

Abstract
Purpose
To determine whether inhibition of mechanistic target of rapamycin (mTOR) kinase-mediated signaling represents a valid therapeutic approach for chronic lymphocytic leukemia (CLL).
Experimental Design
Stratification of mTOR activity was carried out in primary CLL patient samples and an aggressive CLL-like mouse model. The potency of dual mTOR inhibitor AZD8055 to induce apoptosis in primary CLL cells was assessed in the presence/absence of B cell receptor (BCR) ligation. Furthermore, we addressed the molecular and functional impact of dual mTOR inhibition in combination with BTK inhibitor ibrutinib.
Results
Differential regulation of basal mTORC1 activity was observed in poor prognostic CLL samples, with elevated p4EBP1T37/46 and decreased p70S6 kinase activity, suggesting that dual mTORC1/2 inhibitors may exhibit improved response in poor prognostic CLL compared with rapalogs. AZD8055 treatment of primary CLL cells significantly reduced CLL survival in vitro compared with rapamycin, preferentially targeting poor prognostic subsets and overcoming BCR-mediated survival advantages. Furthermore, AZD8055, and clinical analog AZD2014, significantly reduced CLL tumor load in mice. AKT substrate FOXO1, while overexpressed in CLL cells of poor prognostic patients in LN biopsies, peripheral CLL cells, and mouse-derived CLL-like cells, appeared to be inactive. AZD8055 treatment partially reversed FOXO1 inactivation downstream of BCR crosslinking, significantly inhibiting FOXO1T24 phosphorylation in an mTORC2-AKT-dependent manner, to promote FOXO1 nuclear localization, activity and FOXO1-mediated gene regulation. FOXO1 activity was further significantly enhanced on combining AZD8055 with ibrutinib.
Conclusions
Our studies demonstrate that dual mTOR inhibitors show promise as future CLL therapies, particularly in combination with ibrutinib.
Keywords: CLL, mTOR inhibition, BCR signaling, FOXO1
Introduction
Chronic lymphocytic leukemia (CLL) is a malignant lymphoproliferative disorder of mature B lymphocytes, characterized by infiltration and accumulation of leukemic cells in lymphoid organs (1). The pathogenesis of CLL involves genetic aberrations, antigenic stimulation, and microenvironmental stimuli (1, 2). CLL is biologically heterogeneous; while some patients exhibit an indolent disease, others experience a highly chemotherapy-refractory disease (1), emphasizing the need for both prognostic markers and novel therapeutic approaches. A growing number of factors are recognized to have prognostic implications in CLL, with a lack of mutations in gene loci encoding the variable region of the immunoglobulin heavy-chain (IgVH) and ectopic zeta chain-associated protein kinase (ZAP-70) expression conferring poor outcomes (3–5). Cytogenetic aberrations are also important determinants of disease outcome, with deletions in chromosome 13 [del(13q)] conferring a favourable prognosis, while del(11q) [targeting ATM] and del(17p) [targeting TP53] are associated with more progressive disease (6). Indeed, survival rates of del(17p) cohorts had not improved with standard chemotherapy regimens (7).
The BCR has long been implicated in normal B cell development and B cell malignancies, with BCR ligation initiating downstream signaling cascades, via tyrosine kinases Lyn, SYK and BTK, such as the PI3K-AKT signaling pathway, vital for both normal B cell and CLL cell survival (8–10). Targeted inhibition of BCR signaling components, with either ibrutinib (targeting BTK) or idelalisib (targeting PI3Kδ), have demonstrated increased response rates in high-risk patient cohorts (11, 12). However, these treatments are not suitable for all CLL patients, and the development of drug resistance in CLL cells has been demonstrated (13), highlighting the need to develop alternative therapeutic options for CLL patients.
The serine/threonine protein kinase mechanistic target of rapamycin (mTOR), interacts with distinct groups of proteins to form two complexes called mTOR complex-1 (mTORC1: RAPTOR, mLST8, PRAS40 and DEPTOR) and complex-2 (mTORC2: RICTOR, mLST8, mSIN1 and DEPTOR) which regulate essential cellular functions including cell growth, survival and proliferation (14). mTOR signaling is often deregulated in cancer (15), but little is known about its role in CLL pathogenesis. A recent clinical trial showed limited response in relapsed/refractory CLL patients to everolimus, a rapamycin analog (16). This is likely because rapalogs are allosteric inhibitors of mTORC1 only, which uncouple the negative feedback loop mediated by mTORC1 on mTORC2, enabling AKT phosphorylation (pAKTS473) and an elevation in its activity, as is evident in CLL cells (17). The role of PI3K/AKT mediated signaling activated downstream of BCR ligation, is well established in CLL survival (18–20). One mechanism utilized by AKT to promote cell survival and proliferation is through negative regulation of the forkhead box proteins subgroup O (FOXO) family of transcription factors, notably FOXO1, FOXO3a, and FOXO4, through phosphorylation (T24, S256, S319 on FOXO1) leading to their sequestration and inactivity within the cytoplasm (21). This process is key in facilitating normal B cell proliferation, with FOXO family members responsible for regulating a diverse array of cellular functions including cell cycle arrest, apoptosis and DNA-damage repair (21). We sought to address whether inhibition of both mTORC1 and mTORC2 with a dual mTOR kinase inhibitor may represent a valid therapeutic approach for CLL.
Materials and Methods
Animals, primary cells and cell lines
Peripheral blood and LN samples were obtained, after informed consent, from patients with a confirmed diagnosis of CLL that were treatment-naïve or had received treatment but not in the preceding 3 months. The studies were approved by the West of Scotland Research Ethics Service, NHS Greater Glasgow and Clyde (UK) and the Northern Ireland Biobank and all work was carried out in accordance with the approved guidelines. Linked clinical data of prognostic markers of the CLL cohorts were recorded (Suppl Table 1). CLL lymphocytes were isolated and used freshly or cryopreserved (22). CLL cell purity was ≥ 95% in all cases, determined by flow cytometry. CLL cells were cultured at 1x106/mL in RPMI-1640 containing 10% FBS, 50 U/mL penicillin, 50 mg/mL streptomycin, and 2 mM L-glutamine (complete medium; Invitrogen Ltd., Paisley, UK). Buffy coats were obtained after informed consent from the Scottish National Blood Transfusion Service (UK) and mature B cells were isolated by positive selection with magnetic cell sorting using anti-CD19 antibody (ab) bound to magnetic beads (Miltenyi, UK) and density gradient centrifugation using Histopaque (Sigma-Aldrich Co., Poole, UK), and used fresh. MEC-1 cells (a gift from Dr. Joseph Slupsky, University of Liverpool) were authenticated by analysis of morphology and expression of appropriate CD markers by flow cytometry. MEC-1 cells were cultured at 1x106/mL in RPMI-1640 containing 10% FBS, 50 U/mL penicillin, 50 mg/mL streptomycin for up to 20 passages. Donor ICR wildtype mice were purchased from Harlan Laboratories (Oxon, UK) and donor B6.SJL wildtype mice and host recombinase activating gene-2-deficient (RAG-2−/−) and NOD-SCID-γc−/− (NSG; provided by Dr. Karen Blyth, BICR) mice were bred in-house. All mice were maintained at the University of Glasgow Central Research Facilities or the Beatson Research Unit (Glasgow, UK), under standard animal housing conditions in accordance with local and home office regulations. Time-mated mice were generated and liver was extracted at E14.
BCR stimulation assay
CLL cells were incubated at 37°C in the absence (no drug control; NDC) or presence of AZD8055, rapamycin or ibrutinib (Stratech Scientific Ltd., Suffolk, UK) for 30 min. CLL cells were then stimulated with 10 μg/ml F(ab')2 fragment anti-human IgM (Stratech Scientific Ltd.) for a set time as indicated.
Flow Cytometry
Following treatment, CLL cells were harvested, stained with FITC/APC-conjugated Annexin V and 7-amino-actinomycin D (7-AAD). Flow cytometry data were acquired using a FACSCantoII flow cytometer (BD Biosciences) using the FACS Diva software package and analysed using the FlowJo software package (Tree Star, Inc., Ashland, OR) (22). Annexin V−7-AAD− cells were considered viable. For CLL-like mouse cells, anti-IgM-PE, anti-CD45-PerCP, anti-CD23-PE-Cy7, anti-CD5-APC, anti-CD19-APC-Cy7 and anti-CD11b-Pacific Blue abs were used (BD Biosciences). For intracellular staining, surface anti-B220-PE ab staining was followed by fixation with cytofix/cytoperm (BD Biosciences). Cells were then stained with anti-pAKT S473-AF647 and pS6S235/236-V450 abs.
Western blotting
Protein lysates were prepared in lysis buffer (1% Triton, 1 mM DTT, 2 mM EDTA, 20 mM Tris pH 7.5 containing complete protease inhibitor, and PhosStop (Roche)), and Western blotting was carried out (22). All western blotting abs were sourced from Cell Signaling Technology (Hitchin, UK). Densitometry was performed on all the blots using Image Studio Software (LI-COR, USA). Ratios of the intensity levels of phosphorylated proteins over their total counterparts, or reference gene, as indicated were calculated.
IHC and Immunofluorescence (IF)
Sections for IHC were cut at 4 microns on a rotary microtome and dried at 37°C overnight. All IHC was performed on automated immunostainers (Leica BondMaX). Antigen binding sites were detected with a polymer based detection system (Bond cat no. DS 9800). All sections were visualized with DAB, counterstained in haematoxylin and mounted in DPX. All slides were reviewed by an experienced histopathologist. For IF, CLL cells were fixed with 4% (w/v) paraformaldehyde, permeabilized in 0.25% (w/v) Triton X-100 for 10 min and blocked with 1% (w/v) BSA, 0.1% (v/v) Tween 20 in PBS for 30 min. Primary anti-FOXO1 Ab was applied for 1 h at RT and secondary anti-rabbit Ab AF488 (Thermo Fisher Scientific) for 1 h at RT. Nuclei were stained with DAPI (Thermo Fisher Scientific). Z-stack images were acquired on a Zeiss-Axio Imager M1 fluorescence microscope (Carl Zeiss AG, Jena, Germany) and deconvoluted using AxioVision software (Carl Zeiss AG). Object-based quantification of signal colocalisation (Threshold Manders’ Coefficients) between the FOXO1 and DAPI channels was analysed using CellProfiler™ software (Broad Institute of MIT and Harvard, Cambridge, MA, USA). Values for the Manders’ Coefficient range from 0 to 1, expressing the fraction of signal intensity from one channel that exists in pixels above threshold signal intensity in the other channel. Signal thresholds were set by the Costes Auto threshold method. >360 cells were quantified/condition from each CLL patient (n=3).
Cellular Fractionation
Nuclear, cytoplasmic and whole cell fractions were prepared from 1x107 MEC-1 cells per condition, using the Nuclear Extract Kit as per manufacturer’s instructions with modifications (Active Motif, Belgium). Briefly, when collecting the cytoplasmic fraction, cells were resuspended in 100 µl 1 x Hypotonic Buffer for 15 min, with a 1:60 ratio of detergent added before vortexing for 10 sec. Samples were denatured and immunoblotted as described.
In vitro or in vivo B-cell/CLL-like cell generation
Haemopoietic stem and progenitor cells (HSPCs) isolated from E14 liver or adult bone marrow (BM) were retrovirally-transduced using GP+E.86 packaging cells that produce retrovirus-encoding green fluorescent protein (GFP) alone (MIEV-empty vector control) or dominant negative PKCα (PKCα-KR) as described (23). Transduced cells were cultured on OP9 cells that support B cell development in the presence of Flt-3L and IL-7 (10 ng/ml; Peprotech Ltd.) for 7 days and then either further cultured on OP9 cells in the presence of IL-7 only for in vitro cultures, or adoptively transferred (4x105 cells/mouse) into RAG-2-/- or NSG mice to establish CLL-like leukemia in vivo. Blood sampling was performed to track disease development/progression.
In vivo drug treatment
AZD8055 was formulated at 2 mg/mL in 30% Captisol (Ligand Pharmaceuticals, Inc., La Jolla, CA) and administered at 20 mg/kg via oral gavage (OG). Rapamycin was delivered once daily by intraperitoneal (ip) injection at a dose of 4 mg/kg dissolved in Tween-80 5.2% / PEG-400 5.2% (v/v). AZD2014 was prepared at 3 mg/mL in 20% Captisol (Ligand Pharmaceuticals, Inc.) and administered at 15 mg/kg via OG. Ibrutinib was prepared at 2.4 mg/mL in 0.5% methylcellulose (Sigma) and administered at 12 mg/kg via OG. After CLL-like disease confirmation (>0.4% GFP+CD19+ cells in the blood), mice were treated for 2 wk with inhibitors or vehicle control and then sacrificed. BM, spleen and blood were collected for analyses.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen). Inventoried Taqman assays and PCR reagents were purchased from ThermoFisher (Warrington, UK). cDNA synthesis and real-time PCR (RT-PCR) was conducted as described previously (22). Relative gene expression was analyzed by the ΔΔCt method using Glucuronidase Bet (GUSB) as reference control and an assigned calibrator.
FOXO1 activity kit (TRANS-AM)
Nuclear protein lysates were isolated from 1x107 CLL cells per condition using the Nuclear Extract Kit and an ELISA-based method, TransAM (Active Motif) was used to quantify FOXO1 DNA-binding activity according to the manufacturer's protocol.
Statistics
All statistical analysis was performed using GraphPad Prism 6 software (GraphPad Software Inc., CA), p values were determined by two tailed students paired or unpaired t-test or mixed model ANOVA on a minimum of at least 3 biological replicates as indicated. Drug treated values are compared with the NDC control. Mean ± standard error of mean (SEM) is shown. *=p≤0.05; **=p≤0.01; ***=p≤0.001; ****=p≤0.0001.
Results
Dual mTORC1/2 inhibition reduces viability of CLL samples, preferentially targeting poor prognostic cohorts
To determine the basal activity of mTOR in distinct cohorts of CLL patients, we analyzed the phosphorylation status of mTORC1 (S6S235/236; 4EBP1T37/46; RICTORT1135) and mTORC2 (AKTS473) downstream targets in freshly isolated peripheral blood CLL cells, compared with B cells from healthy donors (Figure 1). These data indicated that phosphorylation levels of S6K target proteins S6 and RICTOR were reduced in del(17p) CLL cohort. Interestingly, 4EBP1T37/46 phosphorylation was significantly elevated in the poorer prognostic del(17p) CLL subset indicating that mTORC1-mediated signals are differentially regulated in CLL. AKTS473 phosphorylation was present, but not differentially modulated in peripheral blood CLL cells of the patient subsets (Figure 1). Phosphorylation of S6S235/236 and 4EBP1T37/46 were also differentially regulated when the same CLL patient cohorts were divided by Binet stage, with poorer prognostic (Stage C) patients exhibiting significantly reduced S6 phosphorylation and elevated 4EBP1 phosphorylation (Suppl. Figure 1). These data suggest that primary CLL cells may be preferentially targeted by dual mTORC1/2 inhibitors, rather than rapalogs that are allosteric inhibitors of mTORC1 and unable to elicit a full inhibition of mTORC1 activity (24).
Figure 1. Downstream mTORC1/mTORC2 substrates are differentially regulated in primary CLL cells isolated from distinct prognostic subgroups.

Protein lysates were generated from freshly-isolated B lineage cells (CLL/mature B cells) purified from peripheral blood of CLL patients or healthy donors. CLL patients were stratified based on cytogenetic abnormalities to indicate prognosis: normal/del(13q) (good), del(11q), del(17p) (poor). A. Western blotting was performed to determine the phosphorylated levels of mTORC1 downstream targets (S6S235/236, 4EBP1T37/46 and RICTORT1135) and mTORC2 substrate (AKTS473) together with GAPDH as a loading control. Representative n=2 for each subset shown. B. Densitometry ratios of phosphorylated proteins vs. GAPDH from the blot are shown. n≥6 for each patient subset, n≥4 healthy controls.
Treatment of primary CLL cells with increasing concentrations of AZD8055 for 48 h resulted in a modest but significant reduction in cell viability at clinically achievable concentrations (Figure 2A) (25). Analysis of patient responses to 10 nM rapamycin or 100 nM AZD8055 compared with 1 μM ibrutinib, revealed a significant and comparable reduction in cell viability and an elevation in apoptosis upon AZD8055 or ibrutinib treatment, while rapamycin did not affect CLL cell viability (Figure 2B&C) in agreement with previous results (26). Subdivision of prognostic subsets based on cytogenetics revealed that AZD8055 treatment induced significantly higher levels of apoptosis in poorer prognostic subsets [del(11q) or del(17p)] (Figure 2D). Importantly, the viability of B and T lymphocytes derived from healthy donors was not significantly reduced upon treatment with AZD8055, rapamycin or ibrutinib highlighting the selectivity of inhibitors for CLL cells (Figure 2E).
Figure 2. AZD8055 treatment selectively and significantly reduces primary CLL cell viability preferentially in poor prognostic subsets.

A. Primary CLL cells were incubated with increasing concentrations of AZD8055 or left untreated for 48 h (n=12 CLL patient samples). Cell viability was analyzed by flow cytometry, with the percentage of viable cells assessed as Annexin V-7-AAD-; B. Cell viability was compared upon treatment of primary CLL cells with 100 nM AZD8055 (AZD; blue), 10 nM rapamycin (RAP; red) or 1 μM ibrutinib (IB; pink) for 48 h, compared with NDC (white), (n=15); C. The level of apoptosis (Annexin V+7-AAD-) was compared upon treatment of primary CLL cells with 100 nM AZD8055, 10 nM rapamycin or 1 μM ibrutinib for 48 h, relative to NDC. p values generated by a paired two tailed t test (n=15). Inset: Western blotting was performed to show the levels of PARP cleavage (cPARP) in primary CLL cells treated with 100 nM AZD8055, 10 nM rapamycin or 1 μM ibrutinib for 48 h, or left untreated (NDC). GAPDH is included as a loading control. D. The level of apoptosis induced in primary CLL cells treated with 100 nM AZD8055 minus background (NDC) was compared between cytogenetic subgroups: good (Norm/del(13q)) vs poor (del(11q) or (17p)). p value generated by an unpaired two tailed t test (n≥7 per subgroup). E. Freshly-isolated peripheral blood mononuclear cells from healthy donors, were incubated with 100 or 300 nM AZD8055, 10 nM RAP or 1 μM IB for 48 h, compared with NDC. Cell viability of B (CD19+) and T (CD3+) populations was assessed, as indicated (n=6).
mTOR inhibition overcomes BCR-mediated survival signals
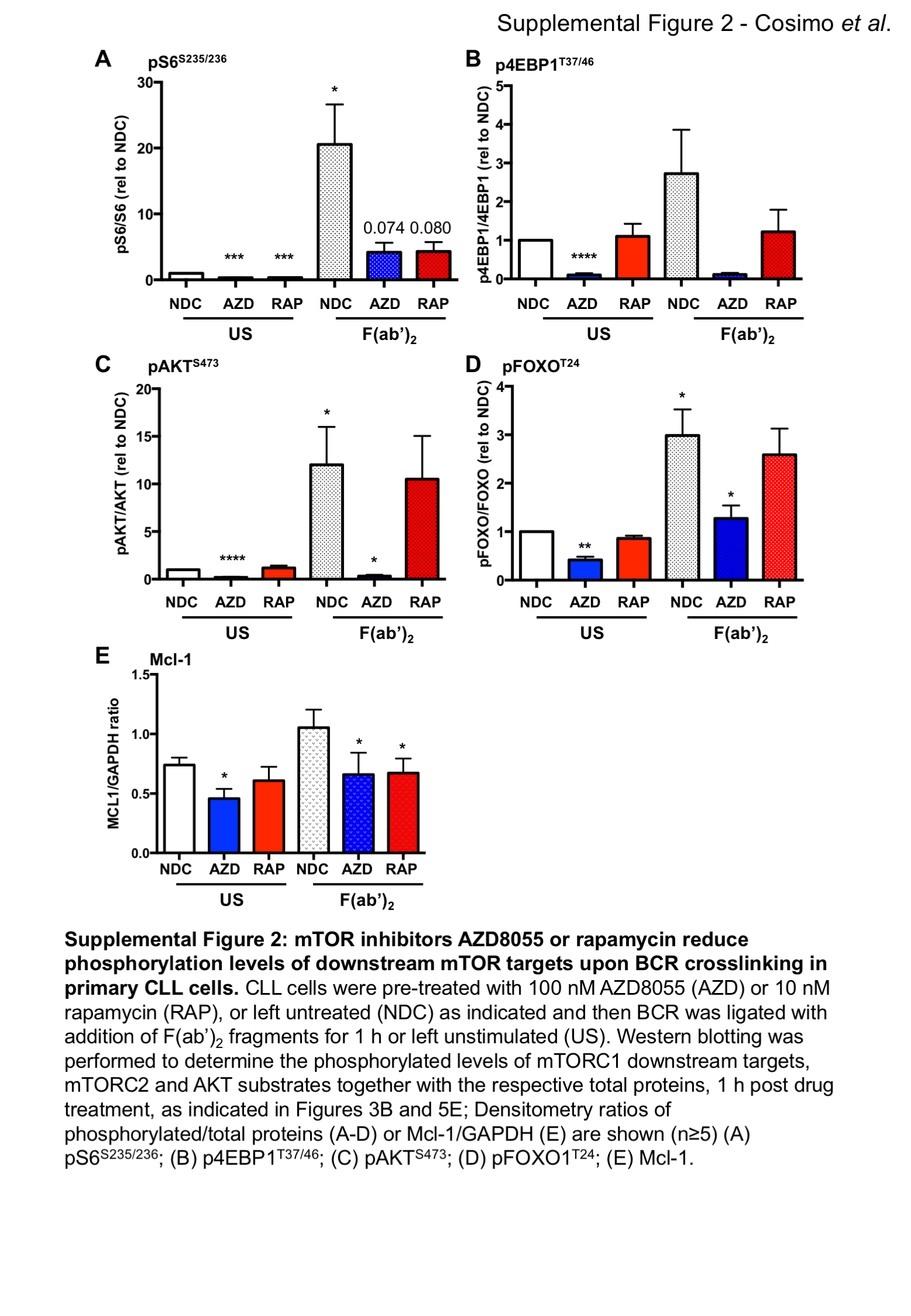
Stimulation of primary CLL cells with F(ab’)2 fragments, to replicate ligation of the BCR with soluble antigen, resulted in a modest elevation in CLL survival and Mcl-1 protein expression, together with an elevation in phosphorylation of downstream targets of mTORC1 (S6S235/236) and mTORC2 (AKTS473), and increased phosphorylation of pERK1/2T202/Y204 (Figure 3A-C; Suppl. Figure 2). 4EBP1T37/46 phosphorylation was not altered downstream of BCR ligation. Treatment of CLL cells with 100 nM AZD8055 or 10 nM rapamycin in F(ab’)2 stimulated CLL cells resulted in a significant inhibition of BCR-mediated survival and a reduction in Mcl-1 expression, which was accompanied by a reduction in S6 phosphorylation with both inhibitors, as expected (Figure 3A-C; Suppl. Figure 2A-C, E). AZD8055 also significantly inhibited 4EBP1T37/46 and AKTS473 phosphorylation (Figure 3C; Suppl. Figure 2B&C). These studies indicate that both mTORC1 and mTORC2 regulate BCR-mediated CLL survival, which can be targeted by mTOR inhibitors.
Figure 3. mTOR inhibition with AZD8055 or rapamycin treatment overcomes the pro-survival effect of BCR crosslinking in primary CLL cells and in a poor prognostic CLL mouse model in vitro.

CLL cells were pre-treated with 100 nM AZD8055 (AZD) or 10 nM rapamycin (RAP), or left untreated (NDC) and then BCR was ligated with addition of F(ab’)2 fragments for 1 or 48 h or left unstimulated (US), as indicated. A. Cell viability was analyzed by flow cytometry after 48 h incubation (n=14). p value generated by a paired two tailed t test; B. Western blotting of the anti-apoptotic protein Mcl-1 was performed after 48 h incubation; C. Western blotting was performed to determine the phosphorylated levels of mTORC1 downstream targets (S6S235/236 and 4EBP1T37/46) and mTORC2 substrate (AKTS473) and pERK (pERK1/2T202/Y204) together with the respective total proteins and a GADPH loading control, 1 h post drug treatment; D. MIEV- or PKCα-KR-HSPC-derived mouse cells from OP9 co-cultures (post d14) were treated for 24 h in the presence of 100 nM AZD8055 or 10 nM rapamycin or left untreated as indicated. Western blotting was performed to determine the phosphorylated levels of mTORC1 downstream targets (S6S235/236 and 4EBP1T37/46) and mTORC2 substrate (AKTS473) together with the respective total proteins or GAPDH. E. 5 x 104 d15 MIEV- or PKCα-KR-HSPC-derived cells were plated on OP9 co-cultures and treated for 48 h in the presence of 200 nM AZD8055 or 10 nM rapamycin or left untreated as indicated, and cells were counted. Data are represented as mean (± SEM) of at least 3 biological replicates. F. Apoptosis was analyzed by flow cytometry after 48 h treatment of MIEV- or PKCα-KR-HSPC-derived cells. Annexin V+7AAD- cells represent early apoptotic cells. Data are represented as mean (± SEM) of at least 4 biological replicates. Significance was determined by a 2-way ANOVA.
Dual mTOR inhibitors reduce tumor load in a CLL-like mouse model in vivo
To address the role of mTOR inhibition in CLL progression, in vitro-generated leukemic cells or mice with established leukemia from a poor-prognostic, proliferating CLL-like mouse model were treated with rapamycin or AZD8055 (23, 27). Analysis of mTOR substrate activation in PKCα-KR-expressing (CLL-like) or MIEV (empty vector control) cells revealed elevated phosphorylation of downstream mTOR targets (S6S235/236, AKTS473) in CLL-like cells (Figure 3D). Moreover, rapamycin and AZD8055 treatment inhibited phosphorylation of the appropriate targets, as noted in CLL patient samples (Figure 3D; Suppl. Figure 3A-C). Of note, rapamycin treatment of MIEV and PKCα-KR-expressing cells resulted in an elevation in AKTS473 phosphorylation, indicating an activation of mTORC2 activity upon S6K inhibition. While rapamycin and AZD8055 inhibited proliferation similarly in CLL-like cells (Figure 3E), AZD8055-induced apoptosis selectively in PKCα-KR-expressing cells, at a significantly higher level than measured in rapamycin-treated cells (Figure 3F), as observed in human CLL samples.
Mice were treated once daily with either AZD8055 (20 mg/kg, OG) or rapamycin (4 mg/kg, ip) or appropriate vehicle after confirmation of CLL-like disease development in blood. An inhibition of S6S235/236 and AKTS473 phosphorylation was demonstrated in CLL-like cells isolated from the spleen and BM of mice 2 h post the first treatment with AZD8055 in vivo, while rapamycin only induced a slight inhibition in S6S235/236 phosphorylation, determined by intracellular flow cytometry (Figure 4A). Flow cytometric analysis of the organs removed from mice after 14 d of treatment revealed that AZD8055-treated mice exhibited a significant reduction in tumor burden (GFP+ CD19+ cells) both in terms of percentage in the spleen and BM, and cell numbers in the BM (Figure 4B-D). These data indicate that inhibition of mTORC1/2 results in a more robust inhibitory response, clearing disease more effectively, likely due to the combined inhibition of proliferation and induction of apoptosis (Figure 3E&F).
Figure 4. Treatment of leukemic mice with AZD8055 leads to a significant reduction in CLL tumor load in vivo.

A. After confirmation of a population of CLL-like cells in the blood (≥ 5% GFP+CD19+), mice were dosed with 20 mg/kg AZD8055, 4 mg/kg rapamycin or respective vehicle controls for 2 h. Thereafter, cells were isolated from BM and spleen (SP) and levels of AKTS473 and S6S235/235 phosphorylation was determined on GFP+B220+ cells with intracellular flow cytometric analysis. Data are represented as mean fluorescence intensity (MFI) ratio of stained sample over isotype control (n=6). p value was generated using the student’s unpaired t-test. B. After confirmation of a population of CLL-like cells in the blood (≥ 0.4% GFP+CD19+), mice were dosed daily with 20 mg/kg AZD8055 (OG), 4 mg/kg rapamycin (IP) or respective vehicle control for up to 14 days. Thereafter, blood, BM and spleen were analyzed for leukemic burden. Representative flow cytometric analyses of vehicle-, AZD8055- or rapamycin-treated mice are shown. Data shown are analyzed for CLL cell markers, CD19 and GFP after live and size (FSC/SSC) and CD45+ gating. The percentage of GFP+ CLL-like cells within the total population is shown; C. Percentage and D. Number of GFP+ CD45+CD19+ cells are shown in treated mice in the organs indicated. All data shown are the mean (± SEM) of 5 individual mice. p values were generated using the student’s unpaired t-test.
mTORC2-mediated negative regulation of FOXO promotes CLL survival
A key function of AKT is the inactivation of the FOXO transcription factor family (21). Analysis of FOXO family member gene expression in freshly isolated CLL cells from peripheral blood revealed that FOXO1 and FOXO4 were significantly upregulated, while FOXO3 expression did not differ significantly compared to normal B cells (Figure 5A). Stratification of FOXO family gene expression in CLL revealed that FOXO1 was significantly upregulated in poorer prognostic subsets, in Binet stage C and patients that have previously received treatment (Suppl. Figure 4). Analysis of FOXO1 expression in patient LN sections, isolated from a cross section of distinct prognostic subsets, revealed FOXO1 upregulation in samples carrying poor prognostic markers compared with LN biopsies from patients with good prognostic markers (Figure 5B). FOXO1 upregulation in CLL patients showed a significant positive correlation with Ki-67 (r2=0.620, p=0.0003; n=16), supporting an upregulation of FOXO1 expression in patients exhibiting progressive disease (Figure 5B).
Figure 5. FOXO1 is reactivated with AZD8055 treatment upon BCR ligation.

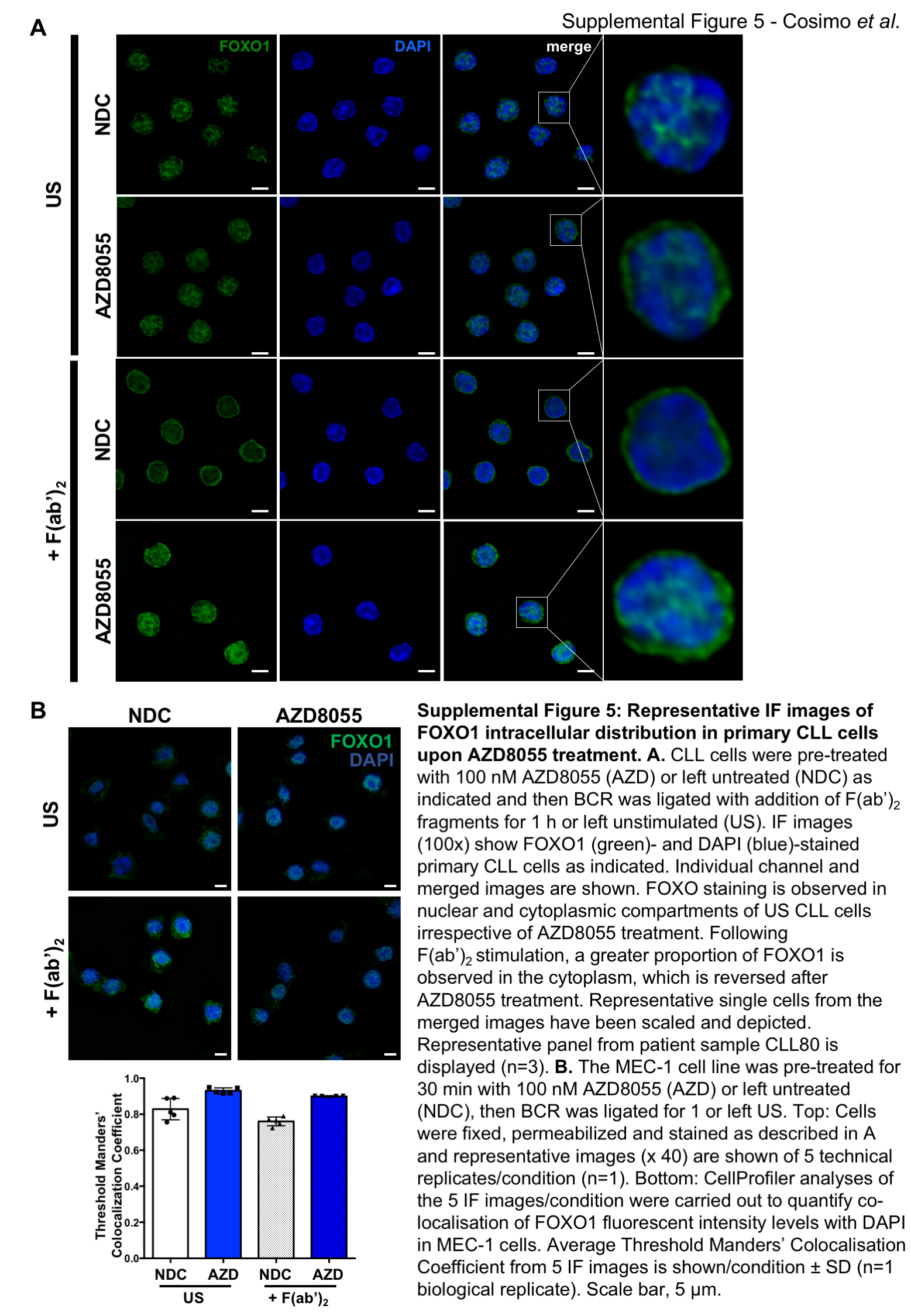
A. RNA was isolated from fresh CLL samples (n=28) and qPCR was performed to measure expression levels of FOXO gene family members. A scatterplot of gene expression is shown, each symbol denotes an individual CLL sample. Bars indicate the mean fold change in gene expression relative to 10 healthy B cell samples ± SEM; B. IHC was performed on LN biopsies from CLL patients of favourable (n=11) and poor prognosis (n=9). Representative images are shown for FOXO1 staining. FOXO1 and Ki-67 stained LN sections were scored and compared between favourable and poor prognostic CLL samples; C. qPCR (from A; n=28) was performed to measure expression levels of FOXO target genes as indicated. Bars indicate the mean fold change in gene expression relative to 10 healthy samples ± SEM; D. Protein lysates were generated from in vitro co-cultures of MIEV- or PKCα-KR-transduced mouse cell cultures. Western blotting compared the phosphorylated and expression levels of FOXO1T24 and total FOXO1 respectively, compared with loading control GAPDH. E-G. CLL cells were pre-treated with 100 nM AZD8055, 10 nM rapamycin, or left untreated (NDC) as indicated. BCR was ligated with addition of F(ab’)2 fragments for 1 h or US. E. Western blotting assessed phosphorylated levels of FOXO1T24 and AKTS473 and expression levels of total FOXO1 and AKT. GAPDH is included as a loading control; F. CLL cells were fixed and permeabilized and stained with anti-FOXO1 Ab (green) and DAPI (blue). Top: A representative 100x deconvoluted image of primary CLL cells is shown (n=3); Scale bar, 5 µm. Bottom: Analyses of the IF images (x40) were performed in CellProfiler to quantify colocalisation of FOXO1 fluorescent intensity levels with DAPI (nuclear stain) in individual cells from individual CLL patients. The average Threshold Manders’ Colocalisation Coefficient for each condition from 3 individual CLL patient samples is shown. G. MEC-1 cells were pre-treated for 30 min with 100 nM AZD8055 (AZD) or left untreated (NDC) as indicated and then BCR was ligated for 1 or left US. Thereafter nuclear and cytoplasmic fractions were prepared and immunoblotted alongside whole cell lysates (WCL). Western blotting shows the levels of FOXO1, lamin A/C (nuclear) and γ-tubulin (cytoplasmic) proteins in each condition. Representative blot shown of n=3 biological replicates.
As FOXOs are widely considered to promote cell cycle arrest, the alignment of FOXO1 upregulation with increased Ki-67, identified morphologically in continuous sections suggests that while FOXO1 expression is increased, it is functionally inactive in CLL cells. Supporting this, established FOXO target genes were deregulated in CLL cells: CDKN1A (p21) and CDKN1B (p27) downregulation and CCND1-3 (cyclin D1-3) upregulation compared to normal controls (Figure 5C) (28). Interestingly, PKCα-KR-CLL-like cells also exhibited elevated phosphorylation and expression levels of FOXO1 compared with MIEV cells (Figure 5D; Suppl. Figure 3D&E). BCR ligation resulted in a significant upregulation of pFOXO1T24, coupled with a concomitant elevation in pAKTS473, phosphorylation events that were reduced by treating CLL cells with AZD8055 but not rapamycin (Figure 5E; Suppl. Figure 2C&D). These data indicate that signals within the tumor microenvironment that stimulate mTORC2-AKT-mediated signaling may inhibit FOXO1 activity, thus priming CLL cells for proliferation within the lymphoid organs. IF analysis revealed that BCR ligation significantly enhanced cytoplasmic localisation of FOXO1, compared to unstimulated CLL cells (Figure 5F; Suppl. Figure 5). However, AZD8055 treatment of F(ab’)2-stimulated cells resulted in a significant increase in nuclear FOXO1 localization compared to untreated (NDC) F(ab’)2-stimulated cells (Figure 5F; Suppl. Figure 5). These data were confirmed by sub-cellular fractionation of MEC-1 cells, with increased FOXO1 levels in the nuclear compartment upon AZD8055 treatment, being mirrored by a reduction of FOXO1 in cytoplasm (Figure 5G). These data indicate that FOXO1 is a target for inactivation downstream of BCR-mediated AKT/mTOR activity that can be reversed by treatment with mTORC1/2 inhibitors.
Dual mTOR inhibitors synergize with ibrutinib to enhance CLL cell death in vitro and in vivo
AKT is unlikely to be fully inhibited in the presence of AZD8055 due to activation of AKT through PI3K/PDK1 activation leading to AKTT308 phosphorylation, as indicated by residual pFOXO1T24 signal detected (Figure 5D). Therefore we investigated the impact of AZD8055 treatment on cell viability and FOXO1 phosphorylation/activity in combination with ibrutinib in CLL cells (29). Initially using the Chou and Talalay method (30), we identified synergy between 100 nM AZD8055 and 1 μM ibrutinib (CI=0.398±0.34). We further demonstrated an enhanced induction of apoptosis, as indicated by an increase in AnnexinV+7-AAD- cells and an elevation in PARP cleavage, with both drugs in combination in the presence and absence of BCR ligation (Figure 6A&B). Furthermore, there was a significant reduction in the expression of Mcl-1 in presence of AZD8055 and ibrutinib alone and in combination (Figure 6B; Suppl. Figure 6).
Figure 6. Combination of AZD8055 with Ibrutinib further increases FOXO1 function upon BCR ligation in primary CLL cells.

CLL cells were pre-treated for 30 min with 100 nM AZD8055 (AZD) or 1 μM ibrutinib (IB) alone or in combination, or left untreated (NDC) as indicated and then BCR was ligated with addition of F(ab’)2 fragments for 1, 24 or 48 h or left US, as indicated. A. Cell viability was analyzed by flow cytometry after 48 h incubation (n=7). p value generated by a paired two tailed t test; B. Western blotting was performed to determine the level of PARP cleavage and Mcl-1 expression after 48 h incubation (n=4). GAPDH is included as a loading control; C&D. Western blotting was performed to determine the phosphorylated levels of FOXO1T24/FOXO1 (C), AKTS473/AKT (C), S6S235/236/S6 (D), 4EBP1T37/45/4EBP1 (D), AKTT308/AKT (D) and pERK1/2T202/Y204/ERK1/2 (D) (n≥4); E. Nuclear lysates were generated from CLL cells and DNA binding activity of FOXO1 was assessed in each condition, as indicated (n=5). Statistical significance calculated using a student’s t test; F. RNA was prepared from CLL cells (n≥8 individual patient samples) treated with AZD8055 (AZ) or ibrutinib (IB) alone or together (CO) in the presence or absence of BCR ligation after 24 h incubation as indicated, and mRNA expression levels of FOXO1, FOXO3, CCND1-2, BCL2L1, and BCL2L11 were determined by qPCR. Each gene is expressed relative to GUSB reference gene and calibrated to ND US or ND F(ab’)2 samples. Data are mean ± SEM, and p values generated by a paired two-tailed t-test. G. After confirmation of a population of CLL-like cells in the blood, mice were dosed daily with 15 mg/kg AZD2014 (OG), 12 mg/kg ibrutinib (OG) alone or in combination, or respective vehicle control for up to 14 days. Thereafter, blood, BM and spleen were analyzed for leukemic burden. The average percentage of GFP+ CLL-like cells in the blood is shown for each treatment arm (top); Cell number of GFP+ CD45+CD19+ population is shown in treated mice in the BM (middle) and SP (bottom). All data shown are the mean (± SEM) of 5 individual mice. p values were generated using the student’s unpaired t-test.
Combination of ibrutinib with AZD8055 enhanced inhibition of S6S235/236, AKTS473 and FOXOT24 phosphorylation in the presence of BCR stimulation (Figure 6C&D; Suppl. Figure 6). As expected there was a more complete inhibition of AKTS473 phosphorylation compared with AKTT308 upon treatment with AZD8055, in the presence or absence of ibrutinib in unstimulated CLL cells (Figure 6C&D; Suppl. Figure 6). However, BCR-mediated stimulation of AKTT308 phosphorylation was reduced to basal levels by AZD8055 or ibrutinib treatments (Figure 6D; Suppl. Figure 6). Interestingly, while ibrutinib was unable to inhibit 4EBP1T37/46 phosphorylation, it reduced phosphorylation of ERK1/2T202/Y204 suggesting that synergy between AZD8055 and ibrutinib occurs through targeting of discrete signaling pathways whilst inducing a more robust inhibition of the same pathways (Figure 6C&D; Suppl. Figure 6). Importantly, BCR ligation resulted in a significant downregulation of FOXO1 activity, which was significantly elevated upon treatment with AZD8055 and ibrutinib (Figure 6E), upregulating FOXO1-target gene regulation: downregulation of CCND2 and BCL2L1 (Bcl-XL) and an upregulation of FOXO1, FOXO3 and BCL2L11 (BIM) (Figure 6E). Combination of AZD8055 with ibrutinib enabled a more robust effect on FOXO activity and FOXO-regulated genes, in the presence of BCR-mediated signals, with BCL2L1, CCND1 and CCND2 showing enhanced responses (Figure 6E).
To test the potential therapeutic effect of combining ibrutinib with dual mTOR inhibitors, mice with confirmed CLL-like disease in blood were treated once daily with a clinical analog of AZD8055, AZD2014 (Vistusertib (31); 15 mg/kg) or ibrutinib (12 mg/kg) alone or in combination, or with the appropriate vehicles. Flow cytometric analysis of the organs removed from mice after 14 days of treatment revealed that all treatment arms exhibited a reduction in tumor burden. Indeed, AZD2014 treatment enhanced ibrutinib treatment in this aggressive CLL mouse model both in terms of percentage of tumor cells in the blood and organs, and cell numbers in the BM (Figure 6G & Suppl Figure 7). Collectively, these studies indicate that the clinical response elicited by ibrutinib in CLL patients may be enhanced in combination with dual mTOR kinase inhibitors.
Discussion
We demonstrate that AZD8055 preferentially decreased cell viability of poor prognostic CLL subsets in vitro generating a more robust inhibitory response in comparison to rapamycin, and significantly reduced tumor load in an aggressive CLL mouse model in vivo. This enhanced inhibitory response by AZD8055 in poor prognostic CLL samples may in part be due to the differential activation status of individual mTOR substrates observed in CLL patient samples from distinct prognostic subsets. In our study, stratification of the basal phosphorylation/activation status of mTOR substrates in fresh CLL samples revealed a differential regulation of mTORC1 downstream substrates, with a decrease in S6 phosphorylation in poorer prognostic del(17p) subset. In addition to phosphorylating S6, S6K is responsible for phosphorylating RICTOR at T1135 and inactivating mTORC2 activity, thus generating a negative feedback loop between mTORC1 and mTORC2 (32). While our findings indicate that the inhibitory influence of S6K activity over mTORC2 is reduced in poorer prognostic CLL, AKT phosphorylation is not elevated in the poor prognostic samples, suggesting that a reduction in mTORC1-S6K activity may prime CLL cells to respond more robustly to microenvironmental stimulation. We also noted an upregulation in 4EBP1 phosphorylation in poorer prognostic CLL patient subsets, which would result in a reduction in binding to the mRNA cap-binding protein eIF4E. This event could enable the translation of proteins including Mcl-1 and c-Myc (33, 34), which are associated with CLL progression (35, 36).
Differential mTOR substrate activation status observed in CLL cells may affect the subsequent response to mTOR inhibitors, with CLL survival of poor prognostic patient samples exhibiting higher sensitivity upon treatment with mTORC1/2 inhibition, compared with rapamycin. Indeed, rapamycin is unable to fully block phosphorylation of 4EBP1 (24), and releases the mTORC1 feedback inhibition on mTORC2 enabling reactivation of AKT (17), suggesting that the efficacy of rapamycin is limited in poor-prognostic patients with reduced mTORC1-S6K activity. This is supported by recent data demonstrating an everolimus-sensitive cohort of CLL patients that carry good prognosis (mutated IgVH), that rely on mTORC1 signaling in a BCR independent manner (37). These studies highlight potential reasons for everolimus only demonstrating modest responses in CLL patients in clinical trial (16), and suggest that dual mTORC1/2 inhibition may provide a more robust inhibitory response, particularly in high-risk CLL patients.
Selectivity of AZD8055 for mTOR was demonstrated in CLL cells, with drug treatment potently inhibiting the mTORC2 phosphorylation site, AKTS473 while not affecting AKTT308 phosphorylation in unstimulated CLL cells. These studies indicate that AZD8055 does not inhibit PDK1 activity. Of note, upon BCR stimulation of CLL cells, treatment with AZD8055 or ibrutinib reduced AKTT308 phosphorylation to basal levels observed in unstimulated CLL cells. Reduction in phosphorylation of AKTT308 has been reported in other cell lines upon treatment with AZD8055 (38), and with additional mTOR kinase inhibitors such as PP242 (39). This is likely due a reduction in the ability of PDK1 to phosphorylate the T308 site on AKT when S473 phosphorylation is inhibited.
Potency of AZD8055 and rapamycin in vivo was tested by assessing the phosphorylation status of S6S235/236 and AKTS473 in the spleen and BM. We show a trend towards significance, more marked with AZD8055 than rapamycin. Referring to clinical trials with AZD8055 or everolimus, the time to maximal serum concentration for both drugs is similar (0.5-2 hr) when dosed orally (25, 40). Rapalogs have a longer half-life than AZD8055, which suggests that given the distinct routes of drug delivery in our model, it may have been appropriate to analyse the activation status of mTOR substrates after >1 dose to enable a steady state to be reached. However, we were concerned that the tumor load would be reduced to the point that it would be difficult to gain a statistically robust read-out of S6S235/236 and AKTS473 phosphorylation status.
One target of mTORC2-AKT-mediated signaling is the FOXO family of transcription factors that play a pivotal role in a multitude of cellular homeostatic processes in specific cell contexts in response to a plethora of environmental stimuli including nutrients, insulin and growth factors (21, 41). FOXO1 has been shown to play a critical role in the regulation of glucose and fatty acid metabolism in skeletal muscle, liver and heart, an exhibits reduced activity in adipocytes isolated from diabetic patients (21, 42). Interestingly, acute mTOR inhibition with AZD8055 treatment results in suppression of glycolysis and a reduction in insulin sensitivity of glucose uptake in muscle (43). While recent studies revealed an essential role for FOXOs in cancer progression, in the maintenance of leukemia-initiating cells in myeloid leukemias (AML and CML), and in promotion of breast tumor invasion (44–46), FOXOs were originally considered to behave as tumor suppressors as conditional triple deletion of FOXO1/3/4 in adult mice resulted in the development of thymic lymphomas and hemangiomas (47). A number of studies support the role of FOXO1 as a tumor suppressor in B cell malignancies: inactivating point mutations in FOXO1 have been identified in non-Hodgkin lymphoma (46, 48); FOXO1 expression is often reduced in Hodgkin lymphoma, mediastinal B cell lymphoma and in 20% of GCB-DLBCL patients compared with normal B lymphocytes (49, 50). These findings indicate a deregulation of FOXO1 by distinct mechanisms in B cell malignancies. We demonstrate that FOXO1 expression is significantly upregulated in primary CLL cells from peripheral blood and LN biopsies of patients exhibiting poor prognosis, positively correlating with Ki-67 in LN biopsies. These findings suggest that FOXO1 may represent a novel target and/or biomarker for progressive disease.
While upregulated in CLL cells, FOXO1 appears to be functionally inactive, as indicated by its cytoplasmic localization and deregulation of FOXO-target genes (CDKN1A, CDKN1B, CCND1-3), downstream of tumor microenvironmental signals generated in vivo. We demonstrate a significant upregulation in FOXO1T24 phosphorylation, and downregulation in FOXO1 activity, in CLL cells upon BCR crosslinking. FOXO1T24 phosphorylation is an AKT-mediated phosphorylation event that enables FOXO binding to 14-3-3 proteins and subsequent export to the cytoplasm. FOXOs are negatively regulated by key oncogenic signals including the PI3K/AKT/mTOR pathways, which are upregulated in CLL cells downstream of BCR- and CD40-ligation within the tumor microenvironment (22, 30). Inactivation of FOXO proteins downstream of these pathways, enabled by the reduced negative regulation of mTORC2 by mTORC1-S6K, may contribute towards CLL pathogenesis. Of note, BCR-mediated inactivation of FOXO1 is partially alleviated by treating CLL cells with AZD8055, through inhibition of mTORC2-AKT-mediated signals, promoting CLL cell death. A recent study using the Syk inhibitor, R406 to inhibit tonic BCR signaling in DLBCL supports the role of FOXO1 in mediating cytotoxic and anti-proliferative activities in B cell malignancies (51).
While targeted therapies, such as ibrutinib, have a positive impact on the survival rates of high-risk CLL patients, resistance mutations either in BTK or PLCγ2 arise in response to treatment, underlining the importance of developing novel treatment strategies for this patient subgroup (13). Combination therapies offer the potential to reduce the ability of the cancer cell to adapt to treatments by targeting distinct signaling components within the cell and mTOR represents an ideal target. We establish that FOXO1 function was further enhanced on combination of AZD8055 with ibrutinib, as indicated by improved inhibition of mTORC1/2-mediated signals. This in turn led to significantly reduced FOXO1T24 phosphorylation, particularly upon BCR ligation and significantly increased FOXO1 activity and the subsequent FOXO-mediated transcription programme. Furthermore, treatment of CLL-bearing mice with AZD2014 (in clinical trial in solid tumors and DLBCL) enhanced the ability of ibrutinib to reduce tumor load. Ibrutinib and AZD2014 have been demonstrated to synergize in DLBCL (ABC subtype) cell lines and subtypes of primary CNS lymphoma, to induce enhanced apoptosis (52, 53). These studies suggest that when BCR signaling plays a central role in disease pathogenesis such as CLL, combinations of BCR antagonists with mTOR kinase inhibitors may represent a promising treatment, due to the enhanced ability to overcome key microenvironmental signals. mTOR inhibitors have also been shown to synergize with BH3 mimetics in small-cell lung cancer by reducing Mcl-1 expression and enabling BIM-induced apoptosis: Venetoclax (ABT-199), which is used to treat relapsed CLL patients that are refractory to BCR antagonists, synergizes with NVP-BEZ235 (PI3K/mTOR inhibitor) and idelalisib (PI3Kδ inhibitor) by downregulating Mcl-1 in DLBCL cell lines (54). Collectively, our studies demonstrate that targeting mTORC1/mTORC2 shows promise as a future therapy for high-risk CLL patients, and highlights the importance of inhibiting mTORC2-AKT-mediated signals to enable activation of FOXO1-mediated molecular braking system. In particular, dual mTORC1/2 inhibitors coupled with current therapies suitable for high-risk CLL patients may offer a rational combination to increase the proportion of minimal residual disease negative remissions, thus reducing the development of CLL clones with resistance mutations.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational Relevance.
Chronic lymphocytic leukemia (CLL) is a heterogeneous disease; some patients exhibit an indolent disease, while others experience a highly chemotherapy-refractory disease, highlighting the need for prognostic markers and novel therapeutic approaches. B cell receptor (BCR) ligation generates signals that play a pivotal role in promoting progression of the leukemic clone: inhibiting the signals that orchestrate these events is key to disrupting disease progression. We establish that the mechanistic target of rapamycin (mTOR) signaling cascade is differentially regulated across prognostic cohorts of CLL patients and that the dual mTORC1/2 inhibitor AZD8055 preferentially decreases viability of cells derived from poor prognostic CLL subsets through inhibition of mTORC2-AKT-mediated signals and subsequent engagement of the FOXO1-regulated molecular braking system. FOXO1 function was further enhanced upon combination of AZD8055 with the BTK inhibitor ibrutinib, indicating that dual mTOR inhibitors may show promise as future CLL therapies, particularly in combination with ibrutinib.
Statement of Significance.
We demonstrate that the mTOR signaling cascade is differentially regulated in distinct prognostic CLL cohorts, and that dual mTORC1/2 inhibitors preferentially induce apoptosis of poor prognostic CLL subsets. Our data establish that mTORC2 inhibition reduces AKT-mediated signals, leading to a reactivation of FOXO1-regulated molecular braking system.
Acknowledgements
The authors thank all CLL patients and blood donors for donating blood samples. AZD8055 and AZD2014 were provided by AstraZeneca.
Financial Support: This study was funded by grants from Medical Research Council UK (MRC) ‘Mechanism of Disease’ grant in collaboration with AstraZeneca awarded to AMM & AMcC (MR/K014854/1) and Bloodwise awarded to AMM, EC & OJS (15041). Cell sorting facilities were funded by the Kay Kendall Leukaemia Fund awarded to AMM (KKL501) and the Howat Foundation. EC and JH are funded by the Bloodwise project grant (15041), AT was funded by a Bloodwise project grant awarded to AMM (13012), MM was funded by a PhD studentship from Friends of Paul O’Gorman Leukemia Research Centre, AKH was supported by a KKLF Clinical training fellowship (KKL838), NM was funded by a MRC-DTP PhD studentship.
Abbreviations
- BCR
B cell receptor
- BM
bone marrow
- CLL
chronic lymphocytic leukemia
- DLBCL
diffuse large B cell lymphoma
- FOXO
forkhead box proteins subgroup O
- GFP
green fluorescent protein
- HSPC
haemopoietic stem and progenitor cells HSPC
- mTOR
mechanistic target of rapamycin
- NDC
no drug control
- NSG
NOD-SCID-γc−/−
- PKCα-KR
dominant negative protein kinase Cα
- RAG-2−/−
recombinase activating gene-2-deficient
Footnotes
Author contributions:
EC designed/performed the majority of experiments, analyzed and interpreted the data, carried out statistical analysis and drafted the manuscript; AT, AMacD, MM, AKH, NM, JH performed some experiments, purified CLL patient samples and carried out data analysis; KD provided technical assistance with the in vivo model and analyzed the data; DO’R & MC performed the IHC of LN biopsies and scored the slides; SG & SC assisted in obtaining funding, provided reagents and supervised the studies; MTL, OS & AMcC obtained funding for the study and supervised the studies; AMM obtained funding for the study, designed the research, supervised the studies, analyzed and interpreted the data and wrote the manuscript. All authors reviewed the manuscript.
Conflict of Interest Statement: At the time of the study, SG and SC were employed by AstraZeneca. Additional authors declare no potential conflicts of interest.
References
- 1.Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Primers. 2017;3 doi: 10.1038/nrdp.2016.96. 16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zenz T, Mertens D, Küppers R, Döhner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10:37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 3.Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–7. [PubMed] [Google Scholar]
- 4.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–54. [PubMed] [Google Scholar]
- 5.Wiestner A, Rosenwald A, Barry TS, Wright G, Davis RE, Henrickson SE, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood. 2003;101:4944–51. doi: 10.1182/blood-2002-10-3306. [DOI] [PubMed] [Google Scholar]
- 6.Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 7.Dreger P, Schetelig J, Andersen N, Corradini P, van Gelder M, Gribben J, et al. Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents? Blood. 2014;124:3841–9. doi: 10.1182/blood-2014-07-586826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rickert RC. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013;13:578–91. doi: 10.1038/nri3487. [DOI] [PubMed] [Google Scholar]
- 9.Petlickovski A, Laurenti L, Li X, Marietti S, Chiusolo P, Sica S, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood. 2005;105:4820–7. doi: 10.1182/blood-2004-07-2669. [DOI] [PubMed] [Google Scholar]
- 10.Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008;111:846–55. doi: 10.1182/blood-2007-05-089037. [DOI] [PubMed] [Google Scholar]
- 11.Brown JR, Byrd JC, Coutre SE, Benson DM, Flinn IW, Wagner-Johnston ND, et al. Idelalisib, an inhibitor of phosphatidylinositol 3-kinase p110δ, for relapsed/refractory chronic lymphocytic leukemia. Blood. 2014;123:3390–7. doi: 10.1182/blood-2013-11-535047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168:960–76. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zent CS, LaPlant BR, Johnston PB, Call TG, Habermann TM, Micallef IN, et al. The treatment of recurrent/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL) with everolimus results in clinical responses and mobilization of CLL cells into the circulation. Cancer. 2010;116:2201–7. doi: 10.1002/cncr.25005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blunt MD, Carter MJ, Larrayoz M, Smith LD, Aguilar-Hernandez M, Cox KL, et al. The PI3K/mTOR inhibitor PF-04691502 induces apoptosis and inhibits microenvironmental signaling in CLL and the Eμ-TCL1 mouse model. Blood. 2015;25:4032–41. doi: 10.1182/blood-2014-11-610329. [DOI] [PubMed] [Google Scholar]
- 18.Ding W, Shanafelt TD, Lesnick CE, Erlichman C, Leis JF, Secreto C, et al. Akt inhibitor MK2206 selectively targets CLL B-cell receptor induced cytokines, mobilizes lymphocytes and synergizes with bendamustine to induce CLL apoptosis. Br J Haematol. 2014;164:146–50. doi: 10.1111/bjh.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011;118:3603–12. doi: 10.1182/blood-2011-05-352492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuang J, Hawkins SF, Glenn MA, Lin K, Johnson GG, Carter A, et al. Akt is activated in chronic lymphocytic leukemia cells and delivers a pro-survival signal: the therapeutic potential of Akt inhibition. Haematologica. 2010;95:110–8. doi: 10.3324/haematol.2009.010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 22.Cosimo E, McCaig AM, Carter-Brzezinski LJ, Wheadon H, Leach MT, Le Ster K, et al. Inhibition of NF-κB-mediated signaling by the cyclin-dependent kinase inhibitor CR8 overcomes prosurvival stimuli to induce apoptosis in chronic lymphocytic leukemia cells. Clin Cancer Res. 2013;19:2393–405. doi: 10.1158/1078-0432.CCR-12-2170. [DOI] [PubMed] [Google Scholar]
- 23.Nakagawa R, Soh JW, Michie AM. Subversion of PKCa signaling in hematopoietic progenitor cells results in the generation of a B-CLL-like population in vivo. Cancer Research. 2006;66:527–34. doi: 10.1158/0008-5472.CAN-05-0841. [DOI] [PubMed] [Google Scholar]
- 24.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naing A, Aghajanian C, Raymond E, Olmos D, Schwartz G, Oelmann E, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br J Cancer. 2012;107:1093–9. doi: 10.1038/bjc.2012.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decker T, Hipp S, Ringshausen I, Bogner C, Oelsner M, Schneller F, et al. Rapamycin-induced G1 arrest in cycling B-CLL cells is associated with reduced expression of cyclin D3, cyclin E, cyclin A, and survivin. Blood. 2003;101:278–85. doi: 10.1182/blood-2002-01-0189. [DOI] [PubMed] [Google Scholar]
- 27.Nakagawa R, Vukovic M, Tarafdar A, Cosimo E, Dunn K, McCaig AM, et al. Generation of a poor prognostic chronic lymphocytic leukemia-like disease model: PKCα subversion induces an upregulation of PKCβII expression in B lymphocytes. Haematologica. 2015;100:499–510. doi: 10.3324/haematol.2014.112276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van der Vos KE, Coffer PJ. The Extending Network of FOXO Transcriptional Target Genes. Antioxid Redox Signal. 2011;14:579–92. doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- 29.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCaig AM, Cosimo E, Leach MT, Michie AM. Dasatinib inhibits B cell receptor signalling in chronic lymphocytic leukaemia but novel combination approaches are required to overcome additional pro-survival microenvironmental signals. Br J Haematol. 2011;153:199–211. doi: 10.1111/j.1365-2141.2010.08507.x. [DOI] [PubMed] [Google Scholar]
- 31.Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HM, Gomez S, et al. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23:1212–6. doi: 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 32.Dibble CC, Asara JM, Manning BD. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol Cell Biol. 2009;29:5657–70. doi: 10.1128/MCB.00735-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 34.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–61. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herishanu Y, Pérez-Galán P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117:563–74. doi: 10.1182/blood-2010-05-284984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pepper C, Lin TT, Pratt G, Hewamana S, Brennan P, Hiller L, et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112:3807–17. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]
- 37.Dietrich S, Oleś M, Lu J, Sellner L, Anders S, Velten B, et al. Drug-perturbation-based stratification of blood cancer. J Clin Invest. 2018;128:427–45. doi: 10.1172/JCI93801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Research. 2010;70:288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 39.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7 doi: 10.1371/journal.pbio.1000038. e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Donnell A, Faivre S, Burris HA, Rea D, Papadimitrakopoulou V, Shand N, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26:1588–95. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 41.Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482–95. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- 42.Rajan MR, Nyman E, Kjølhede P, Cedersund G, Strålfors P. Systems-wide Experimental and Modeling Analysis of Insulin Signaling through Forkhead Box Protein O1 (FOXO1) in Human Adipocytes, Normally and in Type 2 Diabetes. J Biol Chem. 2016;291:15806–19. doi: 10.1074/jbc.M116.715763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleinert M, Sylow L, Fazakerley DJ, Krycer JR, Thomas KC, Oxbøll AJ, et al. Acute mTOR inhibition induces insulin resistance and alters substrate utilization in vivo. Mol Metab. 2014;3:630–41. doi: 10.1016/j.molmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463:676–80. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 45.Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146:697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Gan B, Liu D, Paik JH. FoxO family members in cancer. Cancer Biol Ther. 2011;12:253–9. doi: 10.4161/cbt.12.4.15954. [DOI] [PubMed] [Google Scholar]
- 47.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–23. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJ, et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood. 2013;121:3666–74. doi: 10.1182/blood-2013-01-479865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xie L, Ushmorov A, Leithäuser F, Guan H, Steidl C, Färbinger J, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119:3503–11. doi: 10.1182/blood-2011-09-381905. [DOI] [PubMed] [Google Scholar]
- 50.Xie L, Ritz O, Leithäuser F, Guan H, Färbinger J, Weitzer CD, et al. FOXO1 downregulation contributes to the oncogenic program of primary mediastinal B-cell lymphoma. Oncotarget. 2014;5:5392–402. doi: 10.18632/oncotarget.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szydlowski M, Kiliszek P, Sewastianik T, Jablonska E, Bialopiotrowicz E, Gorniak P, et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B-cell lymphomas. Blood. 2016;127:739–48. doi: 10.1182/blood-2015-06-654111. [DOI] [PubMed] [Google Scholar]
- 52.Ezell SA, Mayo M, Bihani T, Tepsuporn S, Wang S, Passino M, et al. Synergistic induction of apoptosis by combination of BTK and dual mTORC1/2 inhibitors in diffuse large B cell lymphoma. Oncotarget. 2014;5:4990–5001. doi: 10.18632/oncotarget.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grommes C, Pastore A, Palaskas N, Tang SS, Campos C, Schartz D, et al. Ibrutinib Unmasks Critical Role of Bruton Tyrosine Kinase in Primary CNS Lymphoma. Cancer Discov. 2017;7:1018–29. doi: 10.1158/2159-8290.CD-17-0613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015;6:e1593. doi: 10.1038/cddis.2014.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.