

Abstract
A series of 8-hydroxy quinolines were identified as potent inhibitors of catechol O-methyltransferase (COMT) with selectivity for the membrane-bound form of the enzyme. Small substituents at the 7-position of the quinoline were found to increase metabolic stability without sacrificing potency. Compounds with good pharmacokinetics and brain penetration were identified and demonstrated in vivo modulation of dopamine metabolites in the brain. An X-ray co-crystal structure of compound 21 in the S-COMT active site shows chelation of the active site magnesium similar to catechol-based inhibitors. These compounds should prove useful for treatment of many neurological and psychiatric conditions associated with compromised cortical dopamine signaling.
Graphical Abstract
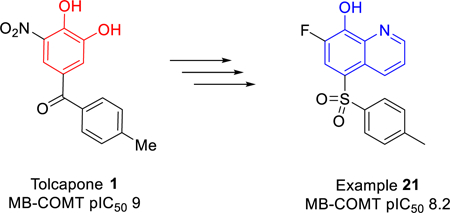
INTRODUCTION
Proper functioning of dopamine signaling in the prefrontal cortex (PFC) is critical for a number of cognitive and behavioral processes that are impaired in a variety of conditions such as ADHD, obsessive-compulsive disorder, traumatic brain injury, and schizophrenia.1 One approach towards selective modulation of dopamine signaling in the PFC is by inhibition of catechol O-methyltransferase (COMT), which is the predominant means of dopamine clearance in the PFC because of the lack of expression of synaptic dopamine transporters in this region,2 in contrast to the striatum.
COMT is a magnesium-containing metalloenzyme that transfers a methyl group from the cofactor S-adenosylmethionine (SAM) to a number of endogenous and exogenous catechols such as dopamine, catecholestrogens, and dietary polyphenols. The COMT enzyme has two isoforms, designated S-COMT for the soluble form and MB-COMT for the membrane-bound form.3 The enzymatic domains of the two forms are identical, with MB-COMT having an extra 50 amino acids at the N-terminus for membrane anchoring that also influences enzymatic activity.4 The expression patterns of the two forms are different, with S-COMT predominating in the periphery and MB-COMT being more prevalent in the brain, although this differential expression is especially pronounced in humans compared to rodents.5, 6 Since the goal is to achieve central COMT inhibition, identification of a compound with MB-COMT selectivity may be theoretically desirable.
Inhibitors of COMT are widely used for treatment of Parkinson’s disease due to their role in inhibiting peripheral metabolism and thereby increasing levels of exogenously administered L-DOPA.7 Notably, the nitrocatechol scaffold has been exploited to provide the clinically used drugs tolcapone 1 and entacapone 2, as well as the recently approved opicapone 3 (Figure 1).8 While they are effective in blocking peripheral COMT activity, entacapone and opicapone have negligible brain penetration, and tolcapone has low but measurable levels in the brain.9 Compounds with improved brain penetration have greater efficacy for the non-motor symptoms of Parkinson’s disease,10 as well as have utility for other psychiatric and neurological conditions such as cognitive impairment in schizophrenia.11 Despite the early clinical success achieved with tolcapone, the drug has been associated with hepatic toxicity.12 Thus, the risk-benefit profile for tolcapone prevents its widespread deployment and new inhibitors of COMT are needed, especially those that are active in the brain as well as those derived from alternate pharmacophores to de-risk any toxicity arising from the nitro-catechol moiety.
Figure 1.
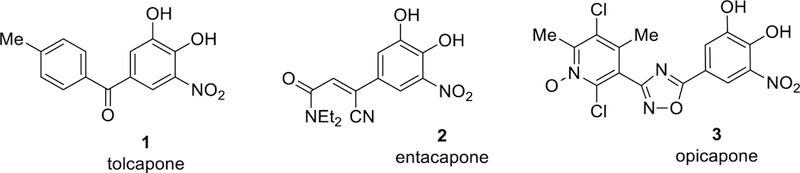
Clinically used COMT inhibitors
Soon after the discovery of COMT by Julius Axelrod in 1957,13 Ross and Haljasmaa investigated a number of compounds for their effects on COMT inhibition and identified 8-hydroxyquinoline (4, Figure 2) as a non-catechol-containing inhibitor.14 Later, Borchardt and colleagues further explored this template with modest success,15 but the field subsequently shifted focus to nitrocatechols given the exquisite potency and high ligand efficiency of the nitrocatechol pharmacophore.7, 16 While 3 was able to overcome the rapid metabolic clearance associated with tolcapone by taking advantage of a slow off rate that extends the pharmacodynamic action beyond its short plasma half-life,17 nitrocatechols generally suffer from poor pharmacokinetics and limited brain penetration. Herein, we describe further exploration of the 8-hydroxyquinoline template and the discovery of potent inhibitors with excellent pharmacokinetics and brain penetration.
Figure 2.
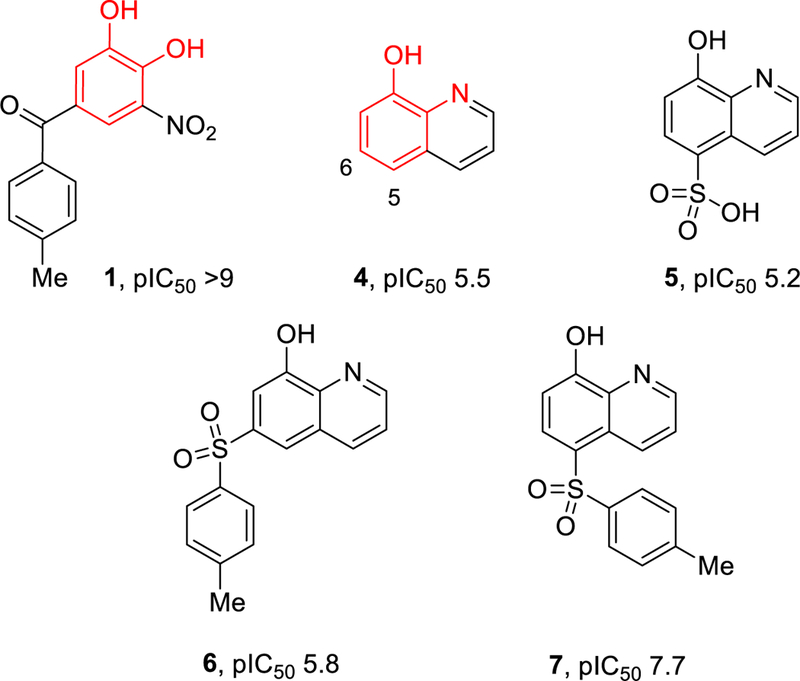
Evolution of the 8-hydroxyquinoline lead. pIC50 values are reported for Human MB-COMT enzyme.
RESULTS AND DISCUSSION
Borchardt’s exploration of 5-substituted 8-hydroxyquinolines showed that sulfonic acid (compound 5, Figure 2) had similar COMT inhibition to other electron withdrawing groups including chloro and nitro.15 In designing additional binding interactions for the 8-hydroxyquinoline 4 (Figure 2) outside the metal chelating motif, modeling suggested substituents on either the 5 or 6-position would overlay with nitrocatechol pharmacophore 1 (Figure 2). We prepared both sulfone isomers 6 and 7 to guide future trajectories for exploration. While the 6-substituted compound had a modest potency increase, the 5-position analog 7 appeared more promising with an in vitro MB-COMT pIC50 of 7.7.
As shown in Table 1, a series of functional groups were appended at the 5-position, with sulfones 7 and 11 as well as sulfonamides 12 and 13 being preferable to ketone 9, amide 10, or direct aromatics like 8. Additional sulfones and sulfonamides of varying sizes and polarity were prepared such as 13; however, no meaningful increases in potency were achieved. A free hydroxyl is required for optimum hydrogen-bonding and magnesium coordination in the COMT active site, as compound 14 exhibited no appreciable inhibition of MB-COMT. Poor metabolic stability in rat hepatocytes was observed for representative compounds 12 and 13, and the 8-hydroxyl moiety was suspected for phase II metabolism. Therefore, a series of sulfone and sulfonamide analogs were prepared that investigated substituents at the 7-position to explore steric and electronic effects on potency and metabolic stability.
Table 1.
Potency and Hepatocyte Clearance of 5-substituted 8-hydroxyquinolines
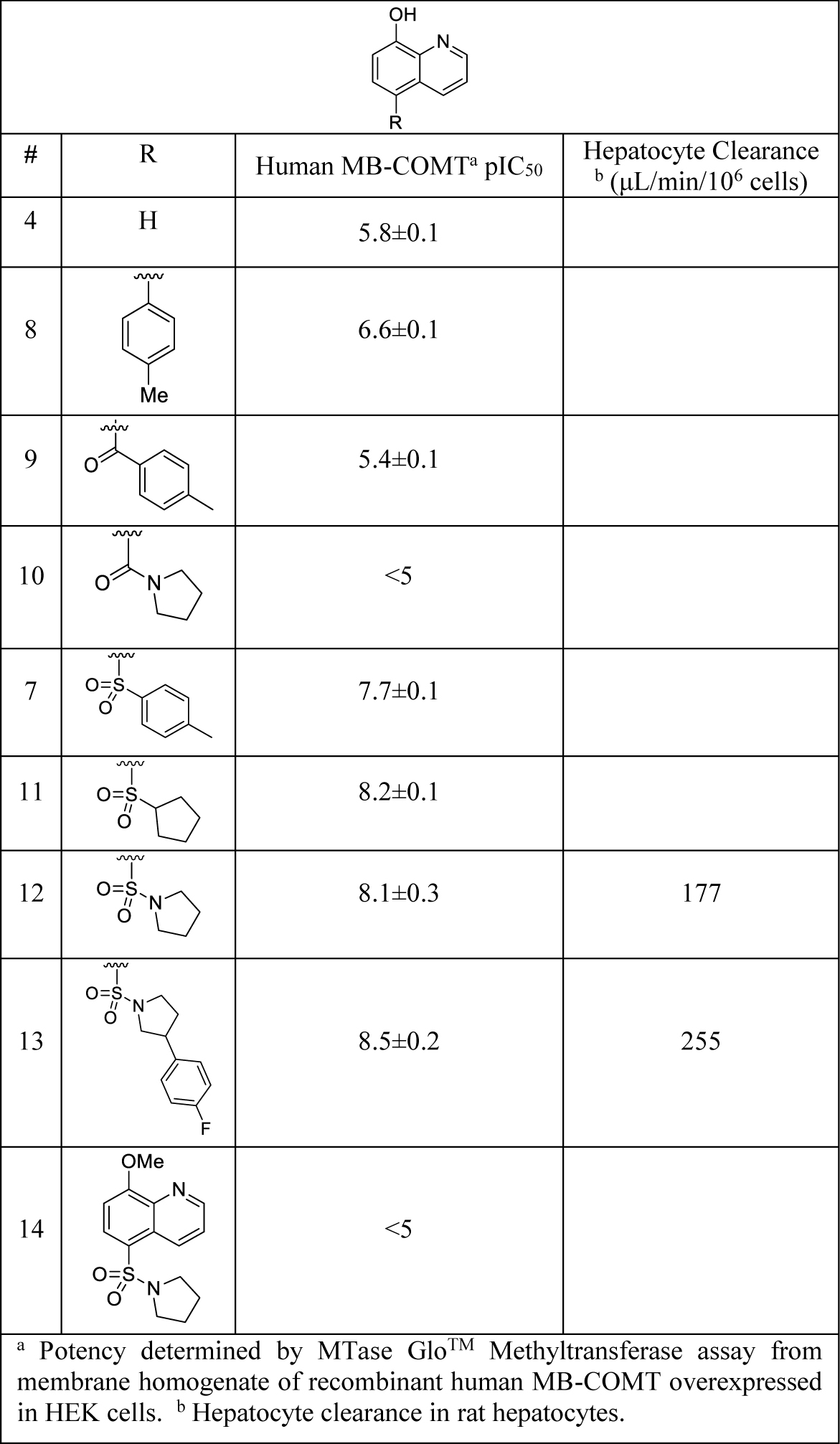 |
Expanding the SAR around 5-substituted sulfone and sulfonamide quinolines provided many potent and brain penetrant examples as described in Table 2. Both aryl and alkyl substitution were tolerated, as well as extension into larger and more diverse chemical space (data not shown, see patent application WO2016123576 for additional examples).18 However, building larger and more expansive sulfone and sulfonamides decreased ligand efficiency, increased lipophilicity and diminished physicochemical properties thereby leaving small substituents with the best combination of potency and physiochemical properties. The clearance profile of both sulfonamides and sulfones is driven by phase II metabolism of the 8-hydroxy group; therefore, screening in hepatocytes was used to capture the impact of metabolism in vitro. Poor metabolic stability was observed with 7-unsubstituted 8-hydroxyquinolines as evident by the high hepatocyte clearance for 15, 16, and 17 (Table 3). A tight correlation between rat hepatocytes and in vivo clearance measured by PK studies was not evident; however, the hepatocyte screen proved useful for triaging compounds with high clearance.
Table 2.
Enzyme Potency of 5,7-substituted 8-hydroxyquinolinesa
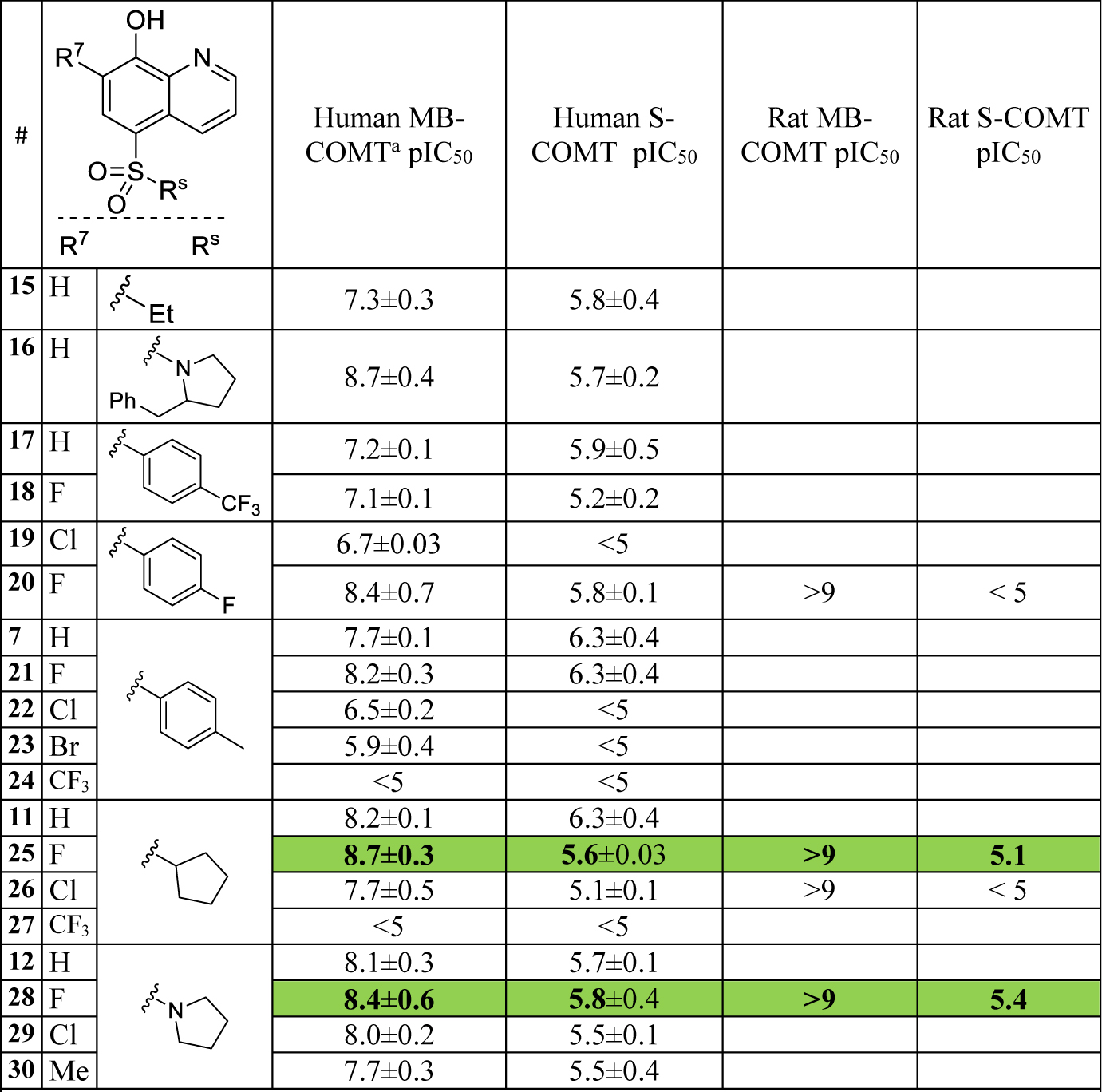 |
Potency determined by MTase Glo™ Methyltransferase assay from recombinant human or rat MB and S-COMT and are the average of three determinations.
Table 3.
ADME and Pharmacokinetic properties of 5,7-substituted 8-hydroxyquinolines
| Example | Hepatocyte Stability (Rat) (μL/min/106 cells) | Rat PKb | |||
|---|---|---|---|---|---|
| %F | T1/2 (h) | Clp (ml/min/kg) | Vss (L/kg) | ||
| 11 | 24 | 0.18 | 26 | 1.8 | |
| 12 | 177 | ||||
| 15 | 39 | ||||
| 16 | 228 | ||||
| 17 | 46 | ||||
| 18 | 12 | 66 | 3.5 | 13 | 3.8 |
| 19 | 4 | 23 | 8.9 | 0.31 | 0.23 |
| 20 | 15 | 100 | 4.5 | 12 | 3.5 |
| 21 | 36 | 34 | 0.26 | 27 | 1.2 |
| 22 | 78 | 45 | 0.31 | 25 | 0.67 |
| 25 | 19 | 32 | 7.7 | 0.62 | 0.41 |
| 26 | 18 | 37 | 11 | 0.3 | 0.3 |
| 28 | 21 | 73 | 2.8 | 11 | 2.2 |
| 29 | 14 | 33 | 8.6 | 1 | 0.8 |
| 30 | 103 | ||||
Rat pharmacokinetics after a 1mpk iv dose (n=3) and 3 or 10 mpk oral dose (n=3)
Improvements in pharmacokinetic properties were observed when installing small groups at the 7-position of the quinoline, presumably by affecting the phase II metabolism of the 8-hydroxy moiety. Substituents to modulate the susceptibility of hydroxyl metabolism with respect to both steric and electronic effects were explored. As shown in Table 3, halogens generally showed improved in vitro and in vivo clearance while larger, more sterically bulky groups reduced MB-COMT potency (e.g. compound 23). Strikingly, a CF3 substituent at the 7-position (compound 4) abolished MB-COMT inhibition. A small methyl substituent was tolerated (compound 30) but proved to have no impact on clearance as measured by rat hepatocytes. Both fluoro and chloro substitution yielded the best profiles with respect to metabolic stability, especially 7-chloro substituted compounds 19, 26 and 29 which exhibited very low clearance and long half-life in rat PK studies (Table 3).
Brain penetration was initially assessed in mice by measuring total brain and plasma levels 5 minutes, 15 minutes, and 1 hour after an intraperitoneal (ip) 10 mg/kg dose in two mice at each timepoint (Supplementary Table 1 for all timepoints and Table 4 for the 1 hour timepoint). Brain penetration was generally rapid, so the 1 hour timepoint was chosen for compound comparison in Table 4. Additionally, free fraction in plasma and brain were measured by equilibrium dialysis. Early examples such as 15 and 16, where hydrogen is in the 7-position exhibited low overall exposure consistent with poor pharmacokinetics. Compounds with either a 7-fluoro or chloro in both sulfone and sulfonamide moieties display good total plasma concentrations but modest brain exposures relative to total plasma. However, most examples in Table 2 have plasma protein binding approaching or greater than 99%, thereby limiting free drug concentrations. Additionally, the examples in Table 4 exhibited higher free fraction in rat brain tissue as compared to plasma which has the expected effect of driving down total brain concentrations.19 Despite the protein/tissue binding differences, when correcting for free drug in both compartments most compounds had reasonable ratios of unbound plasma and unbound brain concentrations. The lower free brain drug concentrations relative to unbound plasma concentrations observed for compounds such as 20 and 22 are outliers to the trend and could not be explained by P-gp efflux as these compounds were not substrates for the transporter (Table 4) and have good apparent permeability (>100 nm/s, data not shown). Despite these two outliers, overall the observations described in Tables 4 and 5 reinforce the notion that optimization of the fraction unbound in the brain does not result in greater brain exposure, rather plasma free fraction is more correlative to drug levels in brain.19
Table 4.
Mouse Brain Exposure of 5,7-substituted 8-hydroxyquinolines at one hour
| Example | P-gp ERa | Plasma Protein Bindingb | Brain Tissue Bindingc | Mouse Brain Penetrationd | |
|---|---|---|---|---|---|
| Total Brain (ng/g) | Total Plasma (ng/ml) | ||||
| 11 | NT | 0.5 (r) | 2.9 | blq | 173 |
| 12 | NT | 3.3 (m) | 4.4 | 10.5 | 68 |
| 15 | NT | 6.9 (m) | 10.3 | 11 | 250 |
| 16 | NT | 0.5 (m) | 0.5 | 109 | 235 |
| 17 | NT | 1.4 (m) | NT | BLQ | 3538 |
| 18 | 1.1 | 0.6 (r) | 3.6 | 1094 | 2542 |
| 19 | 0.8 | 0.5 (r) | 2.6 | 5655 | 16650 |
| 20 | 0.7 | 1.2 (r) | 2.4 | 527 | 5720 |
| 21 | NT | 1.2 (r) | 1.39 | 518 | 1744 |
| 22 | 1.1 | 0.84 (m) | 0.5 | 1686 | 9780 |
| 25 | 0.5 | 0.2 (r) | 4.2 | 1914 | 6160 |
| 26 | NT | 0.5 (r) | 1.6 | 2792 | 14669 |
| 28 | 1.2 | 0.6 (r) | 8 | 110 | 1819 |
| 29 | 0.4 | 0.5 (r) | 3.4 | 1154 | 16250 |
| 30 | NT | 0.5 (m) | 1.8 | 55 | 576 |
A:B/B:A efflux ratio from P-glycoprotein expressing MDCK cells. NT=not tested.
Percent free from either mouse (m) or rat (r) plasma as measured by equilibrium dialysis.
Percent free from rat brain tissue.
Total brain and total plasma concentrations 1 hour after ip dosing 10mg/kg to 2 mice.
Table 5.
Effects of COMT inhibitors on rat biomarkers of dopamine metabolism
| Example | Free Plasma (nM)a | Free Brain (nM)a | Free CSF (nM)b | CSF (nM) | CSF/ Free Plasma Ratio | CSF/ Free Brain Ratio | Free CSF/ Free Plasma Ratio | HVA (%)c | DOPAC (%)c |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 35 | BLQ | NA | BLQ | N/A | N/A | N/A | 28 | 304 |
| 20 | 48 | 7.7 | 59 | 127 | 2.6 | 16 | 1.2 | 86 | 173 |
| 25 | 19 | 12.8 | 139 | 321 | 16.5 | 25 | 7.3 | 66 | 218 |
| 26 | 32 | 5.6 | NA | 358 | 11 | 64 | N/A | 97 | 213 |
| 28 | 23 | 35 | 77 | 157 | 6.8 | 4.4 | 3.3 | 72 | 140 |
Free plasma and brain levels calculated using ratios listed in Table 4 on plasma and brain samples taken immediately after CSF sampling. Total concentrations are given in Supplementary Table 2.
Non-specific binding to CSF was determined by equilibrium dialysis using pooled rat CSF as described in reference 24. Compound 20 showed 53% binding, Compound 25 showed 57% binding, and Compound 28 showed 51% binding.
HVA and DOPAC changes (% of vehicle) 4 hours after 10mg/kg oral dose compared to vehicle (n=5–10 rats per group), except 1 (15mpk ip). BLQ=Below the limit of quantitation.
Central nervous system inhibition of COMT produces measurable changes in dopamine metabolites dihydroxyphenyl acetic acid (DOPAC, increase) and homovanillic acid (HVA, decrease). Assessment of these biomarkers of dopamine metabolism in rat CSF with accompanying measurement of inhibitor concentrations in plasma, brain homogenate, and CSF provides a rapid readout for compound brain penetration and activity. While the region of interest is the cortex, it has been demonstrated that CSF concentrations of HVA and DOPAC are reflective of total brain levels20 and responsive to brain-penetrant COMT inhibitors.21
Table 5 shows the results from a single oral administration (10 mpk) of COMT inhibitors to rats (n=5–10, see Supplementary Table S2 for details). The animals were sacrificed 4 hours post dose where plasma, brain, and CSF were analyzed for dopamine metabolites and drug levels. Values for plasma protein binding and brain tissue binding (Table 4) were used to determine free plasma and brain levels. Changes in HVA and DOPAC in CSF as measured by LC-MS are shown as a difference between compound treated and vehicle treated controls. The positive control, tolcapone (15 mpk, ip injection), exhibited expected biomarker movement where DOPAC was increased and HVA decreased relative to control. Robust increases in DOPAC were observed for all five compounds tested. More modest decreases were observed for HVA, with only 25 and 28 showing a statistically significant difference compared to the vehicle treated group.
All four quinoline analogs produced good peripheral exposure and brain levels as predicted from the mouse brain penetration studies. Measurable drug concentrations for all compounds in brain homogenate extracts exceeded rat MB-COMT IC50 measurements; however, this series has less activity on S-COMT which may contribute to the modest changes in HVA relative to tolcapone. Further studies with these compounds are in progress to obtain a more comprehensive understanding of the effects of COMT inhibition by different classes of inhibitors. An interesting additional observation is the lack of correlation between measured CSF levels and calculated free plasma and free brain levels, which are expected to be similar for highly permeable compounds that are not transporter substrates since the CSF has very low amounts of protein compared to plasma.22 In addition to P-gp (Table 4), compound 28 was also examined for susceptibility to BCRP and MRP2 transporters, as well as for inhibitory activity against P-gp, BCRP, and MRP2. While compound 28 was not found to be a substrate or an inhibitor for these three transporters (data not shown), that does not rule out susceptibility to other transporters involved in blood-brain-CSF homeostasis.23 This prompted measurement of free CSF concentrations using equilibrium dialysis.24 Compounds 20, 25, and 28 showed remarkably high bound concentrations in rat CSF (51–57% bound @ 2 μM testing concentration). Even though proteins like albumin are in CSF at less than 1% compared to plasma,25 there exists sufficient non-specific binding in CSF to effect the distribution of highly bound compounds.
Confirmation of the proposed binding mode of the 8-hydroxyquinolines was confirmed by an X-ray co-crystal structure with 21 and SAM in the rat S-COMT active site (Figure 3). As expected, the hydroxyl and quinoline nitrogen make a bidentate interaction with the catalytic magnesium. A free hydroxyl was observed to be required from the SAR studies (see Table 1, compound 14), and the structure reveals a tight hydrogen-bonding network around the hydroxyl similar to what is seen with nitrocatechols.26 The sulfone oxygens make no clear contacts with the protein, but orient the aromatic ring between tryptophan-38 and tryptophan-143 and over the thioether sidechain of methionine-40. The MB and S isoforms of COMT are identical except for an additional 50 amino acids at the N-terminus of MB-COMT that are responsible for membrane anchoring.27 Without a structure of compound 21 bound to MB-COMT for comparison, it is difficult to rationalize the selectivity for MB-COMT over S-COMT in this series, but it has been proposed that residues 27–50 of MB-COMT lay back over the active site to provide additional binding interactions.28 The close proximity of the leucine-198 sidechain to the quinoline 7-position explains the tolerance for only small substituents at the 7-position. As judged by the potency differences seen in Table 1 between sulfone 7 and sulfonamide 12 versus ketone 9 and amide 10, other effects of the 5-substituent may be at play since those substituents should also be able to “fit” in similar binding modes albeit at a torsional cost. Also notable is that there is no electron density for the transferable methyl group of SAM, and the structure is modeled with SAH despite high purity SAM used in the crystallization conditions.
Figure 3.
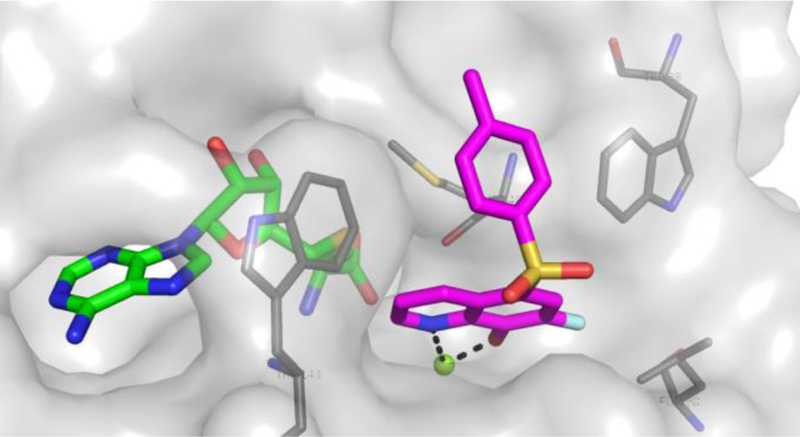
Compound 21 (magenta) bound to the rat S-COMT active site (PDB 6GY1). The hydroxyl and quinoline nitrogen make a bidentate interaction with the catalytic magnesium ion (green sphere). The sulfone oxygens make no clear contacts with the protein, but orient the aromatic ring between trytophan-38 and tryptophan-143 and over the thioether sidechain of methionine-40.
CHEMISTRY
Scheme 1 shows the preparation of unsubstituted and non-fluoro sulfonamides 12, 13 and 16, beginning with chlorosulfonylation of 8-fluoroquinoline 31 followed by treatment of the desired amine and quenching with sodium 2-(trimethylsilyl)ethan-1olate to displace the labile 8-fluoro group to give 33. Removal of the TMSE protecting group followed by treatment with N-chloro succinimide (NCS) gave the desired sulfonamide 29. The corresponding sulfones 7 and 23 were prepared according to Scheme 2 by PMB protection of 5-iodo-8-hydroxyquinoline 35 followed by copper (I) mediated coupling with 4-methylbenzenesulfinate to install the sulfone group.29 The protecting group was removed with TFA at ambient temperature to provide compound 7. Bromination of 7 was achieved with NBS in chloroform to give example 23. Additional 7-chloro sulfone analogs were prepared by chlorination with NCS followed by benzylating 5-bromo-8-hydroxyquinoline 36 (Scheme 3). Palladium catalyzed cross coupling of thiols produced thioethers 35.30 The desired thioethers could be prepared in either one step (compounds 22 and 26) or by use of an intermediary TMS-thioethane (example 19). Oxidation of the sulfur with mCPBA gave sulfones with minimal quinoline N-oxide formation. Acid-catalyzed deprotection of the benzyl group provided the desired analogs 39. Incorporation of a fluorine at the 7-position of the quinoline was accomplished as described in Scheme 4. A Skraup reaction31 was used to prepare the 7-fluoro-8-hydroxyquinoline 41 which could be either chlorosulfonylated and quenched with amines to give sulfonamide 28, or iodinated and protected to give key intermediate 42. Palladium catalyzed coupling of thiols followed by oxidation and deprotection gave the desired sulfones 44. Other quinolines shown in Table 1 could be prepared as shown in Scheme 5, via direct Friedel-Crafts acylation to give ketone 9, or introduction of the 8-hydroxyl as a protected benzyl by SnAr on 8-fluoroquinolines 46 or 48 which led to compounds 10 and 6 respectively.
Scheme 1.
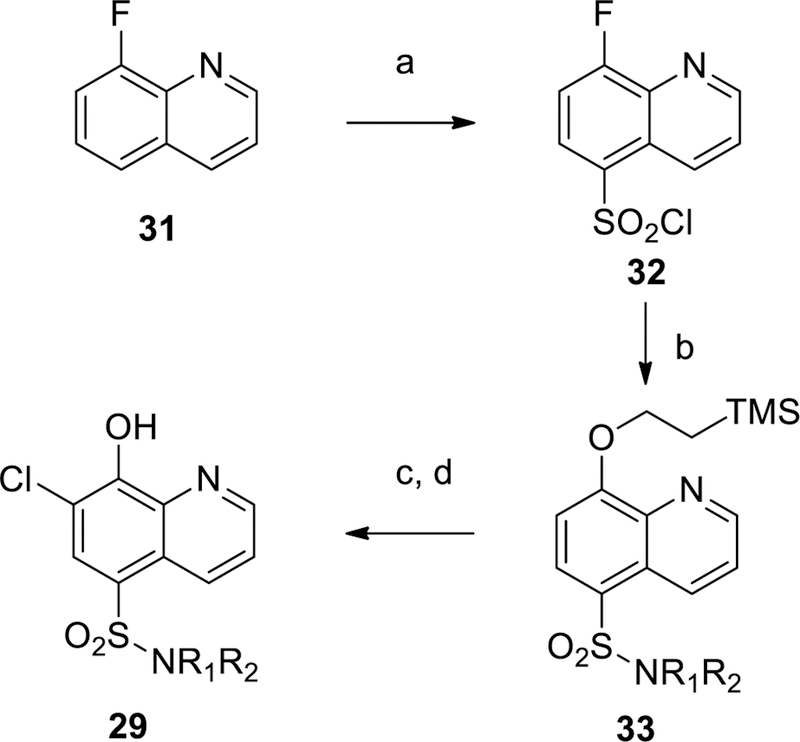
General synthetic prcedures for sulfonamides 12, 13, 16, 29. Reagents and conditions: (a) chlorosulfonic acid, 100 °C, 71%; (b) DIPEA, amine, THF then NaH, 2-(trimethylsilyl)ethanol; (c) CsF, DMF, 60 °C, 84% or TBAF, THF, 34%; (d) NCS, CHCl3, 600C, 24%.
Scheme 2.
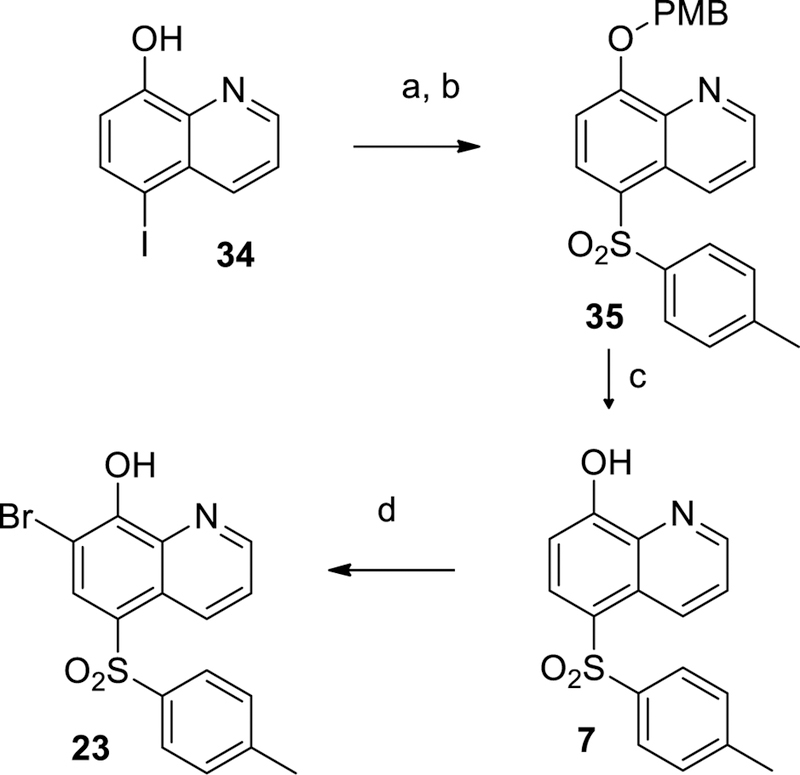
Synthetic protocol for 7 and 23. Reagents and conditions: (a) K2CO3, PMB-Cl, ACN, 22%; (b) CuI, DMSO, 4-methylbenzenesulfinate, 90 °C, 22%; (c) TFA, rt, 92%; (d) NBS, CHCl3, 85%.
Scheme 3.
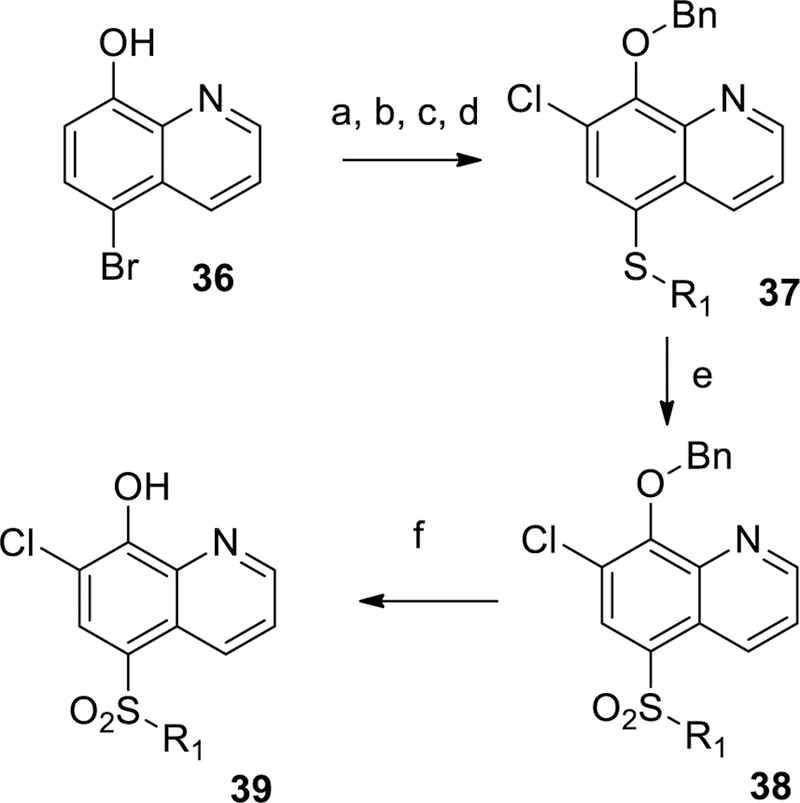
General synthetic procedures for 19, 22, and 26. Reagents and conditions: (a) NCS, CHCl3, 78%; (b) K2CO3, BnBr, ACN, 89%; (c) 2-trimethylsilylethanethiol (example 19) or R1SH (examples 22 and 26), Xantphos, Cs2CO3, Pd2(dba)3, dioxane, 100 °C, 93% example 19; 81% example 22, 93% example 26; (d) for example 19, 4-fluoroiodobenzene, TBAF, Xantphos, Cs2CO3, Pd2(dba)3, dioxane, 90 °C, 93%; (e) mCPBA, DCM; (f) 6M aqueous HCl, 100 °C.
Scheme 4.
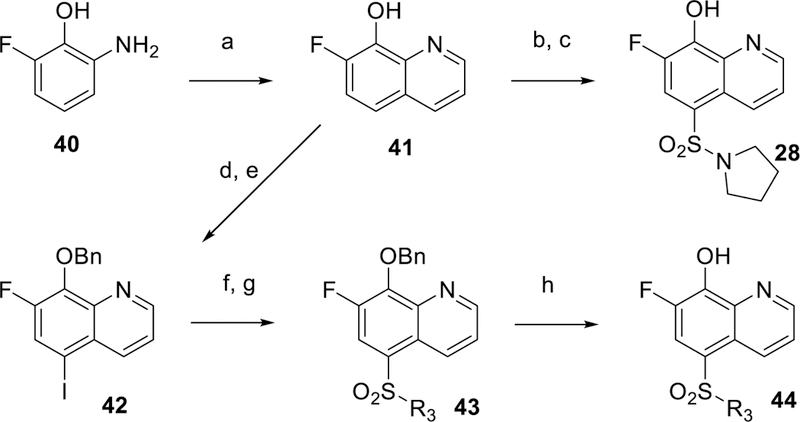
General synthetic protocol for 18, 20, 21, 25, and 28. Reagents and conditions: (a) glycerol, sodium-3-nitrobenzenesulfonate, 70% sulfuric acid, 140 °C, 56%; (b) chlorosulfonic acid, CHCl3, 100 °C, 40%; (c) triethylamine, pyrrolidine, DCM, 0 °C, 98%; (d) NIS, CHCl3, 40 °C, 92%; (e) NaH, BnBr, DMF, 73%; (f) thiol, Xantphos, Cs2CO3, Pd2(dba)3, dioxane, 90 °C; (g) mCPBA, DCM; (h) 6M aqueous HCl, 100 °C.
Scheme 5.
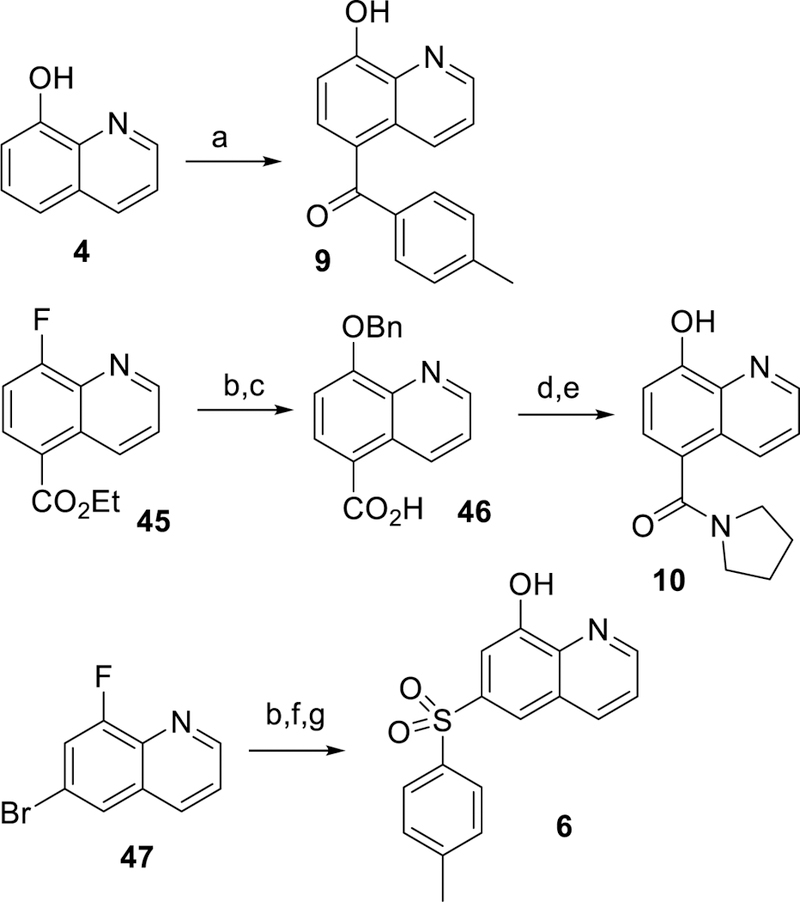
Synthetic protocols for ketone 9, amide 10, and sulfone 6. a) AlCl3, 4-methylbenzoyl chloride, toluene, 110 °C, 5%; b) NaH, BnOH, THF; c) NaOH, THF/H2O, 63%; d) 1-Chloro-N,N,2-trimethyl-1-propenylamine, DCM then pyrrolidine; e) HBr, acetic acid, 63% over 2 steps; f) Cu(I)I, K2CO3, DMEN, DMSO, sodium 4-methylbenzenesulfinate,46%; g) 6M HCl,87%.
CONCLUSIONS
A series of 8-hydroxyquinolines were prepared and evaluated as COMT inhibitors, and several potent MB-COMT inhibitors were identified that demonstrate selectivity over S-COMT. An X-ray co-crystal structure highlights the important chelation of the active site magnesium; however, the interactions responsible for the observed MB-COMT selectivity are not apparent in the S-OMT structure. Small substituents at the 7-position were found to increase metabolic stability without sacrificing potency, and compounds with good pharmacokinetics such as 25 and 28 were identified. This series of compounds generally has good brain penetration, although high plasma protein binding results in high exposures required for efficacy in a rat biomarker assay of COMT activity. Further in vivo characterization of these compounds on central nervous system dopamine metabolism will be reported in due course.
EXPERIMENTAL SECTION
General Procedures.
All commercially available reagents and solvents were used without further purification unless otherwise stated. Automated flash chromatography was performed on an ISCO CombiFlash Rf™ or Biotage Isolera™ using Biotage Flash cartridges with peak detection at 254 nm. Reverse phase purification was accomplished using a Gilson 215 liquid handler equipped with a Phenomenex C18 column (150 × 20 mm I.D., S-5 μm). Peak collection was triggered by UV detection at 214 or 254 nm. 1H NMR spectra were recorded on a Bruker 400 instrument operating at 400 MHz with tetramethylsilane or residual protonated solvent used as a reference. Analytical LC-MS was performed using Agilent 1260 equipped with autosampler (Agilent Poroshell 120 C18 column (50 × 3.0 mm I.D., 2.7 μm); 0.05% TFA in water/acetonitrile gradient; UV detection at 215 and 254 nm) and electrospray ionization. All final compounds showed purity greater than 95% at 215 and 254 nm using this method.
6-Tosylquinolin-8-ol (6).
Step 1.
To a solution of benzyl alcohol (1.1 mL, 10.8 mmol) in THF (30 mL) was added sodium hydride (0.44 g, 10.8 mmol) portionwise over 5 min. The resulting mixture was allowed to stir for 30 min, then a solution of 6-bromo-8-fluoro-quinoline 47 (1.6 g, 7.2 mmol) in THF (15 mL) was added dropwise over 5 min. The contents were stirred at room temperature for 4 days (for convenience). The reaction mixture (which had become gelatinous) was taken up in EtOAc and washed with water (4 X), brine (1 X), dried over MgSO4, filtered, and the solvent removed in vacuo. The desired product crystallised upon standing and was washed with ethyl acetate then air-dried to give 0.53 g of pure desired product. The filtrate was concentrated and purified by automated normal-phase chromatography (0–40% EtOAc/hexanes, 50 g silica gel cartridge). The product-containing fractions were combined and the solvent removed in vacuo to give a white solid, which when combined with the crystallized material to provide a total of 8-benzyloxy-6-bromo-quinoline 48 (1.3 g, 57% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 (dd, J = 4.04, 1.77 Hz, 1 H) 8.31 (dd, J = 8.34, 1.77 Hz, 1 H) 7.83 (d, J = 2.02 Hz, 1 H) 7.60 (dd, J = 8.34, 4.04 Hz, 1 H) 7.53 – 7.57 (m, 2 H) 7.42 – 7.47 (m, 3 H) 7.35 – 7.41 (m, 1 H) 5.34 (s, 2 H). MS (ES+) m/z 314.0 [M+H]+.
Step 2.
A mixture of copper (I) iodide (28 mg, 0.15 mmol), 8-benzyloxy-6-bromo-quinoline 48 (100 mg, 0.32 mmol), sodium 4-methylbenzenesulfinate (170 mg, 0.95 mmol) and potassium carbonate (88 mg, 0.64 mmol) and N,N’-dimethylethylenediamine (6.8 μL, 0.06 mmol) in DMSO (3 mL) was heated to 90°C for 60 min under microwave heating. The contents were diluted with EtOAc, washed with water (3 X), brine, dried over MgSO4, filtered and the solvent removed in vacuo to give a yellow residue. This material was purified by automated normal-phase chromatography (0–100% EtOAc/hexanes, 4 g silica gel cartridge). The product-containing fractions were combined and the solvent removed in vacuo to give 8-benzyloxy-6-(p-tolylsulfonyl)quinoline 49 (57 mg, 46% yield) as a beige solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.07 (dd, J = 4.17, 1.64 Hz, 1 H) 8.22 – 8.27 (m, 1 H) 8.07 (d, J = 1.77 Hz, 1 H) 7.66 – 7.71 (m, 2 H) 7.53 – 7.58 (m, 1 H) 7.47 – 7.52 (m, 2 H) 7.41 (d, J = 2.02 Hz, 1 H) 7.31 – 7.39 (m, 3 H) 7.22 (d, J = 8.08 Hz, 2 H) 5.48 (s, 2 H) 2.39 (s, 3 H). MS (ES+) m/z 390.0 [M+H]+.
Step 3.
8-benzyloxy-6-(p-tolylsulfonyl)quinoline 49 (55 mg, 0.14 mmol) and 6 M hydrochloric acid (0.64 mL, 3.8 mmol) were stirred at 100°C for 4 h, then allowed to cool to room temperature. The solid was collected by filtration, washed with water and dried under vacuum to give 6-(p-tolylsulfonyl)quinolin-8-ol 6 (36 mg, 87% yield) as a beige solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.05 (dd, J = 4.42, 1.64 Hz, 1 H) 8.75 (d, J = 8.34 Hz, 1 H) 8.24 (d, J = 2.02 Hz, 1 H) 7.88 (d, J = 8.34 Hz, 2 H) 7.81 (dd, J = 8.34, 4.29 Hz, 1 H) 7.45 (dd, J = 5.05, 3.03 Hz, 3 H) 2.37 (s, 3 H). MS (ES+) m/z 300.0 [M+H]+.
5-Tosylquinolin-8-ol (7).
Step 1.
To a stirring solution of 5-iodoquinolin-8-ol 34 (4.1 g, 15.3 mmol) in acetonitrile (100 mL) was added potassium carbonate (4.2 g, 30.8 mmol). The suspension was stirred at room temperature for 30 minutes. 4-Methoxybenzyl chloride (2.5 mL, 18.4 mmol) was added and the resultant suspension was heated at 80°C for 4 hours. After cooling to room temperature, the suspension was filtered, the filter cake washed with ethyl acetate and filtrate was concentrated to a residue. The residue was purified by automated normal-phase chromatography and eluted with ethyl acetate/hexanes (0–80%) to give 8-((4-methoxybenzyl)oxy)-quinoline 50 as an off-white solid (1.4 g, 22 % yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.86 (dd, J = 4.04, 1.52 Hz, 1 H) 8.29 (dd, J = 8.46, 1.64 Hz, 1 H) 8.06 −8.11 (m, 1 H) 7.67 (dd, J = 8.59, 4.29 Hz, 1 H) 7.43 – 7.49 (m, 2 H) 7.15 (d, J = 8.34 Hz, 1 H) 6.95 – 7.01 (m, 2 H). MS (ES+) m/z 392.0 [M+H]+.
Step 2.
To a stirring solution of 5-iodo-8-((4-methoxybenzyl)oxy)quinoline 50 (0.20 g, 0.51 mmol) in DMSO (1.5 mL), copper (I) iodide (10 mg, 0.051 mmol), sodium (S)-pyrrolidine-2-carboxylate (14 mg, 0.10 mmol) and sodium 4-methylbenzenesulfinate (109 mg, 0.61 mmol) were added. The solution was heated at 90°C for 24 hrs. Water (2 mL) was added and precipitate isolated by filtration. The crude material was purified by automated normal-phase chromatography, using ethyl acetate/hexanes (0–80%) as an eluent to give 8-((4-methoxybenzyl)oxy)-5-tosylquinoline 35 as an off-white solid (47 mg, 22% yield). 1H NMR (400 MHz, CDCl3) δ ppm 9.00 (s, 2 H) 8.42 (d, J = 8.34 Hz, 1 H) 7.81 (d, J = 8.34 Hz, 2 H) 7.53 (dd, J = 8.84, 4.29 Hz, 1 H) 7.45 (d, J = 8.59 Hz, 2 H) 7.24 – 7.31 (m, 4 H) 7.16 (d, J = 8.59 Hz, 1 H) 6.88 – 6.99 (m, 2 H) 5.45 (s, 2 H) 3.79 – 3.86 (m, 3 H). MS (ES+) m/z 420.0 [M+H]+.
Step 3.
8-((4-Methoxybenzyl)oxy)-5-tosylquinoline 35 (47 mg, 0.11 mmol) was dissolved in TFA (0.86 mL, 11.2 mmol) and stirred at room temperature for 1 hour. The solution was concentrated. The residue was purified by recrystallization in ethyl acetate/hexanes to give 5-tosylquinolin-8-ol 7 as an off-white solid (30 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.94 (dd, J = 4.17, 1.64 Hz, 1 H) 8.89 (dd, J = 8.72, 1.64 Hz, 1 H) 8.39 (d, J = 8.34 Hz, 1 H) 7.80 – 7.86 (m, 2H) 7.73 (dd, J = 8.72, 4.17 Hz, 1 H) 7.35 – 7.41 (m, 2 H) 7.27 (d, J = 8.34 Hz, 1 H) 2.32 (s, 3 H). MS (ES+) m/z 300.0 [M+H]+.
5-(p-Tolyl)quinoline-8-ol (8).
Step 1.
To a solution of 5-bromoquinolin-8-ol 36 (5 g, 22.3 mmol) in DMF (40 mL) and THF (20 mL) was added sodium hydride (1.1 g, 26.7 mmol). After 15 minutes benzyl bromide (3.1 mL, 26.7 mmol) was added slowly dropwise. The reaction was maintained at ambient temperature overnight. The reaction was quenched by addition of water, solids formed and were collected by filtration to yield 7.3 g (assume quantitative yield) of 8-benzyloxy-5-bromo-quinoline 51. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.95 (br. s., 1 H) 8.47 (d, J = 8.08 Hz, 1 H) 7.88 (d, J = 8.34 Hz, 1 H) 7.73 (br. s., 1 H) 7.55 (br. s., 2 H) 7.41 (dd, J = 17.68, 6.82 Hz, 3 H) 7.28 (d, J = 8.08 Hz, 1 H) 5.34 (br. s., 2 H). MS (ES+) m/z 314.0 [M+H]+.
Step 2.
A vial was charged with 8-benzyloxy-5-bromo-quinoline 51 (150 mg, 0.48 mmol), Pd(PPh3)4 (55 mg, 0.05 mmol), 4-methylphenylboronic acid (77 mg, 0.57 mmol), NaHCO3 (0.48 mL, 0.95 mmol) 2M aqueous solution in 1,4-dioxane (5 mL). The vial was sparged with nitrogen for 5 minutes and heated at 90°C overnight. The reaction was concentrated to a residue, resultant residue partitioned between water and ethyl acetate, layers separated, aqueous extracted (2 × 2 mL), organics passed through a phase separator tube and solution concentrated to an oil. Purification was accomplished by automated silica gel chromatography (12 g pre-packed silica cartridge) eluting with 0–100% ethyl acetate/heptane to provide 8-benzyloxy-5-(p-tolyl)quinoline 52 as a yellow glass (150 mg, 97% yield). 1gH NMR (400 MHz, DMSO-d6) δ ppm 2.40 (s, 3 H) 5.36 (s, 2 H) 7.33 – 7.40 (m, 4 H) 7.40 – 7.48 (m, 4 H) 7.51 – 7.61 (m, 4 H) 8.17 (dd, J=8.59, 1.52 Hz, 1 H) 8.89 (dd, J=4.04, 1.52 Hz, 1 H). MS (ES+) m/z 326.0 [M+H]+.
Step 3.
A solution of 8-benzyloxy-5-(p-tolyl)quinoline 52 (150 mg, 0.46 mmol) dissolved in 1,4-dioxane (2 mL) and 6N aqueous hydrochloric acid (3 mL, 18 mmol) was heated at 100°C for 4 hours. The material was concentrated to a residue and purified by automated reversed phase HPLC to yield 85 mg (52%) of 5-(p-tolyl)quinoline-8-ol 8 as a TFA salt. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.40 (s, 3 H) 7.30 (d, J=8.08 Hz, 1 H) 7.33 – 7.37 (m, 4 H) 7.51 (d, J=8.08 Hz, 1 H) 7.73 (dd, J=8.59, 4.55 Hz, 1 H) 8.48 (d, J=8.34 Hz, 1 H) 8.98 (dd, J=4.55, 1.52 Hz, 1 H). MS (ES+) m/z 236.2 [M+H]+.
(8-Hydroxyquinolin-5-yl)(p-tolyl)methanone (9).
To a mixture of 8-hydroxyquinoline 4 (250 mg, 1.7 mmol) dissolved in toluene (3 mL) was added 4-methylbenzoyl chloride (0.23 mL, 1.7 mmol). The resultant solution was cooled to 0°C and aluminum chloride (574 mg, 4.3 mmol) was added in 2 portions. The resultant mixture was allowed to warm to ambient temperature for 2 hours. The material was quenched by pouring over ice and 1M HCl aqueous solution. The acidic solution was extracted with ethyl acetate (2 × 10 mL). The organics were combined, dried over sodium sulfate and concentrated to a residue. Purification was accomplished by automated normal-phase chromatography using 0–5% methanol/dichloromethane as an eluent to provide 24 mg (5% yield) of title compound 9. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.89 (br. s., 1 H) 8.53 (d, J = 7.83 Hz, 1 H) 8.11 (d, J = 8.08 Hz, 2 H) 7.99 (d, J = 6.82 Hz, 1 H) 7.68 – 7.76 (m, 2 H) 7.64 (d, J = 4.55 Hz, 1 H) 7.46 (d, J = 7.58 Hz, 2 H) 2.45 (s, 3 H). MS (ES+) m/z 264.1 [M+H]+.
(8-Hydroxyquinolin-5-yl)(pyrrolidin-1-yl)methanone (10).
Step 1.
To a solution of ethyl 8-hydroxyquinoine-5-carboxylate 45 (270 mg, 1.2 mmol) in dry DMF (10 mL) was added sodium hydride (60 mg, 1.4 mmol) at ambient temperature. After 5 minutes benzyl bromide (0.18 mL, 1.4 mmol) was added in a dropwise manner. The reaction was allowed to stir at ambient temperature. After 15 minutes the reaction was quenched by addition of ice-water. The aqueous mixture was extracted (2 × 20 mL) ethyl acetate, organic washes combined, dried over sodium sulfate and concentrated to a residue. Purification was accomplished automated normal-phase chromatography (40 g silica gel cartridge) eluting 0–100% ethyl acetate/hexanes to provide 259 mg as a 1:1 mixture of ethyl-8-benzyloxyquinoline-5-carboxylate 53 and 8-benzyloxyquinoline. The mixture was taken on in the next step without additional purification. MS (ES+) m/z 308.0 [M+H]+.
Step 2.
To a solution of ethyl-8-benzyloxyquinoline-5-carboxylate 53 mixture (250 mg, 0.81 mmol) in THF (10 mL) was added water (10 mL). The mixture was allowed to stir at 40°C overnight. The pH was adjusted to ~4–5 by addition of 4.5 M aqueous hydrochloric acid. Solids formed and collected by filtration, washed with water and dried in a vacuum oven to yield 8-benzyloxyquinoline-5-carboxylate 46 (144 mg, 63% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 12.99 (br. s., 1 H) 9.42 (dd, J = 8.84, 1.77 Hz, 1 H) 8.91 (dd, J = 4.04, 1.52 Hz, 1 H) 8.26 – 8.31 (m, 1 H) 7.68 (dd, J = 8.59, 4.04 Hz, 1 H) 7.57 (d, J = 6.82 Hz, 2 H) 7.42 – 7.48 (m, 2 H) 7.34 – 7.41 (m, 2 H) 5.39 (s, 2 H). MS (ES+) m/z 280.0 [M+H]+.
Step 3.
To a slurry of 8-benzyloxyquinoline-5-carboxylate 46 (61 mg, 0.22 mmol) in dry dichloroethane (2 mL) was added Ghosez reagent (0.29 mL, 2.1 mmol) at ambient temperature. To drive the reaction into solution additional Ghosez reagent was added (totaling 0.7 mL). After 1 hour, pyrrolidine (0.54 mL, 6.5 mmol) was added. The reaction was stirred at ambient temperature for 1 hour then diluted with dichloromethane and extracted (1 × 3 mL) water. The material was passed through a phase separator tube and organic layer concentrated to a residue. Purification was accomplished by automated normal-phase chromatography using 0–10% methanol/dichloromethane as an eluent. The oily (8-benzyloxy-5-quinolyl)-pyrrolidin-1-yl-methanone solidified upon standing (102 mg). The crude material was taken on without additional purification and or characterization.
A mixture of (8-benzyloxy-5-quinolyl)-pyrrolidin-1-yl-methanone (83 mg, 0.29 mmol) dissolved in acetic acid (0.5 mL) was added hydrobromic acid (0.5 mL). The resultant mixture was heated to 100°C for 3 hours. The material was cooled to ambient temperature and neutralized with 6 M sodium hydroxide. The aqueous solution was extracted (3 × 2 mL) dichloromethane, organics combined, filtered through a phase separator tube and concentrated to a residue. Purification was accomplished by automated normal-phase chromatography using 0–10% methanol/dichloromethane as an eluent. The impure material was concentrated to a residue then purified by automated reversed-phase chromatography eluting 5–95% acetonitrile/water, 0.05% trifluoroacetic acid as modifier. A yellow foam of 8-hydroxyquinolin-5-yl)(pyrrolidin-1-yl)methanone 10 (57 mg, 43% yield) was obtained. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.95 (dd, J = 4.17, 1.39 Hz, 1 H) 8.47 (d, J = 8.59 Hz, 1 H) 7.73 (dd, J = 8.34, 4.55 Hz, 1 H) 7.59 (d, J = 8.08 Hz, 1 H) 7.17 (d, J = 8.08 Hz, 1 H) 3.58 (t, J = 6.82 Hz, 2 H) 3.16 (t, J = 6.44 Hz, 2 H) 1.86 – 1.97 (m, 2 H) 1.73 – 1.84 (m, 2 H). MS (ES+) m/z 243.0 [M+H]+.
5-(Cyclopentylsulfonyl)quinoline-8-ol (11).
Step 1.
8-Hydroxyquinoline-5-sulfonic acid hydrate (10 g, 41.1 mmol) was dissolved in sodium hydroxide solution (1 N, 103 mL, 103 mmol) and heated to 75°C. A solution of benzyl chloride (10.4 ml, 90 mmol) in THF (100 mL) was added and resultant mixture maintained at 75°C overnight. After cooling to room temperature, the solution was extracted with diethyl ether twice, then placed chilled at 4°C for 4 hours. The solid was collected by filtration and dried to give 8-benzyloxyquinoline-5-sulfonic acid 54 (6.3 g, 48% yield) a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.10 – 9.17 (m, 1 H) 8.84 (dd, J = 4.2, 1.6 Hz, 1 H) 7.88 (d, J = 8.1 Hz, 1 H) 7.52 – 7.60 (m, 3 H) 7.39 – 7.46 (m, 2 H) 7.32 – 7.39 (m, 1 H) 7.20 (d, J = 8.1 Hz, 1 H) 5.32 (s, 2 H). MS (ES+) m/z 316.0 [M+H]+
Step 2.
To a stirring solution of 8-benzyloxyquinoline-5-sulfonic acid 54 (6.3 g, 20.0 mmol) and thionyl chloride (100 mL, 20.0 mmol) was added DMF (3 drops). The solution was heated at 80°C for 3 hours. The solution was cooled to room temperature and concentrated. Toluene (50 ml) was added and the resultant mixture concentrated to azeotrope excess thionyl chloride, and the resultant suspension was concentrated to give 8-benzyloxyquinoline-5-sulfonyl chloride 55 (8.9 g) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 5.76 (s, 2 H) 7.30 – 7.45 (m, 13 H) 7.62 – 7.70 (m, 6 H) 8.25 (dd, J = 8.84, 5.05 Hz, 3 H) 8.50 – 8.56 (m, 3 H) 9.59 (dd, J = 5.05, 1.26 Hz, 3 H) 9.66 (dd, J = 8.84, 1.01 Hz, 3 H). MS (ES+) m/z 334.9 [M+H]+
Step 3.
To a stirring solution of 8-benzyloxyquinoline-5-sulfonyl chloride 55 (14 g, 41.9 mmol) in THF (100 mL) was added triphenylphosphine (33 g, 125.8 mmol). An exotherm was observed and reaction mixture turned dark brown. The solution was heated at 60°C for 60 minutes then cooled to room temperature. Diethyl ether (50 ml) was added, a precipitate formed and solids collected by filtration. After dissolving the precipitate in DMF, sodium borohydride (2.0 g, 54.5 mmol) was added. The solution was stirred at room temperature for 30 minutes. Cyclopentyl iodide (8.2 g, 41.9 mmol) was added and the reaction heated to 60°C for two hours. The solution was cooled to room temperature and diluted with water (50 mL). The solution was extracted with ethyl acetate (3 × 50 mL). The combined organic washes were extracted with water (3 × 40 mL), brine (1 × 50 mL), dried over sodium sulfate, filtered and concentrated. The residue was purified automated normal-phase chromatography and eluted with ethyl acetate/hexane (0–50%) to give 8-benzyloxy-5-cyclopentylsulfanyl-quinoline 56 (2.3 g, 16% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ ppm 8.99 – 9.05 (m, 1 H) 8.86 (dd, J = 8.5, 1.6 Hz, 1 H) 7.65 (d, J = 8.1 Hz, 1 H) 7.50 – 7.59 (m, 3 H) 7.29 – 7.43 (m, 3 H) 6.96 – 7.03 (m, 1 H) 5.44 – 5.50 (m, 2 H) 3.36 – 3.47 (m, 1 H) 1.73 – 1.94 (m, 4 H) 1.49 – 1.67 (m, 4 H). MS (ES+) m/z 336.1 [M+H]+.
Step 4.
To a stirring solution of 8-benzyloxy-5-cyclopentylsulfanyl-quinoline 56 (5.3 g, 15.8 mmol) in DCM (150 mL), 3-chlorobenzenecarboperoxoic acid (1.1 g, 4.7 mmol) was added. The solution was stirred at room temperature for 30 minutes. Sodium sulfite solution (1 M, 30 mL) was added. The solution was extracted with sodium hydroxide solution (1.0 N, 2 × 60 mL), brine (1 × 60 mL) and dried over sodium sulfate. The solution was filtered and concentrated. The residue was purified by automated normal-phase chromatography and eluted with methanol/dichloromethane (0–4%) to give 8-benzyloxy-5-cyclopentylsulfonyl-quinoline 57 (3.7 g, 61% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.97 – 9.04 (m, 2 H) 8.22 (d, J = 8.3 Hz, 1 H) 7.75 – 7.81 (m, 1 H) 7.55 – 7.62 (m, 2 H) 7.36 – 7.53 (m, 4 H) 5.42 (s, 2 H) 3.78 – 3.90 (m, 1 H) 1.47 – 1.95 (m, 8 H). MS (ES+) m/z 368.1 [M+H]+.
Step 5.
To a stirring solution of 8-benzyloxy-5-cyclopentylsulfonyl-quinoline 57 (3.7 g, 10.1 mmol) in acetic acid (50 mL), hydrobromic acid (34 mL, 201.9 mmol) was added. The solution was stirred at 100°C for 5 hours. The solution was cooled to room temperature and concentrated to half of its volume. The solution was washed with diethyl ether and etheral wash chilled at 4°C overnight. The white precipitate was collected by filtration and dried to give the desired product as a hydrobromide salt. The solid was dissolved in aqueous sodium hydroxide solution (0.5 N, 50 mL). The solution was filtered to remove particulates and then filtrate adjusted to slightly acidic by adding hydrochloric acid solution (1 N, ~pH 5–6). The solution was triturated with saturated sodium bicarbonate aqueous solution to provide a white precipitate. The precipitate was isolated by filtration, washed with water three times, and air-dried to give 5-cyclopentylsulfonylquinolin-8-ol 11 (1.7 g, 61% yield) as a free base. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.17 (br. s., 1 H) 8.97 – 9.05 (m, 2 H) 8.13 (d, J = 8.3 Hz, 1 H) 7.75 – 7.84 (m, 1 H) 7.24 (d, J = 8.1 Hz, 1 H) 3.81 (quin, J = 7.6 Hz, 1 H) 1.45 – 1.94 (m, 8 H). MS (ES+) m/z 278.0 [M+H]+.
5-(Pyrrolidin-1-ylsulfonyl)quinoline-8-ol (12).
Step 1.
8-Fluoroquinoline 31 (10.5 g, 71.3 mmol) was added dropwise with stirring to chlorosulfonic acid (10 mL, 150.4 mmol). The resulting mixture was stirred at 110°C for ~30 hours. The crude mixture was added dropwise to ice water with stirring, causing solids to precipitate. Solids were collected by filtration, rinsed with a small amount of water and dried under reduced pressure to afford 8-fluoroquinoline-5-sulfonyl chloride 32 (12.5 g, 71% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.36 (d, J = 8.59 Hz, 1 H) 9.04 (d, J = 3.28 Hz, 1 H) 8.02 (dd, J = 8.08, 5.31 Hz, 1 H) 7.82 (dd, J = 8.72, 4.42 Hz, 1 H) 7.62 (dd, J = 10.61, 8.08 Hz, 1 H). MS (ES+) m/z 246.0 [M+H]+.
Step 2.
To a suspension of 8-fluoroquinoline-5-sulfonyl chloride 32 (2.0 g, 8.1 mmol) in THF (25 mL) was added DIPEA (2.8 mL, 16.2 mmol) at ambient temperature. Pyrrolidine (0.67 mL, 8.1 mmol) was added slowly dropwise as a solution in THF (5 mL). Meanwhile to a slurry of sodium hydride (977 mg, 24.4 mmol) in THF (15 mL) was added 2-(trimethylsilyl)ethanol (3.5 mL, 24.4 mmol). After complete addition of pyrrolidine to the 8-fluoroquinoline-5-sulfonyl chloride a HPLC chromatogram confirmed the first step was complete. This sulfonamide mixture was added to the slurry of trimethylsilylethanol sodium salt slowly dropwise. The reaction was allowed to stir at ambient temperature for one hour. The reaction was quenched by addition of water. Ethyl acetate was added and the layers were separated. The aqueous layer was extracted (3 × 50 mL) with ethyl acetate. The organics were combined, dried over sodium sulfate, concentrated to a residue and purified by silica chromatography (100 g) eluting 0–100% ethyl acetate/hexanes. Isolated 2.2 g (70% yield) of trimethyl-[2-[(5-pyrrolidin-1-ylsulfonyl-8-quinolyl)oxy]ethyl]silane 58. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 – 9.08 (m, 2 H) 8.14 (d, J = 8.59 Hz, 1 H) 7.75 (dd, J = 8.84, 4.04 Hz, 1 H) 7.35 (d, J = 8.59 Hz, 1 H) 4.21 – 4.47 (m, 2 H) 3.06 – 3.24 (m, 4 H) 1.59 – 1.79 (m, 4 H) 1.15 – 1.33 (m, 2 H) 0.13 (s, 9 H). MS (ES+) m/z 379.0 [M+H]+.
Step 3.
To a solution of trimethyl-[2-[(5-pyrrolidin-1-ylsulfonyl-8-quinolyl)oxy]ethyl]silane 58 (2.1 g, 5.6 mmol) dissolved in THF (30 mL) was added tetrabutylammonium fluoride (5.6 mL, 5.6 mmol) as a 1M solution in THF. The mixture was allowed to stir for 1 hour. Excess solvent was removed by rotary evaporator and resultant residue partitioned between water and dichloromethane. Extraction of the aqueous material was accomplished by (3 × 30 mL) dichloromethane. The organic layer was dried over sodium sulfate and concentrated to an oil. The material was purified by silica chromatography eluting 0–10% dichloromethane/methanol to provide 5-(pyrrolidin-1-ylsulfonyl)quinoline-8-ol 12, 548 mg (34% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.02 (br. s., 1 H) 9.04 (dd, J = 8.72, 1.64 Hz, 1 H) 8.98 (dd, J = 3.79, 1.26 Hz, 1 H) 8.09 (d, J = 8.34 Hz, 1 H) 7.78 (dd, J = 8.84, 4.29 Hz, 1 H) 7.20 (d, J = 8.34 Hz, 1 H) 3.07 – 3.23 (m, 4 H) 1.71 (dt, J = 6.51, 3.44 Hz, 4 H). MS (ES+) m/z 279.0 [M+H]+.
5-((3-(4-Fluorophenyl)pyrrolidin-1-yl)sulfonyl)quinoline-8-ol (13).
Step 1.
To a suspension of 8-fluoroquinoline-5-sulfonyl chloride 32 (150 mg, 0.61 mmol) in THF (4 mL) was added DIPEA (319 μL, 1.8 mmol) dropwise. After complete addition 3-(4-fluorophenyl)pyrrolidine hydrochloride (123 mg, 0.61 mmol) was added in one portion. The reaction was quenched by addition of water and partitioned between water and dichloromethane. The aqueous was extracted with dichloromethane (2 × 3 mL), organics combined, dried over sodium sulfate and concentrated to a residue. Purification was accomplished by automated silica chromatography eluting with ethyl acetate/heptane at a 0–80% gradient over 20 minutes. 8-Fluoro-5-[3-(4-fluorophenyl)pyrrolidin-1-yl]sulfonyl-quinoline 59 was isolated (210 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.08 – 9.16 (m, 2 H) 8.26 (dd, J = 8.34, 4.80 Hz, 1 H) 7.83 – 7.90 (m, 1 H) 7.78 (dd, J = 9.98, 8.46 Hz, 1 H) 7.18 – 7.27 (m, 2 H) 7.00 – 7.11 (m, 2 H) 3.75 (dd, J = 9.35, 7.58 Hz, 1 H) 3.52 (td, J = 8.97, 3.03 Hz, 1 H) 3.27 – 3.42 (m, 2 H) 3.09 – 3.20 (m, 1 H) 2.14 – 2.28 (m, 1 H) 1.83 – 2.02 (m, 1 H). MS (ES+) m/z 375.0 [M+H]+.
Step 2.
A suspension of sodium hydride (67 mg, 1.6 mmol) in THF (2 ml) was added 2-(trimethylsilyl)ethanol (0.24 mL, 1.6 mmol) slowly, dropwise over 5 minutes. The resultant suspension was added to a solution of 8-fluoro-5-[3-(4-fluorophenyl)pyrrolidin-1-yl]sulfonyl-quinoline 59 (210 mg, 0.56 mmol) in THF (3 mL) and resultant mixture was allowed to stir at ambient temperature for 30 minutes. The reaction was quenched by addition of water and partitioned between water and dichloromethane. The aqueous was extracted with dichloromethane (2 × 3 mL), organics combined, dried over sodium sulfate and concentrated to a residue. Purification was accomplished by automated silica chromatography eluting with ethyl acetate/heptane at a 0–60% gradient over 12 minutes to provide 2-[[5-[3-(4-fluorophenyl)pyrrolidin-1-yl]sulfonyl-8-quinolyl]oxy]ethyl-trimethyl-silane 60 (212 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.02 (dd, J = 8.84, 1.77 Hz, 1 H) 8.95 – 8.99 (m, 1 H) 8.17 (d, J = 8.59 Hz, 1 H) 7.74 (dd, J = 8.72, 4.17 Hz, 1 H) 7.34 (d, J = 8.59 Hz, 1 H) 7.19 (dd, J = 8.72, 5.68 Hz, 2 H) 7.00 – 7.10 (m, 2 H) 4.39 (d, J = 7.58 Hz, 2 H) 3.65 – 3.74 (m, 1 H) 3.41 – 3.52 (m, 1 H) 3.31 (d, J = 8.84 Hz, 2 H) 3.08 (s, 1 H) 2.14 – 2.26 (m, 1 H) 1.80 – 1.95 (m, 1 H) 1.27 (dd, J = 8.72, 7.71 Hz, 2 H) 0.10 – 0.17 (m, 9 H). MS (ES+) m/z 473.0 [M+H]+.
Step 3.
To a solution of 2-[[5-[3-(4-fluorophenyl)pyrrolidin-1-yl]sulfonyl-8-quinolyl]oxy]ethyl-trimethyl-silane 60 (210 mg, 0.44 mmol) in DMF (2 mL) was added cesium fluoride (202 mg, 1.3 mmol). The vial was heated to 60°C for 2 hours then warmed to 75°C for 2 additional hours. The mixture was partitioned between brine and ethyl acetate. The layers were separated and aqueous extracted with ethyl acetate (1 × 5 mL), organics combined, dried over sodium sulfate and concentrated to a DMF suspended residue. To this residue was added diethyl ether and resultant suspension filtered. The filtrate was concentrated, suspended in heptane and collected by filtration to yield 5-((3-(4-fluorophenyl)pyrrolidin-1-yl)sulfonyl)quinoline-8-ol 13, 111 mg (67% yield) as a free base. 1H NMR (400 MHz, DMSO-d6) δ ppm 10.97 – 11.14 (m, 1 H) 9.05 (dd, J = 8.72, 1.64 Hz, 1 H) 8.98 (dd, J = 4.04, 1.52 Hz, 1 H) 8.13 (d, J = 8.34 Hz, 1 H) 7.77 (dd, J = 8.84, 4.04 Hz, 1 H) 7.20 (dt, J = 8.53, 2.81 Hz, 3 H) 7.00 – 7.11 (m, 2 H) 3.68 (dd, J = 9.47, 7.45 Hz, 1 H) 3.40 – 3.50 (m, 1 H) 3.24 – 3.33 (m, 2 H) 3.02 – 3.12 (m, 1 H) 2.17 (d, J = 3.28 Hz, 1 H) 1.85 (dd, J = 12.51, 9.73 Hz, 1 H). MS (ES+) m/z 373.0 [M+H]+.
8-Methoxy-5-(pyrrolidin-1-yulsulfonyl)quinoline (14).
8-fluoroquinoline-5-sulfonyl chloride 32 (50 mg, 0.20 mmol) was dissolved in DCM (1 mL). To this solution was added a solution of pyrrolidine (17 µL, 0.20 mmol) and DIPEA (71 µL, 0.41 mmol) in DCM (0.5 mL). The resulting mixture was stirred at room temperature for 5 min. Added sodium methoxide, 25 wt % in MeOH (200 µL, 3.5 mmol) and stirred at room temperature for 2 h. The reaction mixture was purified directly by automated normal-phase chromatography (20–100% EtOAc/hexanes (7% MeOH additive), 10 g silica gel cartridge). The product-containing fractions were combined and the solvent removed in vacuo to give 8-methoxy-5-pyrrolidin-1-ylsulfonyl-quinoline 14 (47 mg, 79% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.02 (dd, J = 8.72, 1.64 Hz, 1 H) 8.97 (dd, J = 4.17, 1.64 Hz, 1 H) 8.17 (d, J = 8.34 Hz, 1 H) 7.76 (dd, J = 8.84, 4.04 Hz, 1 H) 7.34 (d, J = 8.59 Hz, 1 H) 4.07 (s, 3 H) 3.15 – 3.21 (m, 4 H) 1.69 – 1.75 (m, 4 H). MS (ES+) m/z 293.0 [M+H]+.
5-(Ethylsulfonyl)quinoline-8-ol (15).
Step 1.
To a stirring solution of 8-benzyloxyquinoline-5-sulfonyl chloride 55 (1.5 g, 4.4 mmol) in THF (50 mL), triphenylphosphine (3.5 g, 13.4 mmol) was added. The solution was heated at 60°C for 60 minutes. The solution was cooled to room temperature and diethyl ether (50 mL) added. The precipitate was collected by filtration. The precipitate was dissolved in DMF (20 mL) and to this solution was added sodium borohydride (220 mg, 5.8 mmol). The solution was stirred at room temperature for 30 minutes. Iodoethane (1.0 g, 6.7 mmol) was added and resultant mixture stirred at 60°C for two hours. The solution was cooled to room temperature and water (50 mL) added. The solution was extracted with ethyl acetate (3 × 50 mL). The combined organic solution was extracted with water (3 × 40 mL), brine (1 × 50 mL) and dried over sodium sulfate. The organic solution was filtered and concentrated. The residue was purified by automated normal-phase chromatography and eluted with ethyl acetate/hexane (0–80%) to give 8-benzyloxy-5-ethylsulfanyl-quinoline 61 (580 mg, 43% yield) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ ppm 8.99 – 9.05 (m, 1 H), 8.82 (dd, J = 8.6, 1.8 Hz, 1 H), 7.59 – 7.66 (m, 1 H), 7.49 – 7.58 (m, 3 H), 7.29 – 7.44 (m, 3 H), 6.96 – 7.04 (m, 1 H), 5.43 – 5.50 (m, 2 H), 2.77 – 2.90 (m, 2 H), 1.16 – 1.27 (m, 3 H). MS (ES+) m/z 296.1 [M+H]+.
Step 2.
To a stirring solution of 8-benzyloxy-5-ethylsulfanyl-quinoline 61 (400 mg, 1.3 mmol) in DCM (50 mL), 3-chlorobenzenecarboperoxoic acid (0.84 g, 3.4 mmol) was added. The solution was stirred at room temperature for 30 minutes. Sodium sulfite solution (1 M, 30 mL) was added. The solution was diluted with DCM (30 mL) and extracted with sodium hydroxide solution (1 N, 2 × 30 mL), brine (1 × 30 mL) and dried over sodium sulfate. The solution was filtered and concentrated. The residue was purified by automated normal-phase chromatography and eluted with methanol/DCM (0–8%) to give a white solid. Mixed fractions were pooled and purified by automated reversed-phase chromatography and eluted with 5–95% acetonitrile/water, with 0.05% TFA as modifier to give white solid which was combined with clean, pooled fractions from the previous purification to provide 8-benzyloxy-5-ethylsulfonyl-quinoline 62 (206 mg, 44% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.92 – 9.04 (m, 2 H), 8.21 (d, J = 8.6 Hz, 1 H), 7.80 (dd, J = 8.7, 4.2 Hz, 1 H), 7.35 – 7.63 (m, 6 H), 5.43 (s, 2 H), 3.39 (q, J = 7.4 Hz, 11 H), 1.09 (t, J = 7.3 Hz, 3 H). MS (ES+) m/z 328.1 [M+H]+.
Step 3.
To a stirring solution of 8-benzyloxy-5-ethylsulfonyl-quinoline 62 (200 mg, 0.61 mmol) in acetic acid (1 mL), hydrobromic acid (2.0 mL, 12.2 mmol) was added. The solution was stirred at 100°C for 16 hrs. The solution was cooled to room temperature and diluted with diethyl ether (2 mL). A precipitate formed and was collected by filtration to give 5-ethylsulfonylquinolin-8-ol hydrobromide 15 (132 mg, 67% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.14 (d, J = 8.6 Hz, 1 H), 9.02 – 9.08 (m, 1 H), 8.19 (d, J = 8.3 Hz, 1 H), 7.92 (dd, J = 8.7, 4.4 Hz, 1 H), 7.32 (d, J = 8.3 Hz, 1 H), 3.38 (q, J = 7.3 Hz, 2 H), 1.09 (t, J = 7.3 Hz, 3 H). MS (ES+) m/z 238.1 [M+H]+.
5-((2-Benzylpyrrolidin-1-yl)sulfonyl)quinoline-8-ol (16).
Step 1.
8-fluoroquinoline-5-sulfonyl chloride 32 (50 mg, 0.20 mmol) was suspended in THF (1 mL). To this suspension was added a solution of DIPEA (71 µL, 0.41 mmol) and 2-benzylpyrrolidine (33 mg, 0.20 mmol) in THF (0.5 mL). The resulting mixture was stirred at room temperature for 15 min. To this mixture was added a suspension of 2-(trimethylsilyl)ethanol (29 µL, 0.20 mmol) and sodium hydride (8 mg, 0.20 mmol) in THF (1 mL) and stirred at ambient temperature for 1 hour. A second suspension of 2-(trimethylsilyl)ethanol (29 µL, 0.204 mmol) and sodium hydride (8 mg, 0.20 mmol) in THF (1 mL) was added and stirred at ambient temperature for an additional hour. Treated reaction mixture with saturated NaHCO3 and CHCl3, agitated vigorously and poured into a phase separator tube. The organic layer was concentrated in vacuo and the residue purified by automated normal-phase chromatography (20–100% EtOAc/hexanes, 4 g silica gel cartridge). The product-containing fractions were combined, and the solvent removed in vacuo to provide 2-[[5-(2-benzylpyrrolidin-1-yl)sulfonyl-8-quinolyl]oxy]ethyl-trimethyl-silane 63 (69 mg, 72% yield) as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.06 (dd, J = 8.72, 1.64 Hz, 1 H) 8.97 (dd, J = 4.17, 1.64 Hz, 1 H) 8.23 (d, J = 8.59 Hz, 1 H) 7.76 (dd, J = 8.84, 4.04 Hz, 1 H) 7.36 (d, J = 8.59 Hz, 1 H) 7.22 – 7.28 (m, 2 H) 7.12 – 7.21 (m, 3 H) 4.34 – 4.42 (m, 2 H) 3.84 – 3.92 (m, 1 H) 3.18 – 3.32 (m, 2 H) 2.88 (dd, J = 13.14, 3.79 Hz, 1 H) 2.62 (dd, J = 13.01, 9.73 Hz, 1 H) 1.65 – 1.77 (m, 1 H) 1.41 – 1.60 (m, 3 H) 1.23 – 1.30 (m, 2 H) 0.08 – 0.14 (m, 9 H). MS (ES+) m/z 469.2 [M+H]+.
Step 2.
To a solution of 2-[[5-(2-benzylpyrrolidin-1-yl)sulfonyl-8-quinolyl]oxy]ethyl-trimethyl-silane 63 (46 mg, 0.097 mmol) in DMF (0.5 mL) was added cesium fluoride (44 mg, 0.29 mmol). The contents were stirred at 60°C for 4 h, then at ambient temperature for 48 h (for convenience). Added water and extracted with EtOAc (2 X). The organic layers were combined, washed with brine, dried with MgSO4, filtered and the solvent removed in vacuo to give 5-(2-benzylpyrrolidin-1-yl)sulfonylquinolin-8-ol 16 (30 mg, 84% yield) as a tan solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.05 (br. s., 1 H) 9.09 (dd, J = 8.84, 1.52 Hz, 1 H) 8.98 (dd, J = 4.29, 1.52 Hz, 1 H) 8.18 (d, J = 8.34 Hz, 1 H) 7.79 (dd, J = 8.84, 4.04 Hz, 1 H) 7.10 – 7.31 (m, 6 H) 3.86 (t, J = 8.46 Hz, 1 H) 3.15 – 3.30 (m, 3 H) 2.84 – 2.95 (m, 1 H) 2.62 (dd, J = 13.14, 9.60 Hz, 1 H) 1.63 – 1.76 (m, 1 H) 1.36 – 1.60 (m, 3 H). MS (ES+) m/z 369.0 [M+H]+.
5-((4-(Trifluoromethyl)phenyl)sulfonyl)quinolin-8-ol (17).
Step 1.
A mixture of 8-benzyloxy-5-bromo-quinoline 51 (compound 8, step 1) (5.0 g, 15.9 mmol), 2-trimethylsilylethanethiol (3.8 mL, 23.8 mmol), tris(dba)dipalladium (729 mg, 0.80 mmol), Xantphos (460.4 mg, 0.80 mmol) and cesium carbonate (15.5 g, 47.7 mmol) in dioxane (125 mL) was degassed with nitrogen. The resultant mixture was heated at 100°C overnight. The mixture was passed through a plug of silica eluting with ethyl acetate and concentrated to a residue. The residue was purified by automated normal-phase chromatography eluting 0–30% ethyl acetate/hexanes to yield 5.8 g (99%) of 2-[(8-benzyloxy-5-quinolyl)sulfanyl]ethyl-trimethyl-silane 64 as a golden oil. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.88 – 8.93 (m, 1 H) 8.64 – 8.71 (m, 1 H) 7.62 – 7.72 (m, 2 H) 7.52 – 7.59 (m, 2 H) 7.40 – 7.48 (m, 2 H) 7.38 (d, J = 6.06 Hz, 1 H) 7.27 – 7.33 (m, 1 H) 5.33 (br. s., 2 H) 2.86 – 2.96 (m, 2 H) 0.75 – 0.84 (m, 2 H)- 0.08 – 0.00 (m, 9 H). MS (ES+) m/z 368.2 [M+H]+.
Step 2.
A mixture of 2-[(8-benzyloxy-5-quinolyl)sulfanyl]ethyl-trimethyl-silane 64 (5.2 g, 11.3 mmol), cesium carbonate (11.1 g, 34.1 mmol), Xantphos (329 mg, 0.57 mmol) and tris(dba)dipalladium (521 mg, 0.57 mmol) was suspended in dioxane (100 mL). To this mixture was added 1-bromo-4-(trifluoromethyl)benzene (1.9 mL, 13.6 mmol) followed by tetrabutyammonium fluoride (1 M solution in THF) (13.6 mL, 13.6 mmol). The resultant mixture was degassed (X 2) with nitrogen. With a nitrogen vent afixed, the reaction mixture was heated to 75°C for 2 hours. The reaction was cooled to ambient temperature and filtered through a plug of silica gel. The filtrate was concentrated and purified using silica gel chromatography eluting 0–30% ethyl acetate/hexanes to provide 8-benzyloxy-5-[4-(trifluoromethyl)phenyl]sulfanyl-quinoline 65, 5.1 g (assume quantitative yield). The material was taken on in subsequent reactions without additional purification and or characterization. 1H NMR (400 MHz, CDCl3) δ ppm 9.04 (dd, J=4.17, 1.64 Hz, 1 H) 8.57 (dd, J=8.59, 1.77 Hz, 1 H) 7.83 (d, J=8.08 Hz, 1 H) 7.54 – 7.60 (m, 2 H) 7.50 (dd, J=8.59, 4.04 Hz, 1 H) 7.32 – 7.46 (m, 5 H) 7.11 (d, J=8.08 Hz, 1 H) 7.02 (d, J=8.08 Hz, 2 H) 5.52 (s, 2 H). MS (ES+) m/z 412.0 [M+H]+.
Step 3.
To a stirred solution of 8-benzyloxy-5-[4-(trifluoromethyl)phenyl]sulfanyl-quinoline 65 (5.1 g, 12.5 mmol) in DCM (100 mL), 3-chlorobenzenecarboperoxoic acid (4.5 g, 26.2 mmol) was added. The solution was stirred at room temperature for 3 hours. Additional mCPBA (1 g) was added and the resultant reaction allowed to stir at ambient temperature overnight. Sodium bisulfite (10%, 50 mL) was added and stirred for one hour. The layers were separated. The organic solution was stirred with 10% (2.5 M) NaOH for 1 hour and subsequent layers separated. The organic solution was dried over sodium sulfate and concentrated to a residue. The residue was purified by silica chromatography eluting 0–50% ethyl acetate/hexanes. Isolated a yellow foam. The foam was dissolved in diethyl ether (20 mL). Solids formed and the suspension heated to reflux. The suspension was allowed to cool to ambient temperature and solids collected by filtration, washed with ether and hexanes to yield 8-benzyloxy-5-[4-(trifluoromethyl)phenyl]sulfonyl-quinoline 66, 3.7 g (68% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.96 (dd, J = 4.17, 1.64 Hz, 1 H) 8.85 (dd, J = 8.72, 1.64 Hz, 1 H) 8.57 (d, J = 8.59 Hz, 1 H) 8.20 (d, J = 8.34 Hz, 2 H) 7.97 (d, J = 8.34 Hz, 2 H) 7.72 (dd, J = 8.84, 4.04 Hz, 1 H) 7.53 – 7.62 (m, 3 H) 7.36 – 7.49 (m, 3 H) 5.44 (s, 2 H). MS (ES+) m/z 444.0 [M+H]+.
Step 4.
A solution of 8-benzyloxy-5-[4-(trifluoromethyl)phenyl]sulfonyl-quinoline 66 (3.7 g, 8.5 mmol) and 6 M aqueous hydrochloric acid (30 mL, 180 mmol) was heated to 100°C for 1.5 hours. Dioxane (40 mL) was added to aid solubility. The mixture was heated to 100°C overnight. The reaction was cooled to ambient temperature and pH adjusted to ~5 with 10% NaOH aqueous solution. The slurry was filtered and solids washed with water to yield 5-((4-(Trifluoromethyl)phenyl)sulfonyl)quinolin-8-ol 17, 2.9 g (97% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.35 – 11.51 (m, 1 H) 8.96 (dd, J = 4.30, 1.52 Hz, 1 H) 8.86 (dd, J = 8.84, 1.52 Hz, 1 H) 8.47 (d, J = 8.34 Hz, 1 H) 8.13 – 8.22 (m, 2 H) 7.96 (d, J = 8.84 Hz, 2 H) 7.74 (dd, J = 8.84, 4.04 Hz, 1 H) 7.30 (d, J = 8.59 Hz, 1 H). MS (ES+) m/z 354.0 [M+H]+.
7-Fluoro-5-((4-(trifluoromethyl)phenyl)sulfonyl)quinolin-8-ol (18).
Step 1.
A mixture of 2-amino-6-fluoro-phenol (1.8 mL, 13.9 mmol), glycerol (2.5 mL, 34.7 mmol) and sodium 3-nitrobenzenesulfonate (3.7 g, 16.7 mmol) was treated with 70% sulfuric acid (11.9 mL, 224.5 mmol) and heated to reflux (140°C) for 1 hour. The mixture was cooled, diluted with water (75 mL) and basified with 50% sodium hydroxide aqueous solution, pH~8. Filtration through Celite gave a solid which was washed with dichloromethane. The aqueous filtrate was washed with dichloromethane and combined extracts evaporated to give an oily reside. The oily residue and brown solids were combined and purified by silica chromatography (50 g) eluting 0–20% dichloromethane/methanol to yield 7-fluoroquinolin-8-ol 41 as a pale yellow solid, 1.3 g, 56%. 1H NMR (400 MHz, DMSO-d6) δ ppm 7.42 – 7.49 (m, 1 H) 7.50 – 7.57 (m, 2 H) 8.37 (dd, J=8.34, 1.77 Hz, 1 H) 8.90 (dd, J=4.30, 1.52 Hz, 1 H) 10.26 (br. s., 1 H). MS (ES+) m/z 164.2 [M+H]+.
Step 2.
A solution of 7-fluoroquinolin-8-ol 41 (4.0 g, 24.5 mmol) and N-iodosuccinimide (6.6 g, 29.4 mmol) in chloroform was vigorously stirred at 40°C. After 30 minutes the reaction was diluted with dichloromethane, extracted 10% sodium thiosulfate solution (2 × 20 mL) and dried over sodium sulfate. The 7-fluoro-5-iodo-quinolin-8-ol 67, isolated as pale yellow solids were taken on in subsequent reactions without additional purification. (6.5 g, 92% yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.80 (dd, J = 4.17, 1.39 Hz, 1 H) 8.32 (dd, J = 8.59, 1.52 Hz, 1 H) 7.97 (d, J = 9.85 Hz, 1 H) 7.53 (dd, J = 8.59, 4.29 Hz, 1 H). MS (ES+) m/z 290.0 [M+H]+.
Step 3.
To a solution of sodium hydride (654 mg, 27.2 mmol) in DMF (100 mL) was added 7-fluoro-5-iodo-quinolin-8-ol 67 (6.5 g, 22.7 mmol) as a solution in DMF. After 60 minutes benzyl bromide (3.2 mL, 27.2 mmol) was added slowly dropwise. The reaction was maintained at ambient temperature. The reaction was allowed to stir overnight at ambient temperature. The reaction was quenched by addition of water. Ethyl acetate was added and the layers separated. The aqueous layer was extracted with ethyl acetate (2 × 20 mL), the organics were combined and dried over sodium sulfate. Concentration gave an oily residue that was purified by automated normal-phase chromatography (40 g) eluting 0–20% ethyl acetate/hexanes. Isolated 8-benzyloxy-7-fluoro-5-iodo-quinoline 42 as an orange oil that solidified upon standing (6.3 g, 73% yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.96 (dd, J = 4.04, 1.52 Hz, 1 H) 8.33 (dd, J = 8.46, 1.64 Hz, 1 H) 7.93 (d, J = 10.11 Hz, 1 H) 7.52 – 7.56 (m, 2 H) 7.49 (dd, J = 8.59, 4.04 Hz, 1 H) 7.31 – 7.38 (m, 3 H) 5.54 (s, 2 H). MS (ES+) m/z 380.0 [M+H]+.
Step 4.
A mixture of 8-benzyloxy-7-fluoro-5-iodo-quinoline 42 (3 g, 7.9 mmol), 4-(trifluoromethyl)thiophenol (1.6 mL, 11.8 mmol), tris(dba)dipalladium (362 mg, 0.40 mmol), Xantphos (228 mg, 0.40 mmol) and cesium carbonate (7.7 g, 23.7 mmol) in dioxane (50 mL) was degassed with nitrogen. The resultant mixture was heated at 90°C overnight. The mixture was passed through a plug of silica eluting with ethyl acetate and concentrated to a residue. The residue was purified automated normal-phase chromatography eluting 0–30% ethyl acetate/hexanes to provide 2.0 g (60% yield) of 8-benzyloxy-7-fluoro-5-[4-(trifluoromethyl)phenyl]sulfanyl-quinoline 68 which was taken in in the next step. MS (ES+) m/z 430.0 [M+H]+.
Step 5.
To a mixture of 8-benzyloxy-7-fluoro-5-[4-(trifluoromethyl)phenyl]sulfanyl-quinoline 68 (2.0 g, 4.8 mmol) in DCM (35 mL) at 0°C was added 3-chlorobenzenecarboperoxoic acid (1.6 g, 9.5 mmol). The reaction was stirred at 0°C for 90 minutes. Additional mCPBA was added to drive the reaction to completion. Sodium sulfite (1.0 N solution, 50 mL) was added and stirred for 60 minutes then removed followed by sodium hydroxide solution (1.0 N, 5 mL). The solution was stirred at room temperature overnight. The aqueous phase was removed and organic solution was dried over sodium sulfate. The residue was purified by normal phase chromatography eluting 0–50% ethyl acetate/hexanes to provide 1.3 g (60% yield) of 8-benzyloxy-7-fluoro-5-[4-(trifluoromethyl)phenyl]sulfonyl-quinoline 69 as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.04 (dd, J = 4.04, 1.52 Hz, 1 H) 8.94 (dd, J = 8.84, 1.52 Hz, 1 H) 8.40 (d, J = 11.12 Hz, 1 H) 8.07 (d, J = 8.34 Hz, 2 H) 7.78 (d, J = 8.84 Hz, 2 H) 7.47 – 7.59 (m, 3 H) 7.29 – 7.41 (m, 3 H) 5.72 (s, 2 H). MS (ES+) m/z 462.0 [M+H]+.
Step 6.
A solution of 8-benzyloxy-7-fluoro-5-[4-(trifluoromethyl)phenyl]sulfonyl-quinoline 69 (1.3 g, 2.9 mmol) and 6 M aqueous hydrochloric acid (20 mL, 120mmol) in dioxane (20 mL) to aid solubility, was heated to 90°C for 3 hours. The solution was cooled to ambient temperature and neutralized with 10% sodium hydroxide solution. Solids formed and were collected by filtration. 7-Fluoro-5-((4-(trifluoromethyl)phenyl)sulfonyl)quinolin-8-ol 18 was collected as tan solids (769 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ ppm 9.01 (dd, J = 8.84, 1.52 Hz, 1 H) 8.89 (dd, J = 4.29, 1.52 Hz, 1 H) 8.48 (d, J = 10.36 Hz, 1 H) 8.09 (d, J = 8.34 Hz, 2 H) 7.78 (d, J = 8.34 Hz, 2 H) 7.61 (dd, J = 8.84, 4.29 Hz, 1 H). MS (ES+) m/z 372.0 [M+H]+.
7-Chloro-5-((4-fluorophenyl)sulfonyl)quinolin-8-ol (19).
Step 1.
To a solution of 5-bromoquinolin-8-ol 36 (6.9 g, 31.0 mmol) in chloroform (200 mL), N-chlorosuccinimide (4.5 g, 34.1 mmol) was added. The solution was stirred at 50°C for 20 hours. The solution was concentrated and methanol (150 mL) was added. The solution was stirred for 10 minutes. The precipitate was collected by filtration and dried to give 5-bromo-7-chloro-quinolin-8-ol 70 (6.3 g 78% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 11.08 (br. s., 2 H), 8.99 (br. s., 2 H), 8.46 (br. s., 1 H), 7.99 (br. s., 2 H), 7.77 (br. s., 1 H). MS (ES+) m/z 259.9 [M+H]+.
Step 2.
To a solution of 5-bromo-7-chloro-quinolin-8-ol 70 (6.3 g, 24.3 mmol) and benzyl bromide (3.1 mL, 26.8 mmol) in acetonitrile (100 mL) was added potassium carbonate (5.0 g, 36.5 mmol). The solution was stirred at 50°C for 7 hours then cooled to room temperature. The solution was filtered. Water (50 ml) was added to the filtrate and an off-white precipitate (crystals) formed. The solids were collected by filtration, washed with water and air-dried to give 8-benzyloxy-5-bromo-7-chloro-quinoline 71 (7.6 g, 89% yield) as an off-white solid. 1H NMR (400 MHz, CDCl3) δ ppm 8.99 – 9.05 (m, 1 H), 8.51 (dd, J = 8.6, 1.5 Hz, 1 H), 7.84 – 7.88 (m, 1 H), 7.59 – 7.65 (m, 2 H), 7.52 – 7.59 (m, 1 H), 7.31 – 7.44 (m, 3 H), 5.50 (s, 2 H). MS (ES+) m/z 349.9 [M+H]+.
Step 3.
2-(Trimethylsilyl)ethane-1-thiol (1.7 g, 12.9 mmol) was added to a solution mixture of 8-(benzyloxy)-5-bromo-7-chloro-quinoline 71 (3.0 g, 8.61 mmol), tris(dba)dipalladium(0) (394 mg, 0.43 mmol), Xantphos (249 mg, 0.43 mmol) in 1,4-dioxane (58 mL) at room temperature. Cesium carbonate (8.4 g, 25.8 mmol) was added. The reaction was sealed with a septum and heated to 90°C under nitrogen. Upon completion, the crude material was poured into brine, extracted with ethyl acetate, dried over magnesium sulfate, filtered, concentrated under reduced pressure. The residue was dissolved in a minimal quantity of dichloromethane and purified by automated normal phase chromatography (0–30% ethyl acetate/heptane). The product-containing fractions were combined, to give 8-(benzyloxy)-7-chloro-5-((2-(trimethylsilyl)ethyl)thio)quinoline 72 (3.2 g, 93% yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.99 – 9.04 (m, 1H) 8.66 – 8.73 (m, 1H) 7.61 – 7.69 (m, 2H) 7.54 – 7.59 (m, 1H) 7.47 – 7.52 (m, 1H) 7.33 – 7.44 (m, 3H) 5.45 – 5.52 (m, 2H) 2.94 – 3.04 (m, 2H) 0.91 – 1.01 (m, 2H) 0.03 – 0.09 (m, 9H). MS (ES+) m/z 402.0 [M+H]+.
Step 4.
1,4-Dioxane was degassed by nitrogen displacement for 10 minutes prior to initiating reaction. A solution of 8-(benzyloxy)-7-chloro-5-((2-(trimethylsilyl)ethyl)thio)quinoline 72 (300 mg, 0.75 mmol) in 1,4-dioxane (5 mL) was added to a 50 mL round bottom flask charged with a magnetic stir bar, 4-fluoroiodobenzene (199 mg, 0.90 mmol), tris(dba)dipalladium(0) (34 mg, 0.04 mmol), Xantphos (21 mg, 0.04 mmol). To this solution mixture was added cesium carbonate (729 mg, 2.24 mmol) followed by tetrabutylammonium fluoride (895 µL, 1M THF, 0.89 mmol). The flask was sealed with a rubber septum, and heated to 90°C. Upon completion the reaction was cooled room temperature, poured into brine, extracted with ethyl acetate, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude residue was dissolved in minimal dichloromethane and purified by automated normal phase chromatography eluting with 0–35% ethyl acetate/heptane over a 35 minute gradient. The product attained was 8-(benzyloxy)-7-hloro-5-((4-fluorophenyl)thio)quinoline 73 (275 mg, 93% yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.99 – 9.04 (m, 1 H) 8.61 – 8.67 (m, 1 H) 7.59 – 7.68 (m, 4 H) 7.32 – 7.52 (m, 7 H) 7.19 – 7.27 (m, 2 H) 6.97 – 7.07 (m, 2 H) 5.50 – 5.58 (m, 2 H). MS (ES+) m/z 396.0 [M+H]+.
Step 5.
3-chlorobenzoperoxoic acid (327 mg, 1.46 mmol) was added to a solution of 8-(benzyloxy)-7-chloro-5-((4-fluorophenyl)thio)quinoline 73 (275 mg, 0.69 mmol) in dichloromethane (7 mL) at 0°C. Upon completion the reaction was quenched reaction with dimethyl sulfide (70 µL, 1.53 mmol). The crude mixture was poured into saturated sodium bicarbonate, dried over sodium sulfate, decanted and concentrated under reduced pressure. The residue was dissolved in minimal dichloromethane and purified by automated normal phase chromatography eluting with 0–20% ethyl acetate/heptane over a 20 minute gradient. The product attained was 8-(benzyloxy)-7-chloro-5-((4-fluorophenyl)sulfonyl)quinoline 74 (200 mg, 67% yield). 1H NMR (400 MHz, CDCl3) δ ppm 9.03 – 9.08 (m, 1H) 9.00 (dd, J = 8.8, 1.5 Hz, 1H) 8.53 – 8.56 (m, 1H) 7.96 – 8.02 (m, 2H) 7.53 – 7.60 (m, 3H) 7.34 – 7.41 (m, 3H) 7.17 – 7.24 (m, 2H) 5.61 – 5.66 (m, 2H). MS (ES+) m/z 428.0 [M+H]+.
Step 6.
8-(benzyloxy)-7-chloro-5-((4-fluorophenyl)sulfonyl)quinoline 74 (200 mg, 0.470 mmol) was dissolved in 6 N aqueous hydrochloric acid (5 mL). 1,4-Dioxane (3 mL) was added to solubilize the reaction. The reaction was heated reaction to 80°C and upon completion cooled to room temperature. The mixture was concentrated under reduced pressure, azeotroped with acetonitrile and suspended in chloroform. The precipitate was collected by vacuum filtration and dried under high vacuum. The product attained was 7-chloro-5-((4-fluorophenyl)sulfonyl)quinolin-8-ol 19 (135 mg, 86% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.00 (d, J = 4.11 Hz, 1 H) 8.91 (d, J = 8.72 Hz, 1 H) 8.46 (s, 1 H) 8.11 – 8.18 (m, 2 H) 7.78 (dd, J = 8.72, 4.23 Hz, 1 H) 7.43 (t, J = 8.05 Hz, 2 H). 19F NMR (376 MHz, DMSO-d6) d ppm −199.76 (s, 1 F). MS (ES+) m/z 337.9 [M+H]+.
7-Fluoro-5-((4-fluorophenyl)sulfonyl)quinolin-8-ol (20).
Step 1.
4-fluorobenzenethiol (4.8 mL, 44.7 mmol) was added to a solution mixture of 8-(benzyloxy)-7-fluoro-5-iodoquinoline 42 (11.3 g, 29.8 mmol), tris(dba)dipalladium(0) (1.3 g, 1.5 mmol), Xantphos (862 mg, 1.4 mmol) in 1,4-dioxane (132 mL) at room temperature followed by addition of cesium carbonate (29.1 g, 89.4 mmol). The mixture was heated to 90°C under nitrogen. Upon completion, the mixture was poured into brine, extracted with ethyl acetate, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The crude material was dissolved in a minimal quantity of dichloromethane and purified by automated normal phase chromatography (0–30% ethyl acetate/heptane). The product-containing fractions were combined to give 8-(benzyloxy)-7-fluoro-5-((4-fluorophenyl)thio)quinoline 75 (10.8 g, 95% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.05 (dd, J = 4.20, 1.48 Hz, 1 H) 8.63 (dd, J = 8.59, 1.58 Hz, 1 H) 7.60 – 7.74 (m, 2 H) 7.46 – 7.53 (m, 2 H) 7.30 – 7.42 (m, 6 H) 7.16 – 7.25 (m, 2 H) 5.50 (s, 2 H). MS (ES+) m/z 380.0 [M+H]+.
Step 2.
3-chlorobenzoperoxoic acid (12.7 g, 56.7 mmol) was added to a solution 8-(benzyloxy)-7-fluoro-5-((4-fluorophenyl)thio)quinoline 75 (10.8 g, 28.3 mmol) in dichloromethane (57 mL) at room temperature. The reaction mixture was filtered, solids discarded, filtrate washed with 10% sodium sulfite solution, followed by saturated sodium bicarbonate and brine. The solution was dried over sodium sulfate, decanted and concentrated under reduced pressure. The crude material was dissolved in minimal dichloromethane and purified by automated normal phase chromatography (0–100% ethyl acetate/heptane) to provide 9.0 g (77 % yield) of 8-(benzyloxy)-7-fluoro-5-((4-fluorophenyl)sulfonyl)quinoline 76. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.08 (dd, J = 4.11, 1.33 Hz, 1 H) 8.88 – 8.99 (m, 1 H) 8.50 (d, J = 10.67 Hz, 1 H) 8.17 (dd, J = 8.84, 5.05 Hz, 2 H) 7.73 (dd, J = 8.81, 4.14 Hz, 1 H) 7.25 – 7.57 (m, 7 H) 5.62 (s, 2 H). MS (ES+) m/z 412.0 [M+H]+.
Step 3.
8-(benzyloxy)-7-fluoro-5-((4-fluorophenyl)sulfonyl)quinoline 76 (9.0 g, 21.9 mmol) was suspended in 6 N aqueous hydrochloric acid (120 mL). 1–4-Dioxane (40 mL) was added to solubilize reaction. The mixture was heated to 90°C. Upon completion, the flask was cooled to room temperature, concentrated under reduced pressure and suspended in toluene. The solids were collected via vacuum, filtration. The solids were dissolved in chloroform, poured into 1N sodium hydroxide solution, extracted with chloroform, dried over magnesium sulfate, filtered and concentrated under reduced pressure. The product, 7-fluoro-5-((4-fluorophenyl)sulfonyl)quinolin-8-ol 20 (5.9 g, 84% yield) was isolated as the free base. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.99 (dd, J = 4.17, 1.39 Hz, 1 H) 8.91 (dd, J = 8.84, 1.45 Hz, 1 H) 8.46 (d, J = 10.93 Hz, 1 H) 8.13 (dd, J = 8.87, 5.08 Hz, 2 H) 7.72 (dd, J = 8.78, 4.17 Hz, 1 H) 7.43 (t, J = 8.81 Hz, 2 H). MS (ES+) m/z 322.0 [M+H]+.
7-Fluoro-5-tosylquinolin-8-ol (21).
Step 1.
A mixture of 8-benzyloxy-7-fluoro-5-iodo-quinoline 42 (3.0 g, 7.9 mmol), 4-methylbenzenethiol (1.4 g, 11.8 mmol), tris(dba)dipalladium (362 mg, 0.40 mmol), Xantphos (228 mg, 0.40 mmol) and cesium carbonate (7.7 g, 23.7 mmol) in dioxane (5 mL) was degassed with nitrogen. The resultant mixture was heated at 90°C overnight. The reaction mixture was filtered through a plug of silica gel eluting with ethyl acetate. The filtrate was concentrated and purified automated normal-phase chromatography eluting 0–30% ethyl acetate/hexanes to yield 2.4 g (82%) of 8-benzyloxy-7-fluoro-5-(p-tolylsulfanyl)quinoline 77. 1H NMR (400 MHz, CDCl3) δ ppm 9.01 (dd, J = 4.04, 1.52 Hz, 1 H) 8.62 – 8.67 (m, 1 H) 7.52 – 7.58 (m, 2 H) 7.44 (dd, J = 8.59, 4.29 Hz, 1 H) 7.29 – 7.39 (m, 4 H) 7.09 – 7.20 (m, 4 H) 5.55 (s, 2 H) 2.34 (s, 3 H). MS (ES+) m/z 376.1 [M+H]+.
Step 2.
To a mixture of 8-benzyloxy-7-fluoro-5-(p-tolylsulfanyl)quinoline 77 (2.4 g, 6.4 mmol) in DCM (40 mL) at 0°C was added 3-chlorobenzenecarboperoxoic acid (2.2 g, 12.9 mmol). The reaction was stirred at 0°C for 1 hour. The cooling bath was removed and additional 0.5 g of mCPBA was added to drive the reaction to completion. Sodium sulfite (10% solution, 75 mL) was added and stirred for 60 minutes then removed followed by sodium hydroxide aqueous solution (1 N, 75 mL). The solution was stirred at room temperature overnight. The aqueous phase was removed and organic solution was dried over sodium sulfate. The residue was purified by normal phase chromatography eluting 0–50% ethyl acetate/hexanes. The material was taken on as is without additional purification (oil contained residual ethyl acetate). Isolated 2.4 g (92% yield) of 8-benzyloxy-7-fluoro-5-(p-tolylsulfonyl)quinoline 78 as a viscous, yellow oil. 1H NMR (400 MHz, CDCl3) δ ppm 8.95 – 9.03 (m, 2 H) 8.33 (d, J = 11.12 Hz, 1 H) 7.79 – 7.86 (m, 2 H) 7.47 – 7.54 (m, 3 H) 7.29 – 7.39 (m, 5 H) 5.67 (s, 2 H) 2.40 (s, 3 H). MS (ES+) m/z 408.0 [M+H]+.
Step 3.
A solution of 8-benzyloxy-7-fluoro-5-(p-tolylsulfonyl)quinoline 78 (2.4 g, 5.9 mmol) and 6 M aqueous hydrochloric acid (30 mL, 180 mmol) in dioxane (30 mL) to aid solubility, was heated to 90°C for 3 hours. The solution was cooled to ambient temperature and neutralized with 10% sodium hydroxide solution. Excess dioxane was removed by rotary evaporator and solids formed. The aqueous mixture was neutralized with 10% aqueous sodium hydroxide solution. The orange solids were collected by filtration and washed with water to provide the title compound 21 (1.7 g, 89% yield). 1H NMR (400 MHz, CDCl3) δ ppm 9.04 (dd, J = 8.72, 1.39 Hz, 1 H) 8.86 (dd, J = 4.29, 1.52 Hz, 1 H) 8.42 (d, J = 10.36 Hz, 1 H) 7.82 – 7.87 (m, 2 H) 7.56 (dd, J = 8.72, 4.17 Hz, 1 H) 7.30 (d, J = 8.08 Hz, 2 H) 2.39 (s, 3 H). MS (ES+) m/z 318.0 [M+H]+.
7-Chloro-5-tosylquinolin-8-ol (22).
Step 1.
1,4-Dioxane was degassed by sparging with nitrogen for 10 minutes prior to initiating reaction. A solution of 8-(benzyloxy)-7-chloro-5-((2-(trimethylsilyl)ethyl)thio)quinoline 72 (2.1 g, 5.2 mmol) in 1,4-dioxane (6 mL) was added to a 250 mL round bottom flask charged with a magnetic stir bar, 4-iodotoluene (1.3 g, 5.75 mmol), tris(dba)dipalladium(0) (239 mg, 0.26 mmol), Xantphos (151 mg, 0.26 mmol). To this solution mixture was added cesium carbonate (5.1 g, 15.7 mmol), followed by tetrabutylammonium fluoride (6.3 mL, 1M THF, 6.3 mmol). The reaction was sealed reaction with rubber septum and heated to 90°C. Upon completion the mixture was cooled to room temperature, poured into brine, extracted with ethyl acetate, dried over sodium sulfate, filtered, and concentrated under reduced pressure. The crude mixture was dissolved in minimal dichloromethane and purified by automated normal phase chromatography eluting with 0–40% ethyl acetate/heptane gradient over 35 minutes. The product attained was 8-(benzyloxy)-7-chloro-5-(p-tolylthio)quinoline 79 (1.7 g, 81% yield) was taken on directly in the next step. MS (ES+) m/z 392.0 [M+H]+.
Step 2.
3-Chlorobenzoperoxoic acid (2.0 g, 8.9 mmol) was added to a solution of 8-(benzyloxy)-7-chloro-5-(p-tolylthio)quinoline 79 (1.6 g, 4.0 mmol) in dichloromethane (8.7 mL) at 0 °C. The reaction was warmed to room temperature. Benzoic acid was collected by filtration, the filtrate was washed with 10% sodium sulfite solution, followed by saturated sodium bicarbonate and brine. The solution was dried over sodium sulfate, decanted and concentrated under reduced pressure. The residue was dissolved in a minimal quantity of dichloromethane and purified by automated normal phase chromatography (12 g silica gel cartridge) eluting with 0–100% ethyl acetate/heptane. The product attained was 8-(benzyloxy)-7-chloro-5-tosylquinoline 80 (1.1 g, 64% yield). MS (ES+) m/z 424.0 [M+H]+.
Step 3.
8-(benzyloxy)-7-chloro-5-tosylquinoline 80 (1.1 g, 2.59 mmol) was suspended in of 6 N aqueous hydrochloric acid (5 mL) followed by addition of 1,4-dioxane (6 mL) to solubilize reaction and resultant mixture heated to 65°C. Upon completion the reaction was cooled to room temperature, concentrated under reduced pressure and azeotroped with acetonitrile. The material was taken up in chloroform, product precipitated and solids collected via vacuum filtration. The product attained was 7-chloro-5-tosylquinolin-8-ol 22 (600 mg, 69% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.98 (dd, J = 4.17, 1.39 Hz, 1 H) 8.89 (dd, J = 8.78, 1.39 Hz, 1 H) 8.42 (s, 1 H) 7.91 (d, J = 8.34 Hz, 2 H) 7.77 (dd, J = 8.78, 4.23 Hz, 1 H) 7.39 (d, J = 8.15 Hz, 2 H) 2.33 (s, 3 H). MS (ES+) m/z 334.0 [M+H]+.
7-Bromo-5-tosylquinolin-8-ol (23).
Step 1.
To a slurry of 8-((4-methoxybenzyl)oxy)-5-tosylquinoline 35 (500 mg, 1.2 mmol) in dichloromethane was added trifluoroacetic acid (92 μL, 1.2 mmol). The reaction was maintained at ambient temperature for 1 hour prior to concentration to an oily residue. Water was added and a gooey solid formed. The aqueous solution was neutralized with 6 M sodium hydroxide and diluted with dichloromethane. The layers were separated and organic solution dried over sodium sulfate. Concentration gave a sticky solid that was purified by automated normal-phase chromatography using 0–10% methanol/dichloromethane as an eluent to provide 420 mg (85% yield) of 5-(p-Tolylsulfonyl)quinolin-8-ol 7. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.94 (dd, J = 4.04, 1.52 Hz, 1 H) 8.88 (dd, J = 8.84, 1.52 Hz, 1 H) 8.39 (d, J = 8.34 Hz, 1 H) 7.83 (d, J = 8.34 Hz, 2 H) 7.72 (dd, J = 8.72, 4.17 Hz, 1 H) 7.37 (d, J = 8.34 Hz, 2 H) 7.27 (d, J = 8.34 Hz, 1 H) 3.34 (br. s., 1 H) 2.32 (s, 3 H). MS (ES+) m/z 301.0 [M+H]+.
Step 2.
A solution of 5-(p-tolylsulfonyl)quinolin-8-ol 7 (290 mg, 0.97 mmol) and N-bromosuccinimide (206 mg, 1.1 mmol) in chloroform was vigorously stirred at 40°C for 3 hours. The resulting solution was washed with 10% aqueous sodium thiosulfate solution (1 × 10 mL) and layers separated. The solvent was removed by rotary evaporator and product isolated as a yellow-brown solid. The solids were suspended in dichloromethane and diethyl ether, material collected by filtration and washed with ether to yield pale yellow solids. The filtrate was collected and concentrated to a residue to provide 7-bromo-5-(p-tolylsulfonyl)quinolin-8-ol 23 (131 mg, 36% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.96 (d, J = 4.04 Hz, 1 H) 8.88 (d, J = 7.83 Hz, 1 H) 8.51 (s, 1 H) 7.91 (d, J = 8.34 Hz, 2 H) 7.77 (dd, J = 8.59, 4.29 Hz, 1 H) 7.39 (d, J = 8.59 Hz, 2 H) 2.33 (s, 3 H). MS (ES+) m/z 379.1 [M+H]+.
5-Tosyl-7-(trifluoromethyl)quinolin-8-ol (24).
Step 1.
A mixture of 2-fluoro-3-(trifluoromethyl)aniline (1.8 mL, 13.9 mmol), glycerol (2.5 mL, 34.8 mmol) and sodium 3-nitrobenzenesulfonate (3.7 g, 16.7 mmol) was treated with 70% aqueous sulfuric acid (12 mL, 225.1 mmol) and heated to reflux (140°C) for 2.5 hours. The mixture was cooled to ambient temperature, diluted with water (25 mL) and basified with 50% sodium hydroxide aqueous solution. Filtration through Celite captured a solid which was washed with dichloromethane. The aqueous filtrate was extracted with dichloromethane and combined extracts evaporated to give an oily reside. The oily residue and brown solids were combined and purified by automated normal phase chromatography (50 g silica gel cartridge) eluting with 0–50% ethyl acetate/heptane. Isolated a pale yellow solid (2.1 g, 70% yield) as 8-fluoro-7-(trifluoromethyl)quinoline 81. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.08 – 9.15 (m, 1 H) 8.60 (dd, J = 8.46, 1.39 Hz, 1 H) 8.06 (d, J = 8.59 Hz, 1 H) 7.89 (t, J = 7.58 Hz, 1 H) 7.83 (dd, J = 8.34, 4.04 Hz, 1 H). MS (ES+) m/z 216.2 [M+H]+.
Step 2.
To a suspension of sodium hydride (706 mg, 17.6 mmol) in THF (20 mL) was added 2-(trimethylsilyl)ethanol (2.5 mL, 17.6 mmol). The resultant slurry was stirred for 15 minutes at ambient temperature. The this suspension was added to a solution of 8-fluoro-7-(trifluoromethyl)quinoline 81 (1.0 g, 4.6 mmol) in THF (5 mL) and stirred for 1 hour. The reaction mixture was treated with ice and partitioned between water and ethyl acetate. The layers were separated and the aqueous layer extracted ethyl acetate (1 × 20 mL). The organic washes were combined, extracted with brine (1 × 20 mL) and dried over sodium sulfate. Concentration yielded a pale yellow oil that was purified by automated normal phase chromatography (25 g) eluting 0–40% ethyl acetate/heptane. Trimethyl-[2-[[7-(trifluoromethyl)-8-quinolyl]oxy]ethyl]silane 82 was isolated as a pale yellow oil (1.4 g, 99% yield). 1H NMR (400 MHz, CDCl3) δ ppm 8.94 – 9.05 (m, 1 H) 8.21 (d, J = 8.34 Hz, 1 H) 7.65 – 7.76 (m, 1 H) 7.61 (d, J = 8.59 Hz, 1 H) 7.52 (dd, J = 8.21, 4.17 Hz, 1 H) 4.59 – 4.73 (m, 2 H) 1.34 – 1.47 (m, 3 H) 0.04 – 0.12 (m, 9 H). MS (ES+) m/z 314.0 [M+H]+.
Step 3.
To a solution of trimethyl-[2-[[7-(trifluoromethyl)-8-quinolyl]oxy]ethyl]silane 82 (500 mg, 1.6 mmol) dissolved in 10 mL dioxane was added 37% hydrochloric acid (1.2 mL). The reaction allowed to stir over the 2 days (for convenience) at ambient temperature. The reaction mixture was concentrated to a pale yellow solid. Assumed quantitative conversion to provide 7-(trifluoromethyl)quinolin-8-ol hydrochloride 83. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.99 (dd, J = 4.17, 1.64 Hz, 1 H) 8.47 (dd, J = 8.34, 1.52 Hz, 1 H) 7.74 (dd, J = 8.34, 4.29 Hz, 1 H) 7.68 (d, J = 8.59 Hz, 1 H) 7.54 (d, J = 8.84 Hz, 1 H). MS (ES+) m/z 214.0 [M+H]+.
Step 4.
5-Iodo-7-(trifluoromethyl)quinolin-8-ol 84 was prepared in a similar manner as Compound 18, step 2. The product was prepared from 7-(trifluoromethyl)quinolin-8-ol hydrochloride 83 (400 mg, 1.6 mmol) providing 543 mg (99% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.00 (dd, J = 4.17, 1.39 Hz, 1 H) 8.39 (dd, J = 8.59, 1.52 Hz, 1 H) 8.14 (s, 1 H) 7.86 (dd, J = 8.59, 4.04 Hz, 1 H). MS (ES+) m/z 340.0 [M+H]+.
Step 5.
To a suspension of 5-iodo-7-(trifluoromethyl)quinolin-8-ol 84 (540 mg, 1.5 mmol) in acetonitrile (10 mL) and DMF (5 mL) was added potassium carbonate (242 mg, 1.7 mmol). To this mixture was added 4-methoxybenzyl chloride (0.26 mL, 1.9 mmol) dropwise followed by catalytic sodium iodide. The reaction was allowed to stir at ambient temperature overnight. The reaction was heated to 50°C for 3 hours. The reaction was cooled to ambient temperature and filtered. The filtrate was concentrated to an oil and partitioned between ethyl acetate and brine. The layers were separated and ethyl acetate layer was dried over sodium sulfate. Purification was accomplished by automated normal phase chromatography (25 g silica gel cartridge) eluting 0–35% ethyl acetate/heptane to yield 429 mg (58% yield) of 5-iodo-8-[(4-methoxyphenyl)methoxy]-7-(trifluoromethyl)quinoline 85 as a waxy orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.12 (dd, J = 4.04, 1.52 Hz, 1 H) 8.47 (dd, J = 8.72, 1.64 Hz, 1 H) 8.29 (s, 1 H) 7.87 (dd, J = 8.72, 4.17 Hz, 1 H) 7.40 – 7.50 (m, 2 H) 6.90 – 7.01 (m, 2 H) 5.56 (s, 2 H) 3.72 – 3.80 (m, 3 H). MS (ES+) m/z 482.0 [M+Na]+.
Step 6.
8-[(4-Methoxyphenyl)methoxy]-5-(p-tolylsulfanyl)-7-(trifluoromethyl)quinoline 86 was prepared in a similar manner as Compound 18 step 4. The product was prepared from 5-iodo-8-[(4-methoxyphenyl)methoxy]-7-(trifluoromethyl)quinoline 85 (200 mg, 0.43 mmol) to yield 162 mg (81% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.16 (dd, J = 4.17, 1.64 Hz, 1 H) 8.71 (dd, J = 8.72, 1.64 Hz, 1 H) 7.76 – 7.86 (m, 2 H) 7.43 – 7.51 (m, 2 H) 7.13 – 7.23 (m, 4 H) 6.93 – 7.01 (m, 2 H) 5.60 (s, 2 H) 3.74 – 3.81 (m, 3 H) 2.27 (s, 3 H). MS (ES+) m/z 478.0 [M+Na]+.
Step 7.
To a mixture of 8-[(4-methoxyphenyl)methoxy]-5-(p-tolylsulfanyl)-7-(trifluoromethyl)quinoline 86 (160 mg, 0.35 mmol) in DCM (5 mL) at ambient temperature was added 3-chlorobenzenecarboperoxoic acid (173 mg, 0.70 mmol). The reaction was stirred for 2 hours. The reaction was quenched by addition of dimethyl sulfide. The mixture was extracted with 10% sodium sulfite aqueous solution. The organic layer was dried over sodium sulfate and concentrated in vacuo. Purification was accomplished by automated reversed phase chromatography eluting 20–95% ACN/water with 0.05% TFA as modifier. Isolated 5-(p-tolylsulfonyl)-7-(trifluoromethyl)quinolin-8-ol 24 (109 mg, 85% yield) as a solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.02 (d, J = 4.29 Hz, 1 H) 8.94 (d, J = 8.84 Hz, 1 H) 8.48 (s, 1 H) 7.86 – 7.96 (m, 3 H) 7.39 (d, J = 8.34 Hz, 2 H) 2.34 (s, 3 H). MS (ES+) m/z 368.0 [M+H]+.
5-(Cyclopentylsulfonyl)-7-fluoroquinolin-8-ol (25).
Step 1.
A mixture of 8-benzyloxy-7-fluoro-5-iodo-quinoline 42 (10.4 g, 27.4 mmol), cyclopentanethiol (4.4 mL, 41.1 mmol), tris(dba)dipalladium (1.2 g, 1.3 mmol), Xantphos (0.8 g, 1.3 mmol) and cesium carbonate (26.8 g, 82.2 mmol) in dioxane (100 mL) was degassed with nitrogen for 15 minutes. The resultant mixture was heated at 90°C for 3 hours. The mixture was passed through a plug of silica eluting with ethyl acetate and concentrated to a residue. The residue was purified via silica gel chromatography eluting with ethyl acetate/hexanes (0–30%) to give 8-benzyloxy-5-cyclopentylsulfanyl-7-fluoro-quinoline 87 (9.2 g, 95% yield) as a slightly colored oil. 1H NMR (400 MHz, CDCl3) δ ppm 9.01 (dd, J = 4.2, 1.6 Hz, 1 H) 8.72 (dd, J = 8.6, 1.8 Hz, 1 H) 7.52 – 7.60 (m, 2 H) 7.41 – 7.52 (m, 2 H) 7.30 – 7.40 (m, 3 H) 5.48 – 5.55 (m, 2 H) 3.51 – 3.62 (m, 1 H) 1.95 – 2.10 (m, 2 H) 1.74 – 1.89 (m, 2 H) 1.56 – 1.70 (m, 4 H). MS (ES+) m/z 354.0 [M+H]+.
Step 2.
To a stirring solution of 8-benzyloxy-5-cyclopentylsulfanyl-7-fluoro-quinoline 87 (9.2 g, 26.1 mmol) in DCM (200 mL), 3-chlorobenzenecarboperoxoic acid (13.5 g, 54.9 mmol) was added. The solution was stirred at ambient temperature for 1 hour. The reaction was quenched by addition of dimethyl sulfide (20 drops). The resultant mixture was stirred for 20 minutes. Sodium sulfite (1 N solution, 200 mL) was added. The organic solution extracted and separated. The organic layer was dried over sodium sulfate and concentrated. The residue was purified by normal phase chromatography (110 g silica gel cartridge) eluting with ethyl acetate/hexanes (0–50%) to give 8-benzyloxy-5-cyclopentylsulfonyl-7-fluoro-quinoline 88 (7.8 g, 77% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.06 – 9.15 (m, 2 H) 8.15 (d, J = 10.9 Hz, 1 H) 7.59 (dd, J = 8.6, 4.3 Hz, 1 H) 7.52 (dd, J = 7.7, 1.4 Hz, 2 H) 7.29 – 7.39 (m, 3H) 5.70 (s, 2 H) 3.61 (quin, J = 7.6 Hz, 1 H) 2.01 – 2.16 (m, 2 H) 1.73 – 1.90 (m, 4 H) 1.56 – 1.68 (m, 2 H). MS (ES+) m/z 386.0 [M+H]+.
Step 3.
8-benzyloxy-5-cyclopentylsulfonyl-7-fluoro-quinoline 88 (7.8 g, 20.3 mmol) in hydrochloric acid (6 N, 150 mL, 900 mmol) was stirred at 100°C for two hours. The solution was cooled in ice-water bath for 30 minutes. The yellow precipitate was isolated by filtration and washed with diethyl ether and air-dried to yield a green solid. The solid was free-based by dissolving it in a slightly basic solution (0.05 N NaOH, 20 mL) then the solution was adjusted to slightly acidic by adding hydrochloric acid (1 N). The solution was extracted with dichloromethane (3 × 100 mL). The combined organic solution was washed with brine (2 × 100 mL) and dried over sodium sulfate. The solution was filtered and concentrated to give slightly colored solid. The solid was purified further by crystallization in heptane/chloroform to give 5-cyclopentylsulfonyl-7-fluoro-quinolin-8-ol 25 (4.3 g, 72% yield) as a solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.96 – 9.07 (m, 2 H) 8.09 – 8.18 (m, 1 H) 7.80 (dd, J = 8.7, 4.2 Hz, 1 H) 3.81 – 3.94 (m, 1 H) 1.82 – 1.97 (m, 2 H) 1.61 – 1.82 (m, 4 H) 1.47 – 1.60 (m, 2 H). MS (ES+) m/z 296.0 [M+H]+.
7-Chloro-5-(cyclopentylsulfonyl)quinolin-8-ol (26).
Step 1.
To a stirring solution of 8-benzyloxy-5-bromo-7-chloro-quinoline 71 (7.6 g, 21.8 mmol) in dioxane (100 mL), cyclopentanethiol (3.5 mL, 32.7 mmol), tris(dba)dipalladium (1.0 g, 1.0 mmol), Xantphos (0.63 g, 1.0 mmol), cesium carbonate (21.3 g, 65.4 mmol) were added. Nitrogen was bubbled in for 3 minutes and the resultant solution heated at 90°C overnight. Water (150 mL) was added. The solution was extracted with ethyl acetate (3 × 100 mL). The combined organic solution was extracted with brine (50 mL) and dried over sodium sulfate. The organic solution was filtered and concentrated. The residue was purified by automated normal phase chromatography and eluted with ethyl acetate/hexane (0–30%) to give 8-benzyloxy-7-chloro-5-cyclopentylsulfanyl-quinoline 89 (7.5 g, 93% yield) as a slightly colored oil. 1H NMR (400 MHz, CDCl3) δ ppm 9.01 (dd, J = 4.2, 1.6 Hz, 1 H) 8.71 – 8.80 (m, 1 H) 7.66 – 7.69 (m, 1 H) 7.60 – 7.66 (m, 2 H) 7.46 – 7.54 (m, 1 H) 7.31 – 7.44 (m, 3 H) 5.45 – 5.52 (m, 2 H) 3.52 – 3.63 (m, 1H) 1.96 – 2.10 (m, 2 H) 1.76 – 1.91 (m, 2 H) 1.56 – 1.72 (m, 4 H). MS (ES+) m/z 370.0 [M+H]+.
Step 2.
To a stirring solution of 8-benzyloxy-7-chloro-5-cyclopentylsulfanyl-quinoline 89 (7.5 g, 20.4 mmol) in DCM (100 mL) at 0°C, 3-chlorobenzenecarboperoxoic acid (10.0 g, 40.8 mmol) was added slowly into the solution. The reaction was continued at 0°C for 20 minutes then at room temperature for 20 additional minutes. Sodium sulfite (1.0 N solution, 30 mL) was added. Sodium hydroxide solution (1.0 N, 100 mL) was added. The solution was stirred at room temperature for 20 minutes then the reaction was diluted with DCM (100 mL). The aqueous phase was discarded. The organic solution was extracted with sodium hydroxide solution (1 N, 50 mL), water (100 mL), brine (100 mL) and dried over sodium sulfate. The solution was filtered and concentrated. The residue was purified by automated normal-phase chromatography and eluted with ethyl acetate/hexane (0–60%) to give 8-benzyloxy-7-chloro-5-cyclopentylsulfonyl-quinoline 90 (6.9 g, 84% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.06 – 9.18 (m, 2 H) 8.35 (s, 1 H) 7.54 – 7.67 (m, 3 H) 7.32 – 7.43 (m, 3 H) 5.69 (s, 2 H) 3.56 – 3.68 (m, 1 H) 2.06 – 2.19 (m, 2 H) 1.77 – 1.91 (m, 4 H) 1.60 – 1.71 (m, 2 H). MS (ES+) m/z 402.0 [M+H]+.
Step 3.
8-benzyloxy-7-chloro-5-cyclopentylsulfonyl-quinoline 90 (6.9 g, 17.1 mmol) in aqueous hydrochloric acid (6 N, 81 mL, 489 mmol) was stirred at 100°C for two hours. The solution was cooled in ice-water bath for 30 min. The yellow precipitate was isolated by filtration and washed with diethyl ether and air-dried to give 7-chloro-5-cyclopentylsulfonyl-quinolin-8-ol 26 (5.2 g, 98% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.99 – 9.11 (m, 2 H) 8.14 (s, 1 H) 7.85 (dd, J = 8.6, 4.0 Hz, 1 H) 3.82 – 3.93 (m, 1 H) 1.82 – 1.94 (m, 2 H) 1.62 – 1.82 (m, 4 H) 1.49 – 1.61 (m, 2 H). MS (ES+) m/z 312.0 [M+H]+.
5-(Cyclopentylsulfonyl)-7-(trifluoromethyl)quinolin-8-ol (27).
Step 1.
5-Cyclopentylsulfanyl-8-[(4-methoxyphenyl)methoxy]-7-(trifluoromethyl)quinoline 91 was prepared in a similar manner to Compound 18, step 2. The product was prepared from 5-iodo-8-[(4-methoxyphenyl)methoxy]-7-(trifluoromethyl)quinoline 85 (215 mg, 0.46 mmol) as described in compound 24, step 5 to yield 183 mg (90% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.16 (dd, J = 4.04, 1.52 Hz, 1 H) 8.81 (dd, J = 8.72, 1.64 Hz, 1 H) 7.78 – 7.87 (m, 2 H) 7.43 – 7.51 (m, 2 H) 6.92 – 7.00 (m, 2 H) 5.55 (s, 2 H) 3.76 – 3.79 (m, 3 H) 3.70 – 3.76 (m, 1 H) 1.99 (dd, J = 12.63, 5.56 Hz, 2 H) 1.70 – 1.81 (m, 2 H) 1.48 – 1.65 (m, 4 H). MS (ES+) m/z 456.0 [M+Na]+.
Step 2.
5-Cyclopentylsulfonyl-7-(trifluoromethyl)quinolin-8-ol 27 was prepared in a similar manner to Compound 24, step 7. The product was prepared from 5-cyclopentylsulfanyl-8-[(4-methoxyphenyl)methoxy]-7-(trifluoromethyl)quinoline 91 (180 mg, 0.41 mmol) to provide 97 mg (67% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.11 (dd, J = 6.69, 1.14 Hz, 2 H) 8.20 (s, 1 H) 7.94 – 8.01 (m, 1 H) 3.88 (s, 1 H) 1.84 – 1.95 (m, 2 H) 1.73 – 1.83 (m, 2 H) 1.63 – 1.72 (m, 2 H) 1.49 – 1.60 (m, 2 H). MS (ES+) m/z 346.0 [M+H]+.
7-Fluoro-5-(pyrrolidin-1-ylsulfonyl)quinolin-8-ol (28).
Step 1.
To chlorosulfonic acid (21 mL, 315 mmol) heated to 100°C was added 7-fluoroquinolin-8-ol 41 (7.0 g, 42.9 mmol). A reflux condenser was affixed and the mixture is stirred at 100°C overnight. The mixture is then poured on ice (210 g) with care to limit the exotherm. The aqueous layer was extracted with chloroform (3 × 200 mL). The combined organic phases were dried over sodium sulfate, concentrated under vacuum to give 5.1 g of 7-fluoro-8-hydroxyquinoline-5-sulfonyl chloride 92 as a yellow solid (40% as the HCl salt). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.64 (d, J = 8.34 Hz, 1 H) 9.07 – 9.13 (m, 1 H) 8.01 (dd, J = 8.59, 5.05 Hz, 1 H) 7.97 (d, J = 10.86 Hz, 1 H). MS (ES+) m/z 242.8 [M+H]+ (sulfonic acid).
Step 2.
Triethylamine (5.6 mL, 40.2 mmol) and pyrrolidine (3.3 mL, 40.2 mmol) were dissolved in 80 mL of dichloromethane. A suspension of 7-fluoro-5-pyrrolidin-1-ylsulfonyl-quinolin-8-ol 92 (2.9 g, 10.0 mmol) in 40 mL of dichloromethane was added slowly at 0°C, then the remaining material was dissolved in 40mL of anhydrous THF. The resultant reaction mixture was stirred overnight at ambient temperature. The solvents were evaporated and the collected green solid was washed with a minimum of DCM (10mL), then ethyl acetate (2 × 10 mL) followed by isopropyl alcohol (2 × 10 mL). The solid was dried under high vacuum to provide 2.9 g (98% yield) of 7-fluoro-5-(pyrrolidin-1-ylsulfonyl)quinolin-8-ol 28. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.06 (dd, J = 8.84, 1.52 Hz, 1 H) 9.02 (dd, J = 4.04, 1.52 Hz, 1 H) 8.08 (d, J = 11.12 Hz, 1 H) 7.77 (dd, J = 8.84, 4.29 Hz, 1 H) 3.16 – 3.23 (m, 4 H) 1.67 – 1.74 (m, 4 H). MS (ES+) m/z 297.2 [M+H]+.
7-Chloro-5-(pyrrolidin-1-ylsulfonyl)quinolin-8-ol (29).
Step 1.
To a suspension of 8-fluoroquinoline-5-sulfonyl chloride 32 (2.0 g, 8.1 mmol) in THF (25 mL) was added DIPEA (2.8 mL, 16.2 mmol) at room temperature. Pyrrolidine (0.67 mL, 8.1 mmol) was added dropwise as a solution in THF (5 mL). Separately, to a suspension of sodium hydride (976 mg, 24.4 mmol) in THF (15 mL) was added 2-trimethylsilylethanol (3.5 mL, 24.4 mmol). The crude sulfonamide solution was added slowly to the suspension of trimethylsilylethanol sodium salt, and the resulting mixture was allowed to stir at room temperature for one hour, and was then quenched by addition of water. The resulting mixture was extracted with dichloromethane, and the organic layers were collected, dried over magnesium sulfate, and evaporated under reduced pressure. Purification by automated normal phase chromatography (0–100% ethyl acetate in heptane) afforded trimethyl-[2-[5-pyrrolidin1-ylsulfonyl-8-quinolyl)oxy]ethyl]silane 93 as a white powder (2.1 g, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm: 8.88–9.08 (m, 2H) 8.14 (d, J = 8.59 Hz, 1 H) 7.75 (dd, J = 8.84, 4.04 Hz, 1 H) 7.35 (d, J = 8.59 Hz, 1 H) 4.21–4.47 (m, 2 H) 3.06–3.24 (m, 4 H) 1.59–1.79 (m, 4 H) 1.15–1.33 (m, 2 H) 0.13 (s, 9 H). MS (ES+) m/z 379.0 [M+H]+.
Step 2.
To a solution of trimethyl-[2-[(5-pyrrolidin-1-ylsulfonyl-8-quinolyl)oxy]ethyl]silane 93 (2.1 g, 5.6 mmol) dissolved in THF (35 mL) was added tetrabutylammonium fluoride (5.6 mL, 5.6 mmol) as a 1M solution in THF. The mixture was allowed to stir for one hour. Solvent was removed under reduced pressure and the resultant residue was partitioned between water and dichloromethane. After extracting the aqueous mixture, the organic layers were dried over sodium sulfate and concentrated to an oil. Automated normal phase silica chromatography (0–10% methanol in dichloromethane) afforded 5-Pyrrolidin-1-ylsulfonylquinolin-8-ol 94 (548 mg, 34% yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.02 (br. s., 1 H) 9.04 (dd, J = 8.72, 1.64 Hz, 1 H) 8.98 (dd, J = 3.79, 1.26 Hz, 1 H) 8.09 (d, J = 8.34 Hz, 1 H) 7.78 (dd, J = 8.84, 4.29 Hz, 1 H) 7.20 (d, J = 8.34 Hz, 1 H) 3.07–3.23 (m, 4 H) 1.71 (dt, J = 6.51, 3.44 Hz, 4 H). MS (ES+) m/z 279.0 [M+H]+.
Step 3.
To a suspension of 5-pyrrolidin-1-ium-1-ylsulfonylquinolin-8-ol 94 (1.3 g, 4.6 mmol) in chloroform (15 mL) was added N-chlorosuccinimide (620 mg, 4.6 mmol) and the resulting mixture was stirred at room temperature overnight. Volatiles were evaporated under reduced pressure, and the resulting residue was purified by preparative HPLC (10–100% acetonitrile in water with 0.05% trifluoroacetic acid as modifier) to afford 7-Chloro-5-pyrrolidin-1-ylsulfonyl-quinolin-8-ol 29 as an orange solid (350 mg, 24% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.12–9.20 (m, 1 H) 8.88–8.95 (m, 1 H) 8.27 (s, 1 H) 7.61–7.68 (m, 1 H) 3.33 (s, 4 H) 1.86 (s, 4 H). MS (ES+) m/z 313.0 [M+H]+.
7-Methyl-5-(pyrrolidin-1-ylsulfonyl)quinolin-8-ol (30).
Step 1.
8-Fluoro-7-methyl-quinoline 95 was prepared in a similar manner to Compound 24, step 1. The product was prepared from 2-fluoro-3-methyl-aniline (2.0 g, 15.9 mmol) to provide 2.2 g (88% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 8.92 (dd, J = 4.17, 1.64 Hz, 1 H) 8.39 (dt, J = 8.34, 1.77 Hz, 1 H) 7.73 (d, J = 8.34 Hz, 1 H) 7.57 (dd, J = 8.34, 4.04 Hz, 1 H) 7.51 (dd, J = 8.08, 7.07 Hz, 1 H) 2.46 (d, J = 2.53 Hz, 3 H).
Step 2.
8-Fluoro-7-methyl-quinoline-5-sulfonyl chloride 96 was prepared in a similar manner to Compound 12, step 1. The product was prepared from 8-fluoro-7-methyl-quinoline 95 (500 mg, 3.1 mmol) to provide 509 mg (63% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.48 (dt, J = 8.72, 1.71 Hz, 1 H) 9.09 (dd, J = 4.80, 1.52 Hz, 1 H) 8.01 (d, J = 7.33 Hz, 1 H) 7.90 (dd, J = 8.72, 4.67 Hz, 1 H) 2.51 – 2.52 (m, 3 H). MS (ES+) m/z 260.9 [M+H]+.
Step 3.
8-fluoro-7-methyl-quinoline-5-sulfonyl chloride 96 (50 mg, 0.20 mmol) was dissolved in THF (1.5 mL). To this solution was added DIPEA (106 μL, 0.61 mmol), followed by pyrrolidine (17 μL, 0.20 mmol). The resulting mixture was stirred at room temperature for 15 minutes. To the reaction was added potassium t-butoxide (610 μL, 0.61 mmol) and stirred at ambient temperature for 1 hour. The reaction mixture was treated with ~2 mL saturated NaHCO3 and ~2 mL ethyl acetate, agitated vigorously and the layers separated. Solvent was removed from the organic fraction in vacuo to give a brown residue which was purified by automated normal phase chromatography (0–100%EtOAc/hexanes, 10 g silica gel cartridge) to provide 8-tert-butoxy-7-methyl-5-pyrrolidin-1-ylsulfonyl-quinoline 97, 45 mg (63% yield). 1HNMR (400 MHz, DMSO-d6) δ ppm 8.96 – 9.03 (m, 2 H) 8.06 (s, 1 H) 7.61 – 7.70 (m, 1 H) 3.34 (s, 3 H) 3.17 – 3.27 (m, 4 H) 1.72 (dt, J = 6.51, 3.44 Hz, 4 H) 1.44 (s, 9 H). MS (ES+) m/z 349.0 [M+H]+.
Step 4.
4 M Hydrogen chloride in dioxane (0.5 mL, 2 mmol) was added to 8-tert-butoxy-7-methyl-5-pyrrolidin-1-ylsulfonyl-quinoline 97 (45 mg, 0.12 mmol) and stirred at ambient temperature for 2 hours. To this reaction was added Et2O, collected solid by filtration, washed with Et2O and air-dried to give 7-methyl-5-pyrrolidin-1-ylsulfonyl-quinolin-8-ol hydrochloride 30 (35 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 9.08 (dd, J = 8.59, 1.52 Hz, 1 H) 8.98 (dd, J = 4.29, 1.52 Hz, 1 H) 8.06 (d, J = 0.51 Hz, 1 H) 7.76 (dd, J = 8.84, 4.29 Hz, 1 H) 3.11 – 3.20 (m, 4 H) 2.43 (s, 3 H) 1.65 – 1.73 (m, 4 H). MS (ES+) m/z 293.0 [M+H]+.
COMT Enzyme Purification
C-terminal Hexa-His tagged MB-COMT (human and rat) was constructed in a pCDNA3.1 vector (Invitrogen, Carlsbad, CA) and expressed in HEK293 cells (ATCC, Manassas, VA) using 293 Fectin (Invitrogen, Carlsbad, CA). 300 ml of cells at 83% viability were pelleted (15,000 g, 4 °C, 5 minutes) and re-suspended once in PBS and then in membrane buffer (15 mM Tris, pH 7.5, 1 mM EGTA, 0.3 mM EDTA, 2 mM MgCl2, PIC cocktail (Roche, Diagnostics, Mannheim Germany)). The samples were then homogenized and frozen and defrosted twice in liquid nitrogen. DNase (MilliporeSigma, St. Louis MO) was then added (5 µl per ml) and incubated for 10 minutes at room temperature. The samples were then centrifuged at 40,000 g at 4°C for 25 minutes. The supernatant was discarded, and the pellet was re-suspended in Tris-sucrose buffer (20 mM Tris, pH 7.4, 250 mM sucrose) and homogenized again. The resulting solution was aliquoted and stored at −80 °C until use. MB-COMT protein concentration from the membrane homogenate was estimated using the Bradford Lowry method.
C-terminal Hexa-His S-COMT (human and rat) was constructed in a pTT5 mammalian cell expression vector (Invitrogen, Carlsbad, CA) and expressed in 293–6E cells (ATTC, Manassas, VA) using linear polyethyenimine (PEI, MilliporeSigma, St. Louis, MO). 0.5 L of cells were harvested at 90% viability and pelleted (15,000 g, 4 C, 5 min). Pellets were frozen at −80 °C. The protein was purified from the pellets using Ni-NTA affinity chromatography and size exclusion per the following: Resin: Ni-NTA (Qiagen, Hilden, Germany) equilibrated in Buffer A. Bed volume: 4 mL. Column: 1.0 cm diameter Econo-column (BioRad, Hercules, CA) Buffer A: 50 mM Tris HCl, 150 mM NaCl, 30 mM imidazole, 2 mM MgCl2, 0.1 mM TCEP, pH 8.0 (4 °C) Buffer B: 50 mM Tris HCl, 150 mM NaCl, 300 mM imidazole, 2 mM MgCl2, 0.1 mM TCEP, pH 8.0 (4 °C). The target protein in the soluble lysate fraction was batch bound to 4 mL of Ni-NTA resin at 4 °C for 2 hours. The resin was collected in a 1.0 cm diameter Econo-column, was washed with 20 CV of buffer A, and the protein of interest was eluted in Buffer B. Fractions (1 mL) were collected. The final destination buffer was 100 mM potassium phosphate, 1mM DTT, 5 mM MgCl2, 20% glycerol at pH 7.2.
Compound dilution
All compounds were diluted to a final concentration of 1.5% DMSO from 10 mM stocks in two steps. Stock (7.5 μl) was diluted serially into 15 μl of DMSO. Then 4.5 μl of the previous was diluted into 55.5 μl of COMT assay buffer (50 mM Tris, 5–10 mM MgCl2, 2.5 mM DTT, pH 6.9). 1 μl of that was then deposited into a Corning® low volume 384 well white flat bottom polystyrene NBS™ microplate in triplicate.
For 0 and 100% inhibition controls, DMSO and Tolcapone (synthesized in house) were added to each assay plate. DMSO was added to 12 wells on the plate in a final concentration of 1.5% and Tolcapone was added to 12 wells on the plate with a final concentration of 10 μM.
Tolcapone was diluted to 666.6 μM and then diluted in two steps according to the procedure of the compounds above. DMSO (MilliporeSigma, St. Louis, MO) was also diluted according to the same scheme.
Enzyme reaction
COMT activity was measured using the MTase Glo Methyltransferase Assay (Promega, Madison, WI) according to manufacturer’s instructions. Assays were carried out in Corning® low volume 384 well white flat bottom polystyrene NBS™ microplates with a final volume of 5μL containing approximately 7 ng of human MB-COMT as estimated by the Bradford Lowry method from the membrane homogenate, 4 ng of rat MB-COMT as estimated by the Bradford Lowry method from the membrane homogenate, 1 ng of Human S-COMT or 1 ng of Rat S-COMT respectively. All reactions contained 20 μM high purity S-adenosyl methionine (SAM, CisBio, Bedford, MA) in COMT assay buffer (50 mM Tris, 5–10 mM MgCl2, 2.5 mM DTT, pH 6.9). For MB-COMT, the catechol substrate was 7 μM norepinephrine (MilliporeSigma, St. Louis, MO) and for S-COMT the substrate was 10 μM 7,8-dihydroxy-4-methylcoumarin32 (MilliporeSigma, St. Louis, MO).
Reactions were performed in a 37°C incubator for 1 hr. The plate was removed from the incubator and allowed to cool to room temperature for 15min. MTase reagent A (Promega, Madison, WI) was first diluted 1 to 5 into RO water and 1 μl was then added to the well. The plate was spun down, shaken and allowed to incubate for 30 minutes at room temperature avoiding light. Then 5 μl of MTase reagent B (Promega, Madison, WI) were added to all of the wells. The plate was spun down, shaken and allowed to incubate for 30 minutes at room temperature avoiding light. Luminescence was detected with a Tecan Infinite M100 Pro plate reader.
Standard curve
A standard curve was run on every plate. The amount of S-adenosyl homocysteine (SAH, CisBio, Bedford, MA) produced was determined using a standard curve and a linear back-calculation method. The standard curve comprised of varying concentrations of SAH from 500 nM down to 0 nM while maintaining a final SAM/SAH concentration of 50 μM. In order to correct for background levels present in the enzymatic lysate (MB-COMT), enzyme at assay concentration was added to the standard curve as well.
Determination of Inhibition
Percentage inhibition was calculated by using 10 μM Tolcapone value as 100% inhibition value and the DMSO control as the 0% inhibition value. The dose response curves were constrained at 0% inhibition while keeping the percentage inhibition of the highest compound concentration floating. IC50 was determined by non-linear regressions and curve fitting using a 4-parameter fit with a variable slope in the Dotmatics® studies program.
Potency data presented is an average of three separate experiments in which each data point was run in triplicate (unless otherwise noted) and reported as pIC50 ± SEM in Tables 1 and 2.
X-Ray Crystallography
Following the protocol from M. Ellermann et al.33 1 µM rat S-COMT was incubated overnight at 4 °C with 10 µM compound 21 and 1 mM SAM (Sigma-Aldrich) in 50 mM Tris-HCl pH 7.6, 50 mM NaCl, 10 mM DTT, 2 mM MgCl2, concentrated to 13 mg mL−1 and cleared by centrifugation. Crystallization was set up at 18 °C with hanging drop vapour diffusion and crystals grew in 100 mM Bis-Tris propane pH 5.5–7.0, 100 mM NaCl, 1.2–1.8 M (NH4)2SO4. The crystal was cryo-protected in 25 mM Bis-Tris propane pH 7.3, 40% (w/v) PEG3350, 150 mM sodium formate and flash-cooled in liquid nitrogen for data collection. Diffraction data were collected at 100 K using a MicroMax-007 HF rotating anode X-ray generator (Rigaku) and mar345 image plate detector (marXperts). Data were integrated and scaled using MOSFLM34 and SCALA35. The structure was determined by molecular replacement using PHASER36 (CCP4 program suite37) and the published S-COMT structure (PDB accession code: 3S68)33, without ligand, as search model. Structural refinement was performed with PHENIX38 with altering cycles of manual model building with COOT39. The crystallographic statistics are summarized in Supplementary Table S3. The protein structure was validated using MolProbity40. The coordinates and structure factors have been deposited in the Protein Data Bank (entry code 6GY1). Molecular pictures are prepared using program PyMol41.
Mouse Blood-Brain Barrier (BBB) Assay
Animals
Male, C57BL/6J mice (8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The mice were group-housed (5/cage) and allowed to habituate to the colony room for one week before handling. All mice were handled on alternating days (3X/week) during the following week and again on the day before testing to acclimate the mice to the experimenter. All procedures were approved by the Sobran, Inc. Rangos Facility Animal Care and Use Committee and followed the Guide for the Care and Use of Laboratory Animals.
Drugs
Novel COMT inhibitors (synthesized in-house) were suspended in vehicle (0.1% Tween80, 0.1% 1510 silicone antifoam, 1% methylcellulose 400c/p in water) and administered through intraperitoneal (ip) injection. The administration volume was 10 mL/kg.
BBB Protocol
On the day of testing, mice were moved from the colony room to a holding room where they were weighed. Drug injections were initiated after an hour acclimation period. Each mouse received a single injection of the test compound (10 mg/kg). At the appropriate time point (5, 15, or 60 minutes post injection), mice were moved individually to the procedure table where they were anaesthetized via isoflurane. The chest cavity was opened and blood was collected via cardiac puncture into Lithium-Heparin 1.3mL microtubes (Sarstadt, Numbrecht, Germany). The mouse was then perfused with phosphate buffered saline (PBS) via a 12mL syringe using a 23 gauge needle to clear the brain of any residual blood and the brain was harvested. Blood was then centrifuged at 2000 rpm at 4°C for 15 minutes to separate the plasma. Plasma was then transferred into Thermo Scientific Matrix tubes for storage. Brains were collected in 5mL Thermo Scientific Nunc tubes and homogenized in a 4x (w/v) volume of PBS, pH 7.4. Plasma and brain homogenate samples were stored at −80°C until analysis.
Rat Biomarker Assay
Animals
Male Sprague Dawley rats (176–200 g on arrival; Charles River Laboratories, Wilmington, MA, USA) were used for all studies. Rats were allowed to habituate to the colony room for one week before handling. All rats were handled individually for at least one minute on each of the three days preceding the day of the experiment. All procedures were approved by the Sobran, Inc. Rangos Facility Animal Care and Use Committee and followed the Guide for the Care and Use of Laboratory Animals.
Drugs
Novel COMT inhibitors (synthesized in-house) and tolcapone (synthesized in-house) were suspended in vehicle (0.1% Tween80, 0.1% 1510 silicone antifoam, 1% methylcellulose 400c/p in water) and administered either orally (po) or through intraperitoneal (ip) injection (tolcapone). Administration volumes were 10 mL/kg and 5 mL/kg for po and ip dosing, respectively.
Biomarker protocol
On the day of testing, rats were transferred to a holding room and weighed. After an hour acclimation period, rats received either vehicle or a COMT inhibitor. At the 4 hour time point, rats were moved to a separate procedure room where they were anaesthetized via isoflurane. Once the rats were determined to be unresponsive, fur on their heads was shaved using electric clippers. The rats were positioned in a stereotaxic frame, with their heads pointed down at a 45-degree angle. To collect cerebrospinal fluid (CSF), previously published protocols42 were adapted. Briefly, we used a 23G needle connected via PE50 tubing to a collection syringe to access the cisterna magna according to the protocol. Slight negative pressure was used to ensure the CSF flowed evenly. During collection, we observed the CSF as it flowed through the tubing into the syringe looking for discoloration due to blood contamination. If any blood was observed entering the tubing, a cut was made in the tubing above the blood to minimize the contamination of the CSF sample and only the CSF above the cut was further processed. The CSF was then transferred to previously chilled (dry ice) Eppendorf tubes containing 0.05M perchloric acid (4:1 CSF:perchloric acid ratio). The tubes were put back on dry ice until the end of the procedure. Any samples with visible blood contamination (pink/red color) were not analyzed. Next, the chest cavity was opened, and blood was collected by cardiac puncture. The blood was collected in Lithium-Heparin 1.3mL microtubes (Sarstadt, Numbrecht, Germany) and stored on ice. The rat was then transcardially perfused with ice-cold phosphate buffered saline (PBS) via a peristaltic pump set to a flow rate of 20 mL/min to clear the brain of any residual blood and the brains were collected in 5mL Thermo Scientific Nunc tubes. Blood was then centrifuged at 2000 rpm at 4°C for 15 minutes to separate the plasma. Plasma was then transferred into Thermo Scientific Matrix tubes for storage. CSF, plasma, and brains were stored at −80°C until analysis.
Bioanalysis protocol
Artificial CSF (aCSF) consisted of 125mM NaCl, 5mM potassium chloride, 1mM MgCl2, 5mM D-glucose, 1.25mM sodium phosphase di-basic, 2mM calcium chlroride and 24mM NaHCO3. Triplicate standards and QCs of HVA and DOPAC were prepared in 0.01M perchloric acid in aCSF. Triplicate 10µL standards, QCs, and CSF samples were transferred to a 96-well Corning Axygen ® PCR plate (Corning, NY). 10µL of 100mM sodium tetraborate (MilliporeSigma, St. Louis, MO) in water and 2µL of internal standard consisting of 500 ng/mL each of HVA-d5 and DOPAC-d5 (both C/D/N Isotopes Pointe-Claire, Quebec, CA) in 50% methanol in water (v/v) were added to each sample and the plate was mix for 2 minutes at 1000 rpm. To this was added 10µL of 2% benzoyl chloride in acetonitrile (v/v) to each sample, and the plate was mixed for 4 minutes at 1000 rpm. From the mixture, 5µL of each sample was injected for analysis using an Agilent 6540 QTOF with Jet Stream Electrospray Ionization Source (ESI) and Agilent 1290 UHPLC. Solvents were 10mM ammonium formate in water (A) and 100% acetonitrile (B). Chromatographic separation was achieved over 7.5 minutes using a Phenomenex Luna Omega 2.1 × 100mm, 1.6µm, C18 column with a binary gradient starting 21% B. The flow rate was 500 μl min−1. The autosampler was set at 20 °C. The LC gradient was as follows: 0–4 min, 21% B; 4 – 4.5 min, 21–40% B; 4.5 – 5.0min, 40% B. 5.01 – 6.0 min, 95%B, 6.01 min, 21% B. The mass spec acquisition was performed using full scan MS from m/z 50 to 800 with the following source conditions: Drying and Sheath Gas temperatures at 350°C and 400°C, respectively; Both gas flows at 12L/min; Nebulizer-45psig, VCap, Nozzle and Fragmenter voltages at 3000V, 600V and 100V, respectively. The ammonium adducts of the dual benzoylated DOPAC and DOPAC-d5 (m/z 394.1285 and 399.1599, respectively) and the single benzoylated HVA and HVA-d5 (m/z 304.1179 and 309.1493, respectively) were used for data analysis. Analyte peak areas were determined from the extracted ion chromatograms within a +/− 20ppm window. Concentrations were calculated with linear regression analysis using Agilent Masshunter Quantitative Analysis Software (B.06.00 SP01).
Supplementary Material
ACKNOWLEDGEMENTS
The authors greatfully acknowledge David Boucaut (UCB) for assistance with in vivo studies and Eric Gillent (UCB) and Benoit Culot (UCB) for bioanalysis assistance.
Funding of parts of this work from the National Institutes of Mental Health NIMH R01 MH107126 is gratefully acknowledged.
ABBREVIATIONS USED
- %F
bioavailability
- ACN
acetonitrile
- aCSF
artificial cerebrospinal fluid
- ADHD
attention deficit hyperactivity disorder
- ADME
absorption, distribution, metabolism, and excretion
- AlCl3
aluminum chloride
- BCRP
breast cancer resistance protein
- Bis-Tris
Bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane
- BLQ
below limit of quantitation
- BnBr
benzylbromide
- BnOH
benzyl alcohol
- CHCl3
chloroform
- CLp
plasma clearance
- COMT
catechol O-methyltransferase
- Cs2CO3
cesium carbonate
- CSF
cerebrospinal fluid
- CsF
cesium fluoride
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMDN
N,N-dimethylethylenediamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DNase
deoxyribonuclease
- DOPAC
dihydroxylphenyl acetic acid
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid
- ER
efflux ratio
- ESI
electrospray ionization
- Et
ethyl
- EtOAc
ethyl acetate
- HBr
hydrobromic acid
- HCl
hydrochloric acid
- HEK
human embryonic kidney cells 293
- HPLC
high-performance liquid chromatography
- HVA
homovanillic acid
- ip
intraperitoneal
- iv
intravenous
- K2CO3
potassium carbonate
- Kp u,u
concentration ratio of unbound drug in brain to unbound drug in plasma
- LC
liquid chromatography
- LC-MS
liquid chromatography−mass spectrometry
- L-DOPA
L-3,4-dihydroxyphenylalanine
- M
molar
- m
mouse
- MB-COMT
membrane bound catechol O-methyltransferase
- mCPBA
meta-chloroperoxybenzoic acid
- MDCK
Madin-Darby canine kidney cells
- MeOH
methanol
- MgCl2
magnesium chloride
- MgSO4
magnesium sulfate
- mpk
milligram per kilogram
- MRP2
multidrug resistance-associated protein 2
- MTase
methyltransferase
- N
normal
- N/A
not applicable
- NaCl
sodium chloride
- NaH
sodium hydride
- NaHCO3
sodium bicarbonate
- NaOH
sodium hydroxide
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- Ni-NTA
nickel-nitrilotriacetic acid
- NIS
N-iodosuccinimide
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- Pd(PPh3)4
Tetrakis(triphenylphosphine)palladium(0)
- Pd2(dba)3
tris(dibenzylideneacetone)-dipalladium(0)
- PDB
Protein Data Bank
- PE
polyethylene
- PEG
polyethylene glycol
- PFC
prefrontal cortex
- P-gp
P-glycoprotein 1
- Ph
phenyl
- PIC
cocktail protease inhibitor cocktail
- pIC50
log(IC50) where IC50 represents the compound/substance concentration required for 50% inhibition
- PK
pharmacokinetic
- PMB
p-methoxybenzyl
- PMB-Cl
p-methoxybenzyl chloride
- po
oral
- ppm
parts per million
- QC
quality control standard
- QTOF
quadrapole time of flight
- r
rat
- RO
reverse osmosis
- rpm
revolutions per minute
- SAH
S-adenosyl-L-homocysteine
- SAM
S-adenosyl methionine
- SAR
structure−activity relationship
- S-COMT
soluble catechol O-methyltransferase
- SnAR
neucleophilic aromatic substitution
- T1/2
half-life of the product
- TBAF
tetrabutylammonium fluoride
- TCEP
(tris(2-carboxyethyl)phosphine)
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TM
trademark
- TMS
tetramethylsilane
- TMSE
2-(trimethylsilyl)ethyl
- Tris
tris(hydroxymethyl)aminomethane
- Tris-HCl
tris(hydroxymethyl)aminomethane hydrochloride
- UHPLC
ultra high performance liquid chromatography
- UV
ultraviolet
- Vss
volume of distribution
- Xantphos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
Footnotes
ASSOCIATED CONTENT
Supporting Information
(Tables of raw data for mouse and rat brain penetration, details for the preparation of Rat S-COMT and the crystallographic details, Molecular formula strings (CSV))
Accession Codes
The PDB accession code for the X-ray structure of 21 bound to Rat S-COMT is 6GY1. Authors will release the atomic coordinates and experimental data upon article publication.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Lachman HM; Papolos DF; Saito T; Yu YM; Szumlanski CL; Weinshilboum RM, Human catechol-O-methyltransferase pharmacogenetics: Description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics 1996, 6, 243–250. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kaenmaki M; Tammimaki A; Myohanen T; Pakarinen K; Amberg C; Karayiorgou M; Gogos JA; Mannisto PT, Quantitative role of COMT in dopamine clearance in the prefrontal cortex of freely moving mice. J Neurochem 2010, 114, 1745–1755; [DOI] [PubMed] [Google Scholar]; (b) Yavich L; Forsberg MM; Karayiorgou M; Gogos JA; Mannisto PT, Site-specific role of catechol-O-methyltransferase in dopamine overflow within prefrontal cortex and dorsal striatum. J Neurosci 2007, 27, 10196–10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mannisto PT; Kaakkola S, Catechol-O-methyltransferase (COMT): Biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev 1999, 51, 593–628. [PubMed] [Google Scholar]
- 4.Lotta T; Vidgren J; Tilgmann C; Ulmanen I; Melen K; Julkunen I; Taskinen J, Kinetics of human soluble and membrane-bound catechol O-methyltransferase - a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry 1995, 34, 4202–4210. [DOI] [PubMed] [Google Scholar]
- 5.Papaleo F; Crawley JN; Song J; Lipska BK; Pickel J; Weinberger DR; Chen J, Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci 2008, 28, 8709–8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reenila I; Mannisto PT, Catecholamine metabolism in the brain by membrane-bound and soluble catechol-O-methyltransferase (COMT) estimated by enzyme kinetic values. Medical Hypotheses 2001, 57, 628–632. [DOI] [PubMed] [Google Scholar]
- 7.Learmonth DA; Kiss LE; Soares-da Silva P, The chemistry of catechol O-methyltransferase inhibitors. Int Rev Neurobiol 2010, 95, 119–162. [DOI] [PubMed] [Google Scholar]
- 8.Bonifacio MJ; Palma PN; Almeida L; Soares-Da-Silva P, Catechol-O-methyltransferase and its inhibitors in Parkinson’s disease. CNS Drug Reviews 2007, 13, 352–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Russ H; Müller T; Woitalla D; Rahbar A; Hahn J; Kuhn W, Detection of tolcapone in the cerebrospinal fluid of parkinsonian subjects. Naunyn-Schmiedeberg’s Archives of Pharmacology 1999, 360 (6), 719–720. [DOI] [PubMed] [Google Scholar]
- 10.Müller T, Tolcapone addition improves Parkinson’s disease associated nonmotor symptoms. Ther Adv Neurol Diso 2014, 7, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tunbridge EM; Harrison PJ; Weinberger DR, Catechol-O-methyltransferase, cognition, and psychosis: Val(158)Met and beyond. Biol Psychiat 2006, 60, 141–151. [DOI] [PubMed] [Google Scholar]
- 12.Olanow CW; Watkins PB, Tolcapone. Clinical Neuropharmacology 2007, 30, 287–294. [DOI] [PubMed] [Google Scholar]
- 13.Axelrod J, O-Methylation of epinephrine and other catechols in vitro and in vivo. Science 1957, 126, 400–401. [DOI] [PubMed] [Google Scholar]
- 14.Ross SB; Haljasmaa O, Catechol-O-methyl transferase inhibitors - in vitro inhibition of enzyme in mouse brain extract. Acta Pharmacol Tox 1964, 21, 205–214. [DOI] [PubMed] [Google Scholar]
- 15.Borchardt RT; Thakker DR; Warner VD; Mirth DB; Sane JN, Catechol O-methyltransferase .8. Structure-activity-relationships for inhibition by 8-hydroxyquinolines. J Med Chem 1976, 19, 558–560. [DOI] [PubMed] [Google Scholar]
- 16.Barrow JC, Inhibitors of catechol-O-methyltransferase. CNS & Neurological Disorders-Drug Targets 2012, 11, 324–332. [DOI] [PubMed] [Google Scholar]
- 17.Almeida L; Rocha JF; Falca A; Palma PN; Loureiro AI; Pinto R; Bonifacio MJ; Wright LC; Nunes T; Soares-da-Silva P, Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects. Clin Pharmacokinet 2013, 52, 139–151. [DOI] [PubMed] [Google Scholar]
- 18.Barrow J, Ernst G; Huang Y; Buchler I; Weinberger D COMT Inhibiting Methods and Compositions WO2016123576, 2016.
- 19.Liu XR; Chen CP, Free drug hypothesis for CNS drug candidates. In Blood-Brain Barrier in Drug Discovery: Di L; Kerns EH, Eds. Wiley, Hoboken, New Jersey, 2015, 42–65. [Google Scholar]
- 20.Hutson PH; Curzon G, Dopamine metabolites in rat cisternal cerebrospinal-fluid-major contribution from extrastriatal dopamine neurons. J Neurochem 1986, 46, 186–190. [DOI] [PubMed] [Google Scholar]
- 21.Robinson RG; Smith SM; Wolkenberg SE; Kandebo M; Yao LH; Gibson CR; Harrison ST; Polsky-Fisher S; Barrow JC; Manley PJ; Mulhearn JJ; Nanda KK; Schubert JW; Trotter BW; Zhao ZJ; Sanders JM; Smith RF; McLoughlin D; Sharma S; Hall DL; Walker TL; Kershner JL; Bhandari N; Hutson PH; Sachs NA, Characterization of non-nitrocatechol pan and isoform specific catechol-O-methyltransferase inhibitors and substrates. ACS Chem Neurosci 2012, 3, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin JH, CSF as a surrogate for assessing CNS exposure: An industrial perspective. Curr Drug Metab 2008, 9, 46–59. [DOI] [PubMed] [Google Scholar]
- 23.(a) Banks WA, From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov 2016, 15 (4), 275–292; [DOI] [PubMed] [Google Scholar]; (b) Rankovic Z, CNS drug design: Balancing physicochemical properties for optimal brain exposure. J Med Chem 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- 24.Muller N; Lapicque F; Monot C; Payan E; Gillet P; Bannwarth B; Netter P, Protein-binding of indomethacin in human cerebrospinal-fluid. Biochem Pharmacol 1991, 42, 799–804. [DOI] [PubMed] [Google Scholar]
- 25.Ganrot K; Laurell CB, Measurement of IgG and albumin content of cerebrospinal-fluid, and its interpretation. Clin Chem 1974, 20, 571–573. [PubMed] [Google Scholar]
- 26.Vidgren J; Svensson LA; Liljas A, Crystal-structure of catechol O-methyltransferase. Nature 1994, 368, 354–358. [DOI] [PubMed] [Google Scholar]
- 27.Lundstrom K; Tenhunen J; Tilgmann C; Karhunen T; Panula P; Ulmanen I, Cloning, expression and structure of catechol-O-methyltransferase. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology 1995, 1251, 1–10. [DOI] [PubMed] [Google Scholar]
- 28.(a) Bai HW; Shim JY; Yu J; Zhu BT, Biochemical and molecular modeling studies of the O-methylation of various endogenous and exogenous catechol substrates catalyzed by recombinant human soluble and membrane-bound catechol-O-methyltransferases. Chem Res Toxicol 2007, 20, 1409–1425; [DOI] [PubMed] [Google Scholar]; (b) Magarkar A; Parkkila P; Viitala T; Lajunen T; Mobarak E; Licari G; Cramariuc O; Vauthey E; Rog T; Bunker A, Membrane bound COMT isoform is an interfacial enzyme: general mechanism and new drug design paradigm. Chem Commun 2018, 54, 3440–3443. [DOI] [PubMed] [Google Scholar]
- 29.(a) Suzuki H; Abe H, Copper-assisted displacement reaction of nonactivated iodoarenes with arenesulfinates - convenient alternative synthesis of unsymmetrical diaryl sulfones. Tetrahedron Lett 1995, 36, 6239–6242; [Google Scholar]; (b) Zhu W; Ma DW, Synthesis of aryl sulfones via L-proline-promoted CuI-catalyzed coupling reaction of aryl halides with sulfinic acid salts. J Org Chem 2005, 70, 2696–2700. [DOI] [PubMed] [Google Scholar]
- 30.(a) Mispelaere-Canivet C; Spindler JF; Perrio S; Beslin P, Pd-2(dba)(3)/Xantphos-catalyzed cross-coupling of thiols and aryl bromides/triflates. Tetrahedron 2005, 61, 5253–5259; [Google Scholar]; (b) Mase T; Itoh T, General and practical synthesis of benzothiazoles. Pure Appl Chem 2008, 80, 707–715. [Google Scholar]
- 31.Manske RHF; Kulka M, The Skraup synthesis of quinolines. Org Reactions 1953, 7, 59–98. [Google Scholar]
- 32.Qian XK; Wang P; Xia YL; Dou TY; Jin Q; Wang DD; Hao DC; Bi XL; Ge GB; Yang L, A highly selective fluorescent probe for sensing activities of catechol-O-methyltransferase in complex biological samples. Sensor Actuat B-Chem 2016, 231, 615–623. [Google Scholar]
- 33.Ellermann M; Lerner C; Burgy G; Ehler A; Bissantz C; Jakob-Roetne R; Paulini R; Allemann O; Tissot H; Grunstein D; Stihle M; Diederich F; Rudolph MG, Catechol-O-methyltransferase in complex with substituted 3’-deoxyribose bisubstrate inhibitors. Acta Crystallogr D Biol Crystallogr 2012, 68, 253–260. [DOI] [PubMed] [Google Scholar]
- 34.Battye TG; Kontogiannis L; Johnson O; Powell HR; Leslie AG, iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 2011, 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans P, Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- 36.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ, Phaser crystallographic software. J Appl Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collaborative Computational Project, N., The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- 38.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeLano W, The PyMOL molecular graphics system. Version 12r3pre, Schroedinger, LLC; 2002. [Google Scholar]
- 42.(a) Nirogi R; Kandikere V; Mudigonda K; Bhyrapuneni G; Muddana N; Saralaya R; Benade V, A simple and rapid method to collect the cerebrospinal fluid of rats and its application for the assessment of drug penetration into the central nervous system. J Neurosci Meth 2009, 178, 116–119; [DOI] [PubMed] [Google Scholar]; (b) Mahat MYA; Ahamed NFA; Chandrasekaran S; Rajagopal S; Narayanan S; Surendran N, An improved method of transcutaneous cisterna magna puncture for cerebrospinal fluid sampling in rats. J Neurosci Meth 2012, 211, 272–279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.