

The anti-neoplastic sphingolipid analog SH-BC-893 starves cancer cells to death by down-regulating cell surface nutrient transporters and blocking lysosomal trafficking events. However, the actual mechanism of action giving rise to these phenotypes remains unclear. Here, dynamic phosphoproteomics was used to further understand how the activity of PP2A is affected following cell treatment with SH-BC-893. These analyses combined with functional assays identified the differential regulation of Akt and Gsk3b by SH-BC-893 and C2-ceramide as responsible for the vacuolation of cells by SH-BC-893 but not C2-ceramide.
Keywords: Phosphoproteome, Phosphorylation, Quantification, Cancer Biology*, Cell biology*
Graphical Abstract

Highlights
Quantitative phosphoproteomics of cells treated with sphingolipid analogs or PP2A inhibitor identify novel protein targets of PP2A.
PP2A substrates include several nutrient transporter proteins, GTPase regulators and proteins associated with actin cytoskeletal remodeling.
Differential regulation of Akt and Gsk3b account for the difference in vacuolating phenotype observed between SH-BC-893 and C2-ceramide.
Dynamic phosphoproteomics enabled the correlation of cell signaling with phenotypes to rationalize their mode of action.
Abstract
The anti-neoplastic sphingolipid analog SH-BC-893 starves cancer cells to death by down-regulating cell surface nutrient transporters and blocking lysosomal trafficking events. These effects are mediated by the activation of protein phosphatase 2A (PP2A). To identify putative PP2A substrates, we used quantitative phosphoproteomics to profile the temporal changes in protein phosphorylation in FL5.12 cells following incubation with SH-BC-893 or the specific PP2A inhibitor LB-100. These analyses enabled the profiling of more than 15,000 phosphorylation sites, of which 958 sites on 644 proteins were dynamically regulated. We identified 114 putative PP2A substrates including several nutrient transporter proteins, GTPase regulators (e.g. Agap2, Git1), and proteins associated with actin cytoskeletal remodeling (e.g. Vim, Pxn). To identify SH-BC-893-induced cell signaling events that disrupt lysosomal trafficking, we compared phosphorylation profiles in cells treated with SH-BC-893 or C2-ceramide, a non-vacuolating sphingolipid that does not impair lysosomal fusion. These analyses combined with functional assays uncovered the differential regulation of Akt and Gsk3b by SH-BC-893 (vacuolating) and C2-ceramide (non-vacuolating). Dynamic phosphoproteomics of cells treated with compounds affecting PP2A activity thus enabled the correlation of cell signaling with phenotypes to rationalize their mode of action.
Oncogenic mutations selected during the tumorigenic process rewire the metabolic circuitry to meet the increased anabolic demands of cancer cells. Because oncogenic mutations constitutively drive growth and proliferation, cancer cells depend on a steady influx of nutrients via cell surface transporters and receptors and on the lysosomal degradation of internalized macromolecules into subunits that can be used for biosynthesis and/or the production of ATP (1). Because cancer cells are constitutively anabolic, they are unable to tolerate nutrient stress that causes quiescence and catabolism in normal cells. Restricting nutrient access using sphingolipid-inspired compounds is an appealing therapeutic strategy to impede cancer cell proliferation and survival. Previous reports indicated that endogenous and synthetic sphingolipids starve many different cancer cell types to death by triggering the down-regulation of multiple nutrient transporter proteins and/or blocking lysosomal fusion reactions (2–7).
In mammalian cells, ceramides can function as tumor suppressors, mediating signaling events associated with apoptosis, autophagic responses and cell cycle arrest (8). Several sphingolipids activate protein phosphatase 2A (PP2A)1 and negatively regulate multiple signaling pathways that promote nutrient transporter expression (5, 9–13). Although the mechanism underlying sphingolipid regulation of PP2A activity is not entirely clear, previous reports suggest that ceramides can bind to endogenous protein inhibitors of PP2A to enhance its catalytic activity (13). Interestingly, although Fingolimod (FTY720, Gilenya), pyrrolidine analogs such as SH-BC-893, and ceramide all induce nutrient transporter down-regulation downstream of PP2A activation, only FTY720 and SH-BC-893 produce PP2A-dependent cytoplasmic vacuolation (5). Ceramide, on the other hand, produces distinct effects from FTY720 and SH-BC-893 on the tubular recycling endosome, although whether these effects are PP2A-dependent is less certain (5, 14). These observations suggest that these structurally-related molecules differentially activate PP2A, resulting in distinct patterns of dephosphorylation and different endolysosomal trafficking phenotypes.
To determine how PP2A activity induces nutrient transporter loss and cytosolic vacuolation, we profiled the dynamic changes in protein phosphorylation in the murine prolymphocytic cell line FL5.12 following incubation with SH-BC-893, the specific PP2A inhibitor LB-100, or C2-ceramide. Metabolic labeling and quantitative phosphoproteomics (15–17) identified kinetic profiles that could be correlated with putative PP2A substrates. This approach identified 15,607 phosphorylation sites, of which 958 were dynamically regulated by the treatments. Although 265 putative PP2A sites were common to both PP2A agonists, our analyses also revealed 467 sites uniquely regulated by either SH-BC-893 or C2-ceramide that provided further insights into the SH-BC-893-specific phenotype, vacuolation.
EXPERIMENTAL PROCEDURES
Cell Culture
FL5.12 cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 10 mm HEPES buffer, 55 μm 2-mercaptoethanol, 2 mm l-glutamine, 500 pg/ml murine recombinant IL-3, and antibiotics. HeLa cells were cultured in DMEM with 4.5 g/L glucose and l-glutamine supplemented with 10% FBS and antibiotics. For proteomic analyses FL5.12 cells were grown in triple SILAC S.D.-Media (Thermo Fisher Scientific, Rockford, IL) containing 10% FBS, 500 pg/ml murine recombinant IL-3, 164 μm Lysine (K), 95 μm Arginine (R), 4.3 μm proline (Silantes, Munich, Germany) with additional nutrients consistent with Bendall et al. (18). Cells were incubated at 37 °C and 5% CO2. Cells were counted using a Leica microscope with a 10 × 0.25 objective. Approximately 500 million cells per SILAC channel were grown in 500 ml spinner flasks. Incubation with small molecules was performed by adding 1 ml of small molecule or DMSO (Sigma Aldrich Co., St-Louis, MI) diluted in SILAC RPMI 1640/10% FBS to reach the final concentration. Cells were harvested every 5 min during the first hour of treatment with either 5 μm SH-BC-893 (heavy label) or 50 μm C2-ceramide (medium label) or 10 μm LB-100 (medium label) or DMSO (light label). Drug concentrations used for treatments are based on previously published references (5, 7, 19). Cells were collected by pipetting 75 ml (25 ml per SILAC channel) of culture into 425 ml of −80 °C precooled ethanol.
Flow Cytometry
For quantification of cell surface CD98, 400,000 FL5.12 cells were stained with either PE-conjugated rat IgG2a k isotype control (Biolegend, cat. no. 400508, San Diego, CA) or PE-conjugated rat anti-mouse CD98 (Biolegend, cat. no. 128208) in blocking buffer (PBS with 10% FBS and 0.05% sodium azide) for 30 min on ice. Cells were washed twice in ice-cold wash buffer (PBS with 2% FCS and 0.05% sodium azide), resuspended in wash buffer containing 1 μg/ml DAPI, and returned to ice. Analysis was restricted to live cells (DAPI-negative) and data normalized to the untreated control after background subtraction.
Vacuolation Assay
Vacuoles were quantified as in Perryman et al. (20). Briefly, FL5.12 cells were treated as indicated for 3 h then pelleted by centrifuging at 2000 rpm for 3 min in a microfuge. The cell pellet was resuspended in 10 μl, and 3 μl of the cell suspension was examined under a 0.13 to 0.17 mm thick coverglass (VWR Micro cover glass, square, No.1, cat. no. 48366–045, Radnor, PA) on a microscope slide. FL5.12 cells were evaluated by differential interference microscopy (DIC) using a 100× oil-immersion objective with a Nikon TE2000-S fluorescence microscope. To calculate the vacuolation score, at least 10 different fields of view containing 5–12 cells per field were assessed. Scores were assigned to individual cells as follows: 0 = no vacuoles, 1 = very small vacuoles, 2 = multiple well-defined vacuoles, 3 = multiple large vacuoles. To calculate the vacuolation score, the following formula was used:
Fluorescence Microscopy
To monitor F-actin, FL5.12 cells were fixed with 4% paraformaldehyde for 10 min, washed, and incubated in permeabilization and blocking buffer (PBS with 10% FBS, 0.3% saponin, and 0.05% NaN3) containing Alexa Fluor 488 conjugated phalloidin (Cell Signaling Technologies, 1:100 dilution, Danvers, MA). Cells were then imaged on a Nikon Eclipse Ti spinning-disk confocal microscope using a 100× oil-immersion objective. Images were analyzed using ImageJ; fluorescence intensity was normalized between all images and displayed using the LUT “16 colors.”
Immunoblotting
Cells were lysed in RIPA buffer containing protease (Thermo Fisher Scientific, Cat #88265) and phosphatase inhibitors (Sigma, Cat #4906845001). Protein concentration was quantified using a BCA protein assay (Thermo Scientific, Cat #23223). Samples were prepared in NuPAGE sample buffer (Thermo Fisher Scientific, Cat #NP0007) and 50 mm DTT, run on Invitrogen NuPAGE 4–12% Bis-Tris gels (Cat #NP0336BOX), and transferred to BioTrace NT nitrocellulose membranes (Pall, Cat #66485, Ville St. Laurent, Quebec, Canada). After transfer, membranes were dried at room temperature for 30 min, incubated in blocking solution (5% bovine serum albumin, 0.05% NaN3 in TBST) for 1 h, and then incubated overnight at 4 °C in blocking solution containing primary antibody. Blots were washed three times in TBST (TBS with 0.1% Tween 20), incubated at RT for 1 h in blocking solution containing IRDye-conjugated secondary antibodies (1:10,000), and then washed three times in TBST. Blots were imaged using a LI-COR Odyssey CLx imaging system.
Digestion and Desalting of Cell Extracts
Heavy, medium and light cells from each time point were combined in 500 ml collection tubes (Sorvall centrifuge bottles), separated by centrifugation at 1,200 rpm (Sorvall Legend RT centrifuge) and washed first with 35 ml PBS, then 5 ml PBS, and finally 3 times with 1 ml PBS while transferring from 500 ml collection tubes to 50 ml falcon tubes to 1.7 ml Eppendorf tubes, respectively. Cell lysis was performed by sonication for 15 s with a sonic dismembrator (Thermo Fisher Scientific) after adding 1 ml lysis buffer (8 m Urea, 50 mm TRIS, pH 8, HALT phosphatase inhibitor (Thermo Fisher Scientific). Cells were maintained at 0 °C to prevent protein degradation. Protein concentration was measured by BCA assay (Thermo Fisher Scientific). Protein disulfide bonds were reduced by incubating lysates in 5 mm dithiothreitol (DTT, Sigma-Aldrich) for 30 min at 56 °C while shaking at 1,000 rpm. Alkylation of cysteine residues was achieved by incubation with iodoacetamide (IAA, Sigma-Aldrich) at a concentration of 15 mm for 30 min at RT in the dark. Excess IAA was quenched for 15 min at RT by adding DTT to 5 mm. All samples were diluted 5 times with 20 mm TRIS (Bioshop Burlington, ON, Canada), 1 mm CaCl2, pH 8 and mixed with 2 μg/μl trypsin (Sigma-Aldrich) (protein: trypsin 50:1 w:w) and incubated for 12 h at 37 °C. Trypsin was kept at −80 °C and thawed only once. Desalting was performed on 60 mg solid phase extraction (SPE) reverse phase cartridges (Oasis HLB 3cc cartridge, 60 mg, 30 μm particle size, Waters Mississauga, ON, Canada) previously conditioned with 3 ml methanol, SPE Buffer (50% acetonitrile, ACN, 1% formic acid, FA) and finally with 1% aqueous FA. Peptide samples were loaded, washed with 3 ml 1% FA, eluted in 1 ml SPE buffer and dried on a vacuum centrifuge (Thermo Fisher Scientific) at RT.
Phosphopeptide Enrichment
Phosphopeptide enrichment was performed on 5 μm titansphere particles (Canadian Life Science, Peterborough, ON, Canada) according to published protocols (21, 22). Loading of protein extracts on the titansphere beads, washing, and elution steps were performed using custom spin columns (23) made from 200 μl pipette tip containing a SDB-XC membrane (Empore, 3 m) frit and filled with TiO2 beads. Peptides were desalted in 50 μl of 1% FA and subsequently eluted from spin columns using 50 μl of 50% ACN 0.5% FA.
Offline Strong Cation Exchange Chromatography
To achieve high reproducibility and parallel sample fractionation in high throughput proteomics experiments, we used strong cation fractionation (SCX) on homemade spin columns packed with 18 to 22 mg (for cell lysate of 3 to 8 mg protein extract) of polysulfoethyl A 300Å particle, (Canada Life Science; Peterborough, ON). After equilibrating the SCX particles with each 100 μl of SCX Buffer A (10% ACN/0.2%FA v/v), Buffer B (1 m NaCl in 10% ACN/0.2%FA v/v) and finally 200 μl SCX Buffer A. Peptides were resuspended in 100 μl Buffer A, loaded on the SCX column and eluted in six salt step fractions of 100 μl each with 0, 30, 50, 80, 120 and 500 mm NaCl in SCX Buffer A. Before LC-MS/MS analyses all fractions were dried on a SpeedVac centrifuge at RT (Thermo Fisher Scientific) and resuspended in 4% FA. All centrifugation steps were performed at 4 °C except where indicated.
Mass Spectrometry Analysis
LC-MS/MS analyses were performed on a Q-Exactive HF (SH-BC-893 versus C2-ceramide experiments) or an Orbitrap tribrid Fusion (SH-BC-893 versus LB-100 experiments) mass spectrometer using homemade capillary LC columns (18 cm length, 150 μm ID, 360 μm OD). Capillary LC columns were packed with C18 Jupiter 3 μm particles (Phenomenex, Torrance, CA) at 1,000 psi. Samples were directly injected on LC-columns and separations were performed at a flow rate of 0.6 μl/min using a linear gradient of 5–35% aqueous ACN (0.2% FA) in 150 min. MS spectra were acquired with a resolution of 60,000 using a lock mass (m/z: 445.120025) followed by up to 20 MS/MS data dependent scans on the most intense ions using high energy dissociation (HCD). AGC target values for MS and MS/MS scans were set to 1 × 106 (max fill time 100 ms) and 5 × 105 (max fill time 200 ms), respectively. The precursor isolation window was set to m/z 1.6 with a HCD normalized collision energy of 25. The dynamic exclusion window was set to 20 s.
Data Processing and Analysis
Raw data analysis of SILAC experiments was performed using Maxquant software 1.5.3.8 and the Andromeda search engine (24). The false discovery rate (FDR) for peptide, protein, and site identification was set to 1%, the minimum peptide length was set to 6, and the “peptide requantification” function was enabled. Precursor ions detected as 3D peaks (m/z, intensity and retention time) were considered as peptide features. Precursor mass accuracies were then estimated by MaxQuant in a feature specific fashion as described previously (25). Briefly, mass accuracy of peptide features were determined for each 2D peak (m/z and intensity) across the 3D peak and an intensity weighted bootstrap analysis estimated mass accuracies for each precursor ion. Minimum reporter ion mass accuracy was set to 20 ppm. The option match between runs (2 min time tolerance) was enabled to correlate identification and quantitation results across different runs. Up to 2 missed cleavages per peptide were allowed. To adjust for any systematic quantification errors, SILAC ratios were normalized by time point. All additional parameters are reported in MaxQuant parameters.txt and experimentalDesign.txt provided in supplemental Table S1. The Uniprot mouse proteome database (September 2015 containing 35,281 entries) was used for all database searches. Protein groups were formed by MaxQuant, and all identified peptides were used to make pair-wise comparison between proteins in the database. Proteins containing an equal or overlapping set of peptides were merged together to form a protein group and ranked according to the highest number of peptides.
In addition to an FDR of 1% set for peptide, protein and phosphopeptide identification levels, we used additional criteria to increase data quality. The Andromeda score threshold for the identification of phosphopeptides was set to 40 with a delta score to the second best match of 8, as optimized by Sharma K et al. (27). We selected only peptides for which abundance ratios (FC = Drug/Control) were measured in at least 6 time points. Then we set a cut-off for maximum phosphosite localization confidence across experiments (time points) to 0.75. Next, we distinguished dynamic from static phosphosites by calculating an FDR based on curve fitting as recently described (17). We further performed clustering analysis using the fuzzy means package (28), which is implemented to the R environment (https://cran.r-project.org/).
Gene Ontology (GO) enrichment analyses were performed using the database DAVID version 6.7 (29). Protein interaction networks were defined using the STRING database and interacting proteins were visualized with Cytoscape version 2.8. A protein-protein interaction network was built in STRING version 9.1 for all proteins containing dynamic phosphosites. All interaction predictions were based on experimental evidences with a minimal confidence score of 0.7 (considered as a “high confidence” filter in STRING). Linear motif analysis was done using Motif-x (30, 31). Conservation of phosphorylation sites across species was assessed using the CPhos program (32).
Experimental Design and Statistical Rationale
For time resolved phosphoproteomics experiments, FL5.12 cells were collected from one large cell culture (500 ml) per SILAC channel. Time course experiments (SH-BC-893 and LB-100; SH-BC-893 and C2-ceramide) were conducted on different days. Sample processing for each time course experiment was performed in parallel and from the same batch of materials and chemicals to maximize reproducibility of peptide digestion, phosphopeptide enrichment and fractionation. All time points of each SCX fraction were consecutively loaded onto the LC-MS system to ensure similar peptide coverage for all time points. SCX fractions were injected with increasing salt concentration. All data were analyzed using the same FASTA file (Uniprot Mouse proteome, accessed September 2015, 35,281 entries) and MaxQuant version 1.5.8.3.
Microscopy experiments (e.g. phalloidin staining in Fig. 7A–7C, vacuolation images in Fig. 1C, 7E, and 8A, 8C–8E) were completed in 2–3 biological replicates performed on separate days to account for biological variation. Microscopy images representative of the >100 cells per condition per biological replicate were used in figures. For quantitation of cytoplasmic vacuolation (Fig. 8A, 8C–8E), at least 400 cells per condition from three biological replicates were evaluated and means calculated, error bars show the 95% confidence interval. Statistical significance was evaluated with a Student's t test (two-tailed, paired), as described in each figure legend.
Fig. 7.

C2-ceramide and SH-BC-893 promote actin polymerization. A, FL5.12 cells treated with SH-BC-893 (10 μm) or C2-ceramide (50 μm) for 1 h and then stained for F-actin with Alexa Fluor 488-conjugated phalloidin. B, Same as in A, except cells pre-treated with vehicle or PP2A inhibitors calyculinA (calyA, 5 nm) or LB-100 (100 μm) for 1.5 h before SH-BC-893 addition. C, Same as in A, except cells pre-treated with vehicle or actin polymerization inhibitor latrunculinA (LatA, 1 μm) for 30 min before SH-BC-893 (2.5 μm) or C2-ceramide (30 μm) addition. D, FL5.12 cells pre-treated (30 min) with LatA (1 μm) were treated with SH-BC-893 (2.5 or 5 μm) or C2-ceramide (30 or 50 μm) for 1 h and surface CD98 quantified by flow cytometry. E, FL5.12 cells pre-treated with LatA (30 min, 1 μm) were treated with SH-BC-893 (5 μm) for 2 h and imaged by DIC microscopy to visualize vacuoles. n = 3 in D and n ≥ 30 cells per condition in E. A two-tailed t test was used to compare SH-BC-893 -treated cells to combination of LatA and SH-BC-893, *** = p ≤ 0.001.
Fig. 1.
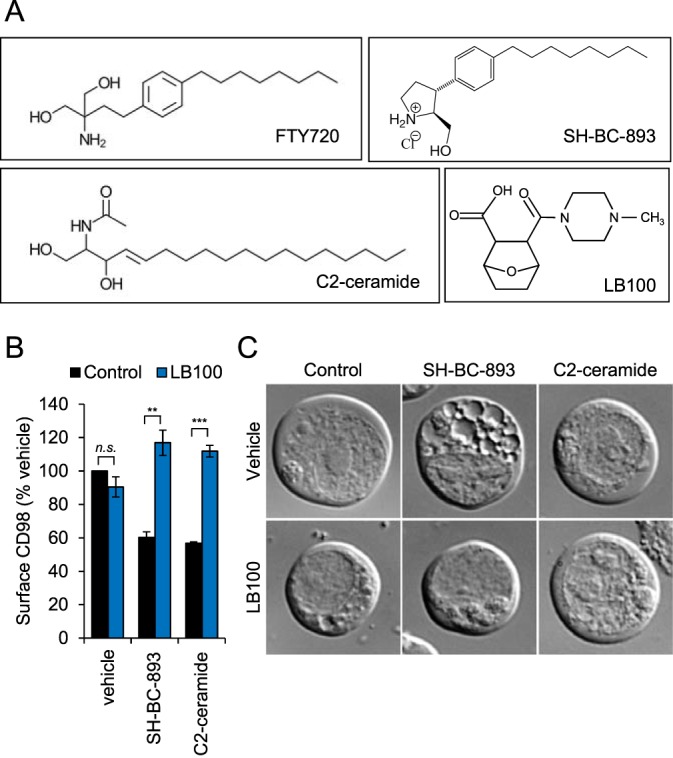
SH-BC-893 and C2-ceramide cause PP2A-dependent disruptions in endolysosomal trafficking. A, Structures of the PP2A activators FTY720 (Fingolimod), C2-ceramide, SH-BC-893 and the PP2A inhibitor LB-100. B, Surface CD98 levels in FL5.12 cells pre-treated with LB-100 (100 μm) for 1.5 h before addition of DMSO, SH-BC-893 (5 μm), or C2-ceramide (50 μm) for 1 h. Using Student's t test (n.s.) not significant; *, p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001 C, DIC microscopy of FL5.12 cells pre-treated with LB-100 (100 μm) for 1.5 h before addition of DMSO, vehicle, SH-BC-893 (5 μm), or C2-ceramide (50 μm) for 2 h.
Fig. 8.

Ceramide inhibits vacuolation by reducing Akt activity. A, FL5.12 cells were treated for 3 h with SH-BC-893 (2.5 μm) and/or C2-ceramide (50 μm) as indicated and imaged by DIC microscopy. B, Western blotting measuring the phosphorylation of the AKT substrate PRAS40 (total and phospho-Thr246) in FL5.12 cells treated for 30 min with SH-BC-893 (5 μm), C2-ceramide (50 μm), and/or MK2206 (1 μm) as indicated. C, FL5.12 cells treated for 3 h with SH-BC-893 (2.5 μm) and/or AKT inhibitor MK2206 (1 μm) as indicated and visualized as in (A). D, Rictor WT and KO murine embryonic fibroblasts (MEFs) treated for 6 h with vehicle or SH-BC-893 (5 μm) then visualized by phase contrast microscopy. E, FL5.12 cells treated for 3 h with SH-BC-893 (2.5 μm) or C2-ceramide (50 μm) and the Gsk3b inhibitor CHIR99021 (10 μm) as indicated; visualized as in (A). In (A, C, E), ≥ 400 cells per condition were analyzed from a total of three independent experiments; using a two-tailed t test to compare either SH-BC-893 or C2-ceramide to the vehicle control (in E) or SH-BC-893 alone to the combination of SH-BC-893 with C2-ceramide (A), MK2206 (C), or CHIR99021 (E), n.s. = not significant, ** = p ≤ 0.01, or *** = p ≤ 0.001.
For flow cytometry experiments quantifying the surface levels of CD98 (Fig. 1B and 7D), at least three biological replicates were performed on different days to account for biological variation. Surface CD98 levels are normalized to vehicle within each experiment, and the normalized values are averaged. Variation is shown as standard error of the mean and statistical significance was assessed by Student's t test (two-tailed, paired), as described in the figure legends.
RESULTS
SH-BC-893 and C2-ceramide Produce Distinct, PP2A-dependent Alterations in Endolysosomal Trafficking
Ceramide is an endogenous tumor suppressor lipid that limits cellular growth and proliferation (7, 8). Despite its antineoplastic actions, ceramide cannot be used to treat cancer patients because it is readily metabolized into inactive forms. Although the synthetic sphingolipid FTY720 overcomes this problem, it also produces dose-limiting bradycardia at the concentrations required for tumor suppression by activating the sphingosine-1phosphate receptor 1(S1PR1) (33, 34). SH-BC-893 is a structurally constrained analog of FTY720 that limits tumor growth in multiple model systems, including prostate and colorectal cancer, without activating S1PR1 (5, 20, 34, 35). Both SH-BC-893 and C2-ceramide (N-Acetyl-sphingosine, Fig. 1A) suppress cell growth and survival in part by down-regulating nutrient transporter proteins through the activation of the cytosolic serine/threonine protein phosphatase PP2A (5, 7, 14, 34). The cell surface antigen 4F2 heavy chain (solute carrier family 3 member 2, Slc3a2 or CD98) is a chaperone required for the surface expression of several associated light chains that transport amino-acids (36). Incubation with either 2.5 μm SH-BC-893 or 50 μm C2-ceramide resulted in a 40% down-regulation of 4F2hc that was reversed by the selective PP2A inhibitor LB-100 (Fig. 1B). These observations are consistent with published studies in multiple cell types (5, 7, 14, 20, 34). In addition to limiting cell surface CD98 expression, SH-BC-893, but not ceramide, prevented the fusion of endocytic vesicles and autophagosomes with lysosomes resulting in enlarged multivesicular bodies, MVB (5); LB-100 reversed this effect (Fig. 1C). These results confirm that disruptions in endolysosomal trafficking that starve cancer cells to death (5) are PP2A-dependent in FL5.12 cells.
Dynamic Phosphoproteomics Deconvolves Signaling Events Associated With Changes in PP2A Activity
To investigate the molecular mechanisms underlying nutrient transporter loss and vacuolation, we profiled the temporal changes in protein phosphorylation in FL5.12 cells treated with SH-BC-893, C2-ceramide, or LB-100 using quantitative phosphoproteomics and metabolic labeling (Fig. 2). Changes in the phosphoproteome associated with SH-BC-893 or LB-100 treatment were analyzed in triple SILAC experiments. LB-100 was selected for these studies as it is more selective toward PP2A than other serine/threonine protein phosphatase inhibitors such as calyculin A or okadaic acid which affect both PP1 and PP2A (37, 38). In a parallel set of experiments, C2-ceramide's effects on the phosphoproteome were compared with those of SH-BC-893 using a similar strategy. Unstimulated cells (0Lys, 0Arg, light), and cells treated with LB-100/C2-ceramide or SH-BC-893 (4Lys, 6Arg, medium or 8Lys, 10Arg, heavy) were collected every 5 min for 60 min and combined together in precooled ethanol (−80 °C) at each time point. Cells were centrifuged, lysed, and digested with trypsin before phosphopeptide enrichment on TiO2 affinity media. The eluted phosphopeptides were subsequently fractionated on strong cation exchange (SCX) spin columns and analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). Phosphopeptide identification and quantification was performed using MaxQuant (25, 39) and kinetic trends and clusters identified using R (http://www.r-project.org/). A total of 15,607 unique phosphopeptides were identified corresponding to an average phosphopeptide enrichment level of 85% for all experiments. SILAC experiments enabled the identification of 12,137 and 9738 unique phosphopeptides on 2949 and 2550 proteins for LB-100/SH-BC-893 and C2-ceramide/SH-BC-893 experiments, respectively (supplemental Table S2). On average, 70% of phosphosites were localized with high confidence (8559 sites for LB-100/SH-BC-893 and 6704 sites for C2-ceramide/SH-BC-893). Closer examination of these data revealed that 3,928 phosphosites in LB-100/SH-BC-893 and 2,919 phosphosites in C2-ceramide/SH-BC-893 experiments were quantified in at least 6 time points and were classified as high-quality kinetic profiles (supplemental Figs. S1 and S2 and supplemental Table S3). Of the 5,029 high quality profiles, 1,818 were common to both experiments (supplemental Fig. S3). A list of phosphopeptides corresponding to these profiles is presented in supplemental Table S3 for both triple SILAC experiments. As described previously, curve fitting with a polynomial model and a false discovery rate (FDR) of 1% distinguished dynamic from static profiles (17). These analyses identified 629 and 428 dynamic profiles corresponding to regulated phosphosites in LB-100/SH-BC-893 and C2-ceramide/SH-BC-893 experiments, respectively (supplemental Table S4). A comparison of these regulated phosphosites revealed that only 54 phosphopeptides were common to all three conditions suggesting that many of the changes in cell signaling taking place were not regulated by all three stimuli (supplemental Fig. S3).
Fig. 2.

Triple SILAC phosphoproteomics workflow. Light cells are treated with vehicle, medium cells were treated with C2-ceramide (50 μm) or LB-100 (10 μm), and heavy cells were treated with SH-BC-893 (5 μm). Cells were collected every 5 min from 0–60 min of stimulation by snap freezing in pre-cooled ethanol. Cells were lysed in urea, digested with trypsin and phosphopeptides were enriched using titanium dioxide. Strong cation fractionation (SCX) was performed before LC-MS/MS analysis. Phosphopeptide identification and quantitation was performed using MaxQuant, and only kinetic profiles of sites with localization confidence > 0.75 and measured in at least 6 time points were selected for further analysis. Polynomial fitting was used to define regulated phosphosites, and only phosphopeptide profiles with FDR < 1% were selected and used for fuzzy c-means clustering and subsequent analyses.
Phosphorylation events that vary in opposite directions in cells treated with SH-BC-893 and LB-100 are likely to be PP2A-dependent. The dynamic profiles showed a progressive broadening of fold change (FC) values with a gradual shift in the median distribution toward a decrease or an increase in phosphorylation for cells treated with SH-BC-893 or LB-100, respectively. The width of this distribution was calculated from the inter quartile range (IQR) of the Log2 (FC) distribution of phosphopeptides at each time point and plotted over the entire cell stimulation period (Fig. 3A). The resulting curve showed a progressive increase in the IQR, in contrast to the trend observed from non-phosphorylated peptides that remained unaffected with time. Also, the extent of changes in protein phosphorylation was more pronounced for cells treated with LB-100 compared with those incubated with SH-BC-893.
Fig. 3.

Phosphoproteomics results from the comparison of treatments with LB-100 (10 μm) and SH-BC-893 (5 μm). A, Inter quartile range (IQR) shows the global change in phosphorylation on treatment. Unmodified peptides (control) do not change over time whereas LB-100 and SH-BC-893 treatments show a progressive increase in IQR with time. B, Heatmap of all 390 phosphosites regulated by SH-BC-893. Different kinetic behaviors are indicated. C, Kinetic trends extracted from LB-100 and SH-BC-893 treatments using fuzzy means clustering; numbers correspond to profiles observed with a particular kinetic trend.
Fuzzy clustering separated these dynamic profiles into those that displayed rapid or progressive increases or decreases in phosphorylation or showed an adaptation-like response (Figs. 3B and 3C). From the 629 dynamic profiles identified in experiments comparing SH-BC-893 and LB-100 treatments, 384 were regulated by SH-BC-893, and are represented in Fig. 3B. Most profiles in SH-BC-893-treated cells showed a progressive dephosphorylation (200 of 384 profiles), consistent with the expected agonist activity of this compound (Fig. 3C). In contrast, profiles from cells treated with the PP2A inhibitor LB-100 displayed a progressive increase in phosphorylation with time (349 of 534 profiles). Fifty-two profiles in cells treated with SH-BC-893 exhibited adaptation characterized by a rapid dephosphorylation over the first 20 min post-stimulation followed by a recovery phase that extended over the remaining time period (Fig. 3C). Interestingly, phosphosites that displayed transient dephosphorylation were more highly conserved than static sites (p < 0.001) or dynamic sites that changed unidirectionally (p < 0.05) suggesting that the adaptation cluster may define a subset of functionally important phosphorylation events (supplemental Fig. S4).
To identify putative PP2A substrates, we compared phosphorylation profiles that changed in opposite directions when FL5.12 cells were treated with LB-100 and SH-BC-893. Out of the 629 dynamic profiles identified in the triple SILAC experiment, 289 profiles were common to both treatments, and 114 of these profiles showed reciprocal (bidirectional) behavior (Fig. 4A). We also noted that a subset of 175 profiles common to both LB-100 and SH-BC-893 displayed comparable trends that were either increasing or decreasing in phosphorylation with time and were referred to as monodirectional. Linear motif analysis of monodirectional or bidirectional phosphopeptides with motif-X revealed that an RxxS motif was mainly represented in bidirectionally regulated sites whereas proline directed phosphorylation (SP and PxSP) was mainly found among monodirectional phosphorylated sites (Fig. 4B). To decipher which phosphorylation events are associated with changes in PP2A activity, we selected phosphosites that exhibited reciprocal trends with SH-BC-893 and LB-100 together with those uniquely regulated by SH-BC-893 (209 sites) and performed Gene Ontology (GO) enrichment using DAVID (Fig. 4C). Proteins associated with guanosine triphosphate hydrolase (GTPase) regulators and actin filament reorganization were significantly enriched; GTPases and actin dynamics both play critical roles in regulating intracellular trafficking.
Fig. 4.

Comparison of dynamic phosphorylation sites following SH-BC-893 and LB-100 treatments. A, Venn diagram showing that 292 phosphosites are common to both treatments whereas 253 and 98 phosphosites are uniquely regulated by LB-100 and SH-BC-893, respectively. Of the 292 common sites, 116 sites showed opposite (Bidirectional) tends and 176 sites are regulated in the same direction (Monodirectional). B, Phosphorylation motifs extracted from motif-X. Monodirectional phosphosites display PxSP and SP motifs. Bidirectional sites that show rapid changes in phosphorylation are present on Serine-rich peptides. C, Gene Ontology enrichment of proteins corresponding to the 214 bidirectional and unidirectional (for SH-BC-893) phosphosites.
A literature analysis was performed to determine whether dynamic profiles had been previously identified as PP2A interactors (40–42). This analysis revealed that 30 proteins (37 phosphosites) identified in our screen were common with PP2A-interactors identified in other reports (40–42) (Fig. 5A, supplemental Table S5). Closer examination of these data indicated that 15 of these proteins are interconnected through a protein-protein interaction network and that 6 of these members (Vim, Pxn, Pak2, Gsk3β, Cfl1, Arhgef2) are known regulators of cytoskeleton organization (Fig. 5B). Phosphorylation profiles of these substrates are shown in Fig. 5C. Previous reports have indicated that Vimentin (Vim) phosphorylation at several residues, including Ser42 and Ser430, is regulated by PP2A and functionally important (43, 44); inactivation of vimentin leads to increased actin stress fiber assembly (45). The cytoskeletal protein Paxillin (Pxn) that regulates the actin-membrane attachment showed a rapid dephosphorylation at site Ser83 on the first 5 min of SH-BC-893 treatment followed by a recovery and a more gradual decrease in phosphorylation that extended over the entire stimulation period. The closely located phosphorylation sites Pxn Thr31 and Thr118 have previously been reported to regulate cell migration in a RhoA dependent manner (46, 47). The cytoskeleton organizing kinase Pak2 showed strong dephosphorylation on Ser141. Furthermore, Arhgef2 Ser122 showed a rapid dephosphorylation on SH-BC-893 treatment. This site is proximal to Ser143 previously described to impact F-actin based morphological changes (48). The actin depolymerizing protein Cofilin1 (Cfl1) showed a progressive increase in phosphorylation at Ser3, a site whose phosphorylation inactivates the actin-depolymerizing activity of cofilin (49). Similarly, SH-BC-893 induces phosphorylation of the glycogen synthase kinase 3 b (Gsk3b) on Ser9, a site that has previously been reported to regulate the reorganization of the microtubule cytoskeleton (50). Except for Cfl1 Ser3, all sites described above showed differential regulation on SH-BC-893 and LB-100 treatments.
Fig. 5.

Identification of putative PP2A targets. A, Comparative analysis of the list of candidate PP2A substrates identified from various proteomic studies. The comparison was done at the protein level because not all data sets reported the position of the phosphorylated sites. B, A network of phosphorylation sites identified as direct PP2A targets based on comparative literature review. Color coding indicates if the proteins are involved in actin reorganization. C, Dynamic profiles of sites from putative PP2A targets involved in actin reorganization.
Dynamic Phosphoproteomics Identifies Distinct Cell Signaling Events Associated with C2-ceramide and SH-BC-893
Although both SH-BC-893 and C2-ceramide down-regulate nutrient transporters in a PP2A-dependent manner, only SH-BC-893 promotes cytoplasmic vacuolation (Fig. 1). Quantitative phosphoproteomics experiments performed on FL5.12 cells treated with SH-BC-893 or C2-ceramide enabled the identification of cell signaling events differentially associated with these compounds. Dynamic profiles again formed three distinct clusters according to fuzzy clustering, displaying rapid or progressive increases or decreases in phosphorylation or an adaptation-like response (Figs. 6A and 6B). In total, we identified 428 dynamic profiles, of which 254 profiles were common to SH-BC-893 and C2-ceramide (Fig. 6C). It is noteworthy that temporal changes in protein phosphorylation for cells treated with SH-BC-893 (Figs. 3B and 6C) yield comparable trends with Pearson coefficient of 0.68 on average between biological replicates (supplemental Fig. S5). Approximately 80% of common profiles between SH-BC-893 and C2-ceramide (203 of 254 profiles) showed a mono-directional trend where progressive changes in protein phosphorylation were similar both in magnitude and direction for these two compounds consistent with the fact that these agents produce largely overlapping phenotypes in cells. Interestingly, 51 of the 254 common profiles showed opposite trends, whereas 126 or 48 profiles were uniquely regulated by C2-ceramide or SH-BC-893, respectively. These differentially regulates sites could be responsible for divergent phenotypes such as vacuolation.
Fig. 6.

Dynamic phosphoproteomics identifies signaling events differently regulated by C2-ceramide and SH-BC-893 treatments. A, Heatmap corresponding to 380 phosphorylation sites that were dynamically phosphorylated on C2-ceramide treatment. Kinetic trends are indicated. B, Dynamic profiles extracted from C2-ceramide and SH-BC-893 treatments using fuzzy means clustering. Numbers correspond to profiles observed for a given cluster. C, Venn diagram of dynamic phosphosites showing that 254 phosphosites are common, 126 and 48 phosphosites are uniquely regulated by C2-ceramide and SH-BC-893, respectively. Of the 254 commonly regulated sites, 51 show opposite change in phosphorylation (bidirectional) and 203 are regulated in the same direction (monodirectional). D, Radar plot showing the enrichment of GO terms associated with specific groups highlighted in different colors. E, Motif-X analyses of bidirectional sites showing an over-representation of basophilic motifs. F, Network of putative Akt substrates and known interacting members.
To correlate phosphorylation changes with the distinct vacuolation phenotypes associated with C2-ceramide and SH-BC-893, dynamic profiles were separated into three groups: (1) Common and mono-directional profiles, (2) Bi-directional profiles or dynamic profiles unique to SH-BC-893, and (3) Dynamic profiles unique to C2-ceramide. GO term enrichment analyses were performed to determine if these groups were related to specific cellular compartments and molecular functions. Results from the corresponding analyses are displayed in the radar plots of Fig. 6D. Most profiles identified belong to group 1 (203 shared monodirectional profiles) and are likely to account for phenotypes induced by both C2-ceramide and SH-BC-893 (e.g. nutrient transporter down-regulation). These profiles correspond to substrates that are largely involved in the regulation GTPase activity and actin cytoskeleton organization. Profiles from group 2 that showed opposite trends or are specific to SH-BC-893 treatment (99 profiles) were associated with the GO terms: endosome, intracellular membrane bound organelle and plasma membrane. Among these we found 11 phosphorylation sites on 9 proteins involved in vesicle trafficking and lysosomal fusion (supplemental Fig. S6). This result is consistent with finding that SH-BC-893 but not C2-ceramide inhibit lysosomal trafficking events (5). Group 3 comprises 126 profiles that are unique to C2-ceramide treatment. Phosphorylated substrates associated with this group are mostly localized in the nucleus with functional GO terms enriched for DNA binding, Chromatin binding and “DNA-directed DNA polymerase activity”. A protein-protein interaction network of the corresponding phosphoproteins highlights several of the corresponding members and regroups proteins involved in DNA replication, DNA repair, nucleotide synthesis, and ribosome assembly (supplemental Fig. S7).
Next, we analyzed dynamic profiles from each group with Motif-X to determine if a specific phosphorylation consensus sequence was over-represented in these data sets (supplemental Fig. S8A). These analyses indicated that a large proportion of dynamic profiles were represented by proline-directed and basophilic phosphorylation motifs. Further analyses performed with the kinase prediction software available from the Group-based Prediction System (GPS) (51) identified Akt as a putative kinase targeting dynamic basophilic motifs. Interestingly, bidirectional sites from group 2 comprised the largest proportion of putative Akt substrates from all groups (Fig. 6E and supplemental Fig. S8B). Among the 21 putative Akt substrates from group 2, we identified 6 proteins (Vim, Slc33a1, Gsk3b, Pikfyve, Acly, Pi4kb) that are known to interact with each other, including 3 proteins (Gsk3b, Pikfyve, Acly) previously reported as Akt substrates (52–54) (Fig. 6F). For convenience, supplemental Fig. S9 presents the kinetic phosphorylation profiles of several Akt substrates. Together, these results suggest that a subset of bidirectional sites are putative Akt substrates associated with vesicle trafficking and could account for the vacuolation phenotype observed only with SH-BC-893.
SH-BC-893 and C2-ceramide Stimulate Actin Polymerization That Is Necessary for Cytoplasmic Vacuolation But Not Surface Nutrient Transporter Loss
To gain a systems-level view of changes in protein phosphorylation observed for C2-ceramide and SH-BC-893, we used GO terms enriched in our data sets (Fig. 6D, supplemental Table S4) combined with manual curation of gene lists to generate a pictogram of regulated phosphosites (supplemental Fig. S10). Protein groups of interest included membrane transport, endocytosis/cell adhesion, actin reorganization, nutrient transport and GTPase regulation (GTPase activators, GAPs and guanidine nucleotide exchange factors, GEFs).
Because dynamic phosphorylation sites similarly affected by SH-BC-893 and C2-ceramide were clustered in proteins that modulate actin dynamics (Fig. 6D), actin polymerization was monitored in SH-BC-893- or C2-ceramide-treated FL5.12 cells using fluorescently-conjugated phalloidin. Both SH-BC-893 and C2-ceramide robustly increased F-actin formation in the cell cortex and in a cytosolic, likely endosomal, compartment (Fig. 7A). Similar effects were observed in murine embryonic fibroblasts (MEFs) and in HeLa cells (data not shown). Consistent with a role for PP2A activation in this phenotype, the PP2A inhibitors LB-100 and calyculinA (CalyA) blocked actin polymerization at both locations (Fig. 7B). The actin polymerization inhibitor Latrunculin A (LatA) also prevented actin polymerization with SH-BC-893 and C2-ceramide (Fig. 7C). However, although LB-100 blocked CD98 down-regulation induced by SH-BC-893 or ceramide (Fig. 1B), LatA did not, suggesting that the loss of surface transporters depends on PP2A activation but not actin polymerization (Fig. 7D). This result was unexpected, as actin polymerization is essential for nutrient permease down-regulation by sphingolipids in yeast (55). Interestingly, LatA reversed vacuolation by SH-BC-893 (Fig. 7E). The actin polymerization stimulator Jasplakinolide did not promote vacuolation on its own (data not shown). Together, these data suggest that both C2-ceramide and SH-BC-893 stimulate actin polymerization in a PP2A-dependent manner and that actin polymerization is necessary but not enough to produce cytoplasmic vacuolation.
Ceramide Inhibits SH-BC-893-induced Vacuolation By Inactivating Akt
The finding that ceramide results in the dephosphorylation of more proteins than SH-BC-893 suggested that, rather than failing to induce vacuolation, ceramide may dephosphorylate and inactivate proteins necessary for vacuolation. In support of this model, co-addition of C2-ceramide inhibited cytoplasmic vacuolation by SH-BC-893 in FL5.12 cells (Fig. 8A). Similar results were obtained in HeLa cells and murine prostate cancer epithelial cells (supplemental Fig. S11A–S11B). As highlighted in supplemental Fig. S9, multiple Akt substrates were dephosphorylated in cells treated with C2-ceramide but not SH-BC-893. That the effect of C2-ceramide was dominant was confirmed in Fig. 8B and supplemental Figs. S11C, S11D by monitoring the Akt-dependent phosphorylation of PRAS40 at threonine 246 (56). These observations suggested that C2-ceramide may block vacuolation by inactivating Akt. Consistent with this model, the allosteric Akt inhibitor MK2206 reduced both Akt activity and vacuolation in FL5.12 cells treated with SH-BC-893 (Figs. 8B, 8C); similar results were obtained in HeLa and mPCE prostate cancer cells (supplemental Figs. S11B, S11E). The deletion of Rictor, an mTORC2 component necessary for Akt activation (57), also blocked vacuolation in the presence of SH-BC-893 again suggesting that Akt activity is essential for this phenotype (Fig. 8D). Together, these data suggest that C2-ceramide does not vacuolate cells because Akt inactivation prevents the manifestation of this phenotype.
Two Akt substrates that are dephosphorylated in the presence of C2-ceramide but not SH-BC-893, Pikfyve and Pik4b, regulate late endocytic trafficking (58, 59). Gsk3b also localizes to MVBs and lysosomes and may regulate their function (60). Consistent with its phosphorylation by Akt, GSK3b Ser9 phosphorylation increases with SH-BC-893 but decreases with C2-ceramide (supplemental Fig. S9). Because Ser9 phosphorylation inhibits Gsk3b activity (61), Gsk3b inhibition may contribute to vacuolation. To test this, SH-BC-893 or C2-ceramide was combined with the Gsk3b inhibitor CHIR99021 and vacuolation monitored. Although CHIR99021 did not vacuolate on its own or induce vacuolation in C2-ceramide-treated cells, CHIR99021 enhanced vacuolation of FL5.12 cells by SH-BC-893 (Fig. 8E). This effect was much more dramatic in HeLa cells where CHIR99021 also sensitized cells to SH-BC-893 induced death despite its complete lack of toxicity as a single agent (Supplemental Figs. S11F, S11G). These data suggest that SH-BC-893, but not C2-ceramide, vacuolates cells because Akt continues to inhibit Gsk3b.
DISCUSSION
In this study, quantitative phosphoproteomics was used to identify phosphoproteins that responded differentially to pharmacological compounds that modulate PP2A activity and to correlate the PP2A-dependent phenotypes that affect intracellular trafficking and nutrient transporter loss. Time-resolved phosphoproteome analyses of FL5.12 cells treated with the PP2A agonist SH-BC-893 or the PP2A inhibitor LB-100 enabled the identification of putative PP2A substrates and phosphorylation events specific to each compound. These analyses confirmed that SH-BC-893 affected PP2A activity as ∼75% of phosphorylation sites (289 out of 384 sites) were also targeted by LB-100. A more detailed analysis of the dynamic changes in protein phosphorylation common to LB-100 and SH-BC-893 revealed that 40% of these profiles displayed the expected reciprocal response and could represent putative PP2A substrates. A significant proportion of these targets are involved in the regulation of cell migration and actin cytoskeleton organization, including previously known PP2A substrates such as Vim, Pxn, Pak2, and Arhgef2.
Gene ontology analysis of phosphoproteins that were regulated similarily by SH-BC-893 and C2-ceramide suggested that changes in actin dynamics contribute to the intracellular trafficking defects observed in sphingolipid-treated cells (Fig. 6D) and led to the discovery that both SH-BC-893 and C2-ceramide promote PP2A-dependent actin polymerization at the cell cortex and in the cytoplasm (Figs. 7A, 7B). Given that actin polymerization plays an important role in sphingolipid-induced nutrient transporter internalization in yeast (55), it was surprising that actin polymerization was not necessary for down-regulation of nutrient transporter proteins by SH-BC-893 and C2-ceramide (Figs. 7C, 7D). Moreover, rather than contributing to the shared phenotype, loss of surface nutrient transporters, actin polymerization was necessary for a phenotype observed uniquely in SH-BC-893-treated cells, vacuolation (Fig. 7E). As vacuolation contributes to the anti-neoplastic activity of SH-BC-893 in vitro and in vivo (5), the molecular mechanism underlying this phenotype is of significant interest.
The differential ability of SH-BC-893 and C2-ceramide to produce vacuolation (Fig. 1C) led us to hypothesize that cell signaling events responsible for vacuolation would be encompassed in phosphosites that varied in opposite manner in SH-BC-893- and C2-ceramide-treated cells. A basophilic phosphorylation motif typical of Akt substrates was enriched among these differentially regulated dynamic sites; multiple Akt substrates were dephosphorylated in C2-ceramide-, but not SH-BC-893-treated, cells (Fig. 6F and supplemental Fig. S8). It has been widely reported that ceramide reduces the phosphorylation and activity of Akt (62). In contrast, Akt Ser473 phosphorylation was maintained in SH-BC-893-treated cells not just in this study (Supplemental Fig. S10A, S10B), but also in vivo in autochthonous prostate tumors (5). Differential regulation of Akt activity by SH-BC-893 and C2-ceramide might result if these sphingolipids activate PP2A through distinct mechanisms. Alternatively, as SH-BC-893 is water soluble to >60 mm whereas C2-ceramide is poorly soluble in aqueous solutions, these compounds may activate PP2A in different subcellular compartments. Membrane-localized C2-ceramide may activate plasma-membrane localized PP2A more effectively than SH-BC-893; Akt is activated by kinases found at the plasma membrane (63). It is also possible that SH-BC-893 and C2-ceramide differentially regulate the phosphorylation of the PP2A regulatory subunit B56β (Ppp2r5b) by Clk2 thereby affecting the interaction between Akt and PP2A (64, 65). Regardless of the mechanism underlying differential Akt activation by these sphingolipids, the reduction in vacuolation when Akt was inhibited by MK2206, Rictor deletion, or C2-ceramide demonstrate that Akt activity is necessary for SH-BC-893-induced vacuolation (Figs. 8A–8D and supplemental Figs. S10A–S10D). In summary, these phosphoproteomics studies led to the unexpected discovery that C2-ceramide was dominant over SH-BC-893 in vacuolation assays (Fig. 8A and supplemental Fig. S10A) and the conclusion that, rather lacking an activity possessed by SH-BC-893, ceramide fails to vacuolate cells because it inactivates Akt.
A detailed mechanistic understanding of how Akt activity supports vacuolation will require additional studies. However, the almost 2-fold increase or decrease in the phosphorylation of the Akt substrate Gsk3b phosphorylation at its inhibitory site Ser9 with SH-BC-893 or C2-ceramide, respectively (supplemental Fig. S8), and the accentuation of vacuolation by the Gsk3b inhibitor CHIR99021 (Fig. 8E and supplemental Fig. S10E) suggest that Gsk3b may suppress vacuolation by positively regulating MVB function. Because CHIR99021 was not sufficient to induce vacuolation in C2-ceramide treated cells (Fig. 8E), it is likely that additional Akt substrates besides Gsk3b contribute to the vacuolation phenotype. Consistent with published studies showing that vacuolation contributes to the anti-neoplastic effects of SH-BC-893 (5), inhibition of Gsk3b also sensitized cells to SH-BC-893-induced death (supplemental Fig. S10F). Gsk3b inhibitors are currently being developed for treatment of acute myeloid leukemia and breast cancer. Considering these findings, there may be a strong rationale for combining Gsk3b inhibitors and SH-BC-893 in cancer treatment. In conclusion, global, kinetic phosphoproteomic screens using PP2A-activating sphingolipids helped to elucidate signaling pathways necessary for cellular starvation by sphingolipids and highlighted a key role for Akt and Gsk3b signaling in regulating lysosomal trafficking events.
Data Availability
All raw LC-MS/MS data and MaxQuant output files can be accessed from the PeptideAtlas (http://www.peptideatlas.org/) with the dataset identifier PASS01168. In addition, all MS/MS spectra of identified peptides and phosphopeptides can be accessed through the MS viewer consortium (http://msviewer.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer) (26). The “SH-BC-893 vs. LB-100” and the “SH-BC-893 vs. C2-ceramide” data sets can be retrieved with the search keys “scuimnsogi” and “emvfcljjhs,” respectively.
Supplementary Material
Acknowledgments
We thank David Sabatini (Whitehead Institute, MIT) for generously providing Rictor knockout MEFs.
Footnotes
* This work was funded in part by the Natural Sciences and Engineering Research Council (NSERC) (P.T. 311598; S.H. 04726), grants to A.L.E. from the NIH (R01 GM089919, R21 CA178230), CDMRP (W81XWH-15-1-0010), the American Cancer Society (RSG-11-111-01-CDD), and the UCI CORCL. The Institute for Research in Immunology and Cancer (IRIC) receives infrastructure support from the Canadian Center of Excellence in Commercialization and Research, the Canadian Foundation for Innovation, and the Fonds de recherche du Québec - Santé (FRQS). Proteomics analyses were performed at the Center for Advanced Proteomic and Chemogenomics Analyses (CAPCA), a Node of the Genomic Technology Platform supported by the Canadian Government through Genome Canada. Imaging was performed in the Optical Biology Core at UCI which is supported in part by NIH P30 CA062203.
 This article contains supplemental material. We declare no competing financial interests.
This article contains supplemental material. We declare no competing financial interests.
1 The abbreviations used are:
- PP2A
- Protein phosphatase 2A
- ACN
- Acetonitrile
- DAPI
- 4′,6-diamidino-2-phenylindole
- FA
- Formic acid
- FBS
- Fetal bovine serum
- FDA
- Food and Drug Administration
- FDR
- False discovery rate
- FL5.12
- Murine prolymphocytic cell line
- GAP
- GTPase activating proteins
- GEF
- Guanine nucleotide exchange factor
- GO
- Gene Ontology
- GTPase
- Guanosine triphosphatase
- HCD
- High collision dissociation
- HeLa
- Cervical cancer cell line
- IAA
- Iodo acetamide
- MVB
- Multivesicular bodies
- PBS
- Phosphate buffered saline
- RPMI
- Roswell Park Memorial Institute medium
- RT
- Room temperature
- SCX
- Strong cation exchange
- SILAC
- Stable isotope labeling of amino acids in cell culture
- SPE
- Solid phase extraction.
REFERENCES
- 1. Selwan E. M., Finicle B. T., Kim S. M., and Edinger A. L. (2016) Attacking the supply wagons to starve cancer cells to death. FEBS Lett. 590, 885–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barthelemy C., Barry A. O., Twyffels L., and Andre B. (2017) FTY720-induced endocytosis of yeast and human amino acid transporters is preceded by reduction of their inherent activity and TORC1 inhibition. Sci. Rep. 7, 13816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guenther G. G., Liu G., Ramirez M. U., McMonigle R. J., Kim S. M., McCracken A. N., Joo Y., Ushach I., Nguyen N. L., and Edinger A. L. (2014) Loss of TSC2 confers resistance to ceramide and nutrient deprivation. Oncogene 33, 1776–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hyde R., Hajduch E., Powell D. J., Taylor P. M., and Hundal H. S. (2005) Ceramide down-regulates System A amino acid transport and protein synthesis in rat skeletal muscle cells. FASEB J. 19, 461–463 [DOI] [PubMed] [Google Scholar]
- 5. Kim S. M., Roy S. G., Chen B., Nguyen T. M., McMonigle R. J., McCracken A. N., Zhang Y., Kofuji S., Hou J., Selwan E., Finicle B. T., Nguyen T. T., Ravi A., Ramirez M. U., Wiher T., Guenther G. G., Kono M., Sasaki A. T., Weisman L. S., Potma E. O., Tromberg B. J., Edwards R. A., Hanessian S., and Edinger A. L. (2016) Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways. J. Clin. Invest. 126, 4088–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Romero Rosales K., Singh G., Wu K., Chen J., Janes M. R., Lilly M. B., Peralta E. R., Siskind L. J., Bennett M. J., Fruman D. A., and Edinger A. L. (2011) Sphingolipid-based drugs selectively kill cancer cells by down-regulating nutrient transporter proteins. Biochem. J. 439, 299–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guenther G. G., Peralta E. R., Rosales K. R., Wong S. Y., Siskind L. J., and Edinger A. L. (2008) Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 17402–17407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morad S. A., and Cabot M. C. (2013) Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 13, 51–65 [DOI] [PubMed] [Google Scholar]
- 9. Chalfant C. E., Kishikawa K., Mumby M. C., Kamibayashi C., Bielawska A., and Hannun Y. A. (1999) Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 274, 20313–20317 [DOI] [PubMed] [Google Scholar]
- 10. Dobrowsky R. T., Kamibayashi C., Mumby M. C., and Hannun Y. A. (1993) Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 268, 15523–15530 [PubMed] [Google Scholar]
- 11. Edinger A. L. (2007) Controlling cell growth and survival through regulated nutrient transporter expression. Biochem. J. 406, 1–12 [DOI] [PubMed] [Google Scholar]
- 12. Habrukowich C., Han D. K., Le A., Rezaul K., Pan W., Ghosh M., Li Z., Dodge-Kafka K., Jiang X., Bittman R., and Hla T. (2010) Sphingosine interaction with acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) regulates PP2A activity and cyclooxygenase (COX)-2 expression in human endothelial cells. J. Biol. Chem. 285, 26825-26831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oaks J., and Ogretmen B. (2014) Regulation of PP2A by Sphingolipid Metabolism and Signaling. Front. Oncol. 4, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Finicle B. T., Ramirez M. U., Liu G., Selwan E. M., McCracken A. N., Yu J., Joo Y., Nguyen J., Ou K., Roy S. G., Mendoza V. D., Corrales D. V., and Edinger A. L. (2018) Sphingolipids inhibit endosomal recycling of nutrient transporters by inactivating ARF6. J. Cell Sci. 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kanshin E., Bergeron-Sandoval L. P., Isik S. S., Thibault P., and Michnick S. W. (2015) A cell-signaling network temporally resolves specific versus promiscuous phosphorylation. Cell. Rep. 10, 1202–1214 [DOI] [PubMed] [Google Scholar]
- 16. Kanshin E., Kubiniok P., Thattikota Y., D'Amours D., and Thibault P. (2015) Phosphoproteome dynamics of Saccharomyces cerevisiae under heat shock and cold stress. Mol. Syst. Biol. 11, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kubiniok P., Lavoie H., Therrien M., and Thibault P. (2017) Time-resolved Phosphoproteome Analysis of Paradoxical RAF Activation Reveals Novel Targets of ERK. Mol. Cell. Proteomics 16, 663–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bendall S. C., Hughes C., Stewart M. H., Doble B., Bhatia M., and Lajoie G. A. (2008) Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol. Cell. Proteomics 7, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dai C., Zhang X., Xie D., Tang P., Li C., Zuo Y., Jiang B., and Xue C. (2017) Targeting PP2A activates AMPK signaling to inhibit colorectal cancer cells. Oncotarget 8, 95810–95823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perryman M. S., Tessier J., Wiher T., O'Donoghue H., McCracken A. N., Kim S. M., Nguyen D. G., Simitian G. S., Viana M., Rafelski S., Edinger A. L., and Hanessian S. (2016) Effects of stereochemistry, saturation, and hydrocarbon chain length on the ability of synthetic constrained azacyclic sphingolipids to trigger nutrient transporter down-regulation, vacuolation, and cell death. Bioorg.. Med. Chem. 24, 4390–4397 [DOI] [PubMed] [Google Scholar]
- 21. Kanshin E., Michnick S. W., and Thibault P. (2013) Displacement of N/Q-rich peptides on TiO2 beads enhances the depth and coverage of yeast phosphoproteome analyses. J. Proteome Res. 12, 2905–2913 [DOI] [PubMed] [Google Scholar]
- 22. Kanshin E., Michnick S., and Thibault P. (2012) Sample preparation and analytical strategies for large-scale phosphoproteomics experiments. Semin. Cell Dev. Biol. 23, 843–853 [DOI] [PubMed] [Google Scholar]
- 23. Ishihama Y., Rappsilber J., and Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 24. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 25. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 26. Baker P. R., and Chalkley R. J. (2014) MS-viewer: a web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharma K., D'Souza R. C., Tyanova S., Schaab C., Wisniewski J. R., Cox J., and Mann M. (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 8, 1583–1594 [DOI] [PubMed] [Google Scholar]
- 28. Kumar L., and MEF. (2007) Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 30. Chou M. F., and Schwartz D. (2011) Biological sequence motif discovery using motif-x. Curr. Protoc. Bioinformatics Chapter 13, Unit 13 15–24 [DOI] [PubMed] [Google Scholar]
- 31. Schwartz D., and Gygi S. P. (2005) An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 23, 1391–1398 [DOI] [PubMed] [Google Scholar]
- 32. Zhao B., Pisitkun T., Hoffert J. D., Knepper M. A., and Saeed F. (2012) CPhos: a program to calculate and visualize evolutionarily conserved functional phosphorylation sites. Proteomics 12, 3299–3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Camm J., Hla T., Bakshi R., and Brinkmann V. (2014) Cardiac and vascular effects of fingolimod: mechanistic basis and clinical implications. Am. Heart. J. 168, 632–644 [DOI] [PubMed] [Google Scholar]
- 34. Chen B., Roy S. G., McMonigle R. J., Keebaugh A., McCracken A. N., Selwan E., Fransson R., Fallegger D., Huwiler A., Kleinman M. T., Edinger A. L., and Hanessian S. (2016) Azacyclic FTY720 analogues that limit nutrient transporter expression but lack S1P receptor activity and negative chronotropic effects offer a novel and effective strategy to kill cancer cells in vivo. ACS Chem. Biol. 11, 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fransson R., McCracken A. N., Chen B., McMonigle R. J., Edinger A. L., and Hanessian S. (2013) Design, Synthesis, and Anti-leukemic Activity of Stereochemically Defined Constrained Analogs of FTY720 (Gilenya). ACS Med. Chem. Lett. 4, 969–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Broer S., and Broer A. (2017) Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 474, 1935–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cohen P., Klumpp S., and Schelling D. L. (1989) An improved procedure for identifying and quantitating protein phosphatases in mammalian tissues. FEBS Lett. 250, 596–600 [DOI] [PubMed] [Google Scholar]
- 38. Ishihara H., Martin B. L., Brautigan D. L., Karaki H., Ozaki H., Kato Y., Fusetani N., Watabe S., Hashimoto K., Uemura D., et al. (1989) Calyculin A and okadaic acid: inhibitors of protein phosphatase activity. Biochem. Biophys. Res. Commun. 159, 871–877 [DOI] [PubMed] [Google Scholar]
- 39. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 40. St-Denis N., Gupta G. D., Lin Z. Y., Gonzalez-Badillo B., Veri A. O., Knight J. D. R., Rajendran D., Couzens A. L., Currie K. W., Tkach J. M., Cheung S. W. T., Pelletier L., and Gingras A. C. (2016) Phenotypic and interaction profiling of the human phosphatases identifies diverse mitotic regulators. Cell Rep. 17, 2488–2501 [DOI] [PubMed] [Google Scholar]
- 41. Wiredja D. D., Ayati M., Mazhar S., Sangodkar J., Maxwell S., Schlatzer D., Narla G., Koyuturk M., and Chance M. R. (2017) Phosphoproteomics profiling of nonsmall cell lung cancer cells treated with a novel phosphatase activator. Proteomics 17, 1700214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wlodarchak N., and Xing Y. (2016) PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 51, 162–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ando S., Tanabe K., Gonda Y., Sato C., and Inagaki M. (1989) Domain- and sequence-specific phosphorylation of vimentin induces disassembly of the filament structure. Biochemistry 28, 2974–2979 [DOI] [PubMed] [Google Scholar]
- 44. Eriksson J. E., He T., Trejo-Skalli A. V., Harmala-Brasken A. S., Hellman J., Chou Y. H., and Goldman R. D. (2004) Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117, 919–932 [DOI] [PubMed] [Google Scholar]
- 45. Jiu Y., Peranen J., Schaible N., Cheng F., Eriksson J. E., Krishnan R., and Lappalainen P. (2017) Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci. 130, 892–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Iwasaki T., Nakata A., Mukai M., Shinkai K., Yano H., Sabe H., Schaefer E., Tatsuta M., Tsujimura T., Terada N., Kakishita E., and Akedo H. (2002) Involvement of phosphorylation of Tyr-31 and Tyr-118 of paxillin in MM1 cancer cell migration. Int. J. Cancer 97, 330–335 [DOI] [PubMed] [Google Scholar]
- 47. Chen H. Y., Shen C. H., Tsai Y. T., Lin F. C., Huang Y. P., and Chen R. H. (2004) Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol. Cell. Biol. 24, 10558–10572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Callow M. G., Zozulya S., Gishizky M. L., Jallal B., and Smeal T. (2005) PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 118, 1861–1872 [DOI] [PubMed] [Google Scholar]
- 49. Arber S., Barbayannis F. A., Hanser H., Schneider C., Stanyon C. A., Bernard O., and Caroni P. (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393, 805–809 [DOI] [PubMed] [Google Scholar]
- 50. Wakatsuki S., Saitoh F., and Araki T. (2011) ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 13, 1415–1423 [DOI] [PubMed] [Google Scholar]
- 51. Xue Y., Ren J., Gao X., Jin C., Wen L., and Yao X. (2008) GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 7, 1598–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., and Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 53. Hill E. V., Hudson C. A., Vertommen D., Rider M. H., and Tavare J. M. (2010) Regulation of PIKfyve phosphorylation by insulin and osmotic stress. Biochem. Biophys. Res. Commun. 397, 650–655 [DOI] [PubMed] [Google Scholar]
- 54. Osinalde N., Mitxelena J., Sanchez-Quiles V., Akimov V., Aloria K., Arizmendi J. M., Zubiaga A. M., Blagoev B., and Kratchmarova I. (2016) Nuclear phosphoproteomic screen uncovers ACLY as mediator of IL-2-induced proliferation of CD4+ T lymphocytes. Mol. Cell. Proteomics 15, 2076–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dickson R. C. (2010) Roles for sphingolipids in Saccharomyces cerevisiae. Adv. Exp. Med. Biol. 688, 217–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kovacina K. S., Park G. Y., Bae S. S., Guzzetta A. W., Schaefer E., Birnbaum M. J., and Roth R. A. (2003) Identification of a proline-rich Akt substrate as a 14–3-3 binding partner. J. Biol. Chem. 278, 10189–10194 [DOI] [PubMed] [Google Scholar]
- 57. Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 58. Kim G. H., Dayam R. M., Prashar A., Terebiznik M., and Botelho R. J. (2014) PIKfyve inhibition interferes with phagosome and endosome maturation in macrophages. Traffic 15, 1143–1163 [DOI] [PubMed] [Google Scholar]
- 59. Clayton E. L., Minogue S., and Waugh M. G. (2013) Mammalian phosphatidylinositol 4-kinases as modulators of membrane trafficking and lipid signaling networks. Prog. Lipid Res. 52, 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taelman V. F., Dobrowolski R., Plouhinec J. L., Fuentealba L. C., Vorwald P. P., Gumper I., Sabatini D. D., and De Robertis E. M. (2010) Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143, 1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sutherland C., Leighton I. A., and Cohen P. (1993) Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J. 296 (Pt 1), 15–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chavez J. A., and Summers S. A. (2012) A ceramide-centric view of insulin resistance. Cell Metab. 15, 585–594 [DOI] [PubMed] [Google Scholar]
- 63. Manning B. D., and Toker A. (2017) AKT/PKB signaling: Navigating the network. Cell 169, 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rodgers J. T., Vogel R. O., and Puigserver P. (2011) Clk2 and B56beta mediate insulin-regulated assembly of the PP2A phosphatase holoenzyme complex on Akt. Mol. Cell 41, 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kuo Y. C., Huang K. Y., Yang C. H., Yang Y. S., Lee W. Y., and Chiang C. W. (2008) Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J. Biol. Chem. 283, 1882–1892 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw LC-MS/MS data and MaxQuant output files can be accessed from the PeptideAtlas (http://www.peptideatlas.org/) with the dataset identifier PASS01168. In addition, all MS/MS spectra of identified peptides and phosphopeptides can be accessed through the MS viewer consortium (http://msviewer.ucsf.edu/prospector/cgi-bin/msform.cgi?form=msviewer) (26). The “SH-BC-893 vs. LB-100” and the “SH-BC-893 vs. C2-ceramide” data sets can be retrieved with the search keys “scuimnsogi” and “emvfcljjhs,” respectively.