

Abstract
Non-Hodgkin’s lymphoma (NHL) is the sixth-most common cancer in the UK, accounting for around 13,700 new cases every year. Until the late 1990s, treatment relied on intensive chemotherapy, such as CHOP (cyclophosphamide–doxorubicin HCl–vincristine [Oncovin]–prednisone). The use of standard CHOP therapy and its variations had resulted in poor five-year survival rates (as low as 26%), particularly in patients with aggressive NHL. Rituximab (Rituxan) was the first chimeric (mouse/human) monoclonal antibody approved for the treatment of NHL. It was approved by the US Food and Drug Administration in 1997 for indolent forms of NHL. It subsequently received EU approval in June 1998, and was licensed under the trade name Mabthera (Roche, Basel, Switzerland). It then went on to be approved for the first-line treatment of aggressive forms of NHL, such as diffuse large B-cell lymphoma (to be used in combination with CHOP or other anthracycline-based chemotherapy) in 2006. It is directed against the CD20 protein, an antigen found on the surface of B-cell lymphomas. With minimal toxicity, activity as a single-agent (for indolent forms of NHL) and safety when combined with chemotherapy (for aggressive forms), it represents great progress in this field. Here, we analyze how this antibody therapeutic was developed from basic molecular and cellular considerations through to preclinical and clinical evaluations and how it came to be a first-line treatment for NHL, and we discuss the impacts the advent of rituximab had on treatment outcomes for patients with DLBCL compared with the pre-rituximab era.
Keywords: non-Hodgkin’s lymphoma, rituximab, monoclonal antibody, B cell
Introduction
The lymphatic system is comprised of a network of vessels that carry lymph fluid containing lymphocytes. These are crucial cells of the adaptive immune system, accounting for 30% of the total white blood cells in the adult circulatory system. Lymphocytes are categorized into T cells and B-cells. In the bone marrow, pluripotent hematopoietic stem cells differentiate into either common lymphoid progenitor cells or myeloid stem cells. Common lymphoid stem cells further differentiate into B-cell, T-cell, and NK-cell lineages.1
In the adaptive immunoresponse, B and T cells are reliant on each other: B cells are responsible for the production of antibodies and function in humoral immunity, whereas T cells are responsible for cell-mediated immunity. In response to a foreign antigen expressed from a pathogen, T cells are activated when the T-cell receptor binds to the antigen-presenting cell via major-histocompatibility-complex glycoproteins. This causes the rapid secretion of cytokines called interleukins that promote the differentiation of B cells into antibody-secreting plasma cells. Antibodies (or immunoglobulins) are directed against the foreign antigen expressed on the pathogen, and are able to block the adhesion of pathogens to the human host cells and enable effector mechanisms that help to prevent the spread of the pathogen.1
The antigens themselves are a series of between five and 15 amino acids that form antigenic determinants (epitopes). An antibody is described as monoclonal when it specifically targets a single epitope, as opposed to more than one. If the antibody targets more than one epitope, then the antibody is polyclonal.2 An antibody molecule is composed of four polypeptide chains – two identical heavy chains and two identical light chains – forming a characteristic Y shape. Light and heavy chains are divided into variable and constant domains. The variable domains of the light (VL) and heavy (VH) chains have a high diversity in amino-acid sequences and determine recognition and specificity of the antibody, whereas the constant domains of the light (CL) and heavy (CH) chains also have functions in Fc-receptor binding for phagocytosis.3
Each B cell (and T cell) is specific for a particular antigen, and hence the diversity of B cells is extraordinary. When activated in response to an antigen, each B cell also has the capability of producing 107–108 antibody molecules. Normally, once the foreign antigen has been neutralized, this antibody production is terminated. When this does not occur, the relentless proliferation of a specific B cell, which may be due to the accumulation of multiple genetic changes, environmental factors, and infectious factors, may result in a cancerous tumor known as a B-cell lymphoma.4
The concept of B-cell-depletion therapy with monoclonal antibodies (MoAbs) is that in essence, antibodies that are specific to a surface antigen of B cells are administered into a patient with a B-cell lymphoma. The antibodies will bind to the surface antigen of the malignant (and normal) B cells and lead to the depletion of B cells and thus destroy the tumor.
Non-Hodgkin’s lymphoma and treatment in the prerituximab era
Five percent of all newly diagnosed malignancies are lymphomas. There are two main types of lymphoma: Hodgkin’s lymphoma and non-Hodgkin’s lymphoma (NHL). These can be distinguished using pathological histology, and initial diagnosis of lymphoma is based on the presence or absence of the Reed–Sternberg cells. If these are present, the lymphoma is classified as Hodgkin’s. If absent, the lymphoma is classified as NHL.5 Ninety percent of lymphomas are accounted for by NHL.6
Globally, it was estimated that NHL had been diagnosed in around 414,772 individuals and accounted for an estimated 215,074 deaths in 2012 alone.7 NHL is a broad term encompassing a range of lymphoproliferative malignancies, whereby the affected lymphocytes proliferate abnormally and accumulate in certain parts of the lymphatic system. It is the most common hematological cancer in adults, with around 13,700 patients being diagnosed in the UK every year.8 The most common symptom of NHL is a painless swelling, typically in a lymph node in the neck, armpits, or groin, and may be accompanied by other general symptoms, including fever, chest pain/pressure, unexplained weight loss, fever, night sweats, shortness of breath, and persistent fatigue. The symptoms depend on the type and location of the NHL, and may be symptomless until the cancer progresses to a more advanced stage.9
NHL can be categorized in a variety of ways. The World Health Organization developed a classification scheme in 1995, which has been updated periodically since its conception.10,11 This system is used worldwide, and is based on pathological, genetic, and clinical factors. It divides NHL according to the cell of origin: an immunophenotypic examination can be carried out to determine whether the NHL derives from B cells (85% of cases) or T cells and natural killer (NK) cells (15% of cases). Classification also considers their architecture (follicular or diffuse) and morphology (small or large cells), seen from pathological histology.12 NHLs can also be categorized according to how they behave clinically. NHL is low-grade or indolent when the lymphoma cells appear to divide slowly, meaning the NHL develops over a prolonged duration. Follicular lymphoma is the most common indolent NHL. On the other hand, NHL is high-grade or aggressive when the lymphoma cells appear to be dividing at a faster rate. The most common aggressive NHL is diffuse large B cell lymphoma (DLBCL).13
DLBCL in particular presents the highest burden on patients and health care providers in Western countries, being the most common high-grade form of NHL. Around a third of all adult lymphomas are of this type.14 As the name suggests, a diffuse growth pattern of larger-than-normal B-cell lymphocytes is observed from histological examination of a biopsy taken from a patient with this form of NHL.15
DLBCL may arise de novo or from the transformation of an indolent lymphoma into an aggressive type. The median age at presentation for DLBCL is 60 years, and if left untreated, death would result within a matter of months. However, if aggressive forms, such as DLBCL, are treated quickly, they have a more positive outlook in terms of curability than indolent NHLs, such as follicular lymphoma.16 DLBCL can be cured, especially if diagnosed at an early stage. However, >70% of patients with DLBCL present to the clinic at an advanced stage.17
The Ann Arbor staging system (I–IV) exists to define how far the disease has spread. Stage I is recognized as early disease, with a single group of lymph nodes being affected. In stage II, known as locally advanced disease, the lymphoma is present in two or more groups of lymph-node regions on one side of the diaphragm. Stage III, advanced disease, entails the lymphoma being above and below the diaphragm. In widespread disease, stage IV, the lymphoma is found in the bone marrow and may also be present in extralymphatic sites (such as the liver).9
Due to the fact that patients presenting with DLBCL are mainly elderly and at an advanced stage of disease, they are seen as a challenging group to treat. Treatment options before the advent of rituximab were severely limited. Depending on the extent of the disease, patients were often treated with chemotherapy or radiation. These conventional yet intense methods of treatment were associated with toxicity and lacked specific antitumor-targeted activity. The go-to treatment for younger and elderly patients with DLBCL was the CHOP regimen (cyclophosphamide–doxorubicin HCl–vincristine [Oncovin]–prednisone).18 CHOP is still used today for DLBCL treatment, but often in conjunction with rituximab (R-CHOP).
The use of standard CHOP therapy and its variations in the prerituximab era was able to achieve complete responses (CRs) in only 40%–50% of elderly patients. Three-year event-free and overall survival (OS) rates were as low as 30% and 35%–40%, respectively.19 Efforts to increase the efficacy of CHOP through the addition of other cytotoxic agents had failed: no significant improvements in disease free survival and OS were found. This may be due to the fact these agents could not be administered unless the cyclophosphamide and doxorubicin dosages were lowered to below that given in the standard CHOP treatment.18,20 In addition, CHOP was more cost-effective and had less toxicity than the more complex CHOP-containing regimens.
It was also identified that altering the standard chemotherapy regimens to a higher degree of intensity could improve outcomes for young patients with poor prognoses, but elderly patients would not have been able to tolerate this intensity of treatment, so did not stand to benefit from it.21,22 Despite failed attempts to increase efficacy, CHOP continued to be the gold standard of DLBCL treatment.
As such, the best therapy option available until the 1990s was unable to cure >50% of patients with aggressive lymphomas (and most patients with low-grade lymphomas).23 This was coupled with the steadily rising incidence of NHL throughout the 1970s and 1980s, particularly in the elderly. By the 1990s, NHL accounted for one in 30 cases of and one in 40 deaths from cancer, with around 2,400 male and 2,200 female deaths.24 Therefore, there was a desperate need for new approaches to treatments with different mechanisms of action and improved toxicity profiles. Here, we examine how rituximab was developed and came to be a first-line treatment for B-cell NHLs, such as DLBCL, and discuss the impact it had on treatment outcomes.
Monoclonal antibody therapy in the prerituximab era
MoAb development began in 1975 by Köhler and Milstein with the use of hybridoma technology, whereby they were able to produce mouse (murine) MoAbs indefinitely from a single clone of B cells in vitro.25 The first therapeutic use of a murine MoAb against lymphoma-associated antigen in a patient (that was not responding to standard chemotherapies) took place in 1980, which was tolerated well without any significant side effects and caused transient clearance of tumor cells from the patient’s circulation.26
It was found that the unique immunoglobulin heavy- and light-chain variable segment (idiotype) of each lymphoma can be exploited as a tumor-specific marker in the patient. Following the preliminary trial carried out by Nadler et al, Miller et al hypothesized and tested a personalized approach to lymphoma therapy that would utilize (murine) MoAbs specific for the unique B-cell receptor expressed on a patient’s lymphoma cells that aimed to eradicate lymphoma cells, but spare healthy cells. This approach is called anti-idiotype therapy.27 Temporary reduction in tumor sizes was achieved in this trial, but immunoresponses to the murine antibody limited the clinical efficacy, as once the immunoresponse had begun, further infusions of antibody failed to reach the tumor. In a further trial, partial clinical responses were achieved in lymphoma patients using high doses (>2 g) of 1F5 (a murine anti-CD20 MoAb). The responses were dose-dependent, and so the higher doses resulted in a higher reduction in lymphoma.28
Although studies by Nadler et al, Miller et al and Press et al established that MoAb use was safe and could lead to a marked antitumor effect, there were obvious limitations to the use of murine MoAbs, as they lacked human-effector functionality.26–28 This rendered them unable to mediate complement-dependent cell lysis in the presence of human complement or lyse target cells by antibody-dependent cellular cytotoxicity (ADCC). As mentioned, immunogenicity was also an issue. When used therapeutically to treat human disease, murine MoAbs do not always have sustained effects.26–28 For instance, in a study by Press et al, therapeutic responses were described as “transient”, and 25% of patients developed a human antimouse-antibody response, leading to clearance of the murine MoAb.28
This antiantibody response limits the clinical efficacy of murine antibody therapy. This is the original rationale behind the generation of chimeric antibodies. A chimeric antibody is a recombinant protein, typically consisting of antigen-binding (Fab) regions from mouse genes and constant regions (Fc) from human genes. Rituximab is a high-affinity genetically engineered chimeric (human/mouse) MoAb. It is made from the binding regions from the original murine antihuman CD20, consisting of the variable regions of the Ig heavy and light chains that are fused with the human Igκ light-chain and γ1 heavy-chain constant regions. The Fc portion of human IgG1 was selected, as it is able to fix complement and activate ADCC.29
Rituximab, a chimeric MoAb, is directed against the CD20 antigen found on the surface of normal and malignant B lymphocytes. It was first approved by the US Food and Drug Administration (FDA) in 1997 and then by the European Medicines Agency in June 1998 for use in selective B-cell-depletion therapy for patients of relapsed, low-grade, or follicular NHL, and gained approval for use in combination with chemotherapies for aggressive forms of NHL in 2006.30,31 A schematic diagram of rituximab is shown in Figure 1.
Figure 1.
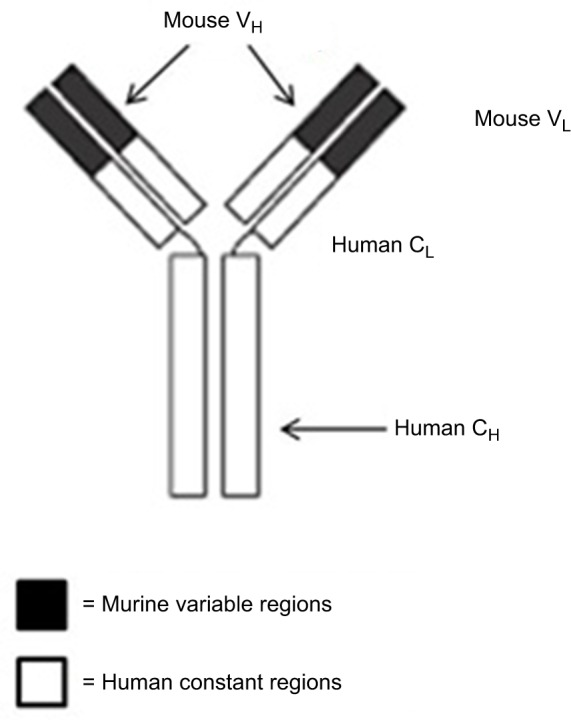
Structure of rituximab, a chimeric monoclonal antibody (~30% mouse origin and ~70% human origin).
Notes: The murine variable regions bind specifically to the CD20 antigen on malignant (as well as normal) B cells. The human constant regions allow human effector mechanisms.
Abbreviations: VL, variable domain (light chain); VH, variable domain (heavy chain); CL, constant domain (light chain); CH, constant domain (heavy chain).
CD20 target identification and validation
Selection of a target antigen is the first step in therapeutic MoAb development. The selection of this target requires extensive knowledge of biological processes involved in specific disease pathology. These processes can then be used to frame an appropriate development strategy to ascertain the mechanism of action, binding specificity, affinity, kinetics, and potency of the target antigen.32 Understanding the role of the antigen is not only important in disease pathology but also in determining if the target antigen has functional redundancy in other systems that may determine likely effects of the MoAb in clinical use.
To date, 86 types of NHL have been classified, each resulting from differing pathogeneses.33 From a molecular perspective, all subtypes have a common B-cell origin, with dysfunction of the B-cell-maturation process at differing stages, which are thereby responsible for the differences seen in each NHL subtype. Cell-surface proteins on B cells can be markers for differentiation and identification. The human B-lymphocyte-restricted differentiation antigen Bp35, later named CD20, is one example of such a marker, and was one of the first to be described.34,35
Biochemical studies were carried out to predict the structural features of CD20. Analysis of data provided by molecular cloning of CD20 revealed a hydrophobic protein of 35 kDa, with four transmembrane regions, intracellular termini, and the existence of an unglycosylated extracellular loop of around 43 residues between the third and fourth transmembrane regions.36–39 It is expressed during early pre-B-cell development, prior to the expression of cytoplasmic p-heavy chains.40 The exact functions have still not been fully confirmed, perhaps because CD20-knockout mice display almost normal phenotypes and because CD20 seems to have no natural ligand.41,42
However, in vitro studies using MoAbs to blockade CD20 resulted in the interruption of B-cell proliferation and differentiation, suggesting CD20 is involved in the cell cycle.39,43 A subsequent study had found that CD20 is implicated in the regulation of intracellular calcium.44 CD20 presented an ideal target for passive immunotherapy, due to its presence on 95% of B-cell lymphomas and nearly all normal B cells (apart from stem cells, pro-B cells, and plasma B cells).45 Other considerations that made it appealing included the accessibility and sensitivity of hematopoietic tumors to lysis by way of immunoeffector mechanisms, the fact it is not normally shed from the cell surface, that there are no detectable serum levels of soluble CD20 (so antibodies target the lymphomas and not free CD20 in solution), and the fact CD20 does not internalize after binding to an antibody.28,37 Rituximab binds to an epitope on the large loop, as shown. Residues important for rituximab recognition are Ala170 and Pro172.45,46
Proposed mechanisms of action
Rituximab binds to the CD20 antigen via its Fab domain, and the Fc domain recruits immunoeffector functions involved in B-cell lysis. The mechanisms of effector-mediated cell lysis that rituximab uses to achieve B-cell depletion in vivo are still not fully understood. However, evidence suggests it stabilizes CD20 on lipid rafts, which promotes ADCC. This is mediated by Fcγ receptors on the surface of granulocytes, macrophages, and NK cells. Other postulated mechanisms include complement-dependent cytotoxicity activated by C1q binding and the subsequent lytic cascade, as well as apoptosis.47,48 The proposed mechanisms of action through which rituximab acts are illustrated in Figure 2.
Figure 2.

The various mechanisms of action rituximab uses to kill B cells associated with NHL through binding of the CD20 antigen.
Note: Binding provokes one or more of the following mechanisms: CDC, ADCC, and/or apoptosis.
Abbreviations: NHL, non-Hodgkin’s lymphoma; CDC, complement-dependent cytotoxicity; ADCC, antibody-dependent cellular cytotoxicity.
Preclinical experiments
Here, we focus on the landmark preclinical study conducted by Reff et al in 1994.29 Their objective was to evaluate the chimeric anti-CD20 antibody binding in vitro. Subsequent in vivo pharmacology and toxicology evaluations aimed to provide data to monitor efficacy and safety parameters of the therapeutic so it could progress onto the clinical stage. The experiment was the first report of a MoAb being utilized in an immunotherapeutic context in the treatment of B-cell lymphoma. In their study, Reff et al identified a high-affinity murine anti-CD20 MoAb (murine MoAb 2B8) and constructed a mouse/human chimeric antibody that was expressed at high levels in mammalian cells. They named this construct C2B8. In vitro experiments confirmed the affinity, in vitro effector function, and immunoreactivity of the chimeric antibodies produced. Following this, in vivo experiments demonstrated marked B-cell depletion in peripheral blood (PB), bone marrow, and lymphatic tissue when the MoAb was administered to the subject animals – cynomolgus monkeys. No toxicity in any of the animals was detected.
Generating the high-affinity murine anti-CD20 MoAb
Reff et al immunized mice from the BALB/c strain weekly with the human lymphoblastoid cell line SB (containing the CD20 antigen) over a 3- to 4-month period. This stimulated the immunoresponse in mice to produce antibodies directed against the CD20 antigen. Mice that displayed high serum titers of anti-CD20 antibodies were identified with the use of known anti-CD20 antibodies. The spleens of said mice were then removed. Spleen cells have only a limited life span, so were fused with mouse myeloma SP2/0 to form a hybrid cell capable of indefinite survival and capable of secreting antibodies. As cell fusion is a random event with three types of hybrid cells being produced, the antibody-producing hybridoma cell is desired and needs to be selected for with a hypoxanthine–aminopterin–thymidine medium. As only hybridoma cells survive and proliferate in the selected medium, the supernatant was tested for the presence of the anti-CD20 antibody.29
Hybridomas were screened by coincubation with ‘251-Bl (IO ng) in 1% BSA, PBS, and 100,000 SB cells. Because the original cultures may have been started with more than one hybridoma cell, single cells from each antibody-positive culture were isolated and then subcultured. After incubation for 1 hour at room temperature, cells were harvested by transferal to 96-well filter plates and washed thoroughly. Duplicate wells containing unlabelled BI and wells containing no inhibiting antibody were used as positive and negative controls, respectively. The supernatant was tested once again for anti-CD20 antibodies. Each positive subculture – having been started from a single cell – represents a clone and its antibodies are monoclonal. That is, each culture secretes a single kind of antibody molecule directed against a single determinant on a preselected antigen. The positive clones provide a continuing source of anti-CD20 antibody. Wells containing >50% inhibition were expanded and cloned. The antibody showing the highest reactivity was derived from the cloned cell line designated 2B8.29
Construction of the chimeric anti-CD20 immunoglobulin DNA-expression vector
The next step Reff et al took was to construct an expression vector to include DNA coding for heavy- and light-chain constant regions from a human source and mouse variable regions from 2B8. This was done so the chimeric 2B8 MoAb could be expressed. The chosen expression vector was a plasmid that when introduced to a target cell would allow for large amounts of the anti-CD20 chimeric antibody protein to be produced using the cell’s own protein-synthesis mechanisms. Recombinant DNA technology was used to construct the expression vector. Tandem Chimeric Antibody Expression 8 (TCAE8) is a widely used vector for chimeric antibody expression, as it permits screening and analysis of the antibodies generated for characteristics, such as binding specificity and epitope-binding regions. As a result of this, the cDNA encoding the light- and heavy-chain variable regions from a preferred or desired anti-CD20 antibody was able to be incorporated into the vector.29
RNA was isolated from the 2B8 mouse hybridoma cell-producing anti-CD20 antibodies, and single-stranded cDNA to the RNA was prepared therefrom using the protocol described by Chomczynski and Sacchi.49 The murine variable region light-chain DNA was isolated and amplified from the cDNA using PCR. Specific DNA primers in the PCR react to isolate the murine variable light-chain region from the cDNA. This DNA fragment was subsequently cloned into the TCAE8 vector in front of the human κ-light-chain constant domain and sequenced. The murine heavy-chain variable region was also isolated using PCR and then cloned in front of the IgG1 constant domains in the plasmid vector.29
Creation of chimeric anti-CD20-producing CHO and SP2/0 transfectomas
The next step Reff et al took was to generate the chimeric anti-CD20-producing CHO and SP2/0 transfectomas.29 A transfectoma is a myeloma cell into which immunoglobulin genes have been transfected and expressed. First, Reff et al used a cotransfection assay for antibody-secreting clones by ELISA. CHO cells were grown in SSFM II in the absence of hypoxanthine and thymidine. Plasmid DNA was inserted into CHO cells by electroporation. Transfectoma clones positive for the secretion of chimeric Ig were identified by an ELISA specific for the human antibody.29
Colonies that produced the highest amount of immunoglobulin were expanded and plated into 96-well plates containing media plus methotrexate, fed every 2–3 days. Then, ELISA was carried out on the supernatants again and colonies producing the highest amount of immunoglobulin identified. Chimeric anti-CD20 antibody was purified from the supernatant of colonies producing the highest amounts using protein A-affinity chromatography. Purified chimeric anti-CD20 was analyzed by electrophoresis in polyacrylamide gels and estimated to be >95% pure. The affinity and specificity of the chimeric antibody were then compared to 2B8.29
Further experiments
After generating C2B8, Reff et al then carried out in vitro experiments to determine the affinity of C2B8, the specificity and immunoreactivity of C2B8, the in vitro effector function of C2B8, the ability of C2B8 to lyse B-lymphoid cell lines, and the ability of C2B8 to achieve ADCC.29
Determination of affinity of C2B8 using a direct and competitive binding assay
The affinity constant (Kap) for the chimeric antibodies was determined through carrying out a direct and competitive binding assay, whereby the direct binding of I125-radiolabeled chimeric anti-CD20 was compared to radiolabeled 2B8 with a Scatchard plot.29 Both types of antibody showed comparable affinity on a number of CD20-positive B-cell lines. Estimated Kap for CHO-produced chimeric anti-CD20 was 5.2×10−9 M and for SP2/0 (mouse myeloma)-produced antibody 7.4×10−9M. Estimated Kap for 2B8 was 3.5×10−9 M. The binding of MoAbs to their specific target antigen CD20 is an absolute requirement for their therapeutic efficacy, and so the fact that the chimeric antibody had comparable affinity to the murine antibody is promising, as having substantially the same specificity and binding capability as the murine anti-CD20 MoAb 2B8 shows therapeutic potential.29
Determination of immunological activity and specificity of C2B8 using radioimmunoassay
Both the specificity and retention of immunoreactivity of the chimeric antibodies produced by the CHO transfectoma were then confirmed using radioimmunoassay. The target antigen CD20 was present on 10,000 B-lymphoid cells. Direct competition by radioimmunoassay compared (unlabeled) C2B8’s ability to compete effectively with 2B8 (I125-labeled) for the CD20 antigen on 10,000 B-lymphoid cells.29 The results showed that substantial and equivalent amounts of chimeric anti-CD20 and 2B8 antibodies were required to produce 50% inhibition of binding of each to CD20 antigens on SB cells. It can be assumed that chimerization was the cause of the minimal loss in inhibition activity of the C2B8 antibodies.29
In vitro characterization of C2B8 using flow cytometry
After the C2B8 antibodies had been deemed to have sufficient affinity and retention of immunoreactivity, the in vitro effector function of C2B8 – specifically, whether C2B8 is able to bind to human C1q – was determined using whole-cell flow cytometry. The C1q complex is involved in the complement system, wherein C1q can bind to IgG (and IgM) when the antibody is bound to the antigen. When C1q is bound, the C1 complex initiates the classical complement pathway, which involves the activation of an enzymatic cascade that results in the assembly of a membrane-attack complex on the B-cell membrane that causes cell lysis.29
The assay used fluorescein-labeled C1q. Equal amounts of C2B8 antibody, human IgG1, K myeloma protein, and 2B8 antibody were incubated with an equivalent number of CD20-positive SB cells, which was followed by a washing step with FACS buffer to remove any unbound antibody. Incubation with fluorescein isothiocyanate-labeled C1q was then carried out. After 30–60 minutes of incubation, cells were then washed again. Analysis was carried out using a FACScan instrument.29
Results from the whole-cell flow cytometry showed evidence of a vast increase in fluorescence with the C2B8 antibodies. This is because the chimeric antibodies binding to the CD20 antigen on the SB cells have human Fc regions that are able to bind to human C1q, causing there to be increased fluorescence compared with the murine antibody. The murine antibody failed to activate complement, and produced the same pattern as the control (fluorescein isothiocyanate-labeled C1q; data not shown). This experiment confirmed that C2B8 had the ability to activate human complement in vitro, and is an important preclinical consideration.29
Complement-dependent cell lysing
Chimeric C2B8 antibodies were tested for their ability to lyse B-lymphoid cell lines in the presence of human serum as a source of complement.29 Labeled CD20-positive SB cells were incubated in the presence of equivalent amounts of human complement and equivalent amounts of either C2B8 or 2B8 antibodies. The results indicated that C2B8 antibodies were able to lyse approximately 50% of the SB target cells. In comparison, no significant lysis of SB cells was seen using HSB cells that contained no CD20 antigen. This was carried out as a negative control.29 The results showed the chimeric antibody produced far more lysis of B-lymphoid cell lines compared to the murine antibody. This is because murine MoAbs lack human-effector functionality and thus are not able to lyse human target cells through such mechanisms as ADCC or Fc-receptor-mediated phagocytosis.29
Antibody-dependent cellular cytotoxicity-effector assay
A potential mechanism used by therapeutic antibodies to kill their target cells is ADCC. The assay is end point-driven in that it analyses target-cell lysis.29 In this ADCC assay, SB and HSB cells were labeled, both with 51 Cr. Analysis was then performed using approved protocols. It was shown that there was substantial chimeric anti-CD20 antibody-dependent cell-mediated lysis of CD20-positive SB target cells at the end of an incubation period of 4 hours at 37°C. The ratio of effector cells:target was 100:1, where effector cells were human peripheral lymphocytes and the target the CD20 antigen.29
The results indicated that the chimeric anti-CD20 antibodies were immunologically active, as there was significant lysis, on approximately 50% of the antigen on SB cells. In comparison, under the same conditions, the murine anti-CD20 MoAb 2B8 had a statistically insignificant effect. The HSB (CD20-negative) cells were not lysed; this was carried out as a control.29
In vitro functional assays (eg, complement-dependent lysis, ADCC) are not sufficient to predict the in vivo capability of a chimeric antibody to destroy/deplete target cells expressing the specific antigen. A 1991 study also illustrated that chimeric mouse/human antibodies have undetectable ADCC activity.50 Therefore, the potential therapeutic efficacy of C2B8 needed to be assessed in vivo. Following the success of the in vitro experiments, monkeys were used as the animal of choice to test for the depletion of PB B lymphocytes.29
In vivo experiment: depletion of peripheral blood B lymphocytes
Reff et al carried out an experiment to ascertain the effect of various doses of immunologically active C2B8 needed for effective depletion of PB B lymphocytes in a nonhuman primate system.29 Cynomolgus monkeys were administered various doses of C2B8 – 0.04, 0.4, 1.6, and 6.4 mg/kg – daily for a period of 4 days, which resulted in a total dose range of 0.04–6.4 mg/kg.29 B-lymphocyte depletion is considered a sufficient indication of C2B8 efficacy. These data showed that there was effective and rapid depletion of B cells in PB where the antibody was in excess, regardless of single- or multiple-dosage levels. Following the final injection, depletion was observed for at least 7 days. Partial recovery of B cells was seen by day 21.29
Clinical evaluations of C2B8
Here, we discuss the key clinical trials that led to the initial approval of rituximab by the FDA in 1997.30
Phase I clinical trial of C2B8: single-dose therapy study
The first phase I clinical trial of single-dose infusions with the chimeric anti-CD20 antibody in patients with relapsed B-cell NHL was conducted in 1994 by Maloney et al following the promising results from Reff et al’s preclinical study.23,29 Maloney et al administered single-dose IDEC-C2B8 (C2B8) to 15 patients who had histologically documented relapsed B-cell NHL. Patients had to have previously undergone at least one prior course of standard therapy. Three patients were treated with a single intravenous dose at each dose level. Doses were 10, 50, 100, or 250 mg/m2 MoAbs. Patients were then evaluated for infusion-related toxicity and effect on PB B cells, T cells, neutrophils, platelets, serum chemistry, Ig, and complement levels. Tumor biopsies were also taken from patients who had been administered doses of 50, 100, or 250 mg/m2 pretreatment and after 2 weeks of treatment, also being examined for antibody binding and B- and T-cell content. Antitumor activity was monitored in all patients.23 All patients completed the therapy. Toxicity ranged from none to severe in one patient. There was no toxicity relating to immunresponse.23 Maloney et al then proceeded to investigate the effect of C2B8 on circulating B cells, serum-antibody pharmacokinetics, and posttreatment biopsy specimens.23
Effect of C2B8 on circulating B cells
To establish the impact of C2B8 on B and T cells, Maloney et al used two-color flow cytometry to quantify the number of B cells in the PB before and after C2B8 therapy. B-cell antigens CD19 and surface Ig were not blocked by the binding of C2B8 to CD20, so these were used to identify B cells. Bound antibodies on B cells from infusion were identified by blocking the binding of a directly labeled anti-CD20 antibody.23 There was a dose-dependent, rapid, and specific depletion of B cells in all but one patient, particularly in those receiving doses >100 mg/m2 C2B8. These depletions endured for 1–>3 months in all except one patient.23
Serum-antibody pharmacokinetics
For serum-antibody concentrations to be determined, ELISA was used. This was based on the capture of C2B8 by poly-clonal antibodies directed against its idiotype.23 Maloney et al reported that antibody levels >10 µg/mL persisted for >14 days in the serum of six of nine patients receiving a single dose of C2B8 (100, 250, or 500 mg/m2). The average half-life of the antibody was 4.4 days, ranging from 1.6 to 10.5 days. It can be concluded that a higher dosage of chimeric antibody leads to a higher serum concentration maintained in vivo, so C2B8 efficacy is influenced by pharmacokinetic factors. The wide range of half-lives may reflect the variable tumor burden among patients. It may also suggest that complex pharmacokinetics were involved, and the factors influencing clearance of 2B8 were largely unknown at the time. The study of serum-antibody pharmacokinetics was limited here, due to the small number of patients used in this trial. Maloney et al also noted it is difficult to establish the half-life of the antibody with patients who have varying degrees of tumor burden and have only received a single nonsaturating dose of antibody.23
Analysis of posttreatment tumor-biopsy specimens
Although patients received only a single nonsaturating dose of C2B8, promising clinical effects were observed in some patients. One such patient achieved partial remission.23 Computed tomography images were taken from a patient with relapsed follicular lymphoma who was treated with 500 mg/m2 C2B8. Images were taken of an abdominal mass pretherapy and 3 months after treatment.23 Regression (shrinkage) of the tumor was seen after treatment.23
In conclusion, the clinical data from this single dose-escalating study showed that C2B8 resulted in no dose-limiting toxicity and demonstrated clinical activity. Antibody infusion caused CD20-positive B-cell depletion in PB at 24–72 hours, continuing for 2–3 months in the majority of patients.23 This trial utilized single-dose infusions of antibody, which is a limitation. This is because a single dose is unlikely to reach a saturating level. It was noted that there was a larger collection of B cells within the lymphomas than in the PB, so it was a possibility the C2B8 could not access the lymphoma cells sufficiently. Maloney et al addressed this in a phase I multiple dose-escalation trial.51
Phase I clinical trial of C2B8: multiple-dose therapy study
Maloney et al further confirmed the safety, pharmacokinetics, and therapeutic effects of C2B8 that they had previously indicated in their previous trial, this time with multiple doses.51 Multiple infusions of C2B8 (125, 250, or 375 mg/m2) were administered to 20 patients with relapsed B-cell lymphoma. In this trial, serum levels of C2B8 were seen to be proportional to the antibody dose infused. It was also observed that higher, more sustained serum levels were attained following multiple doses compared with single doses. Infusion side effects during the initial infusion were generally mild.51 The weekly 375 mg/m2 dose for 4 weeks was selected for further phase II evaluation, and it remains the standard single-agent dose and schedule today.
Phase II clinical trial of C2B8: multiple-dose therapy study
The success of phase I clinical trials boded well for progression of C2B8 onto phase II. McLaughlin et al conducted the pivotal phase II trial that led to FDA approval.30,52,53 It is interesting to note that FDA approval for drugs is normally granted after phase III clinical trials, which are a final confirmation of safety and efficacy.54 However, in this case approval was granted earlier given the nature of the disease.
In this multi institutional trial of C2B8, 166 patients with relapsed low-grade or follicular lymphoma received an intravenous treatment course of C2B8 at a dose of 375 mg/m2 weekly for four doses.52 Of the 166 patients in the intent-to-treat group, there was a 48% overall response rate. Of these, 6% were CRs and the remainder – 42% of patients – achieved partial responses (PRs). The requirement for a CR was the resolution of all symptoms and signs of lymphoma, including bone-marrow clearing, for at least 28 days. For a response to be classified as a PR, ≥50% decrease in the sum of the products of perpendicular measurements of lesions was required, and for there to be no evidence of any progressive disease for a minimum of 28 days.52
Patients who achieved an overall decrease (<50%) in measurable disease were still nonresponders in this study. It is notable that of those who did not achieve a CR or PR, most achieved some net decrease in measurable disease, with a mean decrease of 32% from the nonresponders.52 The median follow-up of patients was 11.8 months. This was the time between the last treatment of C2B8 in patients and the time that data on their outcomes were collected. For patients that were responders, the projected median time to progression was 13.0 months. This was the length of time from the start of C2B8 treatment until the disease started to spread. The pharmacokinetics of the serum antibody and adverse effects were two other major parameters monitored in this trial.52
McLaughlin et al observed that serum-antibody levels were sustained for longer after the fourth infusion than after the first, due to the accumulation of C2B8. Patients who were responders and/or had a lower tumor burden were more likely to accumulate higher serum-antibody concentrations than nonresponders. Nonresponders displayed rapid clearance of the antibody after each infusion. This meant that accumulation to a saturating level could not occur and so there was less B cell depletion, which resulted in a lower decrease in measurable disease.52
Most adverse effects experienced by patients were mild (grade 1). Common adverse effects were fever and chills, mainly experienced after the first infusion. However, 55% of patients experienced no adverse effects/toxicity after the first infusion or for the remainder of the treatment period. Only 3% of patients experienced life-threatening toxicity (grade 4), and only one patient had a microangiopathic hemolytic anemic response to the chimeric antibody.52 Severe and rapid depletion of CD20-positive B cells from the circulation in this therapy would have created anxiety about its possible impacts on immunofunction. If used on a long-term basis, patients would be chronically depleted of B cells and the predicted consequences would be that these patients would have a higher risk of infectious complications. However, the normal B-cell population recovers around 6–9 months following the termination of therapy.52
FDA approval
Initial approval by the FDA was given to Genentech (San Francisco, CA, USA) for the manufacture of IDEC-C2B8 by IDEC Pharmaceuticals. Approval was given for the treatment of relapsed or refractory low-grade or follicular B-cell NHL in November 1997, based on McLaughlin et al’s pivotal phase II trial.30,52 IDEC-C2B8 was renamed rituximab and also assigned the trade names Rituxan and Mabthera.
According to the FDA, guidelines at the time stipulate that in the refractory cancer setting, nonrandomized trials with therapies that show evidence of tolerable treatment toxicity and are reasonably likely to predict a clinical benefit may be approved under accelerated-approval regulations.53 This was the case for rituximab, so McLaughlin et al were permitted to submit the results of their study following FDA approval.30 This also paved the way for further evidence to be gathered to confirm the clinical benefit of rituximab in aggressive forms of NHL.31
Impact on treatment of DLBCL
Currently, we have discussed only the treatment of indolent, low-grade NHL. However, we now consider DLBCL, the most common form of high-grade NHSL. Previously, DLBCL treatment had been severely restricted to CHOP before the advent of rituximab. However, its use had resulted in poor 5-year survival rates for patients with aggressive NHL – as low as 26%.55 Rituximab approval was granted by the FDA for first-line treatment of DLBCL in combination with CHOP or other anthracycline-based chemotherapy regimens in February 2006.31 Approval was granted based on the results of a successful phase III randomized clinical trial comparing R-CHOP to CHOP alone in aggressive lymphomas.
Coiffier et al undertook the first randomized study to compare outcomes with R-CHOP and CHOP in patients with DLBCL in 2002.56 In this, 399 elderly patients 60–80 years old with DLBCL who had been previously untreated and had a poor prognosis were recruited. These patients were randomized into a CHOP (197 patients) or an R-CHOP arm (202 patients). Those in the CHOP arm received eight cycles of the standard dose of CHOP every 3 weeks, while those in the R-CHOP arm received eight cycles of the standard dose of rituximab (375 mg/m2) in combination with CHOP on day 1 of each cycle.56 The CR rate was statistically much higher in the group that received R-CHOP than in the group receiving CHOP alone (76% vs 63%, P=0.005). The median follow-up was 2 years, and both progression-free survival and OS were longer for patients receiving R-CHOP (P<0.001 and P=0.007, respectively). The OS of patients was analyzed with the log-rank test, which calculates a χ2-value for each arm. Log-rank results were used to calculate a P-value, and as this was <0.05, the differences between OS in the R-CHOP and CHOP arms were statistically significant.57 After 2 years, 70% of patients who received R-CHOP were alive, whereas only 57% of patients who had been treated with CHOP alone survived.56 Two other key phase III trials were carried out by Pfreundschuh et al and Habermann et al in 2006.58,59 The results are summarized, along with the results from Coiffier et al’s study, in Table 1.56,58,59
Table 1.
Three key clinical trials with rituximab in combination with CHOP for treatment of aggressive B-cell lymphomas
| Patient population | Regimen | Overall survival | Progression-free survival | |
|---|---|---|---|---|
| Coiffier et al56 | n=399 Previously untreated Aged 60–80 years |
R-CHOP vs CHOP | 70% vs 57% | 57% vs 38% |
| Pfreundschuh et al58 | n=824 Previously untreated Aged 18–60 years |
R-CHOP-like chemotherapy vs CHOP-like chemotherapy | 93% vs 84% (P=0.0001) | 79% vs 59% (P<0.0001) |
| Habermann et al59 | n=632 Previously untreated Aged >60 years |
R-CHOP vs CHOP | Not reached | 53% vs 46% (P=0.04) |
Notes: Trials led to US Food and Drug Administration approval for its use in this context.56,58,59 Adapted from Dotan E, Aggarwal C, Smith MR. Impact of rituximab (Rituxan) on the treatment of B-cell non-Hodgkin’s lymphoma. P T. 2010;35(3):148–157.17
Abbreviations: CHOP, cyclophosphamide–doxorubicin HCl–vincristine [Oncovin]–prednisone; R-CHOP, rituximab with CHOP.
The results from these prospective trials were very promising, as the addition of rituximab to chemotherapy showed a clear benefit in patient outcomes. Until 2014, only a handful of population-based, retrospective analyses had been carried out that assessed rituximab’s impact on survival of patients with DLBCL. These register-based analyses provided data comparing the outcome of patients who had been administered CHOP or R-CHOP, and confirmed that R-CHOP had a positive effect.60–63 However, only patients with advanced stages of DLBCL were included in Sehn et al’s study, and important clinical parameters were lacking.60 Hasselblom et al included only very elderly patients, and Krause et al’s study lacked some clinical information.61,62
Mian et al carried out a real-life analysis of patients with DLBCL who were administered R-CHOP or CHOP. They aimed to evaluate the outcomes of DLBCL patients in the pre- and postrituximab era.64 Between 1999 and 2012, 219 DLBCL patients aged 18–90 years (median 63 years) were investigated: 56% had stage III/IV DLBCL and 26% had a poor prognostic index (a clinical tool to predict prognosis of aggressive NHLs). A total of 219 patients were in the R-CHOP arm and underwent six cycles of R-CHOP. The outcomes of these patients were compared to a historical cohort of 88 patients treated with six cycles of CHOP therapy: 64 of these patients were administered CHOP and 24 CHOP-like chemotherapy alone between 1988 and 2000.64 The median follow-up was 3.5 years, and results showed that the 5-year estimated progression-free survival for the R-CHOP and CHOP arms was 56% and 44% (P=0.002) respectively. Five-year OS was significantly longer for the R-CHOP arm (69%) compared to 40% for the CHOP arm (P<0.001).64 Therefore, there was strong conclusive evidence to confirm that rituximab had an independent impact on the outcomes of patients with DLBCL when combined with CHOP-like therapies compared with CHOP alone. The results from Mian et al’s study were consistent with data published previously.60,63,64
Final remarks
Here, we have largely discussed rituximab in the treatment of DLBCL. However, it has also been shown to improve the prognosis of all B-cell NHL. A meta-analysis of 2,315 patients in seven trials showed that rituximab improved progression-free survival in follicular lymphoma, but the effect on OS was inconclusive.65 Mantle-cell lymphoma is characterized by overexpression of cyclin D1, due to a t(11:14) chromosome translocation. A 2015 meta-analysis of seven studies showed that rituximab improved overall and CR rates in patients with mantle-cell lymphoma.66 However, the small (three) number of randomized clinical trials and the high risk of observational bias in these largely homogeneous studies are limitations to these findings. Finally, a meta-analysis of 13 studies with 237 patients evaluated the efficacy and safety of rituximab treatment in marginal zone lymphoma, showing an overall response rate of 81% and a CR rate of 50%.67
However, although rituximab has changed the treatment paradigms and outcomes for B-cell lymphomas, there are still drawbacks to its use. The cost per course of rituximab infusion is £10,634.95, based on eight cycles at a fixed dose of 1,400 mg per cycle, and thus is very costly.68 The majority of indolent NHLs will eventually relapse and are incurable, driving the search for improved methods of treatment. In 2017, subcutaneous rituximab was approved by the FDA to treat follicular lymphoma and DLBCL following favorable trials.69,70 Toxin-conjugated anti-CD20 therapy initially showed higher response rates as the toxic payload was delivered directly to B-cell malignancies, but was eventually pulled from the market, due to poor sales.71 Current work in this field includes rituximab–doxorubicin therapy; however, this has not been approved by the FDA.72 Furthermore, additional therapeutic anti-CD20 MoAbs have been generated featuring modifications including an alternate binding epitope, additional humanization, or altered glycosylation. Ofatumumab was the first fully humanized MoAb, with unique binding more proximal to the cell membrane, allowing greater complement-dependent cytotoxicity activity compared to rituximab;73 however, it is given at substantially higher doses than rituximab, making a direct comparison of efficacy difficult.
Conclusion
Over 20 years of clinical use later, the R-CHOP chemotherapy (every 14 or 21 days) remains a first-line treatment for DLBCL, and it is probable that it will remain an integral component of therapy regimens. The clear impact rituximab has had on treatment outcomes for B-cell NHLs cannot be disputed. It is safe and well tolerated in all age-groups. Prognosis of DLBCL has improved significantly in the last decade due to these treatments being implemented, although earlier or more precise diagnosis also plays a role. While the advent of rituximab has been one of the most significant milestones in treating lymphoproliferative disorders and most patients will be cured following first-line R-CHOP, there is still room for improvement. Relapse is still an issue for DLBCL patients: over 30% of patients fail to respond to current treatment regimens or suffer relapse.
With improved understanding of the biology of DLBCL in regard to the malignant B cell of origin, the reason that some patients are not cured with current regimes can be inferred. For example, the Lymphoma/Leukemia Molecular Profiling Project discovered that the ABC subtype of DLBCL was associated with much poorer prognosis than other DLBCL subtypes.74 The ABC subtype is associated with upregulated transcriptional activity of the NFκB pathway, which suppresses the effects of cytotoxic therapy regimes in inducing apoptosis (such as R-CHOP). By inhibiting the NFκB pathway with bortezomib in combination with R-CHOP, clinical benefits could be investigated, and clinical trials are currently under way. Unfortunately, recent phase II trials show no significant benefit from the addition of bortezomib to R-CHOP.75
Footnotes
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S3–S23. doi: 10.1016/j.jaci.2009.12.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. ILAR J. 2005;46(3):258–268. doi: 10.1093/ilar.46.3.258. [DOI] [PubMed] [Google Scholar]
- 3.Parham P. Antibody Structure and the Generation of B-Cell Diversity. In: Parham P, editor. The Immune System. 3rd ed. New York: Garland Science, Taylor & Francis Group; 2009. pp. 95–121. [Google Scholar]
- 4.Rui L, Goodnow CC. Lymphoma and the control of B cell growth and differentiation. Curr Mol Med. 2006;6(3):291–308. doi: 10.2174/156652406776894563. [DOI] [PubMed] [Google Scholar]
- 5.Mani H, Jaffe ES. Hodgkin lymphoma: an update on its biology with new insights into classification. Clin Lymphoma Myeloma. 2009;9(3):206–216. doi: 10.3816/CLM.2009.n.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The Leukemia & Lymphoma Society The lymphoma guide. Leuk Lymphoma Soc. 2013:1–48. [Google Scholar]
- 7.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2014;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 8.Non-Hodgkin lymphoma statistics | Cancer Research UK. [Accessed August 24, 2018]. Available from: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/non-hodgkin-lymphoma.
- 9.Gallagher CJ, Lister TASM. Malignant disease. In: Kumar P, Clark M, editors. Kumar and Clark’s Clinical Medicine. Eighth Edi Edinburgh: Saunders/Elsevier; 2012. pp. 461–469. [Google Scholar]
- 10.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swerdlow SH, Campo E, Harris NL. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. World Health Organsiation; 2008. [Google Scholar]
- 12.Frampas E. Lymphomas: basic points that radiologists should know. Diagn Interv Imaging. 2013;94(2):131–144. doi: 10.1016/j.diii.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Harrison AM, Thalji NM, Greenberg AJ, Tapia CJ, Windebank AJ. Rituximab for non-Hodgkin’s lymphoma: a story of rapid success in translation. Clin Transl Sci. 2014;7(1):82–86. doi: 10.1111/cts.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin lymphoma. Lancet. 2017;390(10091):298–310. doi: 10.1016/S0140-6736(16)32407-2. [DOI] [PubMed] [Google Scholar]
- 15.Mey U, Hitz F, Lohri A, et al. Diagnosis and treatment of diffuse large B-cell lymphoma. Swiss Med Wkly. 2012;142(1):1–15. doi: 10.4414/smw.2012.13511. [DOI] [PubMed] [Google Scholar]
- 16.Survival | non-Hodgkin lymphoma | Cancer Research UK. [Accessed September 3, 2018]. Available from: https://www.cancerresearchuk.org/about-cancer/non-hodgkin-lymphoma/survival.
- 17.Dotan E, Aggarwal C, Smith MR. Impact of rituximab (Rituxan) on the treatment of B-cell non-Hodgkin’s lymphoma. P T. 2010;35(3):148–157. [PMC free article] [PubMed] [Google Scholar]
- 18.Fisher RI, Gaynor ER, Dahlberg S, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N Engl J Med. 1993;328(14):1002–1006. doi: 10.1056/NEJM199304083281404. [DOI] [PubMed] [Google Scholar]
- 19.Sonneveld P, De Ridder M, Van der Lelie H, et al. Comparison of doxorubicin and mitoxantrone in the treatment of elderly patients with advanced diffuse non-Hodgkin’s lymphoma using CHOP versus CNOP chemotherapy. J Clin Oncol. 1995;13(10):2530–2539. doi: 10.1200/JCO.1995.13.10.2530. [DOI] [PubMed] [Google Scholar]
- 20.Dixon DO, Neilan B, Jones SE, et al. Effect of age on therapeutic outcome in advanced diffuse histiocytic lymphoma: the southwest Oncology Group experience. J Clin Oncol. 1986;4(3):295–305. doi: 10.1200/JCO.1986.4.3.295. [DOI] [PubMed] [Google Scholar]
- 21.Zinzani PL, Storti S, Zaccaria A, et al. Elderly aggressive-histology non-Hodgkin’s lymphoma: first-line VNCOP-B regimen experience on 350 patients. Blood. 1999;94(1):33–38. [PubMed] [Google Scholar]
- 22.Coiffier B. What treatment for elderly patients with aggressive lymphoma? Ann Oncol. 1994;5(10):873–875. doi: 10.1093/oxfordjournals.annonc.a058722. [DOI] [PubMed] [Google Scholar]
- 23.Maloney DG, Liles TM, Czerwinski DK, et al. Phase I clinical trial using escalating single-dose infusion of chimeric anti-CD20 monoclonal antibody (IDEC-C2B8) in patients with recurrent B-cell lymphoma. Blood. 1994;84(8):2457–2466. [PubMed] [Google Scholar]
- 24.Cartwright R, Brincker H, Carli PM, et al. The rise in incidence of lymphomas in Europe 1985–1992. Eur J Cancer. 1999;35(4):627–633. doi: 10.1016/s0959-8049(98)00401-8. [DOI] [PubMed] [Google Scholar]
- 25.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 26.Nadler LM, Stashenko P, Hardy R, et al. Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen. Cancer Res. 1980;40(9):3147–3154. [PubMed] [Google Scholar]
- 27.Miller RA, Maloney DG, Warnke R, Levy R. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N Engl J Med. 1982;306(9):517–522. doi: 10.1056/NEJM198203043060906. [DOI] [PubMed] [Google Scholar]
- 28.Press OW, Appelbaum F, Ledbetter JA, et al. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood. 1987;69(2):584–591. [PubMed] [Google Scholar]
- 29.Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–445. [PubMed] [Google Scholar]
- 30.FDA Rituximab - Product Approval Information - Licensing Action. [Accessed August 24, 2018]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/1997/ritugen112697L.htm. Published 1997.
- 31.Two New Indications Approved for Rituximab in NHL | Cancer Network. The Cancer Network; [Accessed August 27, 2018]. Available from: http://www.cancernetwork.com/hematologic-malignancies/two-new-indications-approved-rituximab-nhl. Published 2006. [Google Scholar]
- 32.Mould DR, Meibohm B. Drug development of therapeutic monoclonal antibodies. BioDrugs. 2016;30(4):275–293. doi: 10.1007/s40259-016-0181-6. [DOI] [PubMed] [Google Scholar]
- 33.The Lymphoma Research Foundation . Non-Hodgkin lymphoma. The lymphoma Research Foundation; [Accessed August 27, 2018]. Available from: https://www.lymphoma.org/aboutlymphoma/nhl/ [Google Scholar]
- 34.Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol. 1980;125(4):1678–1685. [PubMed] [Google Scholar]
- 35.Clark EA, Shu G, Ledbetter JA. Role of the Bp35 cell surface polypeptide in human B-cell activation. Proc Natl Acad Sci USA. 1985;82(6):1766–1770. doi: 10.1073/pnas.82.6.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oettgen HC, Bayard PJ, van Ewijk W, Nadler LM, Terhorst CP. Further biochemical studies of the human B-cell differentiation antigens B1 and B2. Hybridoma. 1983;2(1):17–28. doi: 10.1089/hyb.1983.2.17. [DOI] [PubMed] [Google Scholar]
- 37.Einfeld DA, Brown JP, Valentine MA, Clark EA, Ledbetter JA. Molecular cloning of the human B cell CD20 receptor predicts a hydrophobic protein with multiple transmembrane domains. EMBO J. 1988;7(3):711–717. doi: 10.1002/j.1460-2075.1988.tb02867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamenkovic I, Seed B. Analysis of two cDNA clones encoding the B lymphocyte antigen CD20 (B1, Bp35), a type III integral membrane protein. J Exp Med. 1988;167(6):1975–1980. doi: 10.1084/jem.167.6.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tedder TF, Boyd AW, Freedman AS, Nadler LM, Schlossman SF. The B cell surface molecule B1 is functionally linked with B cell activation and differentiation. J Immunol. 1985;135(2):973–979. [PubMed] [Google Scholar]
- 40.Valentine MA, Meier KE, Rossie S, Clark EA. Phosphorylation of the CD20 phosphoprotein in resting B lymphocytes. Regulation by protein kinase C. J Biol Chem. 1989;264(19):11282–11287. [PubMed] [Google Scholar]
- 41.O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Mice carrying a CD20 gene disruption. Immunogenetics. 1998;48(2):125–132. doi: 10.1007/s002510050412. [DOI] [PubMed] [Google Scholar]
- 42.Uchida J, Lee Y, Hasegawa M, et al. Mouse CD20 expression and function. Int Immunol. 2004;16(1):119–129. doi: 10.1093/intimm/dxh009. [DOI] [PubMed] [Google Scholar]
- 43.Tedder TF, Forsgren A, Boyd AW, Nadler LM, Schlossman SF. Antibodies reactive with the B1 molecule inhibit cell cycle progression but not activation of human B lymphocytes. Eur J Immunol. 1986;16(8):881–887. doi: 10.1002/eji.1830160802. [DOI] [PubMed] [Google Scholar]
- 44.Bubien JK, Zhou LJ, Bell PD, Frizzell RA, Tedder TF. Transfection of the CD20 cell surface molecule into ectopic cell types generates a Ca2+ conductance found constitutively in B lymphocytes. J Cell Biol. 1993;121(5):1121–1132. doi: 10.1083/jcb.121.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tedder TF, Engel P. CD20: a regulator of cell-cycle progression of B lymphocytes. Immunol Today. 1994;15(9):450–454. doi: 10.1016/0167-5699(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 46.Polyak MJ, Deans JP. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood. 2002;99(9):3256–3262. doi: 10.1182/blood.v99.9.3256. [DOI] [PubMed] [Google Scholar]
- 47.Demidem A, Lam T, Alas S, Hariharan K, Hanna N, Bonavida B. Chimeric anti-CD20 (IDEC-C2B8) monoclonal antibody sensitizes a B cell lymphoma cell line to cell killing by cytotoxic drugs. Cancer Biother Radiopharm. 1997;12(3):177–186. doi: 10.1089/cbr.1997.12.177. [DOI] [PubMed] [Google Scholar]
- 48.Flieger D, Renoth S, Beier I, Sauerbruch T, Schmidt-Wolf I. Mechanism of cytotoxicity induced by chimeric mouse human monoclonal antibody IDEC-C2B8 in CD20-expressing lymphoma cell lines. Cell Immunol. 2000;204(1):55–63. doi: 10.1006/cimm.2000.1693. [DOI] [PubMed] [Google Scholar]
- 49.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 50.Robinson RR, Chartier J, Chang CP, Horwitz AH, Better M. Chimeric mouse-human anti-carcinoma antibodies that mediate different anti-tumor cell biological activities. Human Antibodies. 1991;2(2):84–93. [PubMed] [Google Scholar]
- 51.Maloney DG, Grillo-López AJ, Bodkin DJ, et al. IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J Clin Oncol. 1997;15(10):3266–3274. doi: 10.1200/JCO.1997.15.10.3266. [DOI] [PubMed] [Google Scholar]
- 52.Mclaughlin P, Grillo-López AJ, Link BK, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. 1998;16(8):2825–2833. doi: 10.1200/JCO.1998.16.8.2825. [DOI] [PubMed] [Google Scholar]
- 53.FDA Guidance for Industry FDA Approval of New Cancer Treatment Uses for Marketed Drug and Biological Products Guidance for Industry FDA Approval of New Cancer. 1998. [Accessed August 24, 2018]. Available from: http://www.fda.gov/cber/guidelines.htm.
- 54.Van Norman GA. Drugs, devices, and the FDA: Part 1: an overview of approval processes for drugs. JACC Basic to Transl Sci. 2016;1(3):170–179. doi: 10.1016/j.jacbts.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.International Non-Hodgkin’s Lymphoma Prognostic Factors Project A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med. 1993;329(14):987–994. doi: 10.1056/NEJM199309303291402. [DOI] [PubMed] [Google Scholar]
- 56.Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–242. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- 57.Dudley WN, Wickham R, Coombs N. An introduction to survival statistics: Kaplan-Meier analysis. J Adv Pract Oncol. 2016;7(1):91–100. doi: 10.6004/jadpro.2016.7.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pfreundschuh M, Trümper L, Österborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera international trial (mint) group. Lancet Oncol. 2006;7(5):379–391. doi: 10.1016/S1470-2045(06)70664-7. [DOI] [PubMed] [Google Scholar]
- 59.Habermann TM, Weller EA, Morrison VA, et al. Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. J Clin Oncol. 2006;24(19):3121–3127. doi: 10.1200/JCO.2005.05.1003. [DOI] [PubMed] [Google Scholar]
- 60.Sehn LH, Donaldson J, Chhanabhai M, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol. 2005;23(22):5027–5033. doi: 10.1200/JCO.2005.09.137. [DOI] [PubMed] [Google Scholar]
- 61.Hasselblom S, Stenson M, Werlenius O, et al. Improved outcome for very elderly patients with Diff use large B-cell lymphoma in the immunochemotherapy era. Leuk Lymphoma. 2012;53(3):394–399. doi: 10.3109/10428194.2011.616612. [DOI] [PubMed] [Google Scholar]
- 62.Krause SW, Gerken M, Andreesen R, Hofstädter F, Klinkhammer-Schalke M. Treatment of B cell lymphoma with chemotherapy plus rituximab: a survival benefit can be demonstrated in the routine data of a regional cancer registry. Ann Hematol. 2012;91(4):561–570. doi: 10.1007/s00277-011-1361-6. [DOI] [PubMed] [Google Scholar]
- 63.Lee L, Crump M, Khor S, et al. Impact of rituximab on treatment outcomes of patients with diffuse large B-cell lymphoma: a population-based analysis. Br J Haematol. 2012;158(4):481–488. doi: 10.1111/j.1365-2141.2012.09177.x. [DOI] [PubMed] [Google Scholar]
- 64.Mian M, Augustin F, Kocher F, et al. A success story: how a single targeted-therapy molecule impacted on treatment and outcome of diffuse large B-cell lymphoma. Anticancer Res. 2014;34(5):2559–2564. [PubMed] [Google Scholar]
- 65.Vidal L, Gafter-Gvili A, Salles G, et al. Rituximab maintenance improves overall survival of patients with follicular lymphoma-Individual patient data meta-analysis. Eur J Cancer. 2017;76:216–225. doi: 10.1016/j.ejca.2017.01.021. [DOI] [PubMed] [Google Scholar]
- 66.Vose JM. Mantle cell lymphoma: 2015 update on diagnosis, risk-stratification, and clinical management. Am J Hematol. 2015;90(8):739–745. doi: 10.1002/ajh.24094. [DOI] [PubMed] [Google Scholar]
- 67.Fan F, Wang W, Guanglun L, et al. Efficacy and safety of rituximab in marginal zone lymphoma: a meta-analysis of 13 studies. Int J Clin Exp Med. 2015;8(10):17515–17522. [PMC free article] [PubMed] [Google Scholar]
- 68.NICE Guidelines. Non-Hodgkin’s lymphoma: rituximab subcutaneous injection. 2014. [Accessed August 26, 2018]. Available from: https://www.nice.org.uk/advice/esnm46/chapter/full-evidence-summary.
- 69.Davies A, Berge C, Boehnke A, et al. Subcutaneous rituximab for the treatment of B-cell hematologic malignancies: a review of the scientific rationale and clinical development. Adv Ther. 2017;34(10):2210–2231. doi: 10.1007/s12325-017-0610-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Davies A, Merli F, Mihaljevic B, et al. Pharmacokinetics and safety of subcutaneous rituximab in follicular lymphoma (SABRINA): stage 1 analysis of a randomised phase 3 study. Lancet Oncol. 2014;15(3):343–352. doi: 10.1016/S1470-2045(14)70005-1. [DOI] [PubMed] [Google Scholar]
- 71.Green DJ, Press OW. Whither radioimmunotherapy: to be or not to be? Cancer Res. 2017;77(9):2191–2196. doi: 10.1158/0008-5472.CAN-16-2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin H, Meng T, Shu L, et al. Novel reduction-sensitive micellar nanoparticles assembled from Rituximab-doxorubicin conjugates as smart and intuitive drug delivery systems for the treatment of non-Hodgkin’s lymphoma. Chem Biol Drug Des. 2017;90(5):892–899. doi: 10.1111/cbdd.13010. [DOI] [PubMed] [Google Scholar]
- 73.Cang S, Mukhi N, Wang K, Liu D. Novel CD20 monoclonal antibodies for lymphoma therapy. J Hematol Oncol. 2012;5(1):64. doi: 10.1186/1756-8722-5-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 75.Leonard JP, Kolibaba KS, Reeves JA, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol. 2017;35(31):3538–3546. doi: 10.1200/JCO.2017.73.2784. [DOI] [PubMed] [Google Scholar]