O−/− mouse and when presented with WT peptides, suggest that peptides displayed on WT antigen-presenting cells (APCs) are recognized by H2-O−/− T cells because of the absence of these epitopes in the H2-O−/− mouse.
Fig. 6.

DO control of peptide diversity in a mouse model. A, Equivalent I-Ab expression on splenic WT and H2-O−/− B cells. B, Similar length distribution for peptides eluted from WT and H2-O−/− B cells. C, Species richness. More unique peptides were eluted from WT than H2-O−/− B cells in each of 3 independent experiments. Mean ± S.D. is shown. D, E, F, Diversity indices. Chao2 index (D), Shannon's entropy (E) and Simpson's reciprocal diversity index (F) are greater for WT than for H2-O−/−, calculated for peptides observed in each of 3 biological replicates of both WT and H2-O−/−. G, The number of unique core epitopes observed from WT was significantly greater than for H2-O−/− in each of 3 independent experiments. Paired parametric t test used to calculate p values. H, Rank abundance plot. Fractional intensity of core epitopes from WT (blue) or H2-O−/− (red) in each biological sample is represented as individual lines. I, Histogram of fractional intensities of core epitopes, overlaid with a kernel density plot. J, K, CD4 T cell activation measured by expression of CD25 (J) or CD69 (K) was observed when H2-O−/− mice (host) were immunized with WT splenocytes, but not when WT recipients were immunized with H2-O−/− splenocytes, or with splenocytes from syngeneic controls. Mean ± S.D. shown (n = 8 mice/group, performed in 3 separate experiments with 2–3 mice/group). Unpaired nonparametric t test used to calculate p values.
DISCUSSION
The role of DO in MHC-II antigen presentation has been the subject of numerous biochemical and immunological studies, but understanding of the biological function of DO has been complicated by conflicting reports, in which differing effects have been described depending on the epitope(s) examined (9, 11, 13, 24, 25, 28, 30–33, 72, 73). In addition, previous mass spectrometric studies of DO function were limited by the technology available at the time and only allowed for analysis of qualitative differences in eluted peptides (15, 24, 33). We sought to definitively determine the overall peptidome-wide effect of DO on MHC-II antigen presentation as well as its in vivo effects. We thus performed a comprehensive analysis of the effect of DO on the self-peptide repertoire. We generated DO-KO and WT control cells using CRISPR/Cas9-mediated targeted gene deletion, and we eluted MHC-II-bound peptides from WT and DO-KO cells. We found that while many features of the DO-KO and WT peptidomes were similar, a striking difference was that fewer different peptide sequences were presented in the absence of DO. We ruled out several explanations that could account for these differences in peptide numbers, including different antigen source proteins and lower MHC input. We analyzed DO-KO versus WT peptidomes using unfragmented MS1 parent ion intensities, to avoid sampling issues intrinsic to conventional data-dependent acquisition proteomics, and validated the intensity-based analysis using isotope-labeled peptides. We calculated several measures of peptidome diversity and found that repertoire diversity was significantly reduced in the absence of DO. The picture that emerges from this quantitative analysis is of a steeper abundance profile in the absence of DO, with abundant epitopes presented in even more copies and many of the low abundance epitopes lost. Epitopes presented preferentially in WT exhibited lower binding affinity and increased DM sensitivity, demonstrating that increased DM activity is at least in part responsible for peptide differences in WT versus DO-KO. Here we analyzed the peptidome presented by a single human MHC-II protein HLA-DR1. Other MHC-II proteins with similar DM sensitivities would be expected to behave similarly, but some autoimmune-linked MHC-II proteins have reduced DM sensitivity (74–76) and may show different behavior. To extend our observations to a mouse model and test their immunological relevance in vivo, we performed elutions from I-Ab isolated from mouse B cells and similarly observed reduced peptide numbers in the absence of DO. Immunization experiments showed that H2-O−/− cells were selectively activated by WT APCs and not vice versa, suggesting that epitopes presented on WT cells were not recognized as self-antigens by T cells that were selected or developed in the absence of DO. These data indicate that expression of DO results in an altered MHC-II B cell peptidome and have broad implications regarding the role of DO in immunological processes including thymic selection, peripheral tolerance, and entry and selection of B cells in the germinal center.
This study considerably extends previously reported analyses of the effect of DO on the MHC-II peptidome, in which the authors observed as many peptides presented uniquely in the presence of DO as peptides presented uniquely in the absence of DO (15, 33). Another study concluded that DO expression increases the stringency of DM editing (24), at odds with reports demonstrating that DO is an inhibitor of DM (23–25, 29). These analyses were limited by the available technology to comparing qualitative differences in MALDI spectra, with identification of very few if any individual peptide sequences. With advances in mass spectrometry sensitivity and mass accuracy (60, 77), we were able to characterize essentially the entire peptidomes of intact and of DO-deleted LG-2 cells (we estimate that lowest abundance peptides characterized in our study are present at only a few copies per cell or less). Using quantitative methods, we were able to obtain reliable peptidome comparisons and to characterize the full abundance profile, rather than simply the number of different peptides presented. These results suggest an apparent excess of peptides over MHC-II in loading compartments, as the restricted number of different peptide species in the absence of DO, presumably through increased DM editing, did not result in a lower number of peptide-MHC molecules but did result in decreased peptide diversity. This indicates substantially different constraints on MHC-II as compared with MHC-I processing, where peptide abundance is posited to limit presentation (78). Although DO has been thought to focus antigen presentation by way of restricting presentation to very low pH compartments (26–29), we did not find evidence of differential sampling of intracellular compartments in the absence or presence of DO. Instead, and in contrast to conclusions made in previous MS analyses of DO function (15, 24, 33), we find that DO expression broadens antigen presentation, by promoting presentation of low affinity and/or DM-sensitive antigens. Our results suggest that when DO is downregulated relative to DM, for example upon B cell entry into the germinal center (11, 13) or during DC maturation (10, 12), the diversity of the MHC-II peptidome will be reduced, with increased DM editing leading to presentation of a more focused and limited peptide repertoire.
We attribute much of the effect of DO expression on broadening the MHC-II peptide repertoire to inhibition of DM, with peptides more susceptible to DM-mediated peptide exchange preferentially presented when DM activity is reduced in the presence of DO. DO inhibition of DM activity has been demonstrated in vitro and in vivo (23–25, 29). Two recent studies have reported data on the effect of DM modulation/deletion on MHC-II peptidomes. In a study of DR3-expressing T2 cells transfected with different levels of DM, Alvaro-Benito et al. found that higher DM expression was associated with presentation of MHC-II-bound peptides with lower predicted MHC-II binding affinity (51). In a study comparing the peptide repertoires of 293T cells transfected with HLA-DQ molecules differentially associated with type 1 diabetes, Zhou et al. found that in the absence of DM, peptides with lower binding affinity and faster dissociation were presented (76). These studies agree with our data with respect to DM-sensitive epitopes, in which peptides eluted only in the presence of DO were determined to be more sensitive to DM-mediated exchange.
At the cellular level, the results of this study indicate that DO exerts a selective effect on the MHC-II peptide repertoire, such that certain epitopes are presented only when DO is expressed, whereas other abundant peptide species are presented at lower density. Although this work has delineated the overall effect of DO with respect to the MHC-II peptidome, these data also provide a mechanistic basis for epitope studies in which differing effects of DO have been demonstrated (9, 11, 13, 24, 25, 28, 30–33, 72, 73). Results of our analysis suggest that depending on the specific features of the epitope examined, including whether it is DM-sensitive or -resistant as well as its abundance, DO expression can result in greater presentation, little change, or lesser presentation of a peptide species. In the context of the larger scope of the function of DO in antigen presentation, these data may also explain previous observations observed in settings in which DO expression has been modulated. Mice lacking DO have been shown to bear an autoimmune phenotype (3), and ectopic expression of DO in DCs was shown to result in prevention of diabetes in NOD mice (14). Based on our analysis, we posit that DO expression serves to prevent autoimmunity by allowing for deletion of autoreactive clonotypes in the thymus as well as by mediating presentation of a broad spectrum of self-antigens to promote peripheral tolerance. DO expression is downregulated relative to DM following exposure to inflammatory stimuli (7, 8, 10–12), and the consequent focusing of the MHC-II peptidome on immunodominant (i.e. DM-resistant (19, 21, 22)) epitopes may allow for a more efficient immune response. Such a role for DO regulation has been borne out in studies in which lack of DO has been shown to confer resistance to retrovirus as a result of neutralizing antibody production (1), as well as to enable preferential entry into germinal centers (2). Cell-specific expression of DO, which is then downregulated with the onset of inflammation, could allow for shifts in the MHC peptidome that serve the purpose of promoting tolerance (when DO is expressed) and generating an efficient immune response (when DO is downregulated).
In summary, this work defines a role for DO in regulating the MHC-II peptidome by effectively increasing the breadth of peptides presented to CD4 T cells and by modulating epitope density. Restricted and regulated expression of DO suggest the immune system has evolved to allow for presentation of an optimal MHC-II peptide repertoire to both promote tolerance and initiate an efficient immune response.
Data Availability
All raw files have been deposited at MassIVE data repository (http://massive.ucsd.edu) developed by Center for Computational Mass Spectrometry (University of California, San Diego) with the project accession MSV000082570 (PXD010301).
Supplementary Material
Acknowledgments
We thank Wei Jiang and Elizabeth Mellins (Stanford University School of Medicine) for initial characterization of the DO-KO cell lines; Pia Negroni, Liying Lu, Jeff Colbert, and Peter Trenh for experimental and computational assistance, Sharlene Hubbard for assistance with mouse husbandry, and the efforts of the UMass Medical School Mass Spectrometry Facility and the Broad Institute Genomics Services.
Footnotes
* This work was supported by NIH grants R01-AI38996, R01-AI127869, and T32-AI07349.
 This article contains supplemental Figures and Tables.
This article contains supplemental Figures and Tables.
The authors declare no competing interests.
1 The abbreviations used are:
- DO-KO
- DO-knockout
- sgRNA
- single guide.
REFERENCES
- 1. Denzin L. K., Khan A. A., Virdis F., Wilks J., Kane M., Beilinson H. A., Dikiy S., Case L. K., Roopenian D., Witkowski M., Chervonsky A. V., and Golovkina T. V. (2017) Neutralizing Antibody Responses to Viral Infections Are Linked to the Non-classical MHC Class II Gene H2-Ob. Immunity 47, 310–322 e317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Draghi N. A., and Denzin L. K. (2010) H2-O, a MHC class II-like protein, sets a threshold for B-cell entry into germinal centers. Proc. Natl. Acad. Sci. U.S.A. 107, 16607–16612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gu Y., Jensen P. E., and Chen X. (2013) Immunodeficiency and autoimmunity in H2-O-deficient mice. J. Immunol. 190, 126–137 [DOI] [PubMed] [Google Scholar]
- 4. Hake S. B., Tobin H. M., Steimle V., and Denzin L. K. (2003) Comparison of the transcriptional regulation of classical and non-classical MHC class II genes. Eur. J. Immunol. 33, 2361–2371 [DOI] [PubMed] [Google Scholar]
- 5. Chen X., and Jensen P. E. (2004) The expression of HLA-DO (H2-O) in B lymphocytes. Immunol. Res. 29, 19–28 [DOI] [PubMed] [Google Scholar]
- 6. Douek D. C., and Altmann D. M. (1997) HLA-DO is an intracellular class II molecule with distinctive thymic expression. Int. Immunol. 9, 355–364 [DOI] [PubMed] [Google Scholar]
- 7. Fallas J. L., Yi W., Draghi N. A., O'Rourke H. M., and Denzin L. K. (2007) Expression patterns of H2-O in mouse B cells and dendritic cells correlate with cell function. J. Immunol. 178, 1488–1497 [DOI] [PubMed] [Google Scholar]
- 8. Alfonso C., Williams G. S., Han J. O., Westberg J. A., Winqvist O., and Karlsson L. (2003) Analysis of H2-O influence on antigen presentation by B cells. J. Immunol. 171, 2331–2337 [DOI] [PubMed] [Google Scholar]
- 9. Fallas J. L., Tobin H. M., Lou O., Guo D., Sant'Angelo D. B., and Denzin L. K. (2004) Ectopic expression of HLA-DO in mouse dendritic cells diminishes MHC class II antigen presentation. J. Immunol. 173, 1549–1560 [DOI] [PubMed] [Google Scholar]
- 10. Chen X., Reed-Loisel L. M., Karlsson L., and Jensen P. E. (2006) H2-O expression in primary dendritic cells. J. Immunol. 176, 3548–3556 [DOI] [PubMed] [Google Scholar]
- 11. Chen X., Laur O., Kambayashi T., Li S., Bray R. A., Weber D. A., Karlsson L., and Jensen P. E. (2002) Regulated expression of human histocompatibility leukocyte antigen (HLA)-DO during antigen-dependent and antigen-independent phases of B cell development. J. Exp. Med. 195, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hornell T. M., Burster T., Jahnsen F. L., Pashine A., Ochoa M. T., Harding J. J., Macaubas C., Lee A. W., Modlin R. L., and Mellins E. D. (2006) Human dendritic cell expression of HLA-DO is subset specific and regulated by maturation. J. Immunol. 176, 3536–3547 [DOI] [PubMed] [Google Scholar]
- 13. Glazier K. S., Hake S. B., Tobin H. M., Chadburn A., Schattner E. J., and Denzin L. K. (2002) Germinal center B cells regulate their capability to present antigen by modulation of HLA-DO. J. Exp. Med. 195, 1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yi W., Seth N. P., Martillotti T., Wucherpfennig K. W., Sant'Angelo D. B., and Denzin L. K. (2010) Targeted regulation of self-peptide presentation prevents type I diabetes in mice without disrupting general immunocompetence. J. Clin. Invest. 120, 1324–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perraudeau M., Taylor P. R., Stauss H. J., Lindstedt R., Bygrave A. E., Pappin D. J., Ellmerich S., Whitten A., Rahman D., Canas B., Walport M. J., Botto M., and Altmann D. M. (2000) Altered major histocompatibility complex class II peptide loading in H2-O-deficient mice. Eur. J. Immunol. 30, 2871–2880 [DOI] [PubMed] [Google Scholar]
- 16. Mellins E. D., and Stern L. J. (2014) HLA-DM and HLA-DO, key regulators of MHC-II processing and presentation. Curr. Opin. Immunol. 26, 115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pos W., Sethi D. K., Call M. J., Schulze M. S., Anders A. K., Pyrdol J., and Wucherpfennig K. W. (2012) Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell 151, 1557–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sloan V. S., Cameron P., Porter G., Gammon M., Amaya M., Mellins E., and Zaller D. M. (1995) Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature 375, 802–806 [DOI] [PubMed] [Google Scholar]
- 19. Lovitch S. B., Petzold S. J., and Unanue E. R. (2003) Cutting edge: H-2DM is responsible for the large differences in presentation among peptides selected by I-Ak during antigen processing. J. Immunol. 171, 2183–2186 [DOI] [PubMed] [Google Scholar]
- 20. Pos W., Sethi D. K., and Wucherpfennig K. W. (2013) Mechanisms of peptide repertoire selection by HLA-DM. Trends Immunol. 34, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sant A. J., Chaves F. A., Leddon S. A., and Tung J. (2013) The control of the specificity of CD4 T cell responses: thresholds, breakpoints, and ceilings. Front. Immunol. 4, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin L., Calvo-Calle J. M., Dominguez-Amorocho O., and Stern L. J. (2012) HLA-DM constrains epitope selection in the human CD4 T cell response to vaccinia virus by favoring the presentation of peptides with longer HLA-DM-mediated half-lives. J. Immunol. 189, 3983–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guce A. I., Mortimer S. E., Yoon T., Painter C. A., Jiang W., Mellins E. D., and Stern L. J. (2013) HLA-DO acts as a substrate mimic to inhibit HLA-DM by a competitive mechanism. Nat. Struct. Mol. Biol. 20, 90–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kropshofer H., Vogt A. B., Thery C., Armandola E. A., Li B. C., Moldenhauer G., Amigorena S., and Hammerling G. J. (1998) A role for HLA-DO as a co-chaperone of HLA-DM in peptide loading of MHC class II molecules. EMBO J. 17, 2971–2981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Denzin L. K., Sant'Angelo D. B., Hammond C., Surman M. J., and Cresswell P. (1997) Negative regulation by HLA-DO of MHC class II-restricted antigen processing. Science 278, 106–109 [DOI] [PubMed] [Google Scholar]
- 26. Denzin L. K., Fallas J. L., Prendes M., and Yi W. (2005) Right place, right time, right peptide: DO keeps DM focused. Immunol. Rev. 207, 279–292 [DOI] [PubMed] [Google Scholar]
- 27. Jiang W., Strohman M. J., Somasundaram S., Ayyangar S., Hou T., Wang N., and Mellins E. D. (2015) pH-susceptibility of HLA-DO tunes DO/DM ratios to regulate HLA-DM catalytic activity. Sci. Rep. 5, 17333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liljedahl M., Winqvist O., Surh C. D., Wong P., Ngo K., Teyton L., Peterson P. A., Brunmark A., Rudensky A. Y., Fung-Leung W. P., and Karlsson L. (1998) Altered antigen presentation in mice lacking H2-O. Immunity 8, 233–243 [DOI] [PubMed] [Google Scholar]
- 29. Yoon T., Macmillan H., Mortimer S. E., Jiang W., Rinderknecht C. H., Stern L. J., and Mellins E. D. (2012) Mapping the HLA-DO/HLA-DM complex by FRET and mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 109, 11276–11281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kremer A. N., van der Meijden E. D., Honders M. W., Goeman J. J., Wiertz E. J., Falkenburg J. H., and Griffioen M. (2012) Endogenous HLA class II epitopes that are immunogenic in vivo show distinct behavior toward HLA-DM and its natural inhibitor HLA-DO. Blood 120, 3246–3255 [DOI] [PubMed] [Google Scholar]
- 31. Poluektov Y. O., Kim A., Hartman I. Z., and Sadegh-Nasseri S. (2013) HLA-DO as the optimizer of epitope selection for MHC class II antigen presentation. PLoS ONE 8, e71228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bellemare-Pelletier A., Tremblay J., Beaulieu S., Boulassel M. R., Routy J. P., Massie B., Lapointe R., and Thibodeau J. (2005) HLA-DO transduced in human monocyte-derived dendritic cells modulates MHC class II antigen processing. J. Leukoc. Biol. 78, 95–105 [DOI] [PubMed] [Google Scholar]
- 33. van Ham M., van Lith M., Lillemeier B., Tjin E., Gruneberg U., Rahman D., Pastoors L., van Meijgaarden K., Roucard C., Trowsdale J., Ottenhoff T., Pappin D., and Neefjes J. (2000) Modulation of the major histocompatibility complex class II-associated peptide repertoire by human histocompatibility leukocyte antigen (HLA)-DO. J. Exp. Med. 191, 1127–1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., and Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., and Zhang F. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chicz R. M., Urban R. G., Lane W. S., Gorga J. C., Stern L. J., Vignali D. A., and Strominger J. L. (1992) Predominant naturally processed peptides bound to HLA-DR1 are derived from MHC-related molecules and are heterogeneous in size. Nature 358, 764–768 [DOI] [PubMed] [Google Scholar]
- 37. Gorga J. C., Horejsi V., Johnson D. R., Raghupathy R., and Strominger J. L. (1987) Purification and characterization of class II histocompatibility antigens from a homozygous human B cell line. J. Biol. Chem. 262, 16087–16094 [PubMed] [Google Scholar]
- 38. Yin L., Trenh P., Guce A., Wieczorek M., Lange S., Sticht J., Jiang W., Bylsma M., Mellins E. D., Freund C., and Stern L. J. (2014) Susceptibility to HLA-DM protein is determined by a dynamic conformation of major histocompatibility complex class II molecule bound with peptide. J. Biol. Chem. 289, 23449–23464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolpl A., Halder T., Kalbacher H., Neumeyer H., Siemoneit K., Goldmann S. F., and Eiermann T. H. (1998) Human monoclonal antibody with T-cell-like specificity recognizes MHC class I self-peptide presented by HLA-DR1 on activated cells. Tissue Antigens 51, 258–269 [DOI] [PubMed] [Google Scholar]
- 40. Li B., and Dewey C. N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmead B., and Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karolchik D., Hinrichs A. S., Furey T. S., Roskin K. M., Sugnet C. W., Haussler D., and Kent W. J. (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., and Haussler D. (2002) The human genome browser at UCSC. Genome Res. 12, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 45. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jensen K. K., Andreatta M., Marcatili P., Buus S., Greenbaum J. A., Yan Z., Sette A., Peters B., and Nielsen M. (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Calvo-Calle J. M., Strug I., Nastke M. D., Baker S. P., and Stern L. J. (2007) Human CD4+ T cell epitopes from vaccinia virus induced by vaccination or infection. PLoS Pathog. 3, 1511–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Neilson K. A., Ali N. A., Muralidharan S., Mirzaei M., Mariani M., Assadourian G., Lee A., van Sluyter S. C., and Haynes P. A. (2011) Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 [DOI] [PubMed] [Google Scholar]
- 49. Gamez-Pozo A., Sanchez-Navarro I., Calvo E., Agullo-Ortuno M. T., Lopez-Vacas R., Diaz E., Camafeita E., Nistal M., Madero R., Espinosa E., Lopez J. A., and Fresno Vara J. A. (2012) PTRF/cavin-1 and MIF proteins are identified as non-small cell lung cancer biomarkers by label-free proteomics. PLoS ONE 7, e33752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mueller L. N., Brusniak M. Y., Mani D. R., and Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 [DOI] [PubMed] [Google Scholar]
- 51. Alvaro-Benito M., Morrison E., Abualrous E. T., Kuropka B., and Freund C. (2018) Quantification of HLA-DM-Dependent Major Histocompatibility Complex of Class II Immunopeptidomes by the Peptide Landscape Antigenic Epitope Alignment Utility. Front Immunol. 9, 872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lemeer S., Hahne H., Pachl F., and Kuster B. (2012) Software tools for MS-based quantitative proteomics: a brief overview. Methods Mol. Biol. 893, 489–499 [DOI] [PubMed] [Google Scholar]
- 53. Stern L. J., and Wiley D. C. (1992) The human class II MHC protein HLA-DR1 assembles as empty alpha beta heterodimers in the absence of antigenic peptide. Cell 68, 465–477 [DOI] [PubMed] [Google Scholar]
- 54. Denzin L. K., and Cresswell P. (1995) HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell 82, 155–165 [DOI] [PubMed] [Google Scholar]
- 55. Rudensky A., Rath S., Preston-Hurlburt P., Murphy D. B., and Janeway C. A. Jr. (1991) On the complexity of self. Nature 353, 660–662 [DOI] [PubMed] [Google Scholar]
- 56. Clement C. C., Becerra A., Yin L., Zolla V., Huang L., Merlin S., Follenzi A., Shaffer S. A., Stern L. J., and Santambrogio L. (2016) The Dendritic Cell Major Histocompatibility Complex II (MHC II) Peptidome Derives from a Variety of Processing Pathways and Includes Peptides with a Broad Spectrum of HLA-DM Sensitivity. J. Biol. Chem. 291, 5576–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yin L., and Stern L. J. (2014) Measurement of Peptide Binding to MHC Class II Molecules by Fluorescence Polarization. Curr. Protoc. Immunol. 106, 5 10, 11–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yin L., and Stern L. J. (2014) A novel method to measure HLA-DM-susceptibility of peptides bound to MHC class II molecules based on peptide binding competition assay and differential IC(50) determination. J. Immunol. Methods 406, 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fugmann T., Sofron A., Ritz D., Bootz F., and Neri D. (2017) The MHC Class II Immunopeptidome of Lymph Nodes in Health and in Chemically Induced Colitis. J. Immunol. 198, 1357–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ritz D., Kinzi J., Neri D., and Fugmann T. (2017) Data-Independent Acquisition of HLA Class I Peptidomes on the Q Exactive Mass Spectrometer Platform. Proteomics 17, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stern L. J., and Santambrogio L. (2016) The melting pot of the MHC II peptidome. Curr. Opin. Immunol. 40, 70–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Andreatta M., Karosiene E., Rasmussen M., Stryhn A., Buus S., and Nielsen M. (2015) Accurate pan-specific prediction of peptide-MHC class II binding affinity with improved binding core identification. Immunogenetics 67, 641–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jost L. (2006) Entropy and diversity. Oikos 113, 363–375 [Google Scholar]
- 64. Gotelli N. J., and Colwell R. K. (2010) Estimating species richness. Biological Diversity: Frontiers In Measurement And Assessment, ed Magurran and McGill (Oxford University Press, Oxford: ), 39–54 [Google Scholar]
- 65. Lai X., Wang L., and Witzmann F. A. (2013) Issues and applications in label-free quantitative mass spectrometry. Int. J. Proteomics 2013, 756039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jahnke M., Trowsdale J., and Kelly A. P. (2013) Ubiquitination of HLA-DO by MARCH family E3 ligases. Eur. J. Immunol. 43, 1153–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xiu F., Cote M. H., Bourgeois-Daigneault M. C., Brunet A., Gauvreau M. E., Shaw A., and Thibodeau J. (2011) Cutting edge: HLA-DO impairs the incorporation of HLA-DM into exosomes. J. Immunol. 187, 1547–1551 [DOI] [PubMed] [Google Scholar]
- 68. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., and Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. The Gene Ontology C. (2017) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 45, D331–D338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim A., Hartman I. Z., Poore B., Boronina T., Cole R. N., Song N., Ciudad M. T., Caspi R. R., Jaraquemada D., and Sadegh-Nasseri S. (2014) Divergent paths for the selection of immunodominant epitopes from distinct antigenic sources. Nat. Commun. 5, 5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ferrante A., Anderson M. W., Klug C. S., and Gorski J. (2008) HLA-DM mediates epitope selection by a “compare-exchange” mechanism when a potential peptide pool is available. PLoS ONE 3, e3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Alfonso C., Williams G. S., and Karlsson L. (2003) H2-O influence on antigen presentation in H2-E-expressing mice. Eur. J. Immunol. 33, 2014–2021 [DOI] [PubMed] [Google Scholar]
- 73. Brocke P., Armandola E., Garbi N., and Hammerling G. J. (2003) Downmodulation of antigen presentation by H2-O in B cell lines and primary B lymphocytes. Eur. J. Immunol. 33, 411–421 [DOI] [PubMed] [Google Scholar]
- 74. Hou T., Macmillan H., Chen Z., Keech C. L., Jin X., Sidney J., Strohman M., Yoon T., and Mellins E. D. (2011) An insertion mutant in DQA1*0501 restores susceptibility to HLA-DM: implications for disease associations. J. Immunol. 187, 2442–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nguyen T. B., Jayaraman P., Bergseng E., Madhusudhan M. S., Kim C. Y., and Sollid L. M. (2017) Unraveling the structural basis for the unusually rich association of human leukocyte antigen DQ2.5 with class-II-associated invariant chain peptides. J. Biol. Chem. 292, 9218–9228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhou Z., Reyes-Vargas E., Escobar H., Chang K. Y., Barker A. P., Rockwood A. L., Delgado J. C., He X., and Jensen P. E. (2017) Peptidomic analysis of type 1 diabetes associated HLA-DQ molecules and the impact of HLA-DM on peptide repertoire editing. Eur. J. Immunol. 47, 314–326 [DOI] [PubMed] [Google Scholar]
- 77. Rost H. L., Rosenberger G., Navarro P., Gillet L., Miladinovic S. M., Schubert O. T., Wolski W., Collins B. C., Malmstrom J., Malmstrom L., and Aebersold R. (2014) OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 32, 219–223 [DOI] [PubMed] [Google Scholar]
- 78. Rock K. L., Reits E., and Neefjes J. (2016) Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol. 37, 724–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Denzin L. K., Robbins N. F., Carboy-Newcomb C., and Cresswell P. (1994) Assembly and intracellular transport of HLA-DM and correction of the class II antigen-processing defect in T2 cells. Immunity 1, 595–606 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw files have been deposited at MassIVE data repository (http://massive.ucsd.edu) developed by Center for Computational Mass Spectrometry (University of California, San Diego) with the project accession MSV000082570 (PXD010301).