

Abstract
c‐MYC and the SWI/SNF chromatin remodeling complex act as master regulators of transcription, and play a key role in human cancer. Although they are known to interact, the molecular details of their interaction are lacking. We have determined the structure of the RPT1 region of the INI1/hSNF5/BAF47/SMARCB1 subunit of the SWI/SNF complex that acts as a c‐MYC‐binding domain, and have localized the interaction regions on both INI1 and on the c‐MYC:MAX heterodimer. c‐MYC interacts with a highly conserved groove on INI1, while INI1 binds to the c‐MYC helix‐loop‐helix region. The binding site overlaps with the c‐MYC DNA‐binding region, and we show that binding of INI1 and E‐box DNA to c‐MYC:MAX are mutually exclusive.
Keywords: BAF/PBAF complexes, INI1/hSNF5/BAF47/SMARCB1 subunit, MYC, protein–protein interactions, transcription factors
Abbreviations
- BAF
BRG1‐ or HBRM‐associated factors
- c‐MYC
avian myelocytomatosis virus oncogene cellular homolog
- HSQC
heteronuclear single quantum coherence
- INI1
integrase interactor 1
- MAX
MYC‐associated factor X
- MIZ1
MYC‐interacting zinc finger protein‐1
- PPI
protein–protein interaction
- SMARCB1
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1
- SWI/SNF
SWItch/sucrose nonfermentable
Introduction
The transcription factor c‐MYC (hereafter MYC) acts as a master regulator of genes involved in cell growth, differentiation, metabolism, and apoptosis. Deregulated expression of MYC occurs in the majority of human cancers, playing a pivotal role in tumorigenesis and cancer progression 1, 2. In mouse models, inactivation of MYC dramatically halts tumor cell growth and proliferation, without invoking tumor escape pathways 1, 2, making targeting MYC an attractive approach for anticancer therapy 3. MYC is also an emerging target in other areas of human diseases, such as inflammation and heart disease 4, 5. As MYC dramatically increases the efficiency of somatic cell reprogramming, modulating MYC functions is also important in regenerative medicine 6. Although MYC physiology and pathology have been extensively studied, we still do not know how MYC works at the molecular level, which is a key to be able to target it pharmacologically. MYC coordinates the expression of a large, extremely diverse set of genes in a highly context‐dependent manner. MYC operates within a network of protein–protein interactions (PPIs), crucial for both directing MYC to specific genomic sites and for modulating gene expression 7, 8. However, our knowledge of these interactions is limited.
Several studies have shown that the SWI/SNF chromatin remodeling complex is part of the MYC interactome 9, 10, 11, 12. This multiprotein complex 13 uses ATP to alter chromatin structure by repositioning nucleosomes 13 and plays a key role in regulating gene expression during cell differentiation. Mutations that inactivate SWI/SNF subunits are found in around 20% of human cancers 14. Its role in cancer is complex as it has reported that depending on the type of the tumor the SWI/SNF complex can either inhibit tumor progression, or be required for cancer cell growth. Targeting the SWI/SNF complex is, thus, paradoxically emerging as a potential strategy for anticancer therapy 15.
The interplay between MYC and the SWI/SNF chromatin remodeling complex seems to be multifaceted and very much context‐ and gene dependent 16, 17, 18, 19, 20.
The INI1 (syn hSNF5, BAF47, SMARCB1) subunit 10, 21, 22 has been shown to play a major role in the interaction of the SWI/SNF complex with MYC. This subunit has also been implicated in the recruitment of other transcription factors 23, other chromatin‐associated proteins and a number of viral proteins, such as HIV integrase 24 and EPV EBNA2 25. INI1 was the first subunit of this complex identified to be mutated in cancer. Nonsense mutations and deletions that abolish INI1 expression are present in rhabdoid tumors, the most common malignant CNS tumors of children below 6 months of age. Analysis of the genomes of rhabdoid tumors shows very few other mutational events indicating that epigenetic dysregulation is the central mechanism of oncogenesis 26.
In rhabdoid tumor‐derived cells reintroduction of INI1 appears to suppress MYC functions. In other cancer cell lines, it has been shown instead that INI1 contributes to MYC transcriptional activity 21, and that overexpression of the region of INI1 that binds to MYC blocks MYC transcriptional activation 27.
We set out to determine how these two important transcription regulators interact at the molecular level and have revealed a complex network of competing interactions.
Results
Structure determination of INI1/hSNF5 RPT1
INI1 is a modular protein consisting of an N‐terminal winged‐helix domain 28 followed by two 60‐amino acid imperfect repeats Repeat 1 (RPT1) and Repeat 2 (RPT2), and a C‐terminal coil–coil domain (Fig. 1A). RPT1 has been shown to be required for MYC interaction 21, 27. The expression of residues 184–258 of INI1, which encompasses RPT1, in Escherichia coli provided soluble protein that gave good quality 1H,15N heteronuclear single quantum coherence (HSQC) spectra with chemical shift dispersion typical of a well‐folded protein. INI1 RPT1 behaved as a monomer by NMR and size exclusion chomatography–multi‐angle laser light scattering (SEC‐MALS) analysis. Protein samples were sufficiently stable to allow the collection of the triple‐resonance and nuclear Overhauser enhancement spectroscopy data needed for chemical shift assignment and solution structure determination (Table 1). The NMR solution structure of this fragment was determined (Fig. 1B) (PDB: 5L7B) and based on this a shorter protein construct (184–252) was expressed that readily crystallized. The crystal structure of this protein was also determined using the NMR structure as a molecular replacement model (Fig. 1C and see also Table 2) (PDB: 5L7A). Residues 185–248 of INI1 form a compact folded domain that contains two antiparallel β‐strands linked by a short loop followed by two α‐helices (Fig. 1D). The two strands form a curved base onto which both helices pack and the structure is capped at either end by loops that link the two strands, and strand 2 and helix 1. This topology places the N‐ and C termini of the domain in close proximity. The fold is stabilized by a hydrophobic core that is primarily formed by residues in β1 (I187, I189, L191, M193, I195) and α2 (F233, I237, I241, I245). In addition, the side chain of Q244 in α2 forms hydrogen bonds to the backbone NH and CO groups of R190 in β1, thereby linking the N‐ and C termini of the domain. Comparison of the INI1 RPT1 structure to other known structures using the program dali 29 reveals significant similarities to domains in two kinases involved in the regulation of osmotic stress, the CCT domain of oxidative stress responsive kinase 1 (OSR1) 30 (Fig. 1C,D) and the autoinhibition domain of lysine‐deficient protein kinase 1 (WNK1) 31 (Fig. 1C,D). Both domains share a core ααββ motif with INI1 RPT1 with additional elements of secondary structure at the periphery of their folds (Fig. 1D). The ααββ motifs of the OSR1 and WNK1 domains can be superimposed onto the structure of INI1 RPT1 with an RMSD of 1.9 and 2.0 Å, respectively.
Figure 1.
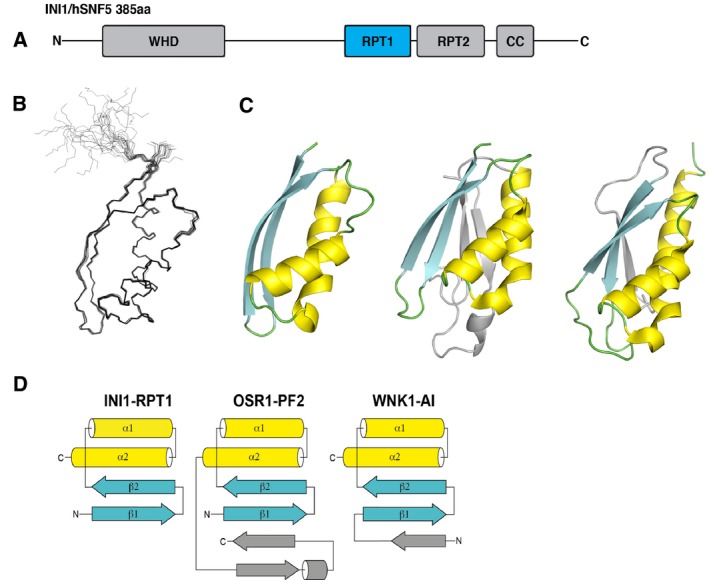
Structure of INI1/hSNF5 RPT1. (A) Representation of the domain structure of INI1. (B) Overlay of the 20 lowest energy NMR structures of RPT1. (C) From left to right: cartoon representations of the X‐ray structure of RPT1, and the structures of the OSR1 CCT domain and the autoinhibitory domain of WNK1 (WNK1‐AI). (D) Schematic of the topologies of the INI1/hSNF5 RPT1, OSR1 CCT, and WNK1‐AI domains.
Table 1.
Summary of conformational constraints and statistics for the 20 accepted NMR structures of the human INI1/SNF5 RPT1 domain
| Structural constraints | |
| Intraresidue | 663 |
| Sequential | 393 |
| Medium‐range (2 < |i − j| < 4) | 421 |
| Long‐range (|i − j| > 4) | 539 |
| Dihedral angle constraints | 21 |
| TALOS constraintsa | 124 |
| Distance constraints for 38 hydrogen bonds | 76 |
| Total | 2237 |
| Statistics for accepted structures | |
| Statistical parameters (±SD) | |
| RMS deviation for distance constraints | 0.0070 ± 0.0004 Å |
| RMS deviation for dihedral constraints | 0.342 ± 0.024° |
| Mean CNS energy term (kcal·mol−1 ± SD) | |
| E (overall) | 80.61 ± 4.31 |
| E (van der Waals) | 19.87 ± 1.35 |
| E (distance constraints) | 7.69 ± 0.91 |
| E (dihedral and TALOS constraints) | 2.07 ± 0.28 |
| RMS deviations from the ideal geometry (SD) | |
| Bond lengths | 0.0015 ± 0.0001 Å |
| Bond angles | 0.356 ± 0.0078° |
| Improper angles | 0.250 ± 0.009° |
| Average atomic RMSD from the mean structure (SD) | |
| Residues 184–249 (backbone)b | 0.237 ± 0.053 Å |
| Residues 184–249 (all heavy atoms)c | 0.720 ± 0.062 Å |
| Structural qualityd | |
| Residues in most favored region of Ramachanran Plot | 88.4% |
| Residues in additional allowed region of Ramachandran Plot | 10.4% |
| Residues in generously allowed region of Ramachandran Plot | 1.2% |
| Residues in disallowed region of Ramachandran Plot | 0.0% |
| PDB code | 5L7B |
a Dihedral angles are estimated using TALOS+‐based chemical shifts of backbone atoms of each amino acid. b Backbone heavy atoms include backbone N, Ca, CO. c Heavy atoms include both backbone and side chain non‐hydrogen atoms. d Statistics are for residues 184–259.
Table 2.
Data collection and refinement statistics
| Native dataset | |
|---|---|
| Data collection | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 43.619, 73.653, 46.460 |
| α, β, γ (°) | 90, 106.596, 90 |
| Resolution (Å)a | 44.5–2.1 (2.22–2.10) |
| Total reflections | 60 325 |
| Unique reflections | 15 711 |
| R sym or R merge (%)b | 0.060 (0.438) |
| I/σI | 14.6 (3.3) |
| Completeness (%) | 95.6 (93.3) |
| Redundancy | 3.8 (3.9) |
| Refinement | |
| Resolution (Å) | 45.0–2.1 |
| No. reflections | 15 656 |
| R work/R free c | 20.0, 0.26.0 |
| Number of atoms | |
| Protein | 2117 |
| Water | 130 |
| B‐factors | |
| Protein | 34.26 |
| Water | 35.40 |
| RMS deviationsd | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 0.918 |
| PDB code | 5L7A |
a Highest resolution shell is shown in parenthesis. b R m:∑h∑i|I/(h, i) − I(h)|/∑h∑i/I(h, i) where I(h, i) are symmetry‐related intensities and I(h) is the mean intensity of the reflection with unique index h. c R‐factor = Σ(|F obs| − k|F calc|)/Σ|F obs| and R‐free is the R value for a test set of reflections consisting of a random 5% of the diffraction data not used in refinement. dRMS deviations from ideal geometry for bond lengths and restraint angles (Engh and Huber).
The MYC:MAX bHLHZip dimer interacts with a conserved site on INI1/hSNF5 RPT1
We next set out to identify which region of INI1 RPT1 interacts with MYC. The C‐terminal part of MYC containing the basic helix‐loop‐helix (bHLH) and leucine zipper (Zip) domains has been shown to be required for interaction with INI1 21 and this interaction has been reported to occur with MYC bound to its heterodimerization‐binding partner MAX 22. We therefore coexpressed the bHLHZip region of MYC with the corresponding region (bHLHZip) of MAX. We then titrated purified unlabeled MYC:MAX bHLHZip dimer into 15N‐labeled INI1 RPT1. This produced significant changes in the 1H,15N HSQC spectrum of INI1 RPT1 (Fig. 2C,D). The bound and free forms of the domain are in fast exchange on the NMR timescale, and analysis of the dependence of the changes in chemical shifts induced on the amount of MYC:MAX dimer added gave a K d of 44 ± 6 μm for the interaction (Fig. 3). The residues that undergo significant chemical shift changes map to an elongated solvent‐exposed groove between β2 and α1 that has properties typical of a PPI interface (Fig. 2B). Within this binding region, there are a number of solvent‐exposed hydrophobic residues (F204, F218, I221, L226), which form two shallow pockets. The binding interface also contains two charged residues that undergo large changes in chemical shift upon binding: D202, which is partially buried, and D225, which is at the edge of the pocket (Fig. 2). The second strand of the sheet (β2) runs along the edge of binding interface. The backbone amides of β2 (i.e., K199, R201, and A203) all undergo significant changes in chemical shift. The resonance of N207 in the loop between β2 and α1 at one end of the binding site changes intensity rather than changing chemical shift upon binding. This suggests that the interaction with the MYC:MAX dimer may affect the dynamic properties of this region of the protein.
Figure 2.
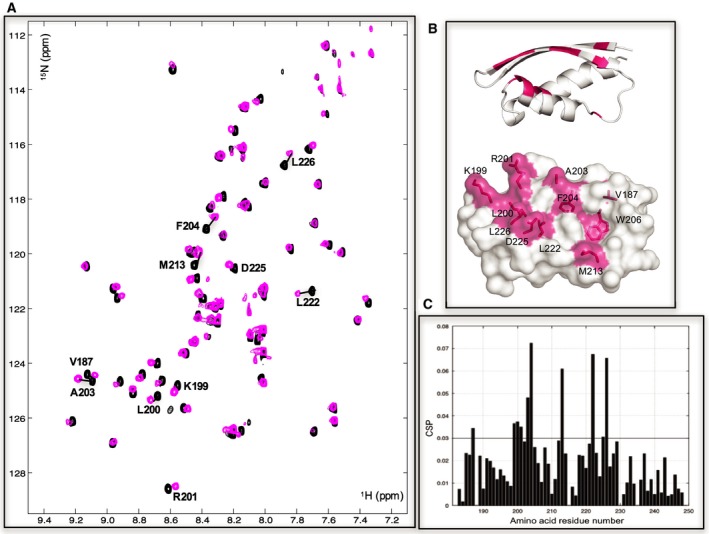
The MYC:MAX bHLHZip dimer binds to a conserved pocket on INI1/hSNF5 RPT1. (A) 1H,15N HSQC spectra of RPT1 without (black) and with (magenta) the addition of unlabeled MYC:MAX bHLHZip dimer (ratio 1 : 2). Residues undergoing chemical shift changes more than the standard deviation (SD) are labeled in black. (B) Cartoon (top) and molecular surface representation (bottom) of RPT1 showing the residues that undergo chemical shift changes more than the standard deviation are highlighted in magenta and labeled. The chemical shift changes of W206 are for the aromatic proton of the side chain (changes in chemical shifts of the backbone are below the threshold). (C) Diagram showing the differences in chemical shifts induced by binding of the MYC:MAX dimer to the 15N‐labeled INI1 RPT1. The black line indicates the calculated standard deviation.
Figure 3.
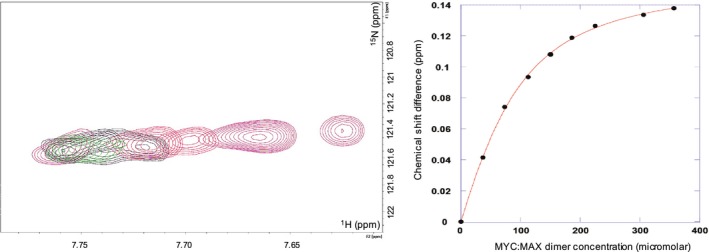
K d determination of the binding of the MYC:MAX dimer to the INI1/hSNF5 RPT1. On the left overlay of 15N HSQC spectra showing the variation of chemical shift changes induced on L222 by the addition of the unlabeled MYC:MAX dimer. The peak moves from right to left with increasing concentration of the dimer; on the right, plot of the induced chemical shifts versus dimer concentration, fitted to a single‐site binding curve. INI1 RPT1 was employed at a concentration of 147 μm.
The domains structurally similar to RPT1 present in the kinases OSR1 and WNK1 mediate PPIs involved in substrate recognition and regulation. An X‐ray structure of the OSR1 CCT domain bound to a peptide from a protein that regulates its activity has been determined (PDB: 2V3S). The peptide binds in an extended shallow pocket that is equivalent to the binding site identified in INI1 RPT1 (Fig. 4). The MYC:MAX dimer therefore appears to bind to a PPI site common to this family of proteins.
Figure 4.
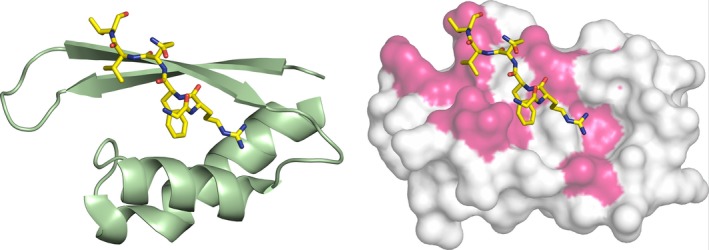
Comparison of the binding site in INI1/hSNF5 RPT1 and OSR1. On the left cartoon representation of the structure of the OSR1 CCT domain (green) in complex with the RFQV‐peptide (yellow). On the right superimposition of the OSR1 CCT domain (not shown) complex with the RFQV‐peptide (yellow) with the RPT1 structure shown as a molecular surface, with the residues that change chemical shifts upon binding to the MYC:MAX complex highlighted in magenta.
The sequence of RPT1 is identical in species ranging from humans to chicken (Fig. 5). The sequences of INI1 homologs in lower eukaryotes are more diverse, although some residues are highly conserved. These include amino acids that form the hydrophobic core as well as Q244, which, as discussed above, also appears to contribute to the stability of the fold. Many of the residues within the binding sites are also highly conserved including the two aspartates and several of the solvent‐exposed hydrophobic residues. In contrast, solvent‐exposed residues on the face opposite to the binding groove are not conserved. The role of residues in the binding site is difficult to probe by mutagenesis as many of them are involved in structurally important hydrogen bonding, or are hydrophobic residues that also contribute to the stability of the fold. We tested the effect of mutating two hydrophilic residues within the binding site, D202 and N207 to alanine. The integrity of the mutant proteins was assessed by recording HSQC spectra. Only small changes were observed for D202A mutant, however, much larger changes were observed for the asparagine mutant suggesting that its mutation alters the structure of the protein. The aspartate mutant showed reduced affinity to the MYC:MAX dimer, such that it was not possible to accurately determine a dissociation constant for its interaction, thus confirming that it forms part of the binding site (Fig. 6). Repeats 1 and 2 present a high degree of sequence similarity (Fig. 5). Many of the residues that stabilize the fold in RPT1 are also conserved in RPT2, which indicates that the structure of the two repeats is very similar. Many of the residues within the binding site identified in RPT1 are also conserved in RPT2, suggesting that this region is functionally important in RPT2. However, some of the amino acids conserved within the binding site of RPT2 differ from RPT1, which is consistent with data showing that the two repeats have different binding specificities 32.
Figure 5.

Sequence alignments of the repeats of family members of the INI1/SNF5. At the top, carton representation of the secondary structure elements of RPT1, followed by sequence alignments of RPT1 (middle) and RPT2 (bottom) for the Homo sapiens (Hs), Drosophila melanogaster (Dm), Caenorhabditis elegans (Ce) and Saccharomyces cerevisiae (Sc) proteins.
Figure 6.
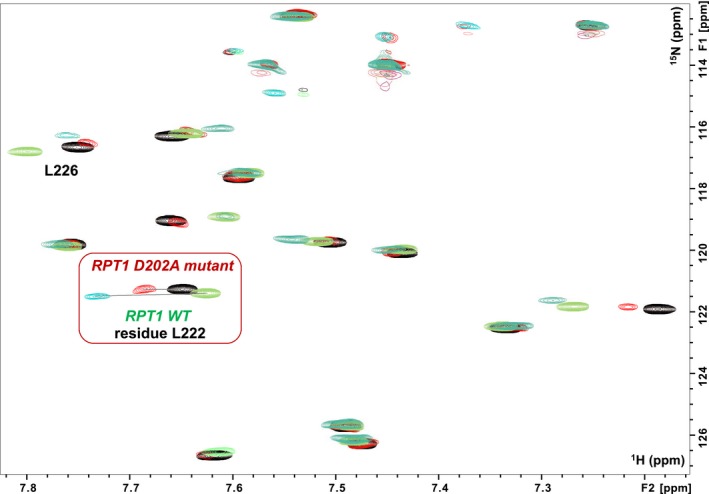
Binding of the MYC:MAX dimer to the INI1 D202A‐mutant. Overlay of a region of the 1H,15N HSQC spectra of RPT1 INI1 WT without (blue gray) and with (green) unlabeled MYC:MAX bHLHZip dimer (ratio 1 : 1), overlaid with the overlay of the same region of the 1H,15N HSQC spectra of RPT1 INI1 D202A‐mutant without (black) and with (red) unlabeled MYC:MAX bHLHZip dimer (ratio 1 : 1). Highlighted in the red square is the residue L222 which undergoes the largest change in chemical shift upon binding of MYC:MAX complex to both INI1 RPT1 WT and the D202A mutant. Comparison of the same ratio (1 : 1) illustrates the lower affinity of the DA mutant as the change in chemical shift of L222 is significantly less.
The INI1/hSNF5 RPT1 interacts with the helix‐loop‐helix region of the MYC:MAX dimer
We next set out to use NMR to map the region of the MYC:MAX dimer that interacts with INI1 RPT1. Assignments for human MYC:MAX were not available so we expressed 15N,13C‐deuterated MYC:MAX bHLHZip dimer and obtained backbone assignments for it using triple resonance NMR experiments (BMRB accession number 27571). Complete assignments were obtained for the leucine zipper (Zip), helix 2 (H2), the loop region and the C‐terminal part of helix 1 (H1) of both MYC and MAX. As was observed for v‐MYC 33 the assignments for the N‐terminal part of H1 in both proteins could not be obtained potentially because of exchange broadening due to fraying of this region. The residues from the basic region of both MYC and MAX are poorly dispersed (causing significant overlap in the central region of spectra), which indicates that they lack a stable structure in the absence of DNA, hampering the full assignment of these regions.
The addition of the 1H,15N MYC:MAX bHLHZip dimer to unlabeled INI1 RPT1 produced changes in the chemical shifts of residues in both MYC and MAX (Fig. 7A,C). Several peaks also undergo significant change in intensity, either decreasing or increasing, to the extent that some peaks are only visible in the bound form, which suggests that binding is accompanied by changes of the dynamic/conformational properties of the heterodimer. The largest changes cluster on a discrete patch on MYC, located at one end of the helix‐loop‐helix (HLH) region (Fig. 7B,C), indicating that this is the main binding site of INI1 RPT1 on the dimer complex. The biggest change overall in chemical shift is on MYC at the start of H2 (i.e., V393) (Fig. 7C).
Figure 7.
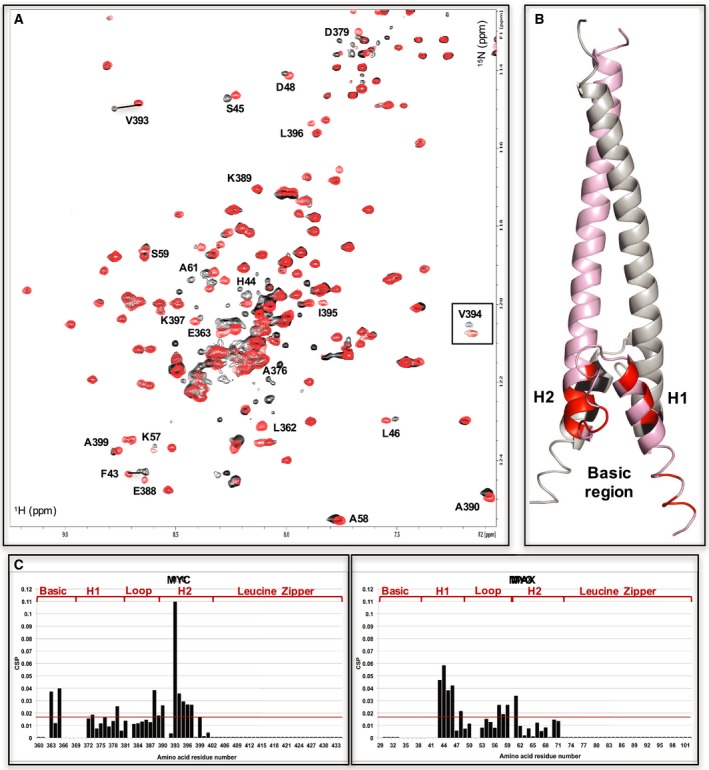
INI1/hSNF5 RPT1 binds to the helix‐loop‐helix region of MYC in the MYC:MAX bHLHZip dimer. (A) 1H,15N BST TROSY spectra of 15N‐labeled MYC:MAX bHLHZip dimer without (red) and with (black, ration 1 : 1) binding to RPT1 that move more than the standard deviation are labeled. The region corresponding to residue V394 is inserted in the spectra in a box to better illustrate the changes of chemical shifts that occur upon binding to INI1. (B) Cartoon representation of the MYC:MAX dimer (PDB = 1NKP, pink: MYC, gray: MAX) with highlighted in red (MYC) and black (MAX) the residues labeled in the spectra. H1 = MYC Helix 1; H2 = MYC Helix 2. (C) Diagrams showing the differences in chemical shifts induced by binding of RPT1 to the 15N‐labeled MYC:MAX dimer (MYC on the left, MAX on the right). Residues 29, 34–42, 52, 60, from MAX, and residues 366–71 from MYC are not assigned. L362 is assigned in the free form, but it could not be assigned in the bound form. Residues 51, 382, 391 are prolines. The red line indicates the standard deviation. H1 = MYC Helix 1; H2 = MYC Helix 2.
Other significant changes in MYC are in the loop that links H1 and H2, other residues at the N terminus of H2, and residues in the basic region (Fig. 7B,C). Some changes can be seen also in H1 (Fig. 7B,C): these changes are from residues that are close in space to the residues affected in H2 and the Loop.
In MAX, the most significant changes in chemical shift are for residues in H1 (Fig. 7B,C) that are across the heterodimer interface from the affected residues on MYC in H2 (Fig. 7B). There are also a few small changes observed at the start of MAX H2. In both proteins, H1 is only partially folded in the free dimer and these changes may result from tightening of the dimer upon forming the complex, rather than a direct effect of the binding to INI1. The chemical shift mapping results are, therefore, consistent with MYC being the main interaction partner of INI1 RPT1.
There are also changes in the basic region of MYC (no changes are observed in the basic region of MAX). Studies on the bHLH‐transcription factors have shown that binding to the HLH region can allosterically induce conformational changes in the basic region 34. Therefore, to understand if the changes in the basic region of MYC are the results of direct binding we then examined the interaction of INI1 with the MYC:MAX HLHZip complex where both basic regions have been removed. As the basic regions in the free MYC:MAX complex are in a disordered conformation removing them also helps with the analysis of the NMR spectra (Fig. 8A). NMR data showed that removing the basic regions does not compromise, or alter the heterodimerization binding interface (Fig. 8A), and biophysical analysis (i.e., DSC) of the HLHZip complex also confirmed that the removal of the basic regions does not destabilize the complex. On the contrary, this complex seems to be more stable having a melting point 7 °C higher than the construct with the basic regions. The NMR spectra of the binding of 15N‐labeled MYC:MAX HLHZip complex to unlabeled INI1 RPT1 showed a comparable footprint on the HLH region of MYC to that observed for binding to the MYC:MAX bHLHZip (Fig. 8B,C). Less chemical shift changes can be seen on MAX possibly (Fig. 8C), because as the structure is more stable, there is less consolidation of heterodimer complex upon binding of INI1. The binding affinity, however, is significantly reduced to the point that is not possible to determine a K d. This suggests that the chemical shift changes observed in the basic region are due to a direct interaction.
Figure 8.
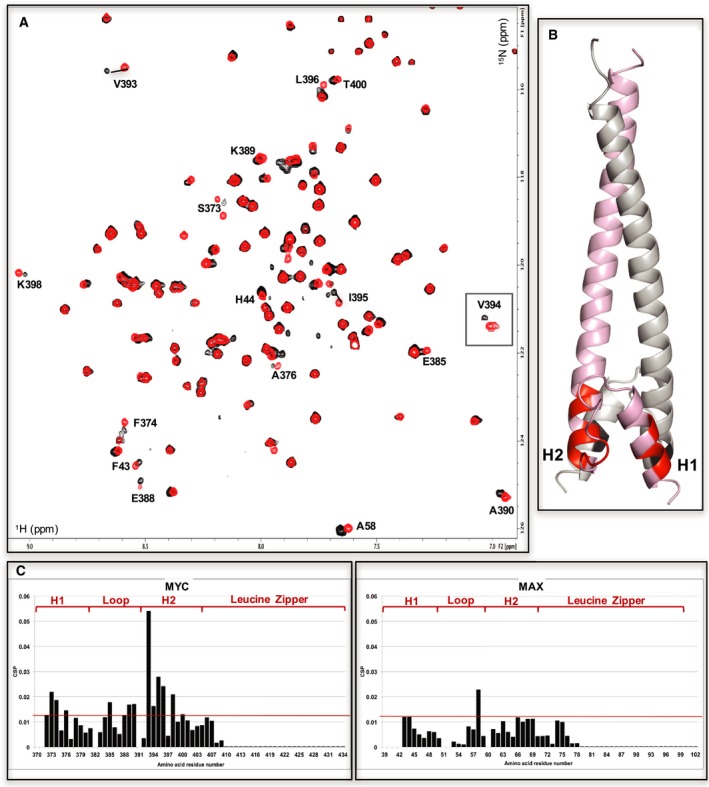
INI1/hSNF5 RPT1 binds to the helix‐loop‐helix region of MYC in the MYC:MAX dimer lacking the basic region. (A) 1H,15N BST TROSY spectra of 15N‐labeled MYC:MAX HLHZip dimer without (red) and with (black, ration 1 : 2) the addition of unlabeled RPT1 (residue 394 is inserted into the spectra as in Fig. 7). Residues implicated in the binding to RPT1 that move more than the standard deviation are labeled. (B) Cartoon representation of the MYC:MAX dimer (pink: MYC, gray: MAX) with highlighted in red (MYC) and black (MAX) the residues labeled in the spectra. H1 = MYC Helix 1; H2 = MYC Helix 2. (C) Diagrams showing the differences in chemical shifts induced by binding of RPT1 to the 15N‐labeled MYC:MAX dimer (MYC on the left, MAX on the right). Residues 39–42, 52, 370–71 are not assigned. Residues 51, 382, 391 are prolines. The red line indicates the standard deviation. H1 = MYC Helix 1; H2 = MYC Helix 2.
The chemical shifts mapping results indicates that INI1 docks onto one face of the heterodimer at one end of the HLH motif of MYC (Fig. 7). This interaction region on MYC does not appear to be an extended linear motif, in contrast to the interaction between the OSR1 CCT domain and the RFQV‐peptide.
INI1 RPT1 binding to MYC:MAX bHLHZip dimer is incompatible with binding to E‐box DNA
The MYC:MAX heterodimer binds to canonical E‐box DNA primarily via the basic regions with an affinity in the nanomolar range 35. INI1 RPT1 binds to a region on MYC that is contiguous to the DNA binding motif, and several of the residues in the putative interaction‐site in the HLH motif make contacts with the DNA in the crystal structure of c‐MYC:MAX bound to E‐box DNA (PDB: 1NKP). Furthermore, there is a strong indication that residues in the basic region itself are involved in the binding to INI1. We, therefore, tested if INI1 RPT1 binding is compatible with the binding of the MYC:MAX bHLHZip complex to canonical E‐box DNA. To this end, we first evaluated the binding of a oligonucleotide containing a canonical E‐box sequence (the same E‐box DNA sequence used in the reported crystal structure) by adding 15N‐labeled MYC:MAX bHLHZip to the DNA. This produced large changes in chemical shift and line broadening, confirming the formation of the MYC:MAX bHLHZip/DNA complex (MW = 34 kDa). An ITC analysis confirmed that the binding is in the nano‐molar range as previously reported 35. Then, we added 15N‐labeled INI1 RPT1 to the unlabeled MYC:MAX bHLHZip/DNA complex. No changes in chemical shifts, or line broadening, were observed in the spectra of the 15N‐labeled INI1 RPT1 (Fig 9: top panel). We also added the DNA to the pre‐formed MYC:MAX/15N‐labeled INI1 RPT1 complex, and observed displacement of the 15N‐labeled INI1 RPT1 by the DNA, as observed by the recovery of a spectrum corresponding to free 15N‐labeled INI1 RPT1. No changes in chemical shifts were also observed in the spectra of 15N‐labeled MYC:MAX complexed with E‐box DNA when unlabeled INI1 was added (Fig. 9: bottom panel).
Figure 9.
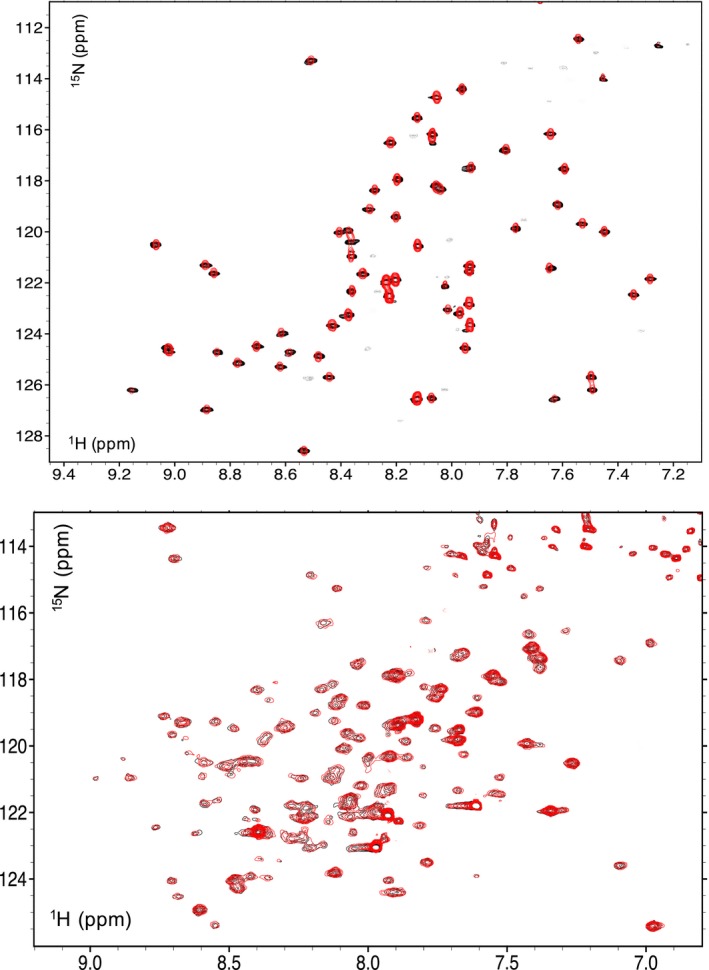
Binding of INI1/hSNF5 RPT1 and DNA to the MYC:MAX bHLHZip dimer are mutually exclusive. Top: overlay of 1H,15N HSQC spectra of RPT1 without (black) and with (red, ratio 1 : 1) unlabeled MYC:MAX bHLHZip dimer bound to DNA. Bottom: overlay 1H,15N BST TROSY spectra of 15N‐labeled MYC:MAX bHLHZip dimer bound to DNA without (black) and with (red, ratio 1 : 1) unlabeled RPT1 INI1. In both spectra, no chemical shift changes are observed, illustrating that INI1 RPT1 does not bind to the MYC:MAX dimer when this is bound to E‐box DNA.
Discussion
Recently several studies have shown that loss of PPIs that are mediated by INI1 is one of the major factors contributing to oncogenesis in malignant rhabdoid tumors 36, 37. Most of the PPIs made by this subunit have been mapped to the repeats (1 and 2) 23, and consistent with this the structure of RPT1 reveals a fold that is an established PPI motif. We have shown that MYC interacts with the canonical binding site of this motif and, therefore, is likely to compete with other binding partners of the SWI/SNF complex. Interestingly, a recent paper has suggested that when MYC is overexpressed it could compete with prodifferentiation transcription factors for binding to the SWI/SNF complex 38. Furthermore, while this study was being completed Yan et al. 39. reported that the MYC‐binding groove of RPT1 interacts with the BAF155 subunit of the SWI/SNF complex. This suggests that MYC and other molecules binding to this site could modulate the interactions between SWI/SNF subunits, and alter the activity of the chromatin remodeling complex.
The highly context dependent functionalities of the SWI/SNF complex make it very difficult to use genetic approaches to determine the role of the interaction between MYC and the INI1. The use of chemical probes 40 could instead be a more productive approach because their effects are tunable, reversible, and most relevant to this interaction, conditional as they can be introduced at any point during cancer development. The MYC binding region in INI1 RPT1 possesses features potentially amenable for small‐molecule binding, and an in silico assessment using LIGSITE 41, which is widely used to evaluate the suitability of proteins to bind small molecules, predicted both the hydrophobic pockets within the MYC binding region as potential small‐molecule binding sites (Fig. 10). Furthermore, targeting domains structurally similar to the INI1 RPT1 has been successfully carried out by fragment‐based screening 42. The biophysical data that we have obtained on INI1 RPT1 will allow the use of both structure‐based design and fragment‐based approaches 43. This strategy could have a dramatic effect on the overall functionality of the SWI/SNF complex, not just on the interaction with MYC. This, though, could provide a means of targeting the SWI/SNF complex itself for therapeutic intervention 15.
Figure 10.
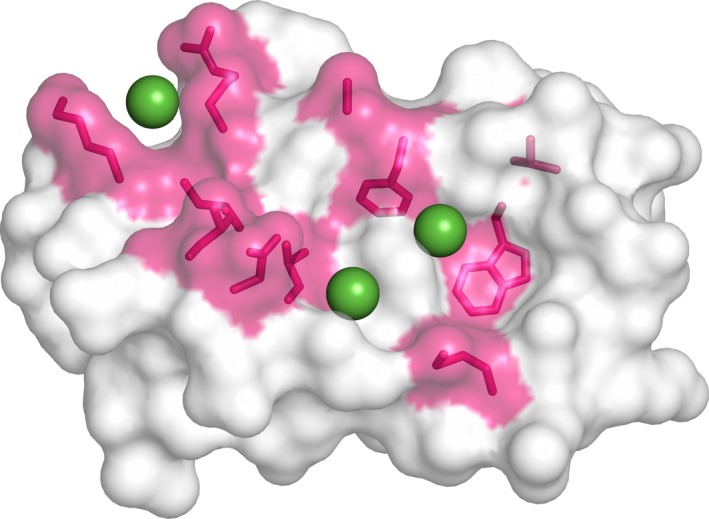
Potential small‐molecule binding pockets in INI1/hSNF5 RPT1. Molecular surface representation of INI1/hSNF5 RPT1 showing the residues that undergo chemical shift more than standard deviation highlighted in magenta, and in green spheres representing three potential small‐molecule binding pockets that were identified by the LIGSITE program.
MYC is an intrinsically disordered protein where the C‐terminal only folds into a HLH‐Zip structural motif upon binding to the obligatory partner MAX 44. Because of their conformational flexibility, disordered proteins such as MYC 45 typically make multiple interactions with their partner proteins: this multivalency allows them to mediate the formation of stable and dynamic complexes 46. MYC has been reported to interact with multiple subunits of the SWI/SNF chromatin remodeling complex 9, and may bind to other regions of INI1 (i.e., RPT2) 22, although these interactions have not been characterized. This suggests that MYC makes multivalent interactions with the SWI/SNF complex. The K d value of the MYC:INI1 RPT1 interaction fits well with a multivalent mode of interaction in which individual components have a relatively modest affinity, but high specificity. In this context, it is relevant to highlight that the only other well‐characterized MYC interaction 47 with a component of chromatin regulatory complexes (i.e., WDR5) has a similar K d value to the one for the MYC:INI1 interaction herein reported. A multivalent mode of binding may allow additional levels of regulation (e.g., post‐translational modification) to play a role in directing what type of SWI/SNF:MYC interaction takes place on target genes. Interestingly, the binding regions identified in both MYC and INI1 RPT1 are subjected to post‐translational modifications, such as phosphorylation, acetylation, and ubiquitination 48.
One striking feature of the binding of INI1 on the MYC is its interplay with the binding of the MYC:MAX dimer to DNA. The interaction maps to a large region involving the HLH motif and the beginning of the basic region. This shows that this region is not only involved in dimerization and DNA binding, but it is also important in mediating PPIs. In fact, our study shows for the first time that the interaction of MYC with a cofactor competes with the MYC:MAX dimer binding to DNA. In particular, we have found that binding of INI1 and DNA to the MYC:MAX complex are mutually exclusive: the interaction of INI1 RPT1 with the MYC:MAX dimer does not take place when MYC is bound to E‐box DNA, which is at first hand an unexpected result as conventionally one would expect that a transcription factor would recruit the SWI/SNF complex to its cognate DNA. This has potentially important functional implications. The factors governing the distribution of MYC on the genome are known to be diverse, and to include49, 50: direct interactions with DNA containing both high‐affinity canonical E‐box sequences and other lower affinity sites, as well as multiple low‐affinity PPIs between the MYC:MAX dimer and other chromatin‐associated protein complexes, and/or transcription factors, such as MIZ1 51, which themselves can bind to DNA. Site distribution has been suggested to depend on the level of MYC in the cell, with lower affinity sites being occupied in tumors with high levels of MYC expression. At sites where MYC:MAX does not directly bind to DNA, the MYC:MAX complex will be available to interact with INI1. At other sites the binding of INI1 to MYC:MAX would compete with DNA with the outcome depending on the relative affinities. As mentioned above, it is possible that MYC makes other contacts with the SWI/SNF complex; the avidity effect of this multivalent mode of binding would then result in a higher affinity interaction, thus enabling the SWI/SNF complex to displace MYC from DNA – especially at lower affinity sites 50. This could explain the observations that the SWI/SNF complex can decrease MYC's binding to some of its target genes in lung cancer cell lines; and that when INI1 is introduced into INI1‐deficient rhabdoid tumor cells, MYC binding is reduced at some of its target genes 22. Our findings about the competition with DNA binding would be, therefore, consistent with INI1 negatively regulating MYC transcriptional activity.
Overall, this work highlights how MYC operates via a complex network of competing interactions, and offers a potential means to manipulate its functions.
Materials and methods
Material
Chemicals were acquired from Sigma‐Aldrich (St. Louis, MO, USA) or Fisher Scientific (Fremont, CA, USA) and were used without further purification. Ni‐NTA resin was from Qiagen (Manchester, UK), Amicon centrifugal units were obtained from Millipore (Billerica, MA, USA). PCR primers were obtained from IDT (Integrated DNA Technologies, Leuven, Belgium).
DNA cloning, and protein expression and purification
INI1/hSNF5
The DNA encoding the residues 184–258 of INI1 was amplified from human cDNA by PCR and cloned into a modified pRSETA (Invitrogen, Life Sciences, Paisley, UK) expression vector that produces proteins fused to N‐terminally His6‐tagged lipoyl domain of Bacillus stearothermophilus dihydrolipoamide acetyltransferase. The resulting plasmids were transformed into E. coli C41 (DE3) cells. Cells were grown in 2XTY media at 37 °C to mid‐log phase and induced with 1 mm IPTG. The temperature was reduced to 22 °C, and the cells were grown for a further 16 h. Isotopically labeled domains were prepared by growing cells in K‐MOPS minimal media containing 15NH4Cl and/or [13C]‐glucose. Cells where lysed by sonication, and the fusion protein was purified by Ni2+‐NTA affinity chromatography. The purified protein was dialyzed overnight in the presence of TEV protease, which cleaves the fusion protein after the lipoyl domain. A second Ni2+‐NTA affinity chromatography step was carried out to remove the lipoyl domain and the protein was further purified by gel filtration using a HiLoad 26/60 Superdex 75 column (GE Healthcare, Little Chalfont, UK).
c‐MYC:MAX bHLHZip (MYC = 352–437, MAX = 22–102) and c‐MYC:MAX HLHZip (MYC = 368–437, MAX = 36–102)
Soluble His6 MYC:MAX bHLHZip, or His6 MYC:MAX HLHzip heterodimer, was produced in E. coli using a pET28a derived plasmid that directs the coexpression of both proteins from a polycistronic mRNA, using a similar approach to Fieber et al. 52. Chemically competent E. coli BL21 (DE3) cells were transformed with this plasmid. Cells were plated on Luria‐Bertani agar supplemented with kanamycin. A single colony was used to inoculate a culture of either 2XTY broth or K‐MOPS minimal media prepared in D2O containing 15NH4Cl and [13C]‐glucose. Cell were grown at 22 °C to an OD 600 of 0.8 and then induced with 1 mm IPTG. Cells were grown for a further 16 h before being collected by centrifugation. Cells were lysed by sonication. For the MYC:MAX bHLHZip complex, this is followed by the addition of DNAse I (150 μL for 1 g of culture pellet) and incubation for 1 h at 37 °C (90 r.p.m.). Centrifugation at 38 000 g at 4 °C for 45 min is then carried out.
The dimer was purified at 25 °C by affinity chromatography using a HisTRAP column (GE Healthcare) according to the standard protocol for Ni2+‐NTA affinity chromatography recommended by the manufacturer, and dialyzed against PBS (with 1–10 mm DTT) overnight at 5 °C. Aliquots of the protein complex were snap‐frozen in liquid nitrogen and stored at −80 °C.
NMR
INI1/hSNF5 samples prepared for NMR spectroscopy experiments were typically 1.5 mm for structural determination in 90% H2O, 10% D2O, containing 20 mm potassium phosphate, pH 6.5, 100 mm NaCl, and 5 mm β‐mercaptoethanol. All spectra were acquired using a Bruker DRX800, DRX600, or DMX500 spectrometer equipped with pulsed field gradient triple resonance at 25 °C, and referenced relative to external sodium 2,2‐dimethyl‐2‐silapentane‐5‐sulfonate for proton and carbon signals, or liquid ammonia for that of nitrogen. Assignments were obtained using standard NMR methods using 13C,15N‐labeled, 15N‐labeled, 10%13C‐labeled, and unlabeled protein samples 53. Backbone assignments were obtained using the following standard set of 2D and 3D heteronuclear spectra: 1H‐15N HSQC, HNCACB, CBCA(CO)NH, HNCACO, HNCO, HBHA(CO)NH, and 1H‐13C HSQC. Additional assignments were made using 2D TOCSY and DQF‐COSY spectra. A set of distance constraints were derived from 2D NOESY spectra recorded from a 1.5 mm sample with a mixing time of 120 ms. Hydrogen bond constraints were included for a number of backbone amide protons whose signals were still detected after 10 min in a 2D 1H‐15N‐HSQC spectrum recorded in D2O at 278 K (pH 5.0). Candidates for the acceptors were identified using the program hbplus for the hydrogen bond donors that were identified by the H–D exchange experiments. When two or more candidates of acceptors were found for the same donor in different structures, the most frequently occurring candidate was selected. For hydrogen bond partners, two distance constraints were used where the distance (D)H–O(A) corresponded to 1.5–2.5 Å and (D)N–O(A) to 2.5–3.5 Å. Torsional angle constraints were obtained from an analysis of C’, N, Cα Hα and Cβ chemical shifts using the program talos 54. The stereospecific assignments of Hβ resonances determined from DQF‐COSY and HNHB spectra were confirmed by analyzing the initial ensemble of structures. Stereospecific assignments of Hγ and Hδ resonances of Val and Leu residues, respectively, were assigned using a fractionally 13C‐labeled protein sample 55. The three‐dimensional structure of the INI1 domain (residues 183–258) was calculated using the standard torsion angle dynamics‐simulated annealing protocol in the program cns 1.2 56. Structures were accepted where no distance violation was greater than 0.25 Å and no dihedral angle violations > 5°. The final coordinates have been deposited in the Protein Data Bank (PDB accession no. 57LB).
Labeled MYC:MAX samples prepared for NMR spectroscopy experiments were typically 200–300 μm in PBS, 10% D2O, pH 7, and 1 mm DTT. All spectra were acquired using a Bruker DRX800 (Bruker BioSpin, Billerica, MA, USA) (binding experiments) or DRX950 (for NMR assignments) spectrometers equipped with pulsed field gradient triple resonance cryoprobe at 25 °C. Backbone assignments were carried using the following standard set of 3D heteronuclear spectra on deuterated samples: HNCO, HN(CA)CO, HNCACB, CBCA(CO)HN. The assignments have been deposited in the Biological Magnetic Resonance Bank (BMRB, accession number 27571).
Chemical shift perturbations were calculated using the root mean square deviation of the changes of the H and N chemical shifts, using a correction factor for the N chemical shifts as discussed by Williamson 57.
Crystallography
Crystals of INI1/hSNF5 (concentration of 6 mg·mL−1) were grown using the vapor diffusion method at 4 °C using a precipitant of 1.4 m tri‐sodium citrate and 100 mm HEPES pH 7.5. Crystals were immersed into the precipitant solution supplemented with 20% (v/v) glycerol prior to vitrification by direct immersion into liquid nitrogen. Native data was collected in‐house on an Fr‐E Superbright rotating anode generator (Rigaku Corp, Tokyo, Japan), equipped with a MarDTB image plate detector (marresearch GmbH, Norderstedt, Germany). Diffraction data were indexed and integrated with xds 58 and scaled and merged with scala [ 59]. The crystal structure of the truncated construct (183–252) of human INI1 was solved by molecular replacement using the NMR structure as a search model. Density modification produced experimental maps that allowed for manually refinement using main and coot 60. The crystal structure was refined to 2.1 Å using phenix 61 and is consistent with the NMR structure (RMSD of 0.77 Å for the backbone atoms of residues 185–245). The crystallographic data are summarized in Table 2. The validity of all models was routinely determined using molprobity (http://molprobity.biochem.duke.edu/) and by using the free R factor to monitor improvements during building and crystallographic refinement. The final coordinates have been deposited in the Protein Data Bank (PDB accession no. 57LA).
Accession numbers
NMR structure of INI1/hSNF5 RPT1‐ PDB ID: 5L7B. Crystal structure of INI1/hSNF5 RPT1‐ PDB ID: 5L7A. NMR assignments of c‐MYC:MAX bHLHZip complex – BMRB accession number 27571.
Conflict of interest
The authors declare that they have no conflict of interest.
Author contributions
GZ conceived and supervised the study; MDA, NH, MB, and GZ performed experiments; SS, MDA, MB, and GZ carried out data analysis; GZ and MB wrote the manuscript with contributions from all authors.
Acknowledgements
The authors thank Dr Stefan M.V. Freund for helping with the setup of NMR experiments, and Dr Chris Johnson for biophysical analysis. The authors also thank the Medical Research Council (MRC) Biomedical NMR Centre at the Francis Crick Institute for access to a 950 MHz NMR spectrometer.
The copyright line for this article was changed on 14 February 2019 following original online publication.
References
- 1. Morton JP & Sansom OJ (2013) MYC‐y mice: from tumour initiation to therapeutic targeting of endogenous MYC. Mol Oncol 7, 248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S & Evan GI (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zinzalla G (2016) Targeting MYC: is it getting any easier? Future Med Chem 8, 1899–1902. [DOI] [PubMed] [Google Scholar]
- 4. McKeown MR and Bradner JE (2014) Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med 4, a014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wolfram JA, Lesnefsky EJ, Hoit BD, Smith MA & Lee H‐G (2011) Therapeutic potential of c‐Myc inhibition in the treatment of hypertrophic cardiomyopathy. Ther Adv Chronic Dis 2, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takahashi K & Yamanaka S (2016) A decade of transcription factor‐mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 17, 183–193. [DOI] [PubMed] [Google Scholar]
- 7. Conacci‐Sorrell M, McFerrin L & Eisenman RN (2014) An overview of MYC and its interactome. Cold Spring Harb Perspect Med 4, a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tu WB, Helander S, Pilstal R, Hickman KA, Lourenco C, Jurisica I, Raught B, Wallner B, Sunnerhagen M & Penn LZ (2015) Myc and its interactors take shape. Biochem Biophys Acta 1849, 469–483. [DOI] [PubMed] [Google Scholar]
- 9. Chan P‐K, Srikumar T, Dingar D, Kalkat M, Penn LZ & Raught B (2014) BioID data of c‐MYC interacting protein partners in cultured cells and xenograft tumors. Data Brief 1, 76–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pal S, Yun R, Datta A, Lacomis L, Erdjument‐Bromage H, Kumar J, Tempst P & Sif S (2003) mSin3A/histone deacetylase 2‐ and PRMT5‐containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol Cell Biol 23, 7475–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR III, Menssen A & Hermeking H (2007) Large‐scale identification of c‐MYC‐associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6, 205–217. [DOI] [PubMed] [Google Scholar]
- 12. Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom‐Cerajewski L, Robinson MD, O'Connor L, Li M et al (2007) Large‐scale mapping of human protein‐protein interactions by mass spectrometry. Mol Syst Biol 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tang L, Nogales E & Ciferri C (2010) Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog Biophys Mol Biol 102, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson BG & Roberts CWM (2011) SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11, 481–492. [DOI] [PubMed] [Google Scholar]
- 15. Zinzalla G (2016) A new way forward in cancer drug discovery: inhibiting the SWI/SNF chromatin remodelling complex. Chembiochem 17, 677–682. [DOI] [PubMed] [Google Scholar]
- 16. Jubierre L, Soriano A, Planells‐Ferrer L, Paris‐Coderch L, Tenbaum SP, Romero OA, Moubarak RS, Almazan‐Moga A, Molist C, Roma J et al (2016) BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene 35, 5179–5190. [DOI] [PubMed] [Google Scholar]
- 17. Buscarlet M, Krasteva V, Ho L, Simon C, Hebert J, Wilhelm B, Crabtree GR, Sauvageau G, Thibault P & Lessard JA (2014) Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood 123, 1720–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi J, Whyte WA, Zepeda‐Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley‐Maslin MA et al (2013) Role of SWI/SNF in acute leukemia maintenance and enhancer‐mediated Myc regulation. Genes Dev 27, 2648–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Q, Madany P, Akech J, Dobson JR, Douthwright S, Browne G, Colby JL, Winter GE, Bradner JE, Pratap J et al (2015) The SWI/SNF ATPases are required for triple negative breast cancer cell proliferation. J Cell Physiol 230, 2683–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romero OA, Setien F, John S, Gimenez‐Xavier P, Gomez‐Lopez G, Pisano D, Condom E, Villanueva A, Hager GL & Sanchez‐Cespedes M (2012) The tumour suppressor and chromatin‐remodelling factor BRG1 antagonizes Myc activity and promotes cell differentiation in human cancer. EMBO Mol Med 4, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J & Kalpana GV (1999) c‐MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 22, 102–105. [DOI] [PubMed] [Google Scholar]
- 22. Stojanova A, Tu WB, Ponzielli R, Kotlyar M, Chan PK, Boutros PC, Khosravi F, Jurisica I, Raught B & Penn LZ (2016) MYC interaction with the tumor suppressive SWI/SNF complex member INI1 regulates transcription and cellular transformation. Cell Cycle 15, 1693–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stojanova A & Penn LZ (2009) The role of INI1/hSNF5 in gene regulation and cancer. Biochem Cell Biol 87, 163–177. [DOI] [PubMed] [Google Scholar]
- 24. Das S (2015) Integrase interactor 1 in health and disease. Curr Protein Pept Sci 16, 478–490. [DOI] [PubMed] [Google Scholar]
- 25. Wu DY, Kalpana GV, Goff SP & Schubach WH (1996) Epstein‐Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF‐SWI complex, hSNF5/Ini1. J Virol 70, 6020–6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR et al (2012) A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Investig 122, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takayama MA, Taira T, Tamai K, Iguchi‐Ariga SM & Ariga H (2000) ORC1 interacts with c‐Myc to inhibit E‐box‐dependent transcription by abrogating c‐Myc‐SNF5/INI1 interaction. Genes Cells 5, 481–490. [DOI] [PubMed] [Google Scholar]
- 28. Allen MD, Freund SM, Zinzalla G & Bycroft M (2015) The SWI/SNF subunit INI1 contains an N‐terminal winged helix DNA binding domain that is a target for mutations in schwannomatosis. Structure 23, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holm L & Laakso LM (2016) Dali server update. Nucleic Acids Res 44, W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Villa F, Goebel J, Rafiqi FH, Deak M, Thastrup J, Alessi DR & van Aalten DM (2007) Structural insights into the recognition of substrates and activators by the OSR1 kinase. EMBO Rep 8, 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moon TM, Correa F, Kinch LN, Piala AT, Gardner KH & Goldsmith EJ (2013) Solution structure of the WNK1 autoinhibitory domain, a WNK‐specific PF2 domain. J Mol Biol 425, 1245–1252. [DOI] [PubMed] [Google Scholar]
- 32. Morozov A, Yung E & Kalpana GV (1998) Structure‐function analysis of integrase interactor 1/hSNF5L1 reveals differential properties of two repeat motifs present in the highly conserved region. Proc Natl Acad Sci USA 95, 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baminger B, Ludwiczek ML, Hoffmann B, Kontaxis G, Bister K & Konrat R (2004) Backbone assignment of the dimerization and DNA‐binding domain of the oncogenic transcription factor v‐Myc in complex with its authentic binding partner Max. J Biomol NMR 30, 361–362. [DOI] [PubMed] [Google Scholar]
- 34. El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Hall‐Ponsele E, Platonova O, Vyas P, Patient R, Porcher C & Mancini EJ (2013) Structural basis for LMO2‐driven recruitment of the SCL:E47bHLH heterodimer to hematopoietic‐specific transcriptional targets. Cell Rep 4, 135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu J, Banerjee A & Goss DJ (2005) Assembly of b/HLH/z proteins c‐Myc, Max, and Mad1 with cognate DNA: importance of protein−protein and protein−DNA interactions. Biochemistry 44, 11855–11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT et al (2017) SMARCB1 is required for widespread BAF complex‐mediated activation of enhancers and bivalent promoters. Nat Genet 49, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ & Roberts CWM (2017) The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romero OA, Verdura S, Torres‐Diz M, Gomez A, Moran S, Condom E, Esteller M, Villanueva A & Sanchez‐Cespedes M (2017) Sensitization of retinoids and corticoids to epigenetic drugs in MYC‐activated lung cancers by antitumor reprogramming. Oncogene 36, 1287–1296. [DOI] [PubMed] [Google Scholar]
- 39. Yan L, Xie S, Du Y & Qian C (2017) Structural insights into BAF47 and BAF155 complex formation. J Mol Biol 429, 1650–1660. [DOI] [PubMed] [Google Scholar]
- 40. Plowright AT, Ottmann C, Arkin M, Auberson YP, Timmerman H & Waldmann H (2017) Joining forces: the chemical biology‐medicinal chemistry continuum. Cell Chem Biol 24, 1058–1065. [DOI] [PubMed] [Google Scholar]
- 41. Huang B & Schroeder M (2006) LIGSITE csc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct Biol 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mori T, Kikuchi E, Watanabe Y, Fujii S, Ishigami‐Yuasa M, Kagechika H, Sohara E, Rai T, Sasaki S & Uchida S (2013) Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. Biochem J 455, 339–345. [DOI] [PubMed] [Google Scholar]
- 43. Scott DE, Ehebauer MT, Pukala T, Marsh M, Blundell TL, Venkitaraman AR, Abell C & Hyvonen M (2013) Using a fragment‐based approach to target protein‐protein interactions. Chembiochem 14, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sammak S & Zinzalla G (2015) Targeting protein‐protein interactions (PPIs) of transcription factors: challenges of intrinsically disordered proteins (IDPs) and regions (IDRs). Prog Biophys Mol Biol 119, 41–46. [DOI] [PubMed] [Google Scholar]
- 45. von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI et al (2003) The F‐box protein Skp2 participates in c‐Myc proteosomal degradation and acts as a cofactor for c‐Myc‐regulated transcription. Mol Cell 11, 1189–1200. [DOI] [PubMed] [Google Scholar]
- 46. Fung HYJ, Birol M & Rhoades E (2018) IDPs in macromolecular complexes: the roles of multivalent interactions in diverse assemblies. Curr Opin Struct Biol 49, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, Olejniczak ET, Clark T, Dey S, Lorey S et al (2015) Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol Cell 58, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang H, Arighi CN, Ross KE, Ren J, Li G, Chen SC, Wang Q, Cowart J, Vijay‐Shanker K & Wu CH (2018) iPTMnet: an integrated resource for protein post‐translational modification network discovery. Nucleic Acids Res 46, D542–D550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sabò A & Amati B (2014) Genome recognition by MYC. Cold Spring Harb Perspect Med 4, a014191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lorenzin F, Benary U, Baluapuri A, Walz S, Jung LA, von Eyss B, Kisker C, Wolf J, Eilers M & Wolf E (2016) Different promoter affinities account for specificity in MYC‐dependent gene regulation. Elife 5, e15161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peukert K, Staller P, Schneider A, Carmichael G, Hanel F & Eilers M (1997) An alternative pathway for gene regulation by Myc. EMBO J 16, 5672–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fieber W, Schneider ML, Matt T, Krautler B, Konrat R & Bister K (2001) Structure, function, and dynamics of the dimerization and DNA‐binding domain of oncogenic transcription factor v‐Myc. J Mol Biol 307, 1395–1410. [DOI] [PubMed] [Google Scholar]
- 53. Bax A, Ikura M, Kay LE, Barbato G & Spera S (1991) Multidimensional triple resonance NMR spectroscopy of isotopically uniformly enriched proteins: a powerful new strategy for structure determination. Ciba Found Symp 161, 108–119; discussion 119–35. [DOI] [PubMed] [Google Scholar]
- 54. Cornilescu G, Delaglio F & Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13, 289–302. [DOI] [PubMed] [Google Scholar]
- 55. Neri D, Szyperski T, Otting G, Senn H & Wuthrich K (1989) Stereospecific nuclear magnetic resonance assignments of the methyl groups of valine and leucine in the DNA‐binding domain of the 434 repressor by biosynthetically directed fractional 13C labeling. Biochemistry 28, 7510–7516. [DOI] [PubMed] [Google Scholar]
- 56. Brunger AT (2007) Version 1.2 of the crystallography and NMR system. Nat Protoc 2, 2728–2733. [DOI] [PubMed] [Google Scholar]
- 57. Williamson MP (2013) Using chemical shift perturbation to characterise ligand binding. Prog Nucl Magn Reson Spectrosc 73, 1–16. [DOI] [PubMed] [Google Scholar]
- 58. Kabsch W (2010) XDS. Acta Crystallogr D 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Emsley P & Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 61. Adams PD, Grosse‐Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK & Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D 58, 1948–1954. [DOI] [PubMed] [Google Scholar]