

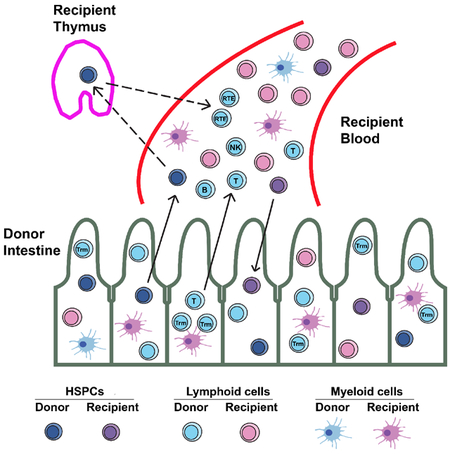
Summary
Human intestinal transplantation often results in long-term mixed chimerism of donor and recipient blood in transplant patients. We followed the phenotypes of chimeric peripheral blood cells in 21 patients receiving intestinal allografts over 5 years. Donor lymphocyte phenotypes suggested a contribution of hematopoietic stem and progenitor cells (HSPCs) from the graft. Surprisingly, we detected donor-derived HSPCs in intestinal mucosa, Peyer’s patches, mesenteric lymph nodes, and liver. Human gut HSPCs are phenotypically similar to bone marrow HSPCs and have multilineage differentiation potential in vitro and in vivo. Analysis of circulating post-transplant donor T cells suggests that they undergo selection in recipient lymphoid organs to acquire immune tolerance. Our longitudinal study of human HSPCs carried in intestinal allografts demonstrates their turnover kinetics and gradual replacement of donor-derived HSPCs from a circulating pool. Thus, we have demonstrated the existence of functioning HSPCs in human intestines with implications for promoting tolerance in transplant recipients.
Graphical Abstract
eTOC Blurb:
This paper demonstrates the presence and multilineage differentiation potential of hematopoietic stem and progenitor cells (HSPCs) carried in human intestinal allografts. These contribute to peripheral blood mixed chimerism in the recipient. Kinetic turnover studies revealed the gradual replacement of intestinal mucosal HSPCs by a circulating pool in humans.
Introduction
Intestinal transplantation (ITx) provides the only long-term option for patients with end-stage intestinal failure (Kubal et al., 2015). ITx may be performed in isolation (iITx) or as part of a multivisceral transplant (MVTx) (Kubal et al., 2015). ITx is associated with high rates of graft failure, approximately 50% at 5 years (Smith et al., 2018) and these rates are reduced in MVTx compared to iITx (Abu-Elmagd et al., 2012; Kato et al., 2006). The large lymphoid load in intestinal allografts results in graft-versus-host disease (GVHD) in 5–9% of patients (Berger et al., 2012). Thus, ITx involves a delicate balance between graft rejection, GVHD, and opportunistic infections due to over-immunosuppression.
Blood chimerism denotes coexistence of hematopoietic cells from both the donor and recipient after a transplant. Blood chimerism, either durable or transient, can be achieved without GVHD, and, even when transient, can promote donor-specific tolerance in animals and humans (Kawai et al., 2011; Kawai et al., 2013; Wekerle et al., 2000). We recently reported (Fu et al., 2017; Zuberet al., 2015) that a high level of donor T cell macrochimerism (denoting ≥4% donor CD3+ T cells) often appears in the blood following ITx, usually without GVHD. This occurs more often in MVTx than in iITx recipients. Macrochimerism correlates with less graft rejection (Zuber et al., 2015) and slower T cell replacement by the recipient within the allograft (Zuber et al., 2016). Long persisting donor blood chimerism can be observed in T/B/NK cells, especially for MVTx patients. Myeloid chimerism is more transient (Zuber et al., 2015).
CD45+CD34+ cells have been reported in adult human small intestine (Lynch et al., 2006) and liver (Golden-Mason and O’Farrelly, 2002; Wang et al., 2012), but detailed analysis of those in the intestine was not reported. We hypothesized that graft-resident hematopoietic stem and progenitor cells (HSPCs) contribute to long-term multilineage blood chimerism after human ITx. Using the current differentiation scheme for human hematopoiesis (Gorgens et al., 2013), we identified HSCs and multiple types of progenitors, including multipotent progenitors (MPPs), lymphoid-primed multipotent progenitors (LMPPs), common lymphoid progenitors (CLPs), myeloid progenitors (MPs), and unclassified progenitors co-expressing CD56 (Golden-Mason and O’Farrelly, 2002; Lynch et al., 2006) in human ileum mucosa, Peyer’s patches (PPs), mesenteric lymph nodes (MLNs), and liver. Colony-forming-cell (CFC) assays, long-term-culture-initiating-cell (LTC-IC) assays and humanized mouse models demonstrated the differentiation potential of gut HSPCs. We further investigated the dynamics of HSPC replacement by the recipient within the intestinal allograft and demonstrate replenishment from a circulating pool.
Results
Long-term multilineage chimerism after ITx
We extended our previous longitudinal prospective study of peripheral blood chimerism (Pts 1–7, 9–10) (Zuber et al., 2015) by recruiting 12 more patients (Pts 13–24) to include a total of 10 MVTx, 1 LITx, and 10 iITx recipients (Table S1) followed up to five years post-ITx. By combining donor- and/or recipient- specific HLA markers with a pan- HLA-ABC mAb, we could reliably distinguish donor from recipient cells as described (Zuber et al., 2015) (Figure S1A and Table S2). High level (>10%) and long-lasting (postoperative day [POD]>90) donor lymphoid (T, B, NK) and myeloid chimerism was more frequently seen in MVTx recipients than iITx recipients (Figure S1, B–D, F–H). A lower level of peak donor T cell chimerism (<4%) in blood was significantly associated with the development of early (POD<90) moderate to severe graft rejection (Figure S1J), strengthening our previous observations (Zuber et al., 2015). Myeloid chimerism was generally at a low level and transient even in MVTx patients (Zuber et al., 2015). However, we observed recurrent donor chimerism among CD14+ monocytes (>10%) late post-Tx in MVTx recipients Pt15 (POD 255) and Pt18 (POD314) and relatively persistent (> 2 months) donor chimerism among CD14+ monocytes in MVTx recipient Pt19 (Figure S1E, I).
Circulating donor-derived T cells are enriched for a recent thymic emigrant (RTE) phenotype and T cell receptor excision circles (TRECs)
Unlike the donor IEL and LPL T cells, which displayed a tissue-resident phenotype (Zuber et al., 2016) and consisted mostly of memory T cells, circulating donor T cells expressed a CD28highCD69−CD103− phenotype consistent with circulating T cells and not tissue-resident T cells (CD28low/− CD69+ CD103+/dim), and included both naïve (CD45RA+) and memory phenotypes (CD45RO+) (Figure 1A, B). Circulating donor CD4 T cells were markedly enriched for the RTE phenotype (Figure 1C) compared with recipient cells in patients ≥5 years old (Figure 1D, G; S2A, B). Younger recipients generally had high recipient and donor RTE levels (Figure 1D; S2B), presumably reflecting robust thymic function. A similar trend was observed for circulating donor CD8 T cells (Figure 1E, F and G; S2A, B). RTEs were significantly enriched in circulating donor compared to recipient CD4 T cells at early (POD≤100), intermediate (POD100–200) and late (POD>200) stages post-Tx (Figure S2C). A similar increase was seen for RTEs among donor vs recipient CD8 T cells from POD100–200. CD31 expression on CD4 T cells is a well-validated RTE marker (Kimmig et al., 2002), whereas its expression on CD8 T cells may be affected by other factors (Tanaskovic et al., 2010). Analysis of TRECs as an indicator of recent thymic origin (Hazenberg et al., 2001) confirmed that CD45RA+ CD4 T cells expressed much higher levels of TRECs than CD45RO+ cells in both donor-derived and recipient T cells in Pt1 and in healthy controls (HCs) (Figure 1H). Both donor and recipient CD45RA+CCR7+ T cells obtained 167–786 days post-Tx from Pts 15, 16, 18 and 19 were markedly enriched for TRECs (Figure 1I). Overall, enrichment of RTE phenotypes and TRECs strongly supports the notion of de novo generation of circulating donor T cells post-Tx. Circulating donor-derived δ1+ T cells and CD19+ B cells in patients with long-term multilineage chimerism were also enriched for naïve populations (Figure S3).
Figure 1. Circulating donor-derived T cells are enriched for RTE phenotypes and TRECs regardless of donor age.

(A) Expression of CD28, CD69 and CD103 on donor CD4 and CD8 T cells from Pt7’s PBMCs or ileum IELs and LPLs isolated on POD127. (B) Expression of CD45RA and CD45RO on donor or recipient CD4 T cells from Pt7’s ileum IELs and LPLs on POD45, and PBMCs on POD45 and POD121. FCM gating of CD4+ RTEs (CD45RA+CD45RO−CD31+) in Pt1 PBMCs POD246 (C) and CD8+ RTEs (CD45RA+CCR7+CD31+) in Pt15 PBMCs POD83 (E). Percentage of RTEs among CD4 (D) or CD8 (F) T cells in healthy control (HC, gray) PBMCs or circulating donor- (red) or recipient-(black) derived CD4 or CD8 T cells post-Tx. (G) Percentage of RTEs among CD4 and CD8 T cells in circulating donor (“D”) or recipient (“R”) T cell populations in the same sample (shown connected with a line) post-Tx of recipients at least 5 years old. Donor and recipient phenotypes in the same sample were compared using a two-tailed paired Student t test. (H) Number of TRECs detected in CD45RA+ or CD45RO+ CD4 T cells from two HCs’ PBMCs and Pt1’s PBMCs POD246/365. (I) Number of TRECs detected in naïve (CD45RA+CCR7+) or non-naïve CD3+ T cells from one HC’s PBMCs and Pts15, 16, 18 and 19’s PBMCs collected on POD255, 786, 314 and 167, respectively. See also Figure S1, S2, S3, S7and Table S1, S2.
Lack of repertoire overlap between pre-Tx or graft donor T cells and circulating donor-derived naïve T cells
Naïve T cells demonstrate high repertoire diversity, making it difficult to detect clonal overlap among different tissues within the same individual (Thome et al., 2016b). Consistently, when we sorted naïve and memory donor T cells from Pt7 PBMCs collected from day 101 to 136 post-Tx and performed high-throughput TCRβ CDR3 sequencing, clones overlapping with those in pre-Tx donor lymphoid tissue were detected only among memory (209/579 unique sequences = 36.1%) but not naïve donor T cells (Figure 2A). Naïve donor T cells in PBMC also showed much less overlap with early ileum biopsy T cells (POD24) compared to their memory counterparts (Figure 2A). Indicative of greater diversity, the clonality (0.061) of naïve circulating donor T cells at POD101–136 was lower than that of their memory counterparts (0.150).
Figure 2. Lack of repertoire overlap between pre-Tx T cells and circulating donor-derivedT cells.

(A) Venn diagram showing TCR clonal overlap among donor memory and naïve T cells sorted from PBMCs POD101–136, pre-Tx donor spleen (including unstimulated [D4U and D8U], and CFSElow stimulated T cells [D4L and D8L]) and post-Tx ileum biopsy (POD24) from Pt7. D4U and D8U represent unstimulated donor CD4 and CD8 T cells, respectively; D4L and D8L repersent CFSElow donor CD4 and CD8 T cell responders in MLRs, respectively. Clonality of each population is shown. (B) Proportional Venn diagram showing TCR clonal overlap among sorted donor T cells in Pt18 PBMCs POD357 (B) or Pt15 PBMCs POD143 (C) and pre-Tx donor spleen or MLN. “Non-mappable” percentage is the percent of TCR sequences in donor PBMC that were not detected in pre-Tx donor spleen or MLN. (D) Abundance plots of nonmappable clones detected from sorted donor T cells in Pt15 PBMCs POD143 (green), and pre-Tx D4U (black) and D8U (red) T cells from spleen or total T cells from MLN (blue), showing relative log abundance of TCR sequences (Y axis) against their log rank in abundance (x axis: low to high frequency). Power-law slopes of abudance plots, whose absolute values vary directly as diversity (DeWolf et al., 2018), are shown. See also Table S1, S2.
Sorting and sequencing donor T cells in Pt18’s and Pt15’s PBMCs on POD357 and POD143, respectively, similarly revealed that a high proportion (96.3% and 98.2%, respectively) of TCRs were undetectable (i.e. “non-mappable”) in the pre-Tx donor lymphoid tissues (Figure 2B, C). The 48,042 non-mappable clones identified from Pt15’s sorted donor PBMC T cells on POD143 showed similarly high diversity as pre-Tx donor unstimulated spleen and MLN (Figure 2C, D) using a new diversity measure (slope) that captures the majority of clones (DeWolf et al., 2018) and by clonality measurements. In sum, the lack of repertoire overlap with pre-Tx donor lymphocytes combined with the enrichment of RTEs and TRECs among donor circulating T cells suggests that these donor circulating naïve T cells developed de novo in the recipient post-Tx.
Long-term (POD>200) circulating donor cells are not GvH-reactive and are functional
We performed MLRs on sorted T cells from Pt15 PBMCs collected on POD83 and POD214, with 27% and 26% of donor T cell chimerism, respectively. In contrast to pre-Tx donor T cells from spleen, which reacted vigorously to recipient antigens, circulating donor T cells from post-Tx PBMCs did not proliferate to recipient antigens but reacted strongly to third party antigens (Figure 3A). However, depletion of CD25+ cells from the sorted T cell responders revealed a donor CD4 T cell response to recipient stimulators (Figure 3A). Similar specific donor T cell hyporesponsiveness to the recipient was observed using PBMCs from Pt7 (POD253, 1.5% donor T cell chimerism) and POD786 splenocytes from Pt16, who still showed 3.7% donor T cell chimerism in the spleen. Hyporesponsivenss of donor T cells in all 3 patients was recipient-specific (Figure 3B), despite equal or even greater HLA mismatching to the recipient than to the 3rd party (Figure 3E)
Figure 3. Long-lasting circulating donor cells show GvH tolerance.

(A) Functional MLR using Pt15 pre-Tx donor splenocytes or post-Tx total or CD25− T cells sorted from recipient PBMCs collected on POD83 and POD214 as responders, and irradiated recipient pre-Tx splenocytes or 3rd party PBMCs as stimulators. CFSE proliferation profile of gated donor CD4 and CD8 T cells is shown. (B) Summary of %CFSElow donor CD4 and CD8 T cells in functional MLRs using pre-Tx donor splenocytes (Pts15, 7 and 16) or post-Tx PBMCs (Pt15 POD214, Pt7 POD253) or splenocytes (Pt16 POD786) as responders against irradiated stimulators. Student t test was used to compare paired data as indicated (* p<0.05, **p<0.01, ***p<0.001; n.s.: no significant difference). Means and standard deviations (SDs) are shown in bar graphs. (C) Donor HLA+CD3+CFSElow cells were sorted on Day 6 of a MLR in which CD25− T cells sorted from POD83 PBMCs were used as responders, and irradiated recipient pre-Tx splenocytes as stimulators. TCRβ CDR3 DNA sequencing was performed on sorted donor HLA+CD3+CFSElow cells, and the cumulative frequency of GvH clones as a percentage of all sequences within donor mappable clones is shown. “Donor mappable clones” refer to clones that were detectable in sequenced pre-Tx spleen, lymph node and/or MLR CFSElow T cell populations from the donor. Four such clones were identified among a total of 124 unique sequences. (D) Total frequencies among all TCR sequences for representative GvH clones that were detected in pre-Tx donor spleen or MLN, post-Tx early (POD<70) and late (POD>140) ileum biopsies (Bx), and early (POD<70) and late (POD>250) PBMCs in Pts 4, 7, 13 and 15. (E) HLA typing (HLA-A, B, C, DRB1 and DQ) and number of HLA mismatches (red) of donor with recipient and 3rd party is shown for Pt15. Pt 15’s donor had a greater number of HLA mismatches to the recipient (8/10) than to the 3rd party (7/10). Pt7 and Pt16’s donors each had equal numbers of HLA mismatches to the recipient and the 3rd party, 5/10 and 8/10, respectively (data not shown). See also Figure S1 and Table S1, S2.
TCR sequencing on sorted donor HLA+CD3+CFSElow cells from Pt15 POD83 MLR indicated that the weak GvH response revealed by depletion of CD25+ cells included a dominant preexisting donor GvH clone (Figure 3C), despite the small sample size and low number of unique clones (124), in which four donor-mappable clones were identified. Additional GvH clones identified in pre-Tx MLR by their CDR3 sequences persisted long-term in both the allograft (POD>140), at high frequency (Zuber et al., 2016), and recipient peripheral blood (POD>250), at low frequency (Figure 3D).
These data suggest that tolerance to the recipient was induced among long-term donor T cells in the recipient lymphoid compartment, consistent with de novo development of the naïve T cells in the recipient thymus and with the possibility that recipient-specific regulatory T cells (Tregs) promote tolerance of GvH T cells from the intestinal allograft.
HSPCs are present in human intestinal mucosa, PPs, MLN and liver
Consistent with the long-term multilineage chimerism in patients with high peak levels (>4%) of donor T cell chimerism in blood (Figure S1), we detected circulating donor HLA+ hematopoietic progenitors (Lin−CD45+CD34+) (Figure 4A) in 7 of 7 patients (Figure 4B). We hypothesized that these originated in the graft, as CD45+CD34+ cells have been reported in adult human small intestine (Lynch et al., 2006) and liver (Golden-Mason and O’Farrelly, 2002; Wang et al., 2012). Using the current differentiation scheme for human hematopoiesis (Gorgens et al., 2013), we further assessed the presence in such tissues of HSCs and progenitors, including MPPs, LMPPs, CLPs, MPs, and CD56+ progenitors (Golden-Mason and O’Farrelly, 2002; Lynch et al., 2006),by flow cytometry (FCM) (Figure 4C; S4). We analyzed HSPCs in intestinal mucosa, PPs, MLNs and BM from multiple deceased organ donors (Figure 4C, D, S4 and Table S3). All of these tissues contained HSPCs at variable levels (Figure 4D). Ileum LPLs included higher percentages of HSCs than IELs or cells from MLN (Figure 4D). Remarkably, the percentages of HSCs and LMPPs were greater among CD45+/dimCD34+ cells of ileum IEL, LPL and PPs than among adult BM or fetal liver cells (Figure 4D). The percentage of Lin−CD45+/dimCD34+ cells or of HSCs among Lin−CD45+/dimCD34+ cells in ileum LPLs was not dependent on donor age or sex (Figure S5). In a small number of transplant patients for whom pre-ITx donor liver biopsies (n=2) and donor organ perfusates (n=8) were available, we were able to detect variable levels of HSPCs (Figure 4E, F). Therefore, both liver and intestines are reservoirs for HSPCs that may partially explain the long-lasting donor mixed chimerism that is more frequently seen in MVTx patients than iITx patients.
Figure 4. Human ileum, Peyer’s patches, MLN, and liver contain HSPCs.

(A) Representative FCM gating of donor HLA+ cells among DAPI−HLA-ABC+Lin−CD45+/dimCD34+ HPs in Pt7 PBMCs POD44. Lin−:CD3−CD5−CD14−CD19−. (B) Percentage (left panel) and concentration (right panel) of donor cells among DAPI−HLA-ABC+Lin−CD45+/dimCD34+ HPs in blood from indicated patients and POD. (C) Representative FCM gating of HSPC among deceased donor D#265 (24 y/o, male) ileum LPLs. (D) Percentage and composition of HSPCs in human ileum (IEL, LPL), PPs, MLN and BM among multiple deceased organ donors. Student t test was used for statistical comparisons between paired tissues, including ileum IEL, LPL, PPs and MLN (* p<0.05, **p<0.01). Means and SDs are shown. Cells from two fetal liver donors are used as reference. Percentages of CD45+/dimCD34+ cells among DAPI−Lin−CD45+/dim cells and percentages of HSC, MPP, LMPP, CLP, MP and CD56+ progenitors among DAPI−Lin-CD45+/dimCD34+ cells in liver biopsies (E) and organ perfusates (F) collected from donors of Pts15, 16, 17, 18, 20, 21, 23 and 24 pre-ITx. See also Figure S4, S5 and Table S1, S2, S3.
Intestinal LPL HSPCs are phenotypically similar to BM HSPCs
We used CyTOF, including barcodes to distinguish BM and ileum LPL-derived CD45+/dim cells from the same organ donor, and analyzed markers of particular hematopoietic lineages (CD3/CD4/CD8/γδ TCR/CD14/CD11c/CD11b/CD56) and progenitors (CD34/CD38/CD10/CD90/CD45RA) and of thymus homing ability (CCR9). Lin−CD45+/dimCD34+ cells from ileum LPL and BM of the same donor demonstrated similar phenotypic properties (localization) in viSNE plots (Figure 5A). Both ileum LPL and BM Lin−CD45+/dimCD34+ cells contained a subpopulation that expressed progenitor thymus homing marker CCR9 (Haddad et al., 2006; Zlotoff and Bhandoola, 2011) (Figure 5B), and T/NK lineage-polarized progenitors that co-expressed CD45RA and CD7 (Haddad et al., 2004; Haddad et al., 2006) (Figure 5C). These phenotypes were observed across a wide age range (26–76 y/o).
Figure 5. Gut HSPCs are phenotypically similar to BM HSPCs in CyTOF analysis.

(A) viSNE plot and density dot plot of CD45+/dimCD34+ cells from lineage-depleted BM cells and ileum LPLs in each individual organ donor (D#259, D#280, D#305 and D#337). CD45+/dimCD34+ cells (gated as shown in dot plots) are indicated in red in viSNE plots. (B) Expression of HP thymus-homing marker CCR9 on viSNE plot of CD45+/dim lineage-depleted BM cells and ileum LPLs in each individual organ donor. CD34+ gating was kept as shown in (A). Overlap between CCR9+ cells (red) and these gated populations is shown in the viSNE plot. (C) Analysis of HSPCs coexpressing CD45RA and CD7 in BM and ileum LPL of adult deceased organ donors D#280, D#305 and D#337. NA: not applicable. See also Table S3.
Multilineage differentiation potential of intestinal LPL HSPCs in vitro and in vivo
Both ileum LPL and BM Lin− CD34-enriched cells formed myeloid/erythroid colonies in CFC assays, although those from ileum were less efficient than BM CD34 cells (Figures 6A, S6A). Long-term LTC-IC assay (Figures 6B, S6B, S6C) using single sorted ileum LPL HSC co-culture with MS5-DL1ind100 feeder cells (Calvo et al., 2012) demonstrated their lymphoid (T/B) differentiation potential. BM and ileum LPL HSCs sorted from the same donor showed similar percentages of colony-forming cells in LTC-IC assays (Figure 6B).
Figure 6. Multilineage differentiation potential of human gut HSPCs in vitro and in vivo.

(A) Representative colonies from short-term CFC assay of Lin− CD34-enriched cells from BM or ileum LPL of D#259 (46 y/o male). Scale bar: 100μm. Bar graph shows normalized colony number per 35mm dish. (B) Representative colonies from long-term LTC-IC assay of single cell sorted HSCs from D#305 (28 y/o female) and D#332 (38 y/o male) BM or ileum LPLs co-cultured with MS5-DL1ind100 stromal cells. CD3+ T cells (green), CD20+ B cells (red) and CD14+ myeloid cells (purple) were detectable within the colony on day 42 by fluorescence microscopy. HC human PBMCs were positive staining controls. Scale bar: 20 μm. Bar graph shows the percentage of colonies forming from single-cell sorted BM or ileum LPL HSC. (C) Sublethally irradiated representative NSG mouse (#110) of 3 mice analyzed with human thymus implant (HLA-A3+A2−A9−) received FACS-sorted CD45+/dimCD34+ cells (1 × 106) from D#293 ileum LPLs (53 y/o female, HLA- A3−A2+A9−) and CD34+ magnetic-activated cell sorting (MACS)-isolated cells (1 × 105) from D#291 BM (26 y/o female, HLA- A3− A2− A9+) at a ratio of 10:1. Representative FCM gating shows T/NK/B/Myeloid lineage reconstitution in mouse blood approximately 19 weeks post-adoptive transfer (second row) and in mouse BM at termination at week 24 (third row). Human HC PBMCs (HLA- A3−A2+A9−) serve as positive controls (first row). Human thymus implant (HLA-A3+A2−A9−) harvested from another NSG mouse (#558) at termination at week 24 (fourth row) contains gut-derived CD4+CD8+ T cells and CD34+ progenitors. Mouse #558 received CD45+/dimCD34+ FACS-sorted cells (4.5 × 105) from D#291 ileum LPLs and CD34+ MACS-isolated cells (2.5 × 105) from D#293 BM at a ratio of 1.8:1. (D) Percentage of ileum (red), BM (purple) or thymus (green)-derived cells among hCD45+mCD45− populations in NSG mouse (#110) blood at different times post-adoptive transfer. (E) Percentage of different lineages repopulating in NSG mouse (#110) blood among HLA- A3−A2+A9− compartment originating from D#293 ileum. See also Figure S6 and Table S3, S4.
We used a humanized mouse model to determine the ability of human intestinal Lin−CD45+/dimCD34+ cells to reconstitute hematopoietic lineages in vivo (Table S4). In one animal (#110), gut HSPC-derived cells of different lineages were identified by FCM (Figure 6, C–E). When residual T cells carried by the human thymus graft had nearly disappeared in NSG recipient (#110) blood (around 12 weeks post-Tx) (Figure 6D), human gut origin cells from D#293 dominated among human CD45+ cells (Figure 6D), demonstrating de novo generation of T/B/NK/Myeloid lineages (Figure 6E). At 24 weeks post-Tx, human gut origin cells (HLAA2+A9−A3−) showed multilineage differentiation in BM harvested from the recipient (#110) (Figure 6C). A small population of gut-derived CD34+ cells remained within the BM (Figure 6C), suggesting that HSCs had engrafted. In another animal with a human fetal thymus implant (#558), gut-derived CD45+/dimCD34+ progenitors and CD4+CD8+ T cells accounted for 0.21% and 60% of gut origin thymocytes (HLA-A2−A3−A9+) in the human thymus graft at 24 weeks (Figure 6C). In two additional mice (#329 and #382) that did not receive a human thymus implant, a 6.4:1 or 4:1 ratio of ileum versus BM origin HSPCs was injected (Figure S6). Human intestine-derived cells from D#238 or D#332 accounted for >90% of hCD45+ cells in mouse PBMCs approximately 2 weeks post-transfer. This proportion declined to 10–50% by week 8 and to <1.5% at week 10 post-transfer, with temporary recovery from week 17 to 22 (Figure S6D, F). Of ten humanized mice in our study (Table S4), four were followed only short-term (2 to 3 weeks). Among six mice with long-term (>24 weeks) follow-up, human gut HSC/HP-derived cells were detectable in the peripheral blood of all and chimerism persisted >12 weeks in four mice. At termination at week 24 or 28 post-adoptive transfer, human gut HSC/HP-derived cells were detectable in one or more tissues (BM/spleen/thymus) of five of the six mice (Table S4). CD4 T cells, NKT cells, B cells and monocytes were the major lineages derived from intestinal HSC/HP. Together, our data indicate that human gut HSPCs have multilineage differentiation potential (T/B/NK/Myeloid) and can likely self-renew, although less than BM HSPCs in some experiments.
Replacement of gut HSPCs from the circulation
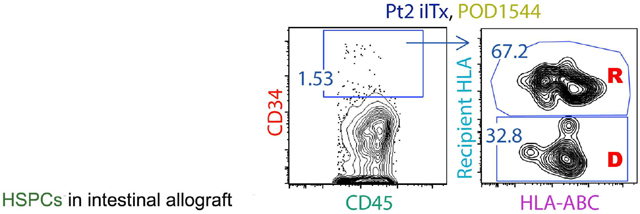
We tracked the origin of HSPCs in intestinal allograft biopsy specimens (jejunum, ileum or colon) collected from 10 ITx recipients whenever sufficient samples were available from stoma revision/closure or small bowel removal (POD28–1606). Representative FCM gating to distinguish donor- versus recipient-derived HSPCs is shown in Figure 7A. A specimen taken from the native colon of Pt14 on POD532 served as a control containing recipient-derived HSPCs (Figure 7 B–D, G). Variable percentages of CD34+ cells were detected among DAPI−CD45+/dim Lin− cells in the intestinal mucosa post-Tx (Figure 7B). Remarkably, as early as one month post-Tx, recipient HSPCs already started to populate some grafts (Pt23 POD28 ileum and Pt19 POD32 ileum, Figure 7C). Early recipient contributions included mainly LMPP, CLP and MP cells in Pt16 (POD47), Pt19 (POD32) and Pt23 (POD37) (Figure 7C). HSCs tended to be more enriched (>80%) among donor than recipient Lin−CD45+/dimCD34+ cells regardless of patients’ ages or early clinical outcomes (Table S1), and this trend was maintained long-term (POD>100) post-Tx in at least 6 patients (Pt20, POD104; Pt19, POD105; Pt17, POD243; Pt16, POD786; Pt15, POD347, 1041; Pt2, POD1544), even beyond 4 years post-Tx (Figure 7C, G). In other patients, the replacement of donor HSCs by the recipient progressed over time (e.g. Pt17, Figure 7C, G) and in others complete or nearly complete replacement of donor HSPCs by the recipient was observed long-term (e.g. Pt4 on POD1606, Pt13 on POD1032, Figure 7C, G). Overall, donor gut HSCs persisted long-term (POD100–1600) in the allograft in 6 out of 8 patients (Figure 7G, right panel). In general, Lin−CD45+/dimCD34+ donor cells in graft LPL included a high percentage of HSCs and MPPs, while the recipient compartment mainly consisted of MPs (Figure 7D). For all specimens from POD>100, donor Lin−CD45+/dimCD34+ cells in the graft LPL (n=9 specimens) included significantly greater proportions of HSCs than those of the recipient (n=13 specimens) (Figure 7E), while the recipient compartment (n=13) contained significantly greater proportions of MPs than the donor (n=9) (Figure 7F).
Figure 7. Dynamics of replacement of donor HSPCs within the graft by recipient cells.

(A) Representative flow cytometric gating strategy using jejunum LPLs taken on POD1544 from Pt2 small bowel resection. Lin−:CD3−CD5−CD14−CD19−. The percentage of CD34+ cells among DAPI−CD45+/dimLin− LPLs (B) and the percentage of each cell type of donor vs recipient origin (C). DAPI−Lin−CD45+/dimCD34+ cells, HSC, MPP, LMPP, CLP, MP and CD56+ progenitors in gut LPLs were collected from different patients during stoma revision/closure or small bowel removal at multiple time points post-Tx (POD28–1606). (D) Composition by percentage of HSC, MPP, LMPP, CLP, and MP among donor or recipient DAPI−Lin−CD45+/dimCD34+ LPLs in different patients at multiple time points post-Tx (refer to Figure 7A). Percentage of HSCs (E) and MPs (F) among donor or recipient Lin−CD45+/dimCD34+ LPLs isolated from patients’ intestinal mucosa after 100 days post-Tx shown in box-and-whisker plots (the line inside the box indicates the median). A two-tailed unpaired student t test was used to compare the donor and recipient compartments. (G) Percentages of donor T cells in peripheral blood (left panel) and donor HSCs in gut (right panel) up to 2000 days post-Tx. (H) Percentage of donor HSCs in the gut and the percentage of donor T cells in the blood at a similar timepoint (POD>100) for individual specimens from patients (circles for MVTx, squares for LITx, and triangles for iITx) who underwent stoma revision/closure or small bowel removal at various time points post-Tx. Chi-square test was performed with a cut-off for donor chimerism of 0.2% in peripheral blood (for T cells) or intestinal allograft (for HSCs). See also Figure S1 and Table S1, S2.
While all patients with persistent T cell chimerism (>0.2%) had persistent intestinal HSC chimerism (Pts 2, 16 and 19), the duration of intestinal HSC chimerism was greater than that of T cell chimerism (Figure 7G). One patient (Pt16) who had early transient donor blood T cell chimerism that became low or undetectable (0–0.33%) during POD23–463 showed resurgent donor T cell chimerism to >4% by POD786, when there were still >50% donor HSCs persisting in the allograft (Figure 7G). Overall, persistent donor chimerism (POD>100) was significantly more common in the allograft than in the peripheral blood (Figure 7H, p=0.0062). Together, our data indicate that graft-derived HSPCs likely contribute to the long-term persistence of donor blood chimerism and that these donor cells are gradually replaced by recipient HSPCs in the circulation that home to the intestine.
Discussion
We demonstrate here that human intestine contains HSPCs with multilineage potentiality and that in the setting of intestinal allotransplantation these cells contribute to circulating T/B/NK/Myeloid cell pools and are gradually replenished from the circulation. Given that lymphoid follicles, PPs and MLN carrying naïve T cells are transplanted with intestinal allografts, and that intestinal mucosa of young donors contains naïve T cells (Thome et al., 2016a), naïve T cells derived from these sources may contribute to donor-derived RTEs in the recipient circulation post-Tx. However, several lines of evidence support the possibility that circulating donor RTE-like cells, especially those detected late post-Tx, developed de novo in the recipient thymus. First, teenaged (Pt3) and adult patients (Pt7) with adult donor grafts showed marked enrichment of RTEs from the donor compared to the recipient, despite the age of the intestinal allografts. Second, any naïve T cells leaving the donor graft would be expected to undergo lymphopenia-driven proliferation (LIP) (Onoe et al., 2010), losing TREC content and acquiring memory markers in the lymphopenic recipient environment.
While the phenotype of circulating donor-derived T cells strongly suggests a recipient thymic origin, other sites of potential T cell development, such as the intestine itself, must be considered. Lin−c-kit+ HPs have been identified in mouse gut cryptopatches and found to give rise to TCR αβ and γδ IEL T cells (Saito et al., 1998). However, αβ T cells are a minor population generated in this site and this pathway is suppressed in the presence of a normal thymus (Guy-Grand et al., 2003). Extrathymic T lymphopoiesis in MLNs and intestinal mucosa was enhanced in lymphopenic conditions (Guy-Grand et al., 2003), and ATG-induced lymphopenia might also promote de novo local generation of T cells from intestinal HSPCs in patients after ITx. This would only be likely to occur early post-Tx, since ATG induction is generally given within the first week in our center.
Evidence for extrathymic T-cell development in human intestine is more limited (Howie et al., 1998; Lynch et al., 2006; Williams et al., 2004). Immature T cells and lymphoid progenitors, along with TdT and RAG gene expression, have been reported in fetal and infant intestines, but disappeared by 18 months of age (Howie et al., 1998; Williams et al., 2004). Many (12/21=57.1%) of our ITx donors were older than 18 months (2–48 y/o, Table S1), arguing that circulating naïve donor T cells did not develop in situ in the graft. In our study, the CD8αα phenotype did not clearly identify cells of intestinal origin (Figure S7), as the vast majority of blood, IEL and LPL CD8 T cells are CD8αβ and few donor or recipient CD8αα T cells were detected in peripheral blood. Thus, while our studies do not rule out an intraepithelial origin for the RTE-like donor T cells detected in recipient blood after ITx, the phenotype, high TREC content and high TCR diversity of these cell populations, as well as their tolerance to recipient stimulators in MLR, favor the hypothesis that these cells develop in the recipient thymus from progenitors carried in the donor graft that enter the circulation and either migrate directly to the thymus or differentiate from earlier progenitors after settling in the recipient BM. Indeed, CyTOF analysis revealed similar phenotypes for Lin−CD45+/dimCD34+ cells obtained from BM vs ileum intestinal LPLs, including subsets expressing the progenitor thymus-homing marker CCR9 (Haddad et al., 2006; Zlotoff and Bhandoola, 2011). We detected donor Lin−CD45+/dimCD34+ progenitors in the circulation of patients with long-term chimerism, supporting the hypothesis that donor HSPCs from the graft migrate into the recipient circulation and into the BM and other sites of potential hematopoiesis, where they differentiate and contribute to the circulating leukocyte pool.
We did not observe any relationship between age and the percentage of Lin−CD45+/dimCD34+ cells or of HSCs among Lin−CD45+/dimCD34+ cells in the ileum of multiple deceased donors aged 9–93 years. In previous studies, T/NK lineage-polarized CD34+ CD45RA+ CD7+ HPs corresponding to candidate prethymocytes were detected in fetal BM and cord blood, found to decline around birth and persisted at a low level in adult BM (Haddad et al., 2004; Haddad et al., 2006). In contrast, we detected high proportions (9–76%) of CD45RA+CD7+ cells among Lin−CD45+/dimCD34+ populations in BM of three donors aged 26 to 76. Lin−CD45+/dimCD34+ CD45RA+CD7+ cells were also detected in ileum LPLs of some but not all donors.
Extramedullary hematopoiesis has been reported in diverse tissues including the gastrointestinal tract, usually in association with pathologic processes, such as myelofibrosis, inflammation, and infection (Granick et al., 2012; Saenz et al., 2010). Thus we cannot rule out the possibility that circulating donor monocytes also emerged from the donor graft, perhaps in response to inflammation, without intermediate residence in the recipient BM. A study in mice demonstrated that lung is a primary site of terminal platelet production (Lefrancais et al., 2017), suggesting that hematopoietic progenitor cells can travel through the bloodstream and differentiate in various organs.
Our recent work has suggested a role for two-way alloreactivity in determining intestinal allograft T cell repopulation by the recipient (Zuber et al., 2016) and blood T cell macrochimersim (Fu et al., 2017). Enrichment of graft-vs-host (GvH) compared to host-vs-graft (HvG) clones in the graft, the absence of de novo Class I donor-specific antibodies (DSA) and freedom from moderate/severe rejection are associated with donor T cell macrochimerism in blood (Fu et al., 2017; Zuber et al., 2016). We hypothesize that early lymphohematopoietic GvH responses (LGVHR; GvH responses that do not induce clinical GVHD (Sykes et al., 1988a, b)) attack recipient hematopoietic cells, creating “space” in the recipient BM that permits engraftment of donor cells, including HSPCs carried within the graft.
We previously demonstrated only low levels and transient lymphoid or myeloid chimerism in recipients of liver transplants alone (Zuber et al., 2015). However, adult human livers (Wang et al., 2012) contain pluripotent HSCs with repopulating potential that may contribute to donor chimerism in MVTx recipients. The large load of GvH-reactive clones that expand locally in the intestinal mucosa (Zuber et al., 2016) may promote the greater and more prolonged chimerism in MVTx compared to LTx recipients (Zuber et al., 2015).
Analyses of serial intestinal biopsy specimens allowed us to measure the dynamics of intestinal HSPC repopulation by circulating recipient cells. Murine studies demonstrated HSC migration to ectopic niches via the circulation (Christensen et al., 2004; Wright et al., 2001). HSCs were detected in the blood of 12- to 18-week human fetuses (Gallacher et al., 2000), and long-term repopulating HSCs were demonstrated in adult human steady state peripheral blood (Bourdieu et al., 2017; Brunet de la Grange et al., 2013). We now demonstrate the turnover of “ectopic” niche HSPCs from the circulating pool in humans.
Our study reveals the underlying mechanism for the persistence of donor mixed chimerism in recipient blood and provides unique information on the dynamics of human HSPCs. Our data suggest that the human gut serves as a significant site of HSPC residence that contributes to circulating leukocytes. Whether this is true under physiological conditions in addition to ITx needs further investigation. The contribution of graft-derived HSPCs to sustained mixed chimerism in the blood suggests the possibility of promoting allograft tolerance in ITx recipients.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Megan Sykes (megan.sykes@columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subject recruitment and clinical protocols
The study was approved by the Columbia University Institutional Review Board (IRB# AAAJ5056). All subjects or legal guardians provided their written, informed consent. Protocol biopsies were obtained in the initial post-ITx period as described previously (Zuber et al., 2016), and additional biopsies were performed for cause. Graft rejection was graded from negative, indeterminate, mild, and moderate until severe rejection based on the pathologic scoring scheme previously reported (Remotti et al., 2012). Blood samples were collected up to four times during the first month post-Tx, then at least once per month if available. Donor liver biopsy and perfusion fluid were collected at the back table in the operation room immediately before ITx. All of the patients (Table S1) received anti-thymocyte globulin (ATG) induction therapy (total dose: 6–10 mg/kg) followed by a maintenance regimen that included tacrolimus and steroids. Ileum (including PPs if available), MLNs, and bone marrow (BM) samples from deceased organ donors (Table S3) were obtained at the time of organ procurement for clinical transplantation through an approved research protocol and material transfer agreement with LiveOnNY as previously described (Carpenter et al., 2017). Donors were free of chronic disease and cancer, were hepatitis B, C and HIV negative. Samples used do not qualify as human subjects, as confirmed by the Columbia University IRB, as tissue is obtained from deceased individuals.
Human fetal thymus and liver tissues (gestational age, 17 to 21 weeks) were obtained from Advanced Bioscience Resources. Fetal thymus fragments were cryopreserved in 10% dimethyl sulfoxide (Sigma-Aldrich, St Louis, MO) and 90% fetal bovine serum (FBS, Thermo Fisher scientific, Waltham, MA). Fetal liver fragments were treated for 20 minutes at 37°C with 100 mg/mL of Liberase (Roche CustomBiotech, Indianapolis, IN) to obtain a cell suspension.
Mice
The study was approved by the Columbia University Institutional Animal Care and Use Committee (IACUC# AC-AAAM8850). Immunodeficient NOD.scid.Il2Rg−/− (NSG) mice have been purchased from The Jackson Laboratory and bred, raised and housed in our specific pathogen-free and helicobacter/pasteurella pneumotropica-free mouse facility with 12-hour light/dark cycle and provided with food and water ad libitum according to Columbia University IACUC. NSG mice used as recipients in this study were all females with 8 weeks of age.
Cell line
The MS5-DL1ind100 cell line was previously established (Calvo et al., 2012) as a conditional tetracycline-inducible system controlling the expression of the human NOTCH ligand Delta-like 1 (DL1) in murine stromal MS5 cells. The sex of MS5-DL1ind100 cell line is unknown as this aspect of their provenance is unclear in the literature. We obtained the MS5-DL1ind100 cell line from Drs. Francoise Pflumio and Julien Calvo at Paris Diderot University. MS5-DL1ind100 cells were cultured in alpha-MEM media (Gemini Bio-Products, Broderick, CA) with 10% FBS and 1% PenStrep/L-Glutamine.
METHOD DETAILS
Intraepithelial lymphocyte (IEL) and lamina propria lymphocyte (LPL) isolations
IEL and LPL were separated either from graft biopsy specimens or surgically obtained graft specimens at the time of stoma closure/revision, or from ileal mucosa of deceased donors, according to a protocol adapted from previous reports (Binda et al., 2009) and described previously (Zuber et al., 2016). In brief, the specimens were treated for 20 minutes at 37°C with 2 mmol/L dithiothreitol followed by two 30-minutes incubations with 0.5 mmol/L EDTA with continuous stirring in a water bath at 37°C. LPLs were isolated from the remaining tissue, digested and stirred in a collagenase-containing medium (RPMI 1640, 1mg/mL Collagenase D, 100 I.U/mL penicillin-streptomycin). DNAse (0.1 mg/mL) was added to EDTA and collagenase media, each time large intestinal mucosa specimens were processed.
Human leukocyte antigen (HLA)-specific staining and cellular staining
Candidate monoclonal HLA class I allele-specific antibodies (mAb) were screened for the ability to discriminate donor and pre-transplant (pre-Tx) recipient cells, based on molecular HLA typing information. FITC, PE or biotin-conjugated HLA-specific mAb were purchased from One Lambda or BD Biosciences. Each HLA-specific mAb (Table S2) was used in combination with pan-HLA-ABC-APC or PE (G46–2.6) antibody and quality control tested for specificity. Those that readily distinguished donor from the pre-Tx recipient peripheral blood mononuclear cells (PBMCs) were included in lineage-specific panels of antibodies, as previously reported (Zuber et al., 2015). Briefly, the multicolor T-cell panel included anti-CD45-V500 (HI30), CD3-PerCPCy5 (UCHT1), γδ TCR-PE-Cy7 (immu510), CD4-AF700 (OKT4), CD8- APC-Cy7 (SK1), CD56-BV605 (HCD56), CD19 (HIB19)/CD28 (V-CD28.05)-Pac Blue, CD69-BV650 (FN50), CD103-PE/FITC (Ber-ACT8), CD45RO (UCHL1)/CD161 (DX12)/HLA-DR (L243)-BV711, NKG-2D biotin (1D11)-Strep-AF594 and DAPI. Myeloid lineage panel included anti-CD45-V500, CD33-AF700 (WM53), CD11c-PECy5 (B-ly6), CD14-Pac Blue (M5E2), HLA-DRBV711 (L243), CD141-BV711 (1A4), CD123-biotin (9F5)-Strep-AF594, CD1c-PE-Cy7 (L161), CD15 (HI98)/Sirpα/β (SE5A5) -PE, CD45RA-AF700 (HI100) and DAPI. HSPC panel included anti-CD3 (OKT3)/CD19 (HIB19)/CD56 (HCD56)-BV650, CD5-BV711 (UCHT2), CD14 (M5E2)/CD19 (HIB19)-Pac Blue, CD45-PE-CF594 (HI30), CD34-PE (QBEnd10), CD38-PECy7 (HIT2), CD45RA-BV510 (HI100), CD90-PerCP-Cy5.5 (eBio5E10), CD10-APC-Cy7 (HI10A), and DAPI. Naïve-memory T cell panel included anti-CD3-PerCP-Cy5.5 (UCHT1), CD4-AF700 (OKT4), CD8-APC-Cy7 (SK1), CD45RA-BV510 (HI100), CCR7-PE-Cy7 (G043H7), CD31-BV605 (WM59), CD28-Pac Blue (V-CD28.05), HLA-ABC-APC (G46–2.6) and DAPI. Naïve B cell panel included anti-CD45-V500 (HI30), CD19-BV650 (HIB19), CD38-PE-Cy7 (HIT2), CD24-PE (ML5), CD27-BV711 (O323), IgM-PE-CF594 (G20–127), IgA-biotin (G20–359)-Strep-AF594, IgG-V450 (G18–145) and DAPI. Naïve γδ T cell panel included anti-CD3-PerCP-Cy5.5 (UCHT1), CD4-AF700 (OKT4), CD8-APC-Cy7 (SK1), CD45RA-BV510 (HI100), CD27 BV711 (O323), CD28-Pac Blue (V-CD28.05), CD11a-PE (HI111), δ2-FITC (immu389), γδ TCR-PE-Cy7 (immu510) and DAPI. Data was acquired using an LSR II flow cytometer (BD Biosciences, San Jose, CA) using DIVA software. Analysis was carried out using FlowJo software (TreeStar, Inc, Ashland, OR).
Carboxyfluorescein succinimidyl ester (CFSE)-MLR, cell sorting and TCRβ CDR3 DNA sequencing
These were performed as described (Morris et al., 2015; Zuber et al., 2016). Briefly, graft-versus-host (GvH) and host-versus-graft (HvG) mixed lymphocyte reactions (MLRs) were set up using thawed pre-Tx donor and recipient cells. Two hundred thousand CFSE-labeled responder cells and two hundred thousand violet-dye-labeled irradiated (35 Gy) stimulators were plated in each well of a round-bottom 96-well plate in MLR medium (AIM-V supplemented with 5% AB heat-inactivated human serum, 0.01M Hepes, and 50μm 2-mercaptoethanol). MLR cultures were incubated at 37°C for 6 days, at the end of which MLR were harvested. Cells were stained with anti-CD3, CD4 and CD8, before FACS sorting on a BD™ Influx cell sorter to isolate two discrete violet dye-negative cell populations (CD3+CD4+CFSElo, CD3+CD8+CFSElo), representing the CD4+ and CD8+ recipient-anti-donor (or donor-anti-recipient) reactive T cells (“stims”), respectively. For unstimulated cell populations, pre-Tx donor and recipient cells harvested from spleen or MLN were thawed and stained with anti-CD3, CD4 and CD8, and then FACS sorted into CD3+CD4+ and CD3+CD8+ populations (“unstims”). Genomic DNA was isolated from sorted cell populations using the QIAGEN DNeasy Blood and Tissue Kit (Germantown, MD). DNA was frozen down at −20°C and shipped on dry ice to Adaptive Biotechnologies (Seattle, WA) for high-throughput TCR sequencing. The TCR sequencing data were retrieved from Adaptive’s Immunoseq software.
PCR amplification, read sequencing, and mapping, with bias correction and internal controls, were performed by Adaptive Biotechnologies, returning tabulated read counts corresponding to unique clonal CDR3 DNA sequences across all samples. From these, sample read counts across clones were normalized to frequency of detection. CD8 versus CD4 sorting error was corrected for by removing ambiguous sequences present in both populations at a high to low frequency ratio less than 5:1. Donor and recipient shared CDR3s or ambiguous sequences were removed. After this, separate CD4 and CD8 tables containing clonal expression frequencies in pre-Tx reference sample, CFSElow stimulated cells, and biopsies were compiled and re-normalized. Alloreactive clones were defined by two-fold or greater expansion in stimulated compared to un-stimulated pre-Tx cells, and by minimum frequency of 0.001% in CFSElow, which serves to ensure 85% repeatability, as determined by power analysis (Morris et al., 2015). Clonality, which ranges from 0 to 1, is primarily used as a measure of diversity, such that higher clonality indicates less diversity (Langerak et al., 2007). Our recent work has established a new approach for quantitative population diversity comparisons by measuring power-law slopes of abundance plots (DeWolf et al., 2018).
In some experiments, functional MLR was set up using pre-Tx donor splenocytes, or post-Tx total or CD25− T cells sorted from recipient PBMCs or splenocytes (isolated from splenectomy pre-2nd Tx) as responders, and irradiated recipient pre-Tx splenocytes or 3rd party PBMCs as stimulators. Additionally, donor HLA+CD3+CFSElow cells were sorted on Day 6 of a MLR in which CD25− T cells sorted from post-Tx PBMCs were used as responders and irradiated recipient pre-Tx splenocytes were used as stimulators. TCRβ CDR3 DNA sequencing was performed on sorted donor HLA+CD3+CFSElow cells, and the cumulative frequency of GvH clones as a percentage of all sequences within donor mappable clones was analyzed. “Donor mappable clones” refer to clones that were detectable in sequenced pre-Tx spleen, lymph node and/or MLR CFSElow T cell populations from the donor.
Quantification of human T cell receptor excision circles (TRECs)
Naïve (CD45RA+) and memory (CD45RO+) CD4+ T cells were sorted from PBMCs of Pt1 (POD246/365) and two healthy control donors. Naïve (CD45RA+CCR7+) and non-naive (CD45RA−CCR7+ or CD45RA−CCR7− or CD45RA+CCR7−) CD3+ T cells were sorted from one healthy control donor. Naïve (CD45RA+CCR7+) CD3+ T cells were sorted from PBMCs of Pt15 (POD255), Pt16 (POD786), Pt18 (POD314) and Pt19 (POD167). Cell pellets were stored at −80°C before analysis. Signal joint TREC (sjTREC) analysis was performed in the Immunology Unit of the Duke Regional Biocontainment Laboratory (RBL). TREC content was quantified using an established real-time PCR approach with a standard curve of known molecules of human TREC (Geenen et al., 2003; Sempowski et al., 2002). The following primer and probe sequences were used: 5′ primer, 5′-CACATCCCTTTCAACCATGCT- 3′; 3′ primer, 3′ GCCAGCTGCAGGGTTTAGG-3′; probe, 5′−6-FAMCAGGGCAGGTTTTTGTAAAGGTGCTCACTT-3′BHQ1 (Black Hole Quencher).
Mass cytometry (CyTOF)
BM and ileum LPL cells isolated from the same deceased organ donor were ficolled to collect the mononuclear cell layer. Lineage depletion (biotin-labeled antibody cocktail included CD3, CD14, CD19, CD33, CD11b, CD326) was performed to enrich CD34 cells using a customized MACS kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Lin− BM and ileum LPLs were stained and examined by means of mass cytometry (CyTOF) (Bendall et al., 2011) at the Human Immune Monitoring Center of the Icahn School of Medicine at Mt. Sinai. In brief, cells were washed with PBS containing 0.2% BSA and incubated in RPMI medium containing 1μM Cell-ID Intercalator-103Rh (Fluidigm, South San Francisco, CA) to label dead cells. After washing, cells were barcoded with CD45–165Ho and CD45–175Lu for BM and ileum LPLs, respectively. The barcoded cells were then combined and stained together with antibodies against selected surface markers for 30 minutes on ice. Antibodies (CD45–89Y, CD326–141Pr, CD19–142Nd, CD117–143Nd, CD38–144Nd, CD4–145Nd, CD8–146Nd, CD7–147Sm, CD127–149Sm, γδ TCR-152Sm, CD62L-153Eu, CD3–154Sm, CD10–156Gd, CCR4–158Gd, CD90–159Tb, CD14–160Gd, CD11c-162Dy, HLA-ABC-163Dy, CD34–166Er, CCR9–168Er, CD45RA-170Er, CD11b-172Yb, HLA-DR-174Yb and CD56–176Yb) were obtained either in preconjugated form from Fluidigmor were conjugated in-house by using MaxPar X8 conjugation kits (Fluidigm). Cells were then washed and fixed, resuspended in diH2O containing EQ Four Element Calibration Beads (Fluidigm) and acquired on a CyTOF2 Mass Cytometer (Fluidigm). Data files were normalized by using a bead-based normalization algorithm (CyTOF software, Fluidigm) and debarcoded using CD45 gating. The gated populations were visualized in lower dimensions using viSNE (Amir el et al., 2013) in Cytobank (http://www.cytobank.org/) to identify the CD45+/dimCD34+ cells, for which the expression levels of additional markers, including CCR9, was further analyzed.
Colony Forming Cell (CFC) assay
BM and ileum LPL cells isolated from deceased organ donors were ficolled and lineage-depleted (CD3, CD14, CD19, CD33, CD11b, CD326) to enrich CD34 cells using a customized MACS kit (Miltenyi Biotec). CFC assay of CD34-enriched BM and ileum mononuclear cells was performed as described (Kaufman et al., 2001; Pereira et al., 2007). Cell Resuspension Solution and methylcellulose-based medium containing recombinant human SCF, GM-CSF, IL-3 and EPO was purchased from R&D Systems (Minneapolis, MN). Colonies consisting of at least 40 cells were counted at the end of the incubation period (14–16 days), and the type of colony was determined by the morphology of cells (Kaufman et al., 2001; Pereira et al., 2007). CFU-E: Colony forming unit-erythroid; CFU-G: Colony forming unit-granulocyte; BFU-E: Burst forming unit-erythroid; CFU-M: Colony forming unit-macrophage; CFU-GEMM: Colony forming unit-granulocyte, erythrocyte, macrophage, megakaryocyte.
Long-term Culture-Initiating-Cell (LTC-IC) assay
Individual HSCs (Lin−CD45+/dimCD34+CD38−/lowCD45RA−CD90+) were sorted on a BD™ Influx cell sorter from human ileum LPLs into 96 well plates in 50 μl of complete medium, co-cultured in contact with MS5/DL1ind100 cells in reconstituted alpha-MEM supplemented with 10% FBS (Gemini Bio-Products, Broderick, CA) and 10% human AB serum (Gemini Bio-Products, Broderick, CA) in the presence of recombinant human stem cell factor (50 ng/ml, PeproTech, Rocky Hill, NJ), rhFlt3-ligand (20 ng/ml, PeproTech, Rocky Hill, NJ), Insulin (20 nM, Sigma-Aldrich, St Louis, MO) and rhIL-7 (10 ng/ml, R&D Systems, Minneapolis, MN). Medium was half changed twice a week during the first 21 days. At 21 days, the cells were placed in fresh medium supplemented with doxycycline (1 μg/ml, Sigma-Aldrich, MO). Cultures were continued for 21 days with refreshment of medium two to three times a week. At 42 to 45 days, cells were harvested and labelled with DAPI and three anti-human antibodies (Alexa 488-conjugated mouse anti-human CD3, purified rabbit anti-human CD20 with secondary Alexa 555-conjugated donkey anti-rabbit Ab, and Alexa 647-conjugated mouse anti-human CD14) for immunofluorescent imaging analysis using a Leica DMI 6000B fluorescent microscope (Leica Microsystems, Buffalo Grove, IL).
Humanized mouse model
BM and ileum LPL cells isolated from deceased organ donors were centrifuged through ficoll to collect the mononuclear cell layer. BM cells were further enriched for CD34 using a positive selection MACS kit (Miltenyi Biotec). Ileum LPLs were lineage-depleted (CD3, CD14, CD19, CD33, CD11b, CD326) to enrich CD34 cells using a customized MACS kit (Miltenyi Biotec), then FACS sorted to collect CD45+/dimCD34+ populations. BM CD34-enriched cells obtained from donor “A”, combined with gut Lin−CD45+/dimCD34+ cells from donor “B” at ratios between 1.5:1 and 10:1 were injected intravenously into irradiated (1Gy/mouse on day-1) NSG mice. In some experiments, mice were also transplanted with human fetal thymus tissue (harvested at 20–21 weeks of gestation) measuring about 1mm3 from donor “C” under the kidney capsule on day-1 to allow T cell development within a human thymus (Kalscheuer et al., 2012) (Table S4). The NSG mice were treated anti-CD2 Ab at 0.4mg/mouse for 2−3 doses during the first two weeks post-Tx. Donors “A”, “B” and “C” were distinguished by their different HLA-A locus expression using allele group-specific mAbs in FCM (for example, donor “A”: A9+A2−A3−, donor “B”: A2+A9−A3−, donor “C”: A3+A2−A9−). Tail bleeding was performed at 2 to 4 weeks and thereafter every two to four weeks until the end of the experiment around week 24 or 28. Development of T, B, NK cell and monocyte chimerism in peripheral blood of humanized mice was monitored by FCM. At the end of experiment, peripheral blood, BM, spleen and implanted thymus were harvested. Lineage markers, CD34 and donor-specific HLA-A loci were stained and analyzed using FCM.
QUANTIFICATION AND STATISTICAL ANALYSIS
Analysis of TCRβ repertoire data was performed in R and Rstudio using standard commands. EulerAPE (Micallef and Rodgers, 2014) was used to generate proportional Venn diagrams. Additional statistics and figures were generated using GraphPad Prism (GraphPad Software, La Jolla, CA). Student’s t test was used for statistical comparisons between paired groups. Chi-square test was performed with a cut-off for donor chimerism of 0.2% in peripheral blood (for T cells) or intestinal allograft (for HSCs). Log-rank (Mantel-Cox) test was performed for Kaplan-Meier plot of freedom from moderate to severe rejection of patients with >4% and <4% of donor T cell peak chimerism in blood. P<0.05 is considered to be a statistically significant difference.
DATA AND SOFTWARE AVAILABILITY
Raw TCR sequences data are freely accessible through https://doi.org/10.21417/Fu112018. The codes used to analyze TCR sequences are available and can be downloaded from a public GitHub repository (https://github.com/Aleksobrad/TCR-analysis).
Supplementary Material
Highlight:
Human intestine contains hematopoietic stem cells and multiple types of progenitors
Donor graft HSPCs contribute to multilineage blood chimerism in the recipient
Long-term circulating donor T cells are tolerant to the recipient but functional
Intestinal HSPCs undergo replacement by the recipient from a circulating pool
Acknowledgments
The study was funded by the National Institute of Allergy and Infectious Diseases grant P01 AI106697. Research reported here was performed in the Columbia Center for Translational Immunology (CCTI) Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health awards S10RR027050 and S10OD020056. We thank Ms. Nicole Casio for assistance with the submission of the manuscript and Drs. Isabelle André-Schmutz and Joji Fujisaki for helpful review of the manuscript. We also thank Monica Velasco, Peter Liou, Tamas Gonda, and Shilpa Ravella for their care of intestinal transplant recipients; Alina Iuga for her pathology reviews; and Constantin Aschauer for assistance with sample processing. We thank Francoise Pflumio and Julien Calvo for kindly providing the MS5-DL1in100 cell line and for their guidance in optimizing co-culture conditions for LTC-IC. We thank the Flow Cytometry Core at Columbia Center for Translational Immunology (CCTI) and Human Immune Monitoring Center at Mt. Sinai and the Immunology Unit of the Regional Biocontainment Laboratory at Duke Medical Center for their excellent services. This work was made possible in part by samples made available through the support of the V. Segal and S. Segal CCTI Biobank Core. We thank transplant coordinators and Dr. Amy Friedman from LiveOnNY for procurement of organ donor tissues. We gratefully acknowledge the generosity of the donor families, our ITx patients, and their families for making this study possible. The study was funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH) (P01 AI106697). D.L.F. was supported by NIH grants AI106697 and AI128949. Research reported here was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, NIH (S10RR027050 and S10OD020056).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: The authors declare no competing interests.
References
- Abu-Elmagd KM, Kosmach-Park B, Costa G, Zenati M, Martin L, Koritsky DA, Emerling M, Murase N, Bond GJ, Soltys K, et al. (2012). Long-term survival, nutritional autonomy, and quality of life after intestinal and multivisceral transplantation. Annals of surgery 256, 494–508. [DOI] [PubMed] [Google Scholar]
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature biotechnology 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science (New York, NY) 332, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, Zeevi A, Farmer DG, and Abu-Elmagd KM (2012). Immunologic challenges in small bowel transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 12 Suppl 4, S2–8. [DOI] [PubMed] [Google Scholar]
- Binda E, Erhart D, Schenk M, Zufferey C, Renzulli P, and Mueller C (2009). Quantitative isolation of mouse and human intestinal intraepithelial lymphocytes by elutriation centrifugation. Journal of immunological methods 344, 26–34. [DOI] [PubMed] [Google Scholar]
- Bourdieu A, Avalon M, Lapostolle V, Ismail S, Mombled M, Debeissat C, Guerinet M, Duchez P, Chevaleyre J, Vlaski-Lafarge M, et al. (2017). Steady state peripheral blood provides cells with functional and metabolic characteristics of real hematopoietic stem cells. Journal of cellular physiology. [DOI] [PubMed] [Google Scholar]
- Brunet de la Grange P, Vlaski M, Duchez P, Chevaleyre J, Lapostolle V, Boiron JM, Praloran V, and Ivanovic Z (2013). Long-term repopulating hematopoietic stem cells and “side population” in human steady state peripheral blood. Stem cell research 11, 625–633. [DOI] [PubMed] [Google Scholar]
- Calvo J, BenYoucef A, Baijer J, Rouyez MC, and Pflumio F (2012). Assessment of human multi-potent hematopoietic stem/progenitor cell potential using a single in vitro screening system. PloS one 7, e50495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DJ, Granot T, Matsuoka N, Senda T, Kumar BV, Thome JJC, Gordon CL, Miron M, Weiner J, Connors T, et al. (2017). Human immunology studies using organ donors: Impact of clinical variations on immune parameters in tissues and circulation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JL, Wright DE, Wagers AJ, and Weissman IL (2004). Circulation and Chemotaxis of Fetal Hematopoietic Stem Cells. PLOS Biology 2, e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWolf S, Grinshpun B, Savage T, Lau SP, Obradovic A, Shonts B, Yang S, Morris H, Zuber J, Winchester R, et al. (2018). Quantifying size and diversity of the human T cell alloresponse. JCI insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Zuber J, Shonts B, Obradovic A, Lau S. p., Savage T, Xia A, Simpson M, Yang S, and Miron M (2017). Differing Mechanisms for Early Versus Persistent Donor T cell Chimerism in Peripheral Blood of Human Intestinal Transplant Recipients. Transplantation 101, S63–S64. [Google Scholar]
- Gallacher L, Murdoch B, Wu D, Karanu F, Fellows F, and Bhatia M (2000). Identification of novel circulating human embryonic blood stem cells. Blood 96, 1740–1747. [PubMed] [Google Scholar]
- Geenen V, Poulin JF, Dion ML, Martens H, Castermans E, Hansenne I, Moutschen M, Sekaly RP, and Cheynier R (2003). Quantification of T cell receptor rearrangement excision circles to estimate thymic function: an important new tool for endocrine-immune physiology. The Journal of endocrinology 176, 305–311. [DOI] [PubMed] [Google Scholar]
- Golden-Mason L, and O’Farrelly C (2002). Having it all? Stem cells, haematopoiesis and lymphopoiesis in adult human liver. Immunology and cell biology 80, 45–51. [DOI] [PubMed] [Google Scholar]
- Gorgens A, Radtke S, Horn PA, and Giebel B (2013). New relationships of human hematopoietic lineages facilitate detection of multipotent hematopoietic stem and progenitor cells. Cell cycle (Georgetown, Tex) 12, 3478–3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granick JL, Simon SI, and Borjesson DL (2012). Hematopoietic stem and progenitor cells as effectors in innate immunity. Bone marrow research 2012, 165107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy-Grand D, Azogui O, Celli S, Darche S, Nussenzweig MC, Kourilsky P, and Vassalli P (2003). Extrathymic T cell lymphopoiesis: ontogeny and contribution to gut intraepithelial lymphocytes in athymic and euthymic mice. The Journal of experimental medicine 197, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad R, Guardiola P, Izac B, Thibault C, Radich J, Delezoide AL, Baillou C, Lemoine FM, Gluckman JC, Pflumio F, et al. (2004). Molecular characterization of early human T/NK and B-lymphoid progenitor cells in umbilical cord blood. Blood 104, 3918–3926. [DOI] [PubMed] [Google Scholar]
- Haddad R, Guimiot F, Six E, Jourquin F, Setterblad N, Kahn E, Yagello M, Schiffer C, Andre-Schmutz I, Cavazzana-Calvo M, et al. (2006). Dynamics of thymus-colonizing cells during human development. Immunity 24, 217–230. [DOI] [PubMed] [Google Scholar]
- Hazenberg MD, Verschuren MC, Hamann D, Miedema F, and van Dongen JJ (2001). T cell receptor excision circles as markers for recent thymic emigrants: basic aspects, technical approach, and guidelines for interpretation. Journal of molecular medicine (Berlin, Germany) 79, 631–640. [DOI] [PubMed] [Google Scholar]
- Howie D, Spencer J, DeLord D, Pitzalis C, Wathen NC, Dogan A, Akbar A, and MacDonald TT (1998). Extrathymic T cell differentiation in the human intestine early in life. Journal of immunology (Baltimore, Md : 1950) 161, 5862–5872. [PubMed] [Google Scholar]
- Kalscheuer H, Danzl N, Onoe T, Faust T, Winchester R, Goland R, Greenberg E, Spitzer TR, Savage DG, Tahara H, et al. (2012). A model for personalized in vivo analysis of human immune responsiveness. Science translational medicine 4, 125ra130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Tzakis AG, Selvaggi G, Gaynor JJ, David AI, Bussotti A, Moon JI, Ueno T, DeFaria W, Santiago S, et al. (2006). Intestinal and multivisceral transplantation in children. Annals of surgery 243, 756–764; discussion 764–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DS, Hanson ET, Lewis RL, Auerbach R, and Thomson JA (2001). Hematopoietic colony-forming cells derived from human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America 98, 10716–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Cosimi AB, and Sachs DH (2011). Preclinical and clinical studies on the induction of renal allograft tolerance through transient mixed chimerism. Current opinion in organ transplantation 16, 366–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Sachs DH, Sykes M, Cosimi AB, and Immune Tolerance N (2013). HLA-mismatched renal transplantation without maintenance immunosuppression. The New England journal of medicine 368, 1850–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmig S, Przybylski GK, Schmidt CA, Laurisch K, Mowes B, Radbruch A, and Thiel A (2002). Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. The Journal of experimental medicine 195, 789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubal CA, Mangus RS, and Tector AJ (2015). Intestine and multivisceral transplantation: current status and future directions. Current gastroenterology reports 17, 427. [DOI] [PubMed] [Google Scholar]
- Langerak AW, Groenen PJ, Jm van Krieken JH, and van Dongen JJ (2007). Immunoglobulin/T-cell receptor clonality diagnostics. Expert opinion on medical diagnostics 1, 451–461. [DOI] [PubMed] [Google Scholar]
- Lefrancais E, Ortiz-Munoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, Thornton EE, Headley MB, David T, Coughlin SR, et al. (2017). The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch L, O’Donoghue D, Dean J, O’Sullivan J, O’Farrelly C, and Golden-Mason L (2006). Detection and characterization of hemopoietic stem cells in the adult human small intestine. Journal of immunology (Baltimore, Md : 1950) 176, 5199–5204. [DOI] [PubMed] [Google Scholar]
- Micallef L, and Rodgers P (2014). eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PloS one 9, e101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris H, DeWolf S, Robins H, Sprangers B, LoCascio SA, Shonts BA, Kawai T, Wong W, Yang S, Zuber J, et al. (2015). Tracking donor-reactive T cells: Evidence for clonal deletion in tolerant kidney transplant patients. Science translational medicine 7, 272ra210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoe T, Kalscheuer H, Chittenden M, Zhao G, Yang YG, and Sykes M (2010). Homeostatic expansion and phenotypic conversion of human T cells depend on peripheral interactions with APCs. Journal of immunology (Baltimore, Md : 1950) 184, 6756–6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C, Clarke E, and Damen J (2007). Hematopoietic colony-forming cell assays. Methods in molecular biology (Clifton, NJ) 407, 177–208. [DOI] [PubMed] [Google Scholar]
- Remotti H, Subramanian S, Martinez M, Kato T, and Magid MS (2012). Small-bowel allograft biopsies in the management of small-intestinal and multivisceral transplant recipients: histopathologic review and clinical correlations. Archives of pathology & laboratory medicine 136, 761–771. [DOI] [PubMed] [Google Scholar]
- Saenz SA, Siracusa MC, Perrigoue JG, Spencer SP, Urban JF Jr., Tocker JE, Budelsky AL, Kleinschek MA, Kastelein RA, Kambayashi T, et al. (2010). IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464, 1362–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Kanamori Y, Takemori T, Nariuchi H, Kubota E, Takahashi-Iwanaga H, Iwanaga T, and Ishikawa H (1998). Generation of intestinal T cells from progenitors residing in gut cryptopatches. Science (New York, NY) 280, 275–278. [DOI] [PubMed] [Google Scholar]
- Sempowski GD, Gooding ME, Liao HX, Le PT, and Haynes BF (2002). T cell receptor excision circle assessment of thymopoiesis in aging mice. Molecular immunology 38, 841–848. [DOI] [PubMed] [Google Scholar]
- Smith JM, Weaver T, Skeans MA, Horslen SP, Harper AM, Snyder JJ, Israni AK, and Kasiske BL (2018). OPTN/SRTR 2016 Annual Data Report: Intestine. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 18 Suppl 1, 254–290. [DOI] [PubMed] [Google Scholar]
- Sykes M, Sheard MA, and Sachs DH (1988a). Effects of T cell depletion in radiation bone marrow chimeras. II. Requirement for allogeneic T cells in the reconstituting bone marrow inoculum for subsequent resistance to breaking of tolerance. The Journal of experimental medicine 168, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M, Sheard MA, and Sachs DH (1988b). Graft-versus-host-related immunosuppression is induced in mixed chimeras by alloresponses against either host or donor lymphohematopoietic cells. The Journal of experimental medicine 168, 2391–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaskovic S, Fernandez S, Price P, Lee S, and French MA (2010). CD31 (PECAM-1) is a marker of recent thymic emigrants among CD4+ T-cells, but not CD8+ T-cells or gammadelta T-cells, in HIV patients responding to ART. Immunology and cell biology 88, 321–327. [DOI] [PubMed] [Google Scholar]
- Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, Granot T, Griesemer A, Lerner H, Kato T, et al. (2016a). Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nature medicine 22, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJ, Grinshpun B, Kumar BV, Kubota M, Ohmura Y, Lerner H, Sempowski GD, Shen Y, and Farber DL (2016b). Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, Lo CM, Chen L, Cheung CK, Yang ZF, Chen YX, Ng MN, Yu WC, Ming X, Zhang W, et al. (2012). Hematopoietic chimerism in liver transplantation patients and hematopoietic stem/progenitor cells in adult human liver. Hepatology (Baltimore, Md) 56, 1557–1566. [DOI] [PubMed] [Google Scholar]
- Wekerle T, Kurtz J, Ito H, Ronquillo JV, Dong V, Zhao G, Shaffer J, Sayegh MH, and Sykes M (2000). Allogeneic bone marrow transplantation with co-stimulatory blockade induces macrochimerism and tolerance without cytoreductive host treatment. Nature medicine 6, 464–469. [DOI] [PubMed] [Google Scholar]
- Williams AM, Bland PW, Phillips AC, Turner S, Brooklyn T, Shaya G, Spicer RD, and Probert CS (2004). Intestinal alpha beta T cells differentiate and rearrange antigen receptor genes in situ in the human infant. Journal of immunology (Baltimore, Md : 1950) 173, 7190–7199. [DOI] [PubMed] [Google Scholar]
- Wright DE, Wagers AJ, Gulati AP, Johnson FL, and Weissman IL (2001). Physiological migration of hematopoietic stem and progenitor cells. Science (New York, NY) 294, 1933–1936. [DOI] [PubMed] [Google Scholar]
- Zlotoff DA, and Bhandoola A (2011). Hematopoietic progenitor migration to the adult thymus. Annals of the New York Academy of Sciences 1217, 122–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Rosen S, Shonts B, Sprangers B, Savage TM, Richman S, Yang S, Lau SP, DeWolf S, Farber D, et al. (2015). Macrochimerism in Intestinal Transplantation: Association With Lower Rejection Rates and Multivisceral Transplants, Without GVHD. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 15, 2691–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shonts B, Lau SP, Obradovic A, Fu J, Yang S, Lambert M, Coley S, Weiner J, Thome J, et al. (2016). Bidirectional intragraft alloreactivity drives the repopulation of human intestinal allografts and correlates with clinical outcome. Science Immunology 1, eaah3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw TCR sequences data are freely accessible through https://doi.org/10.21417/Fu112018. The codes used to analyze TCR sequences are available and can be downloaded from a public GitHub repository (https://github.com/Aleksobrad/TCR-analysis).