

Avian colibacillosis, caused by strains of avian pathogenic Escherichia coli, is a major bacterial disease of severe economic significance to the poultry industry worldwide. The virulence mechanisms of APEC are not completely understood. This study investigated the influence of an antioxidant protein, SodA, on the phenotype and pathogenicity of APEC O2 strain E058. This is the first report demonstrating that SodA plays an important role in protecting a specific APEC strain against hydrogen peroxide-induced oxidative stress and contributes to the virulence of this pathotype strain. Identification of this virulence factor will enhance our knowledge of APEC pathogenic mechanisms, which is crucial for designing successful strategies against associated infections and transmission.
KEYWORDS: avian pathogenic Escherichia coli, mutant, sodA, virulence
ABSTRACT
Strains of avian pathogenic Escherichia coli (APEC), the common pathogen of avian colibacillosis, encounter reactive oxygen species (ROS) during the infection process. Superoxide dismutases (SODs), acting as antioxidant factors, can protect against ROS-mediated host defenses. Our previous reports showed that the sodA gene (encoding a Mn-cofactor-containing SOD [MnSOD]) is highly expressed during the septicemic infection process of APEC. sodA has been proven to be a virulence factor of certain pathogens, but its role in the pathogenicity of APEC has not been fully identified. In this study, we deleted the sodA gene from the virulent APEC O2 strain E058 and examined the in vitro and in vivo phenotypes of the mutant. The sodA mutant was more sensitive to hydrogen peroxide in terms of both its growth and viability than was the wild type. The ability to form a biofilm was weakened in the sodA mutant. The sodA mutant was significantly more easily phagocytosed by chicken macrophages than was the wild-type strain. Chicken infection assays revealed significantly attenuated virulence of the sodA mutant compared with the wild type at 24 h postinfection. The virulence phenotype was restored by complementation of the sodA gene. Quantitative real-time reverse transcription-PCR revealed that the inactivation of sodA reduced the expression of oxidative stress response genes katE, perR, and osmC but did not affect the expression of sodB and sodC. Taken together, our studies indicate that SodA is important for oxidative resistance and virulence of APEC E058.
IMPORTANCE Avian colibacillosis, caused by strains of avian pathogenic Escherichia coli, is a major bacterial disease of severe economic significance to the poultry industry worldwide. The virulence mechanisms of APEC are not completely understood. This study investigated the influence of an antioxidant protein, SodA, on the phenotype and pathogenicity of APEC O2 strain E058. This is the first report demonstrating that SodA plays an important role in protecting a specific APEC strain against hydrogen peroxide-induced oxidative stress and contributes to the virulence of this pathotype strain. Identification of this virulence factor will enhance our knowledge of APEC pathogenic mechanisms, which is crucial for designing successful strategies against associated infections and transmission.
INTRODUCTION
As a subset of extraintestinal pathogenic Escherichia coli (ExPEC), avian pathogenic Escherichia coli (APEC) strains cause a wide range of localized and systemic infections collectively called avian colibacillosis, leading to great economic losses in poultry (1–3). APEC strains use virulence factors for colonizing and invading the host, including adhesins, invasins, outer membrane proteins, iron acquisition systems, and others (4–11).
APEC strains usually enter the host by crossing the respiratory epithelia and penetrate deeply into the mucosa and submucosa to reach the bloodstream, causing septicemia (12, 13). Phagocytic cells, such as macrophages and heterophils, are elicited into the respiratory tract and play a key role in the initial host defense against APEC (14, 15). An important killing mechanism of phagocytes involves the production of highly microbicidal reactive oxygen metabolites during the so-called oxidative burst, which is generally induced by the engulfment of bacteria (16). Thus, within the host, APEC faces reactive oxygen species (ROS) from the oxidative burst of macrophages. ROS, including superoxide anions (O2−), hydroxyl radicals (OH·−), and hydrogen peroxide (H2O2), are known to damage DNA, RNA, proteins, and lipids (16, 17).
Superoxide dismutases (SODs) are metalloenzymes that can directly convert deleterious superoxide to molecular oxygen and hydrogen peroxide; hydrogen peroxide is in turn metabolized by catalases and/or peroxidases (18). Hence, these enzymes play an important role in the pathogenesis of some pathogens (19–22). SODs are classified into the following three types depending on the metal cofactor used: Cu,ZnSOD (SodC), MnSOD (SodA), and FeSOD (SodB). Previously, we used an analysis of global gene expression profiles to show that the expression levels of the sodA gene in APEC strain E058 were clearly increased during infection in vivo (23). However, the role of sodA in the pathogenesis of APEC is not fully understood. Therefore, in this work, we constructed a sodA deletion mutant of the virulent APEC O2 strain E058 and evaluated its phenotype. The effects of sodA on serum and environmental stress resistance, biofilm formation, intracellular survival, virulence, and gene expression were evaluated to fully identify the contribution of SodA to the virulence of APEC O2 strain E058.
RESULTS
Bactericidal effects of serum and environmental stress on sodA mutant.
Serum resistance and environmental stress adaptation correlate with the virulence of APEC during infection in vivo (5, 24). In a serum bactericidal assay, the bacterial numbers of the sodA mutant inoculated in serial serum dilutions were similar to those of wild-type strain E058. The control strain DH5α was sensitive to serum, which demonstrated that the serum was active and bactericidal (Fig. 1A). Thus, the deletion of sodA did not appear to affect serum resistance.
FIG 1.
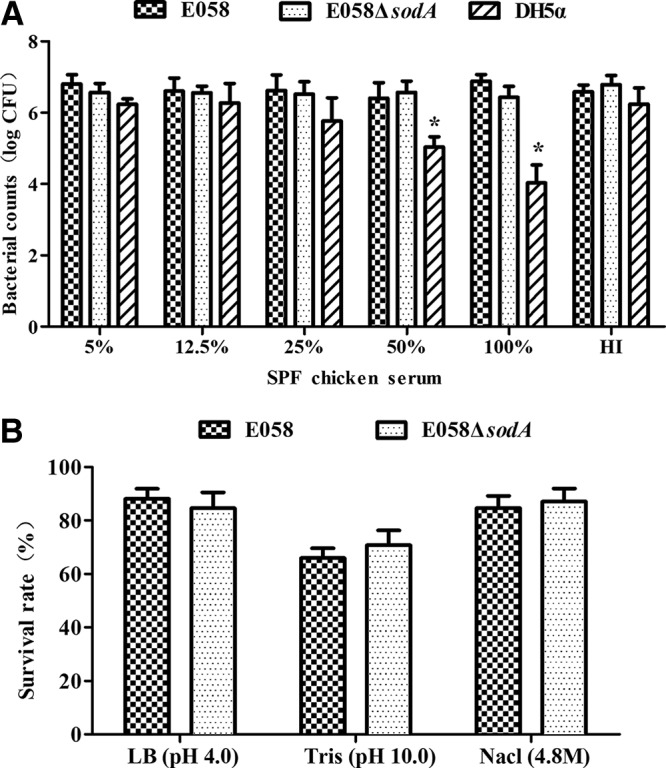
Bacterial resistance to SPF chicken serum and environmental stress. (A) Resistance to SPF chicken serum. Bacteria were incubated with the diluted SPF chicken serum at 37°C for 30 min. Bacteria were counted by plating on LB agar at serial dilutions. HI, heat-inactivated SPF chicken serum. (B) Resistance to environmental stress. Bacteria were examined for adaptation to environmental stresses, including alkalinity, acidity, and high osmolarity. Survival was calculated as the number of bacteria remaining after the stress exposure divided by the initial number of bacteria. Data represent averages of the results from three independent assays (*, P < 0.05).
To identify whether SodA plays a role in adapting to environmental stresses, including alkalinity, acidity, and high osmolarity, we compared the resistance phenotype of the sodA mutant and its wild-type strain under these stressful conditions. The results showed that the survival of the sodA mutant resembled that of the wild-type strain (Fig. 1B), indicating that SodA was not required for resistance to these environmental stresses.
Effect of sodA mutation on sensitivity to oxidative stress.
We examined the protective effects of SodA in APEC E058 against the oxidant hydrogen peroxide. Growth rate and survival in the presence of 10 mM H2O2 were tested in the sodA mutant, complementation, and wild-type strains. The growth of the sodA mutant was similar to that of the wild-type in Luria-Bertani broth (LB) medium but was severely inhibited in LB medium containing 10 mM H2O2 (Fig. 2A). The sodA mutant was poorly viable during exposure to 10 mM H2O2 compared to the wild-type and complementation strains (Fig. 2B).
FIG 2.

Effect of sodA deletion from APEC strain E058 on sensitivity to oxidative stress. (A) Growth curves of sodA mutant, complementation, and wild-type strains. Strains E058 (wild type), E058 ΔsodA (sodA deletion mutant), and ReE058 ΔsodA (complementation strain) were grown in Luria-Bertani broth with or without 10 mM H2O2 at 37°C, and their growth was determined by measurement of the optical density at 600 nm (OD600). (B) The survival rates of strains in 10 mM H2O2 for 30 min at 37°C. Data represent the averages of the results from three independent assays (**, P < 0.01).
Ability of sodA mutant to form biofilm.
To determine the effect of sodA deletion on biofilm production, the biofilm formation ability of the sodA mutant, complementation, and wild-type E058 strains was examined using the microtiter plate method. Crystal violet staining of bacteria grown in LB rich or M9 minimum medium showed that the sodA mutant produced a decreased amount of biofilm compared with the wild-type and the complementation strains (Fig. 3), even though their growth rates were similar. Higher production of biofilm was observed under conditions using M9 minimal medium (Fig. 3). These results indicate that sodA is involved in biofilm formation by APEC E058.
FIG 3.
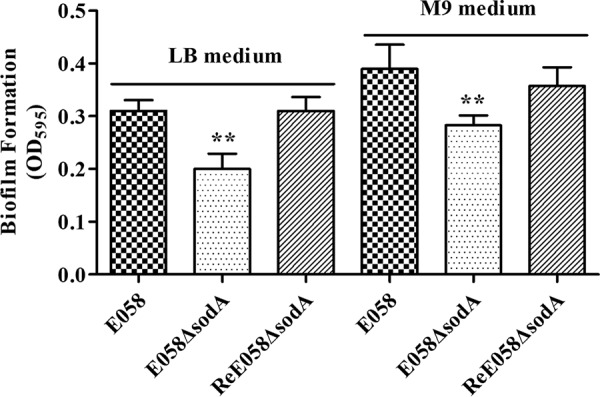
Quantification of the biofilms produced by the sodA mutant, the complementation strain, and wild-type APEC strain E058. Biofilm formation was measured for bacteria inoculated in LB rich and M9 minimum media in 96-well microtiter plates for 24 h. OD595 values for stained biofilm are the mean ± standard deviation (SD) of the results from three independent biological replicates (**, P < 0.01).
SodA promotes the ability of APEC E058 to survive within chicken macrophages.
An ingestion assay was used to analyze the ingestion of the sodA mutant by avian macrophage HD-11 cells. The inactivation of sodA rendered the bacteria more vulnerable to macrophages. The sodA mutant showed a highly increased ingestion ratio compared to the wild-type and complementation strains (P < 0.01) (Fig. 4A).
FIG 4.

Survival of bacteria in HD-11 chicken macrophages. (A) Ingestion of wild-type strain E058, E058 ΔsodA isogenic mutant, and ReE058 ΔsodA complementation strain by HD-11 cells. The values represent the average data from three independent experiments. The error bars indicate standard deviations. (B) Intracellular growth of bacteria in chicken macrophage HD-11 cells. The intracellular growth of wild-type strain E058, the E058 ΔsodA isogenic mutant, and the ReE058 ΔsodA complementation strain were compared over a 12-h period. Standard errors of the mean for the results from three independent experiments are shown. The asterisks indicate statistically significant differences (**, P < 0.01).
To determine whether SodA affects bacterial survival within macrophages, we compared the bacterial yields of sodA mutant, complementation, and wild-type strains recovered at 2, 4, 6, 8, and 12 h postinfection (beginning at time zero [T0]). The proportions of wild-type bacteria recovered from macrophages were 0.19%, 0.18%, 0.22%, 0.17%, and 0.15% of the inoculum at 2, 4, 6, 8, and 12 h postinfection (h p.i.), which were 1.1- to 1.4-fold higher than those of the primary ingested bacteria (T0) (Fig. 4B). However, the sodA mutant survived poorly in macrophages, with proportions of 0.17%, 0.16%, 0.12%, 0.07%, and 0.04% of the inoculum recovered at 2, 4, 6, 8, and 12 h p.i., which were 1.2- to 4.8-fold lower than those of the primary ingested bacteria (T0) (Fig. 4B). Moreover, the survival ability within macrophages was restored for the complementation strain. Thus, it can be concluded that sodA is involved in the survival of APEC E058 within macrophages.
Competition between the wild-type strain E058 and the sodA mutant in vitro and in vivo.
The results of competition experiments comparing mutant and wild-type strains reflect the effects of the mutation on the bacterial growth or virulence. In vitro, in LB medium, the sodA mutant competed equally with the wild-type strain (data not shown). However, during coinfection in vivo, the sodA mutant was weakened and showed a significantly reduced competitive index (CI), with mean decreases of 33-fold, 24-fold, 22-fold, and 36-fold compared to the wild-type strain in blood, liver, lung, and spleen, respectively (all P < 0.01) (Fig. 5). Meanwhile, complementation of the mutation restored competitive fitness. These results indicate that SodA confers APEC E058 a survival advantage in an in vivo competitive colonization model.
FIG 5.
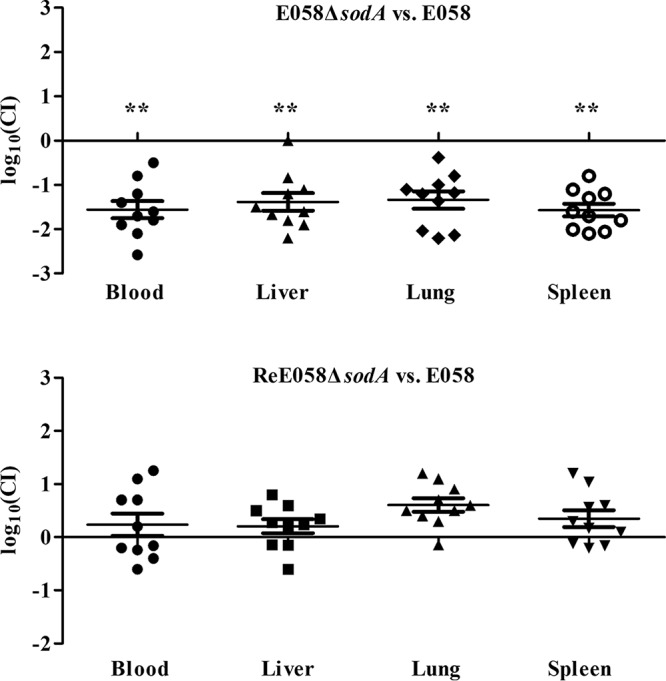
In vivo competition assays. Comparative colonization levels, presented as the log10 competitive index, in the blood, liver, lung, and spleen of SPF chickens 24 h after inoculation with a mixture of wild-type APEC strain E058 and the sodA mutant or complementation strain. Horizontal bars indicate the mean log10 CI values. Statistically significant decreases in CI values are indicated with asterisks (**, P < 0.01).
SodA contributes to bacterial colonization during systemic infection in vivo.
To investigate the involvement of sodA in APEC E058 virulence, colonization by the sodA mutant and its complementation strain, as well as the wild-type strain, was determined in challenged chickens. Bacterial loads in various tissues were compared between groups of challenged chickens at 24 h postinfection. The sodA mutant colonized poorly in vivo in comparison to the corresponding parental strain, with the bacterial loads recovered significantly lower than those of the wild-type strain E058 (P < 0.01) (Fig. 6). Complementation of the sodA mutant restored bacterial colonization capacity, with bacterial loads reaching the level of the wild-type strain. These results showed that inactivation of sodA may lead to reduced bacterial numbers in the internal organs and limit systemic invasion, indicating that SodA is involved in APEC E058 virulence and contributes to survival during systemic infection in the chicken infection model.
FIG 6.

Colonization by the wild-type strain E058, E058 ΔsodA mutant, and ReE058 ΔsodA complementation strain during systemic infection of chickens. The data are presented as log10 CFU per milliliter of heart blood or log10 CFU per gram of tissue. The horizontal bars indicate the mean values. Each data point represents a sample from an individual chicken. Statistically significant differences, as determined by the Mann-Whitney test, are indicated by asterisks (**, P < 0.01).
Expression profile of genes involved in oxidant resistance.
The transcription levels of several oxidant resistance genes were analyzed by quantitative real-time reverse transcription-PCR (qRT-PCR) in the sodA mutant. The expression of the oxidative stress response genes katE, perR, and osmC was significantly reduced in the sodA mutant strain, while the sodB and sodC genes were not affected compared to the wild-type strain. The expression levels of the tested genes were restored in the complementation strain (Fig. 7).
FIG 7.
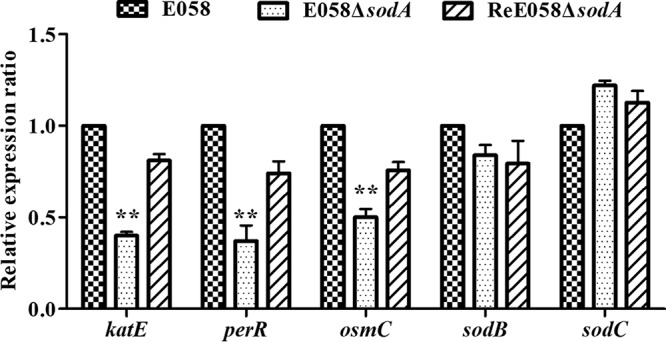
Quantitative real-time reverse transcription-PCR (qRT-PCR) analysis of gene expression. Expression of genes involved in oxidative resistance in APEC was measured by qRT-PCR for each strain. Data were normalized to the housekeeping gene gapA. Results are relative expression ratios compared to wild-type strain E058 (**, P < 0.01). The error bars indicate standard deviations.
DISCUSSION
APEC, like most pathogens, may experience oxidative stress caused by ROS derived from aerobic metabolism, environmental sources, and the host immune response (16, 25). Bacterial antioxidants play a critical role in the detoxification of endogenously and host-derived oxidative radicals during host-pathogen interactions (26, 27). Mn-cofactor-containing superoxide dismutase SodA can destroy the toxic superoxide anion, which is produced both endogenously and within the oxidative burst of host phagocytes. For a comprehensive understanding of the influence of sodA on the phenotype and pathogenicity of APEC, the sodA-deficient mutant and complementation strains from the highly virulent APEC O2 strain E058 were constructed in this study. The deletion of sodA did not affect the growth or serum or environmental stress resistance of APEC E058. However, the sodA mutant displayed an increased susceptibility to killing by exogenous hydrogen peroxide (Fig. 2). The sensitivity of the sodA mutant to H2O2 may be due to increased cellular damage from hydroxyl radicals generated by iron-mediated Fenton reaction (28). This phenotype displayed by the sodA mutant is what would be expected of a bacterial strain that has lost an important cytoplasmic defense against oxidative stress and is similar to the phenotypes of other bacterial sodA mutants (19, 29).
Biofilms play an important role in protecting pathogens from the host immune system (30). Previous research found that a higher level of expression of the sodA gene was induced during the transition to biofilm growth both in E. coli and Staphylococcus aureus (31, 32). This prompted us to study the effect of the sodA deletion on biofilm production by APEC E058. We tested the biofilm formation ability of the bacteria under nutrient-rich or -poor culture conditions (i.e., LB rich or M9 minimum medium, respectively). The results showed that a mutation of sodA decreased biofilm formation in both LB and M9 media (Fig. 3). Additionally, biofilm formation in M9 minimal medium provides greater amounts of biofilm, which is probably caused by cell starvation, which triggers biofilm formation. These results suggest that the sodA gene involved in the bacterial oxidative stress responses may play a significant role in the development of biofilm.
In view of the role of sodA in bacterial resistance to the oxidative burst of host phagocytes, we tested the ability of the sodA mutant to survive within chicken macrophage HD-11 cells. The sodA mutant was more easily ingested by the macrophages, while the wild-type and complemented bacteria appeared to resist ingestion. The deletion of sodA also resulted in decreased intracellular growth of APEC E058 in macrophages (Fig. 4), while no differences were found between the survival within macrophages of the wild-type and complemented strains. Thus, the lack of a functional sodA gene alters the interaction of APEC E058 with macrophages. Our results indicate that Mn-cofactor-containing SOD (MnSOD), by its ability to scavenge the primary product of the oxidative burst, is able to effectively impair the host response to APEC infection and thus may represent an important virulence factor in APEC E058.
To determine the contribution of sodA to APEC infection, we performed avian experimental infection tests. The sodA mutant was significantly outcompeted by the wild-type strain during in vivo coinfection (Fig. 5). Coinfection experiments can show fitness defects as well as decreased virulence, depending on the type of mutation. To test whether or not the drop in competitive fitness also occurred in vitro or if it was only seen during infection, we also conducted in vitro competition test. The sodA mutant competed equally with the wild-type strain in LB medium, which excluded the possibility of a fitness defect and demonstrated the decreased virulence phenotype of the mutant. As we have shown a potential role for SodA in survival in a macrophage cell line, we further investigated the survival of the mutant strain in the blood. The decreased numbers for the sodA mutant in the blood correlate with less of a capacity to survive in phagocytic cells. Moreover, the sodA mutant showed significantly decreased colonization compared with the wild-type strain in various organs tested in a single-strain challenge model (Fig. 6), implying that SodA plays an important role in the virulence of APEC E058 in chickens. The complementation strain showed restored virulence and colonized the internal organs of inoculated chickens to the same extent as the wild-type strain. Thus, the disruption of the sodA gene in APEC E058 leads to attenuation of virulence in systemic infection in chickens.
A previous report described a lack of a role for MnSOD in an extraintestinal pathogenic E. coli O18 strain in a newborn rat meningitis model (33). Their results indicated, however, that the elimination of sodA had little or no effect on either gastrointestinal tract colonization or the transition to bacteremia following oral inoculation in neonatal rats. Their results seem to be contradiction to ours, since APEC strains are closely related to the ExPEC strains that are associated with human neonatal meningitis. The contradiction might be due to the differences in the strains (APEC O2 versus NMEC O18) or the different inoculation routes (chickens infected via the air sac versus neonatal rat infected via oral administration). In their study, they determined the proportions of E. coli bacteremia in neonatal rats after oral inoculation but did not quantify the bacterial loads in the bloodstream, which may not fully reflect the virulence difference between the wild type and isogenic mutant. As the authors of that study indicated, their results did not rule out the possibility that SOD activity is important for survival in the bloodstream. Alternatively, previous work has shown that the virulence mechanisms of ExPEC strains can vary. Some may be invasive and internalize within cells and survive in phagocytes, whereas others may be highly refractory to phagocytosis and remain mainly extracellular. Such differences in pathogenic lifestyle could also result in differences in the importance of certain genes or pathways that would be dependent on the pathogenic mechanisms of the particular APEC strain or ExPEC strain. Thus, the previous results on ExPEC suggest that SodA may not always be important for certain types of ExPEC strains.
We also measured the effects of sodA deletion on the expression of other oxidation resistance genes. All of the katE, perR, and osmC genes, which are known to participate in the antioxidant defense mechanism against H2O2-induced stress (34–36), had lower transcriptional levels in the sodA mutant (Fig. 7) than in the wild type, which may be due to the loss of function of SodA that results in decreased production of H2O2 from intracellular superoxide radicals. The transcription of sodB or sodC was not affected by the disruption of sodA, suggesting that no compensation effect exists between these isozymes. Our previous array data showed that the sodA gene was highly expressed in vivo, while the other two metal-cofactor-containing SODs (sodB and sodC) were normally expressed (23), which implied that SodA may play a more important role than the other two SODs in the extraintestinal infectious process of APEC.
Taken together, our results suggest that sodA plays an important role in protecting against hydrogen peroxide (H2O2)-induced oxidative stress and that it contributes to the virulence of APEC strain E058.
MATERIALS AND METHODS
Bacterial strains, plasmids, primers, and growth conditions.
The bacterial strains and plasmids used in this study are described in Table 1. Oligonucleotide primers obtained from Sangon (Shanghai, China) are listed in Table 2. APEC O2 serotype E058 (iss+ iut+ iroN+ tsh+ irp+ cvaC+ sit+ mntH+) is a highly virulent strain, isolated from a chicken with a respiratory infection (37). Bacteria were routinely grown at 37°C in Luria-Bertani broth (LB) with aeration. When necessary, ampicillin and chloramphenicol were added at 60 and 30 μg/ml, respectively.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain, plasmid, or cell line | Characteristicsa | Source or reference |
|---|---|---|
| Strains | ||
| E058 | Wild-type avian E. coli serotype O2 | 37 |
| E058 ΔsodA | E058 ΔsodA::cat | This study |
| ReE058 ΔsodA | Complementation of E058 ΔsodA | This study |
| DH5α | endA1 hsdR17(rK− mK+) supE44 thi-1 recA1 gyrA (Nalr) RelA1 Δ(lacIZYA-argF)U169 deoR [ϕ80dlacΔ(lacZ)M15] | Invitrogen |
| Plasmids | ||
| pACYC184 | Medium-copy-number vector, p15A ori, Cmr Tcr | Gifted |
| pACYC-sodA | pACYC184 carrying sodA ORF and its putative native promoter | This study |
| pKD46 | Ampr; expresses λ Red recombinase | 38 |
| pKD3 | cat gene, template plasmid | 38 |
| Cell line HD-11 | Chicken macrophage line, chicken myelomonocytic transformed by the myc-encoding MC29 virus | 42 |
Nalr, nalidixic acid resistance; Cmr, chloramphenicol resistance; Tcr, tetracycline resistance; ORF, open reading frame; Ampr, ampicillin resistance.
TABLE 2.
Primers designed and used in this study
| Primer by use | Primer sequence (5′–3′) | Target gene |
|---|---|---|
| Deletion | ||
| AF | ATGAGCTATACCCTGCCATCCCTGCCGTATGCTTACGATGCCCTGGAACCGCACTTCGATAAGCAGACCATTGTGTAGGCTGGAGCTGCT | sodA |
| AR | TTATTTTTTCGCCGCAAAACGTGCCGCTGCTTCGTCCCAGTTCACCACGTTCCAGAACTCTTTAATGTAGATGGGAATTAGCCATGGTCC | |
| Complementation | ||
| RAF | CTCAAGCTTCCATCGTAATCGCGTTACa | sodA |
| RAR | CTCTCTAGATTATTTTTTCGCCGCAAAACGb | |
| RT-qPCR | ||
| katE RT-F | TTGTGGGAAGCCATTGAA | katE |
| katE RT-R | GCGATTGAGCACCATTTT | katE |
| perR RT-F | AGATGACGCCACCCAATA | perR |
| perR RT-R | GGGCATACCAGTTTACCG | perR |
| osmC RT-F | CGGGATTCACGCCAACAT | osmC |
| osmC RT-R | CGGCACCGCAACTTCACT | osmC |
| sodB RT-F | TATCACTACGGCAAGCACC | sodB |
| sodB RT-R | CAGGCAGTTCCAGTAGAAAGTA | sodB |
| sodC RT-F | ACATGAAGGGCCAGAAGG | sodC |
| sodC RT-R | ATATTATCGCCGCCAACG | sodC |
HindIII restriction site is in bold.
XbaI restriction site is in bold.
Construction of the sodA mutant and its complementation strain.
The sodA mutant was constructed using gene replacement methods based on the lambda Red recombinase system (38). The chloramphenicol resistance cassette, flanked by 5′ and 3′ sequences of sodA, was obtained by amplifying template plasmid pKD3 using primers AF and AR (Table 2). The PCR product was transformed into strain E058 containing pKD46. The mutant (E058 ΔsodA) was confirmed by PCR and verified by sequence analysis.
To construct the complementation strain, the sodA open reading frame and its upstream promoter were amplified and subcloned into pACYC184 using primer pairs RAF and RAR (Table 2). The purified recombinant plasmid pACYC-sodA was transformed into the sodA mutant strain to complement the sodA gene deletion to generate the ReE058 ΔsodA strain.
Oxidative stress experiments.
Overnight (18-h) cultures of the sodA mutant, complemented strain, and wild-type strain E058 were diluted 1:100 into LB broth and incubated until the optical density at 600 nm (OD600) reached 0.4. The bacteria were challenged in LB containing 10 mM H2O2 for 30 min at 37°C. Viable cells were counted after the oxidative stress challenge, and the percent survival was calculated as the number of CFU per milliliter remaining after the oxidant treatment divided by the initial number of CFU per milliliter times 100.
To determine bacterial growth curves under oxidative stress, overnight cultures were washed twice in phosphate-buffered saline (PBS) and standardized to an OD600 of 1.0, and approximately 106 CFU were inoculated into 5 ml LB with or without 10 mM H2O2. Bacterial growth was measured every 30 min by spectrophotometry (OD600). The experiments were performed in triplicate.
Adaptation to chicken serum and environmental stress.
Complement-sufficient specific-pathogen-free (SPF) chicken serum was prepared and pooled from 10 SPF chickens. A bactericidal assay was performed in a 96-well plate, as described previously, but with the following modifications (39). SPF chicken serum was diluted to 5, 12.5, 25, 50, and 100% in PBS (pH 7.2). Bacteria (10 μl containing 106 CFU) were inoculated into wells containing 190 μl of the diluted SPF chicken serum and heat-inactivated SPF chicken serum and then incubated at 37°C for 30 min. Bacteria were counted by plating on LB agar at serial dilutions. E. coli DH5α was used as the control strain.
Bacterial adaptation to environmental stress was examined as described previously (5). The bacteria were suspended in PBS and diluted to 108 CFU/ml. For the acidic and alkaline adaptation, 100-μl bacterial suspensions were exposed to 900 μl LB broth (pH 4.0) or 100 mM Tris (pH 10.0) for 30 min. For high-osmolarity stress, bacteria were mixed equally with 4.8 M NaCl and incubated for 1 h at 37°C. After environmental stress exposure, bacteria were diluted serially and plated on LB agar. Survival was calculated as the number of bacteria remaining after the stress exposure divided by the initial number of bacteria.
Biofilm assay.
The biofilm assay was carried out as previously described but with a slight modification (40). Overnight cultures were inoculated at 108 CFU/ml in LB or M9 minimal medium (6 g Na2HPO4, 3 g KH2PO4, 0.5 g NaCl, 1 g NH4Cl, 2 mM MgSO4, and 0.1 mM CaCl2 per liter of water [pH 7.0]) supplemented with 0.4% glucose. Then, 200 μl of bacterial cultures was incubated in 96-well plates at 37°C for 24 h. After three washes with PBS, the samples were stained for 30 min with 0.1% crystal violet, rinsed with sterile PBS, and air dried. Finally, the dye was dissolved in 95% ethanol. The solubilized biofilm was determined by measuring the OD595. The experiment was performed in triplicate.
Ingestion and intracellular survival assay.
Ingestion and intracellular survival assays were performed as previously described (41). The avian macrophage cell line HD-11 was cultured in Dulbecco’s modified Eagle’s medium (Gibco, NY) with 10% fetal bovine serum (PAA, Pasching, Australia). Cells were maintained at 37°C in a 5% CO2 environment with 2 × 105 cells per well in 24-well cell culture plates. Bacteria were added to the cells with a multiplicity of infection of 100 for 1 h to allow ingestion. Wells were washed with PBS, and the appropriate cell culture medium containing 100 μg/ml gentamicin was added for 1.5 h to kill extracellular bacteria. At this time (T0) and after different incubation periods (2, 4, 6, 8, and 12 h), the macrophages were washed with PBS, lysed with 0.1% Triton X-100, diluted in PBS, and plated onto LB agar plates for APEC CFU determination. The ingestion ratio was determined by dividing the number of ingested bacterial cells at T0 by the initial bacterial inoculation number. Intracellular growth was expressed as the fold change in the bacterial number at an additional incubation time point (i.e., 2, 4, 6, 8, or 12 h) relative to the number of ingested bacteria at T0.
In vitro and in vivo competition assays.
For the in vitro competition assay, cultures of wild-type strain E058 and the sodA mutant were mixed in a ratio of 1:1, incubated in LB broth for 4 h at 37°C, and then plated onto LB medium with or without chloramphenicol. For the in vivo competition assay, 5-week-old SPF chickens (White Leghorn; Jinan SPAFAS Poultry Co. Ltd., Jinan, China) were inoculated with cultures of wild-type strain E058 and its E058 ΔsodA mutant or ReE058 ΔsodA complemented strain, mixed in a ratio of 1:1 (1 × 108 CFU for each strain), via the left air sac. The chickens were provided with food and water ad libitum and treated in accordance with the regulations of the Administration of Affairs Concerning Experimental Animals; the protocol was approved by the Animal Care and Use Committee of Yangzhou University (approval ID SYXK [Su] 2007–0005, 21 September 2016). At 24 h after infection, cardiac blood samples (0.2 ml) were collected and suspended in 0.8 ml of PBS. Tissue samples (0.25 g) from livers, lungs, and spleens of the chickens were collected, weighed, triturated in 1 ml of PBS, and homogenized. Serial dilutions were plated on LB medium with or without chloramphenicol to select for the mutants and total bacteria, respectively. The results are shown as the log10 competitive index (CI). The CI was calculated for each mutant by dividing the output ratio (mutant/wild type) by the input ratio (mutant/wild type).
Bacterial colonization ability during systemic infection.
Five-week-old SPF chickens were infected with the wild-type, mutant, and complementation strains via the left thoracic air sac, with each bacterial suspension containing 108 CFU. After 24 h, 10 chickens from each group were euthanized by carbon dioxide asphyxiation, and organs were removed aseptically. Cardiac blood samples (0.2 ml) were collected and suspended in 0.8 ml of PBS. Tissue samples (0.25 g) from livers, lungs, and spleens of the chickens were collected, weighed, triturated in 1 ml of PBS, and homogenized. Tenfold serial dilutions of homogenates were plated onto LB agar plates to determine the numbers of bacteria.
Quantitative real-time reverse transcription-PCR.
To analyze how gene expression was affected by the disruption of sodA, we performed qRT-PCR analyses for various oxidation resistance genes in APEC E058. Overnight cultures of strains were diluted 1:100 in fresh LB medium and grown until logarithmic phase. Following RNA isolation and cDNA synthesis, qRT-PCR was performed to determine the transcription levels of the selected genes using SYBR premix Ex Taq and gene-specific primers (Table 2), and the data were normalized to the gapA transcript.
Statistical analysis.
Differences between groups were analyzed using the Statistical Package for the Social Sciences (SPSS version 15.0; SPSS, Chicago, IL). P values of <0.05 were considered significant.
ACKNOWLEDGMENTS
We thank James Allen from Liwen Bianji, Edanz Group China, for editing the English text of a draft of this paper.
This work was supported by the National Key R&D Program of China (grants 2017YFD0500203 and 2017YFD0500705), the National Natural Science Foundation of China (grants 31602059 and 31672553), and the Special Fund for Agroscientific Research in the Public Interest (grant 201303044). The work was also supported by a general financial grant from the China Postdoctoral Science Foundation (grant 2015M580477), the Natural Science Foundation of Jiangsu Province (grants BK20140485 and BK20151308), the Jiangsu Postdoctoral Science Foundation (grant 1501076C), and by a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
REFERENCES
- 1.Johnson TJ, Kariyawasam S, Wannemuehler Y, Mangiamele P, Johnson SJ, Doetkott C, Skyberg JA, Lynne AM, Johnson JR, Nolan LK. 2007. The genome sequence of avian pathogenic Escherichia coli strain O1:K1:H7 shares strong similarities with human extraintestinal pathogenic E. coli genomes. J Bacteriol 189:3228–3236. doi: 10.1128/JB.01726-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Younis G, Awad A, Mohamed N. 2017. Phenotypic and genotypic characterization of antimicrobial susceptibility of avian pathogenic Escherichia coli isolated from broiler chickens. Vet World 10:1167–1172. doi: 10.14202/vetworld.2017.1167-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dziva F, Stevens MP. 2008. Colibacillosis in poultry: unravelling the molecular basis of virulence of avian pathogenic Escherichia coli in their natural hosts. Avian Pathol 37:355–366. doi: 10.1080/03079450802216652. [DOI] [PubMed] [Google Scholar]
- 4.Kariyawasam S, Johnson TJ, Nolan LK. 2006. The pap operon of avian pathogenic Escherichia coli strain O1:K1 is located on a novel pathogenicity island. Infect Immun 74:744–749. doi: 10.1128/IAI.74.1.744-749.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Bao Y, Meng Q, Xia Y, Zhao Y, Wang Y, Tang F, ZhuGe X, Yu S, Han X, Dai J, Lu C. 2015. IbeR facilitates stress-resistance, invasion and pathogenicity of avian pathogenic Escherichia coli. PLoS One 10:e0119698. doi: 10.1371/journal.pone.0119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hejair HMA, Ma J, Zhu Y, Sun M, Dong W, Zhang Y, Pan Z, Zhang W, Yao H. 2017. Role of outer membrane protein T in pathogenicity of avian pathogenic Escherichia coli. Res Vet Sci 115:109–116. doi: 10.1016/j.rvsc.2017.01.026. [DOI] [PubMed] [Google Scholar]
- 7.Hejair HMA, Zhu Y, Ma J, Zhang Y, Pan Z, Zhang W, Yao H. 2017. Functional role of ompF and ompC porins in pathogenesis of avian pathogenic Escherichia coli. Microb Pathog 107:29–37. doi: 10.1016/j.micpath.2017.02.033. [DOI] [PubMed] [Google Scholar]
- 8.Sabri M, Caza M, Proulx J, Lymberopoulos MH, Bree A, Moulin-Schouleur M, Curtiss R III, Dozois CM. 2008. Contribution of the SitABCD, MntH, and FeoB metal transporters to the virulence of avian pathogenic Escherichia coli O78 strain χ7122. Infect Immun 76:601–611. doi: 10.1128/IAI.00789-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vidotto MC, Navarro HR, Gaziri LC. 1997. Adherence pili of pathogenic strains of avian Escherichia coli. Vet Microbiol 59:79–87. doi: 10.1016/S0378-1135(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 10.Janben T, Schwarz C, Preikschat P, Voss M, Philipp HC, Wieler LH. 2001. Virulence-associated genes in avian pathogenic Escherichia coli (APEC) isolated from internal organs of poultry having died from colibacillosis. Int J Med Microbiol 291:371–378. [DOI] [PubMed] [Google Scholar]
- 11.Caza M, Lepine F, Milot S, Dozois CM. 2008. Specific roles of the iroBCDEN genes in virulence of an avian pathogenic Escherichia coli O78 strain and in production of salmochelins. Infect Immun 76:3539–3549. doi: 10.1128/IAI.00455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ackermann MR, Cheville NF. 1991. Ultrastructural studies of the lung of turkeys (Meleagris gallopavo) inoculated intratracheally with Escherichia coli. Vet Pathol 28:183–191. doi: 10.1177/030098589102800301. [DOI] [PubMed] [Google Scholar]
- 13.Pourbakhsh SA, Boulianne M, Martineau-Doizé B, Fairbrother JM. 1997. Virulence mechanisms of avian fimbriated Escherichia coli in experimentally inoculated chickens. Vet Microbiol 58:195–213. doi: 10.1016/S0378-1135(97)00163-6. [DOI] [PubMed] [Google Scholar]
- 14.Mellata M, Dho-Moulin M, Dozois CM, Curtiss R III, Lehoux B, Fairbrother JM. 2003. Role of avian pathogenic Escherichia coli virulence factors in bacterial interaction with chicken heterophils and macrophages. Infect Immun 71:494–503. doi: 10.1128/IAI.71.1.494-503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Geus ED, Vervelde L. 2013. Regulation of macrophage and dendritic cell function by pathogens and through immunomodulation in the avian mucosa. Dev Comp Immunol 41:341–351. doi: 10.1016/j.dci.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 16.Miller RA, Britigan BE. 1997. Role of oxidants in microbial pathophysiology. Clin Microbiol Rev 10:1–18. doi: 10.1128/CMR.10.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayir H. 2005. Reactive oxygen species. Crit Care Med 33:S498–S501. doi: 10.1097/01.CCM.0000186787.64500.12. [DOI] [PubMed] [Google Scholar]
- 18.Hassan HM. 1984. Determination of microbial damage caused by oxygen free radicals, and the protective role of superoxide dismutase. Methods Enzymol 105:404–412. doi: 10.1016/S0076-6879(84)05056-4. [DOI] [PubMed] [Google Scholar]
- 19.Tang Y, Zhang X, Wu W, Lu Z, Fang W. 2012. Inactivation of the sodA gene of Streptococcus suis type 2 encoding superoxide dismutase leads to reduced virulence to mice. Vet Microbiol 158:360–366. doi: 10.1016/j.vetmic.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 20.Esteve-Gassent MD, Elliott NL, Seshu J. 2009. sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Mol Microbiol 71:594–612. doi: 10.1111/j.1365-2958.2008.06549.x. [DOI] [PubMed] [Google Scholar]
- 21.Bakshi CS, Malik M, Regan K, Melendez JA, Metzger DW, Pavlov VM, Sellati TJ. 2006. Superoxide dismutase B gene (sodB)-deficient mutants of Francisella tularensis demonstrate hypersensitivity to oxidative stress and attenuated virulence. J Bacteriol 188:6443–6448. doi: 10.1128/JB.00266-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Battistoni A. 2003. Role of prokaryotic Cu, Zn superoxide dismutase in pathogenesis. Biochem Soc Trans 31:1326–1329. doi: 10.1042/bst0311326. [DOI] [PubMed] [Google Scholar]
- 23.Gao Q, Xia L, Liu J, Wang X, Gao S, Liu X. 2016. DNA microarray-mediated transcriptional profiling of avian pathogenic Escherichia coli O2 strain E058 during its infection of chicken. Microb Pathog 100:1–9. doi: 10.1016/j.micpath.2016.08.032. [DOI] [PubMed] [Google Scholar]
- 24.Mellata M, Dho-Moulin M, Dozois CM, Curtiss R III, Brown PK, Arne P, Bree A, Desautels C, Fairbrother JM. 2003. Role of virulence factors in resistance of avian pathogenic Escherichia coli to serum and in pathogenicity. Infect Immun 71:536–540. doi: 10.1128/IAI.71.1.536-540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang FC. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat Rev Microbiol 2:820–832. doi: 10.1038/nrmicro1004. [DOI] [PubMed] [Google Scholar]
- 26.Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. 2001. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun 69:4980–4987. doi: 10.1128/IAI.69.8.4980-4987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards KM, Cynamon MH, Voladri RK, Hager CC, DeStefano MS, Tham KT, Lakey DL, Bochan MR, Kernodle DS. 2001. Iron-cofactored superoxide dismutase inhibits host responses to Mycobacterium tuberculosis. Am J Respir Crit Care Med 164:2213–2219. doi: 10.1164/ajrccm.164.12.2106093. [DOI] [PubMed] [Google Scholar]
- 28.Takeda K, Sato J, Goto K, Fujita T, Watanabe T, Abo M, Yoshimura E, Nakagawa J, Abe A, Kawasaki S, Niimura Y. 2010. Escherichia coli ferredoxin-NADP+ reductase and oxygen-insensitive nitroreductase are capable of functioning as ferric reductase and of driving the Fenton reaction. Biometals 23:727–737. doi: 10.1007/s10534-010-9339-8. [DOI] [PubMed] [Google Scholar]
- 29.Poyart C, Pellegrini E, Gaillot O, Boumaila C, Baptista M, Trieu-Cuot P. 2001. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect Immun 69:5098–5106. doi: 10.1128/IAI.69.8.5098-5106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valle J, Solano C, Garcia B, Toledo-Arana A, Lasa I. 2013. Biofilm switch and immune response determinants at early stages of infection. Trends Microbiol 21:364–371. doi: 10.1016/j.tim.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Resch A, Rosenstein R, Nerz C, Gotz F. 2005. Differential gene expression profiling of Staphylococcus aureus cultivated under biofilm and planktonic conditions. Appl Environ Microbiol 71:2663–2676. doi: 10.1128/AEM.71.5.2663-2676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schembri MA, Kjaergaard K, Klemm P. 2003. Global gene expression in Escherichia coli biofilms. Mol Microbiol 48:253–267. doi: 10.1046/j.1365-2958.2003.03432.x. [DOI] [PubMed] [Google Scholar]
- 33.Bloch CA, Thorne GM, Ausubel FM. 1989. General method for site-directed mutagenesis in Escherichia coli O18ac:K1:H7: deletion of the inducible superoxide dismutase gene, sodA, does not diminish bacteremia in neonatal rats. Infect Immun 57:2141–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JW, Helmann JD. 2006. The PerR transcription factor senses H2O2 by metal-catalysed histidine oxidation. Nature 440:363–367. doi: 10.1038/nature04537. [DOI] [PubMed] [Google Scholar]
- 35.Jha V, Chelikani P, Carpena X, Fita I, Loewen PC. 2012. Influence of main channel structure on H2O2 access to the heme cavity of catalase KatE of Escherichia coli. Arch Biochem Biophys 526:54–59. doi: 10.1016/j.abb.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 36.Saikolappan S, Das K, Sasindran SJ, Jagannath C, Dhandayuthapani S. 2011. OsmC proteins of Mycobacterium tuberculosis and Mycobacterium smegmatis protect against organic hydroperoxide stress. Tuberculosis (Edinb) 91:S119–S127. doi: 10.1016/j.tube.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L, Gao S, Huan H, Xu X, Zhu X, Yang W, Gao Q, Liu X. 2009. Comparison of virulence factors and expression of specific genes between uropathogenic Escherichia coli and avian pathogenic E. coli in a murine urinary tract infection model and a chicken challenge model. Microbiology 155:1634–1644. doi: 10.1099/mic.0.024869-0. [DOI] [PubMed] [Google Scholar]
- 38.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dozois CM, Dho-Moulin M, Bree A, Fairbrother JM, Desautels C, Curtiss R III.. 2000. Relationship between the Tsh autotransporter and pathogenicity of avian Escherichia coli and localization and analysis of the tsh genetic region. Infect Immun 68:4145–4154. doi: 10.1128/IAI.68.7.4145-4154.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luo G, Huang L, Su Y, Qin Y, Xu X, Zhao L, Yan Q. 2016. flrA, flrB and flrC regulate adhesion by controlling the expression of critical virulence genes in Vibrio alginolyticus. Emerg Microbes Infect 5:e85. doi: 10.1038/emi.2016.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gahring LC, Heffron F, Finlay BB, Falkow S. 1990. Invasion and replication of Salmonella typhimurium in animal cells. Infect Immun 58:443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beug H, von Kirchbach A, Doderlein G, Conscience JF, Graf T. 1979. Chicken hematopoietic cells transformed by seven strains of defective avian leukemia viruses display three distinct phenotypes of differentiation. Cell 18:375–390. doi: 10.1016/0092-8674(79)90057-6. [DOI] [PubMed] [Google Scholar]