

Summary
The genomic landscape of metastatic castration-resistant prostate cancer (mCRPC) differs from that of the primary tumor and is dynamic during tumor progression. The real-time and repeated characterization of this process via conventional solid tumor biopsies is challenging. Alternatively, circulating cell-free DNA (cfDNA) containing circulating tumor DNA (ctDNA) can be obtained from patient plasma using minimally disruptive blood draws and is amenable to sequential analysis. ctDNA has high overlap with the genomic sequences of biopsies from metastases and has the advantage of being representative of multiple metastases. The availability of techniques with high sensitivity and specificity, such as next-generation sequencing (NGS) and digital PCR, has greatly contributed to the development of the cfDNA field and enabled the detection of genomic alterations at low ctDNA fractions. In mCRPC, a number of clinically relevant genomic alterations have been tracked in ctDNA, including androgen receptor (AR) aberrations, which have been shown to be associated with an adverse outcome to novel antiandrogen therapies, and alterations in homologous recombination repair (HRR) genes, which have been associated with a response to PARP inhibitors. Several clinical applications have been proposed for cfDNA analysis, including its use as a prognostic tool, as a predictive biomarker, to monitor tumor response and to identify novel mechanisms of resistance. To date, the cfDNA analysis has provided interesting results, but there is an urgent need for these findings to be confirmed in prospective clinical trials.
Keywords: prostate cancer, liquid biopsy, cell-free DNA, circulating tumor DNA, biomarker
Introduction
Prostate cancer is one of the leading causes of cancer death in males in the western world1, which occurs following the development of castration resistance. However, metastatic prostate cancers show heterogeneity in their clinical behavior, ranging from slowly progressing disease with long-lasting responses to antiandrogen therapy to rapidly lethal tumors characterized by resistance to available therapies. With the increasing number of options for the treatment of advanced prostate cancer2,3, we hypothesize that further improvements in patient outcomes require molecularly directed treatments paired to clinically implementable biomarker tests. This approach requires a stepwise process from biomarker discovery and assay development to clinical qualification, first retrospectively and via the prospective analysis of retrospective collections and then, importantly, in prospective trials where the biomarker defines treatment randomization. The molecular landscape of metastatic castration-resistant prostate cancer has been characterized and includes several recurrent molecular pathways4,5 that are putatively involved in tumor progression and resistance. These pathways include AR mutations and amplifications, TP53 mutations and loss, DNA repair gene deletions and PTEN loss.
Intrapatient tumor heterogeneity has been associated with the reduced efficacy of systemic therapies in many forms of cancer6–8. Advanced prostate cancers are no exception; these cancers have been shown to be driven by multiple competing clones that display intra and intertumor heterogeneity9. Evolutionary diversity and clonal evolution might encounter several bottlenecks, including the development of metastasis and selective treatment pressure, that might limit the individual diversity of the main oncogenic drivers10. Treatment pressure induces the emergence of multiple clones either by selection or by divergent differentiation9,11–13 (Figure 1).
Figure 1. Prostate cancer is heterogeneous.

A. Interpatient heterogeneity. Every patient has different genomic drivers of prostate cancer progression and resistance. The drivers are represented separately for simplicity, but multiple drivers can be present simultaneously in the same patient. B. Intrapatient heterogeneity. Distant metastases are composed of different metastatic clones. Circulating cell-free DNA has an advantage over tumor biopsy to capture genomic events from distant clones that are driving tumor progression. mCRPC= metastatic castration-resistant prostate cancer; HR = homologous recombination; MMR = mismatch repair.
Taking a biopsy of a metastasis is the gold standard approach for analyzing the genomic landscape of advanced disease. However, biopsy is invasive, carries a risk of morbidity, and can be technically challenging in bone or deep lymph nodes, which are the most frequent locations of prostate cancer metastases3. In addition, although the individual diversity of most oncogenic drivers seems to be limited in advanced metastatic disease, a single metastatic tumor might not capture the total burden of molecular alterations. Most likely, due to these limitations, unlike metastases of other advanced tumors that are biopsied for therapeutic decision-making, recurrent prostate cancer metastases are not routinely biopsied in most centers. An ideal method for personalized therapy needs to be able to, in real-time, identify the most relevant oncogenic drivers of the disease, accurately capture tumor heterogeneity, be minimally invasive and provide the possibility to periodically reassess the molecular phenotype of the disease14.
Liquid biopsy
The term “liquid biopsy” describes the analysis of the molecular features of tumors in samples obtained from the blood and other body fluids such as urine and cerebrospinal fluid. Liquid biopsy includes the analysis of cfDNA15, other nucleic acids15, circulating tumor cells (CTCs) 16,17 and exosomes18.
The presence of cell-free nucleic acids in human blood was first described by Mandel and Métais in 194819. cfDNA can be found in varying quantities in blood, usually at low levels in healthy volunteers and at increased levels in patients after events such as exercise20, myocardial infarction21 or trauma22. To date, the most widespread translational research has been on the prenatal assessment of fetal DNA23–25 and cancer15,26,27.
The innovative approach of liquid biopsies for the analysis of components such as cfDNA enables the comprehensive characterization of the evolutionary processes that occur following treatment response and subsequent progression. Being minimally invasive, cfDNA collection facilitates the repeated sampling in real-time needed to capture tumor evolution and the molecular dynamics of the disease28.
The origin of tumor DNA in the circulation has been related to the properties of apoptosis and necrosis29. As a result of cell death, tumor DNA can be released from primary and metastatic tumors as well as circulating tumor cells. cfDNA is usually fragmented in small 70-200 base pair fragments with a peak of approximately 166 base pairs30 corresponding roughly to the length of DNA wrapped around a single nucleosome31. Multiple studies have shown that DNA wrapped around nucleosomes is protected from the nuclease activity in blood and that even the tissue of origin can be estimated due to tissue-specific nucleosome positioning32,33.
The presence of cfDNA in plasma is usually higher in cancer patients than in noncancer patients34,35, with concentrations that range from 0 to more than 1000 ng/ml of blood, with an average of 19 ng/ml in first-line metastatic CRPC patients36. This concentration depends on several factors, including tumor metastatic volume, metastasis sites, and tumor progression. There is still a need for a better understanding of the differential contribution of tumor location and tumor microenvironment to the release of DNA. For example, for patients with tumors of the central nervous system, the molecular profile might be more accurately represented in the cerebrospinal fluid than in the bloodstream37. Likewise, for urological cancers, urine might be an attractive source for cfDNA. However, for cfDNA analyses in urine, the expected size of cfDNA is smaller, as cfDNA has to pass through the glomerular filtration system; downstream assays thus needs to be designed accordingly38. The concentration of cfDNA is also influenced by the clearance and degradation of DNA in the liver and the kidney and by nuclease activity in the blood. cfDNA has a relatively short half-life that ranges from minutes to several hours39. The short half-life gives cfDNA an advantage over protein-based biomarkers usually used in the clinic to monitor tumor progression, such as serum PSA, which have longer half-lives and need several weeks to undergo changes representative of tumor dynamics40. In addition, cfDNA is pathway-agnostic, since it covers a broad range of molecular alterations and, unlike PSA, can be used in tumors that are not dependent on the AR pathway.
The proportion of tumor DNA in the pool of cfDNA is called the tumor fraction or tumor content (TC) and varies greatly from almost undetectable to more than 90%41. The presence of low cfDNA levels and a low tumor fraction have been limitations for cfDNA analyses for solid tumors. However, the introduction of new genomic technologies with high sensitivity and specificity, including next-generation sequencing42 and digital PCR43, has greatly contributed to the study of ctDNA44,45. While digital PCR is a relatively inexpensive and quick method of measuring the allelic frequency of rare mutations in a mixture of tumoral and nontumoral DNA and has increased the sensitivity for detecting allelic frequencies to as low as 0.01%36, it does not provide a comprehensive profile of the molecular events that can be detected in cfDNA. Another limitation of digital PCR is that the sensitivity, particularly for identifying gene copy number variations, can be limited if the TC is low41,46. Next-generation sequencing (NGS), on the other hand, is capable of providing such information, and whole-genome, exome and targeted sequencing have all been successfully applied to cfDNA41,46,47. While NGS-based studies are still significantly more expensive than digital PCR, the number of questions that can be asked is higher for the same cfDNA input, but the tradeoff is a lower sensitivity than that of digital PCR for detecting specific alterations48. One important limitation for the identification of deletions using NGS is that NGS is limited to plasma with a high TC. Analysis of heterozygous single nucleotide polymorphisms (SNPs) provides an opportunity to overcome this limitation. If several gene aberrations are found to demonstrate predictive value, as seems very likely, NGS will be preferred. Indeed, NGS has already been implemented as a discovery tool in many clinical studies of novel agents49–51.
Molecular alterations in cfDNA from advanced prostate cancer patients
The molecular landscape of advanced lethal prostate cancer has been defined based on tumor material obtained from tumor biopsies and warm autopsies5,10,52,53 and possesses features absent or rarely found in primary tumors54,55. The accumulation of these aberrations follows a highly dynamic process directly related to the selective pressure imposed by the treatment of a genetically unstable disease. In advanced prostate cancer, the most frequently observed genomic alterations are the amplification or mutation of the AR, mutations of DNA repair genes and the loss or mutation of the tumor suppressor genes TP53 and PTEN (Figure 2).
Figure 2. The dynamic genomic landscape of advanced metastatic prostate cancer.

The most relevant genomic events become more frequent during tumor progression and castration resistance. ctDNA can be studied in sequential liquid biopsies during tumor progression to capture the changing genomic landscape. Several additional molecular alterations are usually observed during progression. Many molecular events can be observed simultaneously in the same patient. The graph represents the genomic landscape of prostate cancer at different stages of the disease. A patient does not necessarily need to develop all the stages; some patients never develop metastasis, and some are diagnosed with de novo metastases. The relative frequencies of the aberrations are based on publicly available data5,54,55. Doce = docetaxel; Abi = abiraterone; HR = homologous recombination, MMR mismatch repair.
AR aberrations
The androgen receptor (AR) is the master regulator of prostate cancer 56,57,58 and castration is the backbone treatment upon which other treatments are supplemented. Intriguingly, molecular aberrations in the AR are seldom observed before castration55,59, whereas lethal prostate cancers are frequently associated with AR aberrations, including AR gain, AR mutations and AR genomic structural rearrangements; furthermore, the detection of these aberrations increases during tumor progression, being observed in 10-15% of samples at castration resistance, 30-40% of samples from patients in second and later lines of advanced CRPC therapy36 and up to 60%10 of samples harvested at warm autopsy.
Analyses of cfDNA in sequential liquid biopsy samples have allowed the interrogation of the AR genomic landscape in CRPC and have greatly contributed to the understanding of the complex dynamics observed in the AR under the selective pressure of androgen deprivation and antiandrogen therapy during tumor progression and evolution.
Progression to bicalutamide therapy is associated with the emergence of a point mutation in the AR, c.2226G>T (p.W742C). This mutation has been associated with promiscuous AR activation and has been proposed to be involved in the observed PSA response to antiandrogen withdrawal12. Interestingly, the treatment of these patients with a new generation antiandrogen, enzalutamide, is able to induce tumor responses and the regression of this clone in preclinical models60. In addition, there is evidence of the clinical efficacy of enzalutamide and abiraterone in patients harboring this mutation. Annala et al.61 randomized 202 treatment-naïve CRPC patients to either abiraterone or enzalutamide. This mutation was detected before treatment in three patients, specifically, one on abiraterone and two on enzalutamide. A PSA decline was observed in all patients, and in two patients, the decline was greater than 50%, thus meeting the criteria for a PSA response. The AR mutation c.2623C>T (p.T875Y) has been associated with a broadened specificity in vitro to ligands such as estrogens, progesterone and adrenal androgens62,63 and has been observed in patients receiving abiraterone41,64. This mutation was observed before treatment in nine patients in the study by Annala et al., specifically, three patients on abiraterone and six patients on enzalutamide. All patients on abiraterone had a PSA decline, and in two of the patients, the decline was greater than 50% and met the PSA response criteria. Five out of six patients on enzalutamide had a PSA response. These results suggest that the novel antiandrogens abiraterone and enzalutamide retain clinical activity in patients with these AR mutations detectable in plasma DNA.
The sequential analysis of cfDNA using NGS has identified two point mutations in the AR associated with resistance to abiraterone and prednisone: c.2105T>A (p.L702H) and c.2632A>G (p.T878A), which were previously associated with AR activation by prednisone and progesterone, respectively36,41,46,62,65. Another point mutation, c.2629T>C (p.F877L), has been associated with resistance to enzalutamide 66,67, although this mutation seems to be very uncommon68.
The detection of AR gain in cfDNA has been associated with resistance to enzalutamide and abiraterone in chemotherapy-naïve and post-docetaxel CRPC36,41,46,64,68–70 patients and has prognostic significance independent of other prognostic factors71–73. On the other hand, preliminary data suggest that tumors with AR gain are not resistant to taxane treatment74, which if prospectively confirmed could poise plasma AR as one of the first predictive biomarkers in prostate cancer. AR gain was previously defined based on the method used for detection: healthy volunteer data was used to estimate noise when analyzing cfDNA in plasma using NGS46, and for ddPCR modeling, studies were used that estimated the likelihood of the AR CN cutoff value that best predicted the association with outcome36. A recent study that used a different method for AR gain estimation adjusted by the TC showed a worse outcome in univariate analysis 61, but the independent prognostic value of AR gain was unable to be confirmed. Differences in the method used for AR gain estimation preclude direct comparison with previous studies 75. The analytical validation of new methodologies used to define AR gain, as well as a meta-analysis of AR CN data acquired from multiple trials at different institutions, is needed to identify the best cutoff value for considering AR CN gain clinically relevant.
AR genomic structural rearrangements (AR-GSRs) have been identified in one-third of CRPC patients and have been proposed to be potential drivers of resistance to antiandrogen therapies. These molecular alterations are highly heterogeneous in breakpoint location, rearrangement class and subclonal enrichment. However, most AR-GSRs are associated with the expression of splice variants lacking the ligand-binding domain76. These alterations can also be detected in cfDNA using whole-genome sequencing (WGS) and have been associated with the expression of AR splice variants in CTCs47. Although heterogeneous, AR-GSRs appear to be associated with a poorer outcome to antiandrogen therapy and an adverse prognosis. The correlation between both AR-GSRs and other AR aberrations and the clinical outcome needs to be further studied. One of the potential advantages of the study of AR-GSRs via cfDNA is that cfDNA analysis can be used in most patients, whereas other methods, such as tumor biopsy or CTC analyses, are limited by accessibility to the material.
cfDNA analyses can identify tumor heterogeneity and monitor clonal evolution during treatment. If clinically validated in advanced prostate cancer, cfDNA analysis could provide biomarkers to select patients more likely to benefit from antiandrogen therapies and those who should be switched to alternative therapies such as taxanes.
Homologous recombination repair genes (HRR)
Prostate cancer patients with germline mutations are at higher risk of developing metastases, and germline mutations are associated with poor survival77. In lethal prostate cancer, tumor biopsies demonstrate increased germline HRR gene mutations with a frequent loss of heterozygosity (LOH)5. The loss of function of HRR genes predicts a response to PARP-inhibitors such as olaparib78,79 and to platinum therapies80. The sequencing of cfDNA enables the detection of germline as well as somatic genomic alterations81. Moreover, next-generation cfDNA sequencing has shown a near-perfect correlation with biopsy samples for the detection of somatic mutations in HRR genes 82,83.
cfDNA analysis enables the detection and monitoring of HRR gene alterations during treatment50,51. A response to olaparib in prostate cancer patients with HRR germline mutations was associated with a decrease in the allelic frequency in cfDNA to approximately 50%, suggesting an abrogation of the LOH. For patients with somatic mutations in HRR genes, a response to PARP inhibitors was associated with a significant decrease in the allelic frequency of the mutations to less than 5%, confirming the presence of a molecular response50.
BRCA1/2-deficient ovarian and breast carcinomas, which are initially sensitive to platinum and PARP inhibitors, develop treatment resistance associated with secondary mutations that restores the DNA repair function of BRCA1/284,85. Interestingly, in prostate cancer, cfDNA sequencing has been shown to be a powerful tool to study the mechanisms of resistance to PARP inhibitors50,51. For example, in patients with BRCA2 germline mutations who responded to PARP inhibitors, additional somatic BRCA2 frameshift mutations were observed at progression. These additional mutations were associated with the restoration of the open reading frame. Similar findings were observed in patients with BRCA2 somatic mutations and patients with PALB2 mutations, in whom additional alternative somatic deletions were associated with the restoration of the open-reading frame. These findings have great implications for the understanding of the mechanisms underlying the sensitivity and resistance to PARP inhibitors and likely to platinum compounds. PARP inhibitors have been demonstrated to exert a profound selective pressure associated with divergent subclonal evolution that restores HRR function, both in patients with germline mutations and in those with somatic mutations. It remains to be seen whether these clones behave as dominant clones after PARP inhibitor treatment is stopped. This knowledge could open the door to dynamic personalized therapies that include smart sequencing or a combination of anticancer therapies and treatment rechallenge depending on the molecular drivers controlling progression.
The efficacy of novel antiandrogens in HRR-deficient tumors is a matter of controversy. Whereas some authors suggest that enzalutamide or abiraterone treatment is associated with a poorer outcome 61,81,86, others either do not find a difference 87 or show a better outcome88,89. These studies are limited by the relatively low frequency of the mutations. Further studies are needed to clarify whether BRCA2, ATM and other mutations influence the treatment response differently; how LOH modifies this effect; in what ways frequently associated mutations in genes such as TP53 contribute; and how prior therapies influence the development of resistance. The comprehensive and dynamic analysis of cfDNA might be helpful in solving these conundrums.
TP53 aberrations
Aberrations in TP53 are observed in half of all lethal prostate cancers5,10,52 and have been associated with castration resistance and the activation of androgen-independent pathways90–92. These aberrations are also found in plasma DNA61,68,93. Annala et al. identified mutations, rearrangements and deletions in more than fifty percent of patients with ctDNA above the threshold defined for detection in treatment-naïve CRPC (TC >30%)61. The presence of TP53 inactivation was associated with worse outcome to abiraterone or enzalutamide in multivariate analysis independent of other clinical and genetic factors. Interestingly, patients who harbored two or more TP53 defects had an even earlier time to progression than those who had only one TP53 defect. Although some patients harboring TP53 aberrations benefited from the treatment, most patients with primary resistance to abiraterone or enzalutamide and ctDNA above the threshold for detection (TC>30%) harbored mutations resulting in TP53 inactivation. Although further studies are needed to use TP53 as a predictive biomarker, a closer evaluation of these patients is recommended.
PTEN and PI3K pathway defects
The PI3K pathway is altered in almost half of all advanced CRPC patients 5. PI3K pathway alterations involve the loss or mutation of PTEN and aberrations in PIK3CA/B and AKT. These alterations have also been detected with a high concordance in ctDNA68,82. The loss of PTEN, which leads to PI3K activation, is known to mediate resistance to antiandrogen therapy94 and predicts a response to AKT inhibitors95. PI3K pathway defects have been associated with a poor response to the novel antiandrogens enzalutamide and abiraterone61 and suggests the possibility for potential treatment selection.
Other genomic alterations
A number of other alterations have been described to have a role in metastatic prostate cancer and treatment response. The loss of RB1 is frequently observed in lethal prostate cancers and has been correlated with lineage plasticity and the activation of AR-independent pathways when associated with P53 loss. ctDNA analysis was able to detect losses of RB1 associated with a poorer response to enzalutamide and abiraterone61,68,82. Many other genomic alterations, including gains of MYC, MET and CCND1, have also been detected in cfDNA 64,68,93,96. In addition, cfDNA has the potential to indicate the mutational load, a sentinel for mismatch repair (MMR)-deficient tumors, and to identify patients who could benefit from immunotherapy97. The possibility to characterize, in real time and serially, all these molecular alterations could expand the possibilities for the design and conduct of clinical trials, including umbrella and basket trials 92,98,99.
cfDNA analyses need to follow a qualification process
All biomarkers, including cfDNA analyses, need to follow a predefined analytical and clinical qualification process before being approved by the regulatory agencies and being implemented in the clinic. As an example, a plasma AR study is in the process of following a predefined roadmap for biomarker development and qualification (Figure 3). This roadmap includes the development of an accurate and reproducible assay to measure plasma AR46 and a retrospective evaluation of the association of plasma AR with outcome in a prospectively collected set of patients41. After the analytical qualification was conducted, it was clinically validated in two homogeneous sets of patients with mCPRC treated with novel antiandrogenic agents36. The next step to clearly demonstrate the clinical utility of plasma AR will be to confirm these results in a clinical trial randomized by plasma AR status. Via a method analogous to that used in drug development, patients with the desired biomarker, in this case AR gain, would be randomized to the available treatments. Alternatively, randomized prospective trials including investigational therapies would need to be stratified and adequately powered to assess the predictive value of predefined biomarkers. These strategies would allow us to assess the predictive rather than the prognostic value of the biomarkers. All other cfDNA analyses need to follow a similar pathway of qualification before they can be routinely used in the clinic.
Figure 3. Roadmap for the qualification of plasma AR as a biomarker.
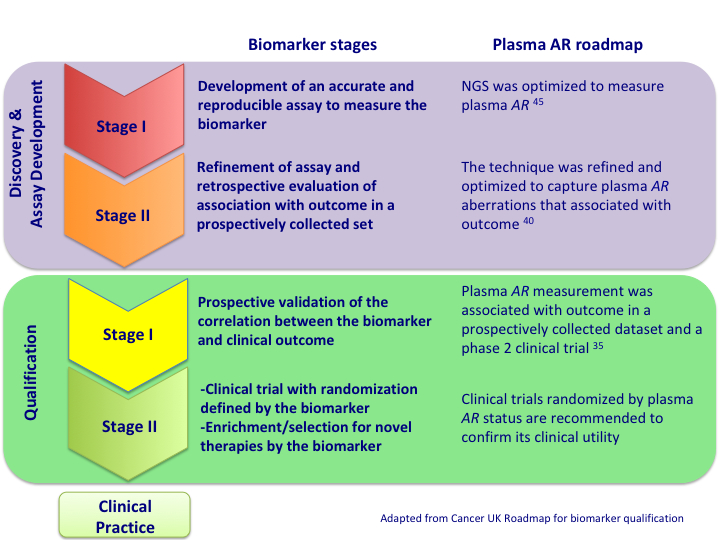
All biomarkers need to follow an analytical and clinical qualification process before they can be approved and implemented in the clinic. As an example, this figure shows the roadmap followed for plasma AR qualification and the future directions for the development of plasma AR as a biomarker.
The association among different molecular alterations in mCPRC is not fully understood, and the relative contribution of each mutation to the clinical outcome needs to be carefully dissected and prospectively validated96.
Potential clinical applications of cfDNA in advanced prostate cancer
Along with the improvement in molecular techniques and the increase in the knowledge about genetics and drug development, several clinical applications of cfDNA analysis can be foreseen in advanced prostate cancer:
As a prognostic tool. Circulating-free DNA, tumor fraction DNA and several molecular alterations, including the most relevant clinical prognostic factors in mCRPC, are associated with independent prognostic significance in multivariable analyses36,41,71–73,96. It will be interesting to design new prognostic nomograms that include both clinical and molecular features. cfDNA can be easily obtained at different time points and is a perfect candidate to improve currently available prognostic tools. It is tempting to study the clinical significance of cfDNA in localized disease and the association of cfDNA with tumor relapse and distant metastases100.
As a predictive marker. It is very likely that some molecular alterations, including HRR and AR aberrations, are associated with the differential efficacy of anticancer therapies. The detection of AR gain is associated with an adverse outcome to novel antiandrogen therapies. However, the predictive value of AR gain needs to be qualified in prospective studies before it can be used to make treatment decisions. In addition, some AR mutations that emerge during antiandrogen therapy are associated with bicalutamide or abiraterone-prednisone treatment and are known to be sensitive to other therapies. The identification of these aberrations can be helpful to guide treatment therapy. HRR gene mutations are associated with increased sensitivity to PARP inhibitors and to platinum therapies. The detection of HRR gene mutations in plasma is highly correlated with the detection of these mutations in molecular analyses of biopsy tissues from metastases. If clinically qualified, cfDNA analysis could potentially become a new standard method to assess this feature.
To monitor tumor response. The sequential analysis of cfDNA allows a dynamic measure of its quantity and molecular alterations. Changes in the quantity of cfDNA during treatment closely correlate with the tumor response50. cfDNA has a relatively short half-life and can be more sensitive and specific than other protein markers such as PSA, particularly in challenging cases where the PSA level is very low or there is early rising PSA. As genomic analysis becomes more readily available for clinicians and methods are standardized, it can become part of the laboratory information available for the clinician. In addition, the molecular characterization of cfDNA allows the allelic frequency of mutations in key driver genes to be measured. The sequential analysis of cfDNA allows clinicians to closely follow the dynamics of these mutations. The clinical response to anticancer therapy in tumors that harbor somatic mutations is associated with molecular responses that include significant decreases in the allelic frequency. In contrast, the appearance of resistant clones can precede clinical or radiographic progression by several months and could provide the opportunity to introduce early changes to the anticancer treatment regimen in order to improve patient outcome.
To identify novel mechanisms of resistance. The analysis of circulating cfDNA provides an unprecedented opportunity for the identification of new mechanisms of resistance. Since the collection of circulating cfDNA is minimally invasive and allows us to serially obtain a comprehensive molecular analysis, cfDNA analysis gives us the opportunity to identify novel gene alterations associated with tumor progression. Moreover, cfDNA analysis allows us to better understand the mechanisms underlying tumor progression and provides us with the opportunity to design novel drugs and treatment strategies, as it has allowed us to identify the methods underlying the acquisition of resistance to PARP inhibitors via the selection of mutations that revert the open reading frame of BRCA2 50,51.
Limitations of plasma DNA
However, cfDNA analysis is subject to relevant limitations: A) The sensitivity of the sequencing techniques, although improving along with the development of technology, is limited by the availability of an adequate amount of ctDNA. This limitation is particularly evident at earlier stages of disease and for the identification of gene copy losses, although it might also be relevant at later stages of disease and for the identification of other genomic aberrations. The restriction of the genomic analysis to the TC has partially alleviated this limitation. The estimation of the TC is usually defined by the presence of known tumor genomic aberrations. A restriction or adjustment for the TC, although appreciably improving the sensitivity, enriches the population with a cohort of patients with a poorer outcome and might neglect genomic events occurring in tumor clones lacking the predefined aberrations. This type of analysis might be relevant for the interpretation of the results and should be taken into consideration when comparing different studies; B) The specificity of plasma DNA analysis is limited by the quality of the DNA and by the sequencing technique. The identification of genomic aberrations in plasma DNA needs to be cautiously interpreted, since not all mutations or copy number variations are biologically meaningful. A comprehensive biological and clinical approach is required to validate new biomarkers; and C) The analysis of plasma DNA does not capture additional layers of biological complexity, such as differences in RNA expression or protein levels that might be clinically relevant. A study restricted to plasma DNA might be overlooking important information regarding the expression of AR splice variants, which might have prognostic and/or predictive value101,102.
In summary, cfDNA analysis is very promising in advanced prostate cancer. cfDNA analysis is minimally invasive, is broadly applicable and provides a comprehensive molecular analysis of the tumor that would be difficult to obtain by other means. Precision medicine needs to integrate all this information in order to personalize the treatment of metastatic prostate cancer. The qualification of cfDNA-derived biomarkers is urgently needed before such biomarkers can be implemented in the clinic.
Footnotes
Conflict of Interest Disclosures
G.A. is on the ICR list of rewards to inventors for abiraterone. G.A. has received honoraria, consulting fees, or travel support from Astellas, Medivation, Janssen, Millennium Pharmaceuticals, Ipsen, Ventana, ESSA Pharmaceuticals, and Sanofi-Aventis and grant support from Janssen, AstraZeneca, and Arno. E.G.B. has received speaker honoraria or travel support from Astellas, Janssen-Cilag and Sanofi- Aventis. The other authors have no conflicts to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Gillessen S, Attard G, Beer TM, Beltran H, Bossi A, Bristow R, et al. Management of Patients with Advanced Prostate Cancer: The Report of the Advanced Prostate Cancer Consensus Conference APCCC 2017. Eur Urol. 2017:1–34. doi: 10.1016/j.eururo.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Attard G, Parker C, Eeles RA, Schröder F, Tomlins SA, Tannock I, et al. Prostate cancer. Lancet. 2016;387:70–82. doi: 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- 4.Grasso CS, Wu Y-M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012 doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitt MW, Loeb LA, Salk JJ. Vol. 13. Nature Publishing Group; 2015. The influence of subclonal resistance mutations on targeted cancer therapy; pp. 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N Engl J Med. 2017 doi: 10.1056/NEJMoa1616288. NEJMoa1616288–13. [DOI] [PubMed] [Google Scholar]
- 9.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520:353–357. doi: 10.1038/nature14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat Med. 2016;22:369–378. doi: 10.1038/nm.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taplin ME. Androgen Receptor Mutations in Androgen-Independent Prostate Cancer: Cancer and Leukemia Group B Study 9663. Journal of Clinical Oncology. 2003;21:2673–2678. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 13.Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Comms. 2016;7:1–12. doi: 10.1038/ncomms13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perez-Gracia JL, Sanmamed MF, Bosch A, Patiño-Garcia A, Schalper KA, Segura V, et al. Strategies to design clinical studies to identify predictive biomarkers in cancer research. Cancer Treatment Reviews. 2017;53:79–97. doi: 10.1016/j.ctrv.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 15.Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–437. doi: 10.1038/nrc3066. [DOI] [PubMed] [Google Scholar]
- 16.Lorente D, Mateo J, de Bono JS. Molecular characterization and clinical utility of circulating tumor cells in the treatment of prostate cancer. Am Soc Clin Oncol Educ Book. 2014;34:e197–203. doi: 10.14694/EdBook_AM.2014.34.e197. [DOI] [PubMed] [Google Scholar]
- 17.Haber DA, Velculescu VE. Blood-Based Analyses of Cancer: Circulating Tumor Cells and Circulating Tumor DNA. Cancer Discov. 2014;4:650–661. doi: 10.1158/2159-8290.CD-13-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nawaz M, Camussi G, Valadi H, Nazarenko I, Ekström K, Wang X, et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat Rev Urol. 2014;11:688–701. doi: 10.1038/nrurol.2014.301. [DOI] [PubMed] [Google Scholar]
- 19.Mandel P. Les acides nucleiques du plasma sanguin chez l'homme. CR Acad Sci Paris. 1948 [PubMed] [Google Scholar]
- 20.Beiter T, Fragasso A, Hudemann J, Niess AM, Simon P. Short-term treadmill running as a model for studying cell-free DNA kinetics in vivo. Clinical Chemistry. 2011;57:633–636. doi: 10.1373/clinchem.2010.158030. [DOI] [PubMed] [Google Scholar]
- 21.Antonatos D, Patsilinakos S, Spanodimos S, Korkonikitas P, Tsigas D. Cell-free DNA levels as a prognostic marker in acute myocardial infarction. Annals of the New York Academy of Sciences. 2006;1075:278–281. doi: 10.1196/annals.1368.037. [DOI] [PubMed] [Google Scholar]
- 22.Swaminathan R, Butt AN. Circulating nucleic acids in plasma and serum: recent developments. Annals of the New York Academy of Sciences. 2006;1075:1–9. doi: 10.1196/annals.1368.001. [DOI] [PubMed] [Google Scholar]
- 23.Lo YMD, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, et al. Presence of fetal DNA in maternal plasma and serum. The Lancet. 1997;350:485–487. doi: 10.1016/S0140-6736(97)02174-0. [DOI] [PubMed] [Google Scholar]
- 24.Lo YM. Fetal DNA in maternal plasma: biology and diagnostic applications. Clinical Chemistry. 2000;46:1903–1906. [PubMed] [Google Scholar]
- 25.Bianchi DW. From prenatal genomic diagnosis to fetal personalized medicine: progress and challenges. Nat Med. 2012;18:1041–1051. doi: 10.1038/nm.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early stage lung cancer evolution. Nature. 2017;61:69. doi: 10.1038/nature22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diaz LA, Bardelli A. Liquid Biopsies: Genotyping Circulating Tumor DNA. Journal of Clinical Oncology. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2007;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–1665. [PubMed] [Google Scholar]
- 30.Jiang P, Chan CWM, Chan KCA, Cheng SH, Wong J, Wong VW-S, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA. 2015;112:E1317–25. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo YMD, Chan KCA, Sun H, Chen EZ, Jiang P, Lun FMF, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2:61ra91–61ra91. doi: 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 32.Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivanov M, Baranova A, Butler T, Spellman P, Mileyko V. Non-random fragmentation patterns in circulating cell-free DNA reflect epigenetic regulation. BMC Genomics. 2015;16(Suppl 13):S1. doi: 10.1186/1471-2164-16-S13-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarzenbach H, Müller V, Milde-Langosch K, Steinbach B, Pantel K. Evaluation of cell-free tumour DNA and RNA in patients with breast cancer and benign breast disease. Molecular BioSystems. 2011;7:2848–2854. doi: 10.1039/c1mb05197k. [DOI] [PubMed] [Google Scholar]
- 35.Sozzi G, Conte D, Leon M, Cirincione R, Roz L, Ratcliffe C, et al. Quantification of Free Circulating DNA As a Diagnostic Marker in Lung Cancer. J Clin Oncol. 2003;21:3902–3908. doi: 10.1200/JCO.2003.02.006. [DOI] [PubMed] [Google Scholar]
- 36.Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol. 2017:1–9. doi: 10.1093/annonc/mdx155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Mattos-Arruda L, Mayor R, Ng CKY, Weigelt B, nez-Ricarte FMI, Torrejon D, et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat Comms. 2015;6:1–6. doi: 10.1038/ncomms9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu T, Li J. Clinical applications of urinary cell-free DNA in cancer: current insights and promising future. Am J Cancer Res. 2017;7:2318–2332. [PMC free article] [PubMed] [Google Scholar]
- 39.Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer—A survey. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2007;1775:181–232. doi: 10.1016/j.bbcan.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Dawson S-J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S-F, et al. Analysis of Circulating Tumor DNA to Monitor Metastatic Breast Cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 41.Romanel A, Tandefelt DG, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7:312re10. doi: 10.1126/scitranslmed.aac9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murtaza M, Dawson S-J, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 43.Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA. 1999;96:9236–9241. doi: 10.1073/pnas.96.16.9236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. Development of Personalized Tumor Biomarkers Using Massively Parallel Sequencing. Sci Transl Med. 2010;2:20ra14–20ra14. doi: 10.1126/scitranslmed.3000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, et al. Detection of Chromosomal Alterations in the Circulation of Cancer Patients with Whole-Genome Sequencing. Sci Transl Med. 2012;4:162ra154–162ra154. doi: 10.1126/scitranslmed.3004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, et al. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6:254ra125–254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Laere B, van Dam P-J, Whitington T, Mayrhofer M, Diaz EH, Van den Eynden G, et al. Comprehensive Profiling of the Androgen Receptor in Liquid Biopsies from Castration-resistant Prostate Cancer Reveals Novel Intra-AR Structural Variation and Splice Variant Expression Patterns. Eur Urol. 2017:1–9. doi: 10.1016/j.eururo.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 48.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nature Biotechnology. 2016;34:547–555. doi: 10.1038/nbt.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seed G, Yuan W, Mateo J, Carreira S, Bertan C, Lambros M, et al. Gene Copy Number Estimation from Targeted Next-Generation Sequencing of Prostate Cancer Biopsies: Analytic Validation and Clinical Qualification. Clin Cancer Res. 2017;23:6070–6077. doi: 10.1158/1078-0432.CCR-17-0972. [DOI] [PubMed] [Google Scholar]
- 50.Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017:1–13. doi: 10.1158/2159-8290.CD-17-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017:1–8. doi: 10.1158/2159-8290.CD-17-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grasso CS, Wu Y-M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol. 2017;2017:1–16. doi: 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Network TCGAR. Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541:359–364. doi: 10.1038/nature20788. [DOI] [PubMed] [Google Scholar]
- 56.Richter E, Srivastava S, Dobi A. Androgen receptor and prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:114–118. doi: 10.1038/sj.pcan.4500936. [DOI] [PubMed] [Google Scholar]
- 57.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 58.Heinlein CA, Chang C. Androgen Receptor in Prostate Cancer. Endocrine Reviews. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 59.Network TCGAR. Abeshouse A, Ahn J, Akbani R, Ally A, Amin S, et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018;8:444–457. doi: 10.1158/2159-8290.CD-17-0937. [DOI] [PubMed] [Google Scholar]
- 62.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332:1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 63.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, et al. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Mol Endocrinol. 1997;11:450–459. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- 64.Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clinical Cancer Research. 2015;21:2315–2324. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 65.Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, et al. Abiraterone Treatment in Castration-Resistant Prostate Cancer Selects for Progesterone Responsive Mutant Androgen Receptors. Clinical Cancer Research. 2015;21:1273–1280. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013;2 doi: 10.7554/eLife.00499.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, et al. A Clinically Relevant Androgen Receptor Mutation Confers Resistance to Second-Generation Antiandrogens Enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 68.Wyatt AW, Azad AA, Volik SV, Annala M, Beja K, McConeghy B, et al. Genomic Alterations in Cell-Free DNA and Enzalutamide Resistance in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016:1–9. doi: 10.1001/jamaoncol.2016.0494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salvi S, Casadio V, Conteduca V, Lolli C, Gurioli G, Martignano F, et al. Circulating AR copy number and outcome to enzalutamide in docetaxel-treated metastatic castration-resistant prostate cancer. Oncotarget. 2016;7:37839–37845. doi: 10.18632/oncotarget.9341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salvi S, Casadio V, Conteduca V, Burgio SL, Menna C, Bianchi E, et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br J Cancer. 2015;112:1717–1724. doi: 10.1038/bjc.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molina A, Rathkopf DE, Fizazi K, Kantoff PW, Li J, Azad AA, et al. A prognostic index model for predicting overall survival in patients with metastatic castration-resistant prostate cancer treated with abiraterone acetate after docetaxel. Ann Oncol. 2016;27:454–460. doi: 10.1093/annonc/mdv594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Halabi S, Lin CY, Kelly WK, Fizazi KS, Moul JW, Kaplan EB, et al. Updated Prognostic Model for Predicting Overall Survival in First-Line Chemotherapy for Patients With Metastatic Castration-Resistant Prostate Cancer. Journal of Clinical Oncology. 2014;32:671–677. doi: 10.1200/JCO.2013.52.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scher HI, Heller G, Molina A, Attard G, Danila DC, Jia X, et al. Circulating Tumor Cell Biomarker Panel As an Individual-Level Surrogate for Survival in Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2015;33:1348–1355. doi: 10.1200/JCO.2014.55.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conteduca V, Wetterskog D, Jayaram A, Tandefelt DG, Salvi S, Lolli C, et al. Clinical utility of digital droplet PCR to track plasma AR aberrations in castration-resistant prostate cancer patients. London: 2016. pp. 1–1. [Google Scholar]
- 75.Jayaram A, Wetterskog D, Attard G. Plasma DNA and Metastatic Castration-Resistant Prostate Cancer: The Odyssey to a Clinical Biomarker Test. Cancer Discov. 2018;8:392–394. doi: 10.1158/2159-8290.CD-18-0124. [DOI] [PubMed] [Google Scholar]
- 76.Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Comms. 2016;7:1–12. doi: 10.1038/ncomms13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, et al. Germline BRCA Mutations Are Associated With Higher Risk of Nodal Involvement, Distant Metastasis, and Poor Survival Outcomes in Prostate Cancer. Journal of Clinical Oncology. 2013;31:1748–1757. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 79.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cheng HH, Pritchard CC, Boyd T, Nelson PS, Montgomery B. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur Urol. 2016;69:992–995. doi: 10.1016/j.eururo.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Annala M, Struss WJ, Warner EW, Beja K, Vandekerkhove G, Wong A, et al. Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair–deficient Prostate Cancer. Eur Urol. 2017;72:34–42. doi: 10.1016/j.eururo.2017.02.023. [DOI] [PubMed] [Google Scholar]
- 82.Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of Circulating Tumor DNA and Matched Metastatic Tissue Biopsy in Prostate Cancer. J Natl Cancer Inst. 2017;110:1–9. doi: 10.1093/jnci/djx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017;7:1006–1017. doi: 10.1158/2159-8290.CD-17-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary Somatic Mutations Restoring BRCA1/2Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J Clin Oncol. 2011;29:3008–3015. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Afghahi A, Timms KM, Vinayak S, Jensen KC, Kurian AW, Carlson RW, et al. Tumor BRCA1Reversion Mutation Arising during Neoadjuvant Platinum-Based Chemotherapy in Triple-Negative Breast Cancer Is Associated with Therapy Resistance. Clin Cancer Res. 2017;23:3365–3370. doi: 10.1158/1078-0432.CCR-16-2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Castro E, Laorden NR, Rodriguez JMP, del Pozo A, Sáez MI, Colmenero AM, et al. LBA32PROREPAIR-B: A prospective cohort study of DNA repair defects in metastatic castration resistant prostate cancer (mCRPC) Annals of Oncology. 2017;28 doi: 10.1093/annonc/mdx440.025. [DOI] [Google Scholar]
- 87.Mateo J, Cheng HH, Beltran H, Dolling D, Xu W, Pritchard CC, et al. Clinical Outcome of Prostate Cancer Patients with Germline DNA Repair Mutations: Retrospective Analysis from an International Study. Eur Urol. 2018:1–7. doi: 10.1016/j.eururo.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hussain M, Daignault-Newton S, Twardowski PW, Albany C, Stein MN, Kunju LP, et al. Targeting Androgen Receptor and DNA Repair in Metastatic Castration-Resistant Prostate Cancer: Results From NCI 9012. J Clin Oncol. 2018;36:991–999. doi: 10.1200/JCO.2017.75.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Antonarakis ES, Lu C, Luber B, Liang C, Wang H, Chen Y, et al. Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur Urol. 2018:1–8. doi: 10.1016/j.eururo.2018.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen C-C, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ku S-Y, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lunardi A, Ala U, Epping MT, Salmena L, Clohessy JG, Webster KA, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2013 doi: 10.1038/ng.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ulz P, Belic J, Graf R, Auer M, Lafer I, Fischereder K, et al. Whole-genome plasma sequencing reveals focal amplifications as a driving force in metastatic prostate cancer. Nat Comms. 2016;7:12008–12. doi: 10.1038/ncomms12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Bono JS, De Giorgi U, Massard C, Bracarda S, Rodrigues DN, Kocak I, et al. PTEN loss as a predictive biomarker for the Akt inhibitor ipatasertib combined with abiraterone acetate in patients with metastatic castration-resistant prostate cancer (mCRPC) Annals of Oncology. 2016;27 doi: 10.1093/annonc/mdw372.02. [DOI] [Google Scholar]
- 96.Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018 doi: 10.1158/2159-8290.CD-17-0937. CD-17-0937-31. [DOI] [PubMed] [Google Scholar]
- 97.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gonzalez Billalabeitia E, Seitzer N, Song SJ, Song MS, Patnaik A, Liu X-S, et al. Vulnerabilities of PTEN-TP53-deficient prostate cancers to compound PARP-PI3K inhibition. Cancer Discov. 2014;4:896–904. doi: 10.1158/2159-8290.CD-13-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Patnaik A, Swanson KD, Csizmadia E, Solanki A, Landon-Brace N, Gehring MP, et al. Cabozantinib Eradicates Advanced Murine Prostate Cancer by Activating Antitumor Innate Immunity. Cancer Discov. 2017;7:750–765. doi: 10.1158/2159-8290.CD-16-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.CHUN FKH, MULLER I, LANGE I, FRIEDRICH MG, Erbersdobler A, Karakiewicz PI, et al. Circulating tumour-associated plasma DNA represents an independent and informative predictor of prostate cancer. BJU International. 2006;98:544–548. doi: 10.1111/j.1464-410X.2006.06352.x. [DOI] [PubMed] [Google Scholar]
- 101.Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Zhu Y, et al. Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J Clin Oncol. 2017;35:2149–2156. doi: 10.1200/JCO.2016.70.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016;2:1441–10. doi: 10.1001/jamaoncol.2016.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]