

Abstract
Objectives
Non-randomised clinical trial designs involving comparisons against external controls or specific standards can be used to support regulatory submissions for indications in diseases that are rare, with high unmet need, without approved therapies and/or where placebo is considered unethical. The objective of this review was to summarise the characteristics of non-randomised trials submitted to the European Medicines Agency (EMA) or Food and Drug Administration (FDA) for indications in haematological cancers, haematological non-malignant conditions, stem cell transplants or rare metabolic diseases.
Methods
We conducted systematic searches of EMA databases of conditional approvals, exceptional circumstances, or orphan drug designations and FDA inventories of orphan drug designations, accelerated approvals, breakthrough therapy, fast-track and priority approvals. Products were included if reviewed by at least one agency between 2005 and 2017, the primary evidence base was non-randomised trial(s) and the indication was for haematological cancers, stem cell transplantation, haematological conditions or rare metabolic conditions.
Results
We identified 43 eligible indication-specific products using non-randomised study designs involving comparisons with external controls, submitted to the EMA (n=34) and/or FDA (n=41). Of the 43 indication-specific products, 4 involved matching external controls to the population of a non-randomised interventional study using individual patient-level data (IPD), 12 referred to external controls without IPD and 27 did not explicitly reference external controls. The FDA approved 98% of submissions, with 56% accelerated approvals; most required postapproval confirmatory randomised controlled trials (RCT). The EMA approved 79% of submissions, with a quarter of approvals conditional on completion of a postapproval RCT or additional non-randomised trials.
Conclusions
There has been a large increase in submissions to the EMA and FDA using non-randomised study designs involving comparisons with external controls in recent years. This study demonstrated that regulators may be willing to approve such submissions, although approvals are often conditional on further confirmatory evidence from postapproval studies.
Keywords: systematic review, external controls, regulatory submissions, non-randomised studies
Strengths and limitations of this study.
Our review was conducted using a systematic and transparent approach, and involved in-depth extractions and validations of the submission summaries.
We were limited to regulators’ summary documents, rather than the manufacturers’ original submissions.
The Food and Drug Administration (FDA) does not publicly provide information on non-approvals. However, we did identify several FDA non-approvals by searching for press releases.
We limited our scope to products with prespecified designations, with prespecified indications and reviewed between 2005 and 2017. Within these, we found a high overall approval rate; however, this is not intended to generalise to all submissions based on non-randomised evidence.
Background
Both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) recognise well-designed and executed randomised controlled trials (RCT) as the gold standard approach in confirming efficacy of a new product.1 However, both agencies acknowledge situations when an RCT may be operationally and ethically infeasible. Conducting an adequately powered phase III RCT in rare diseases can be challenging, and the absence of suitable comparators can affect RCT feasibility. For example, when there is no established standard of care, and randomisation to placebo or to no treatment could carry a risk of serious or irreversible harm, it may be considered medically unethical to conduct a traditional RCT.2
Regulators acknowledge these difficulties and under some circumstances may be willing to accept non-RCT evidence.3 This may be the case when evidence from early non-randomised clinical trials (eg, a single-arm phase II trial) suggests that a new medicine might offer substantial benefits over currently available therapies, particularly where no other licensed products are available.3 If approved by regulators, this can allow patients access to potentially life-saving medicines prior to confirmatory evidence from traditional phase III RCTs.
Both the EMA and FDA have approval pathways and designations that align with circumstances where the evidentiary standards deviate from the typical RCT as an adequate and well-controlled investigation of new products.1 The EMA’s exceptional circumstances designation applies to products for which comprehensive data cannot be provided,4 and its conditional approval mechanism enables expedited conditional approval in situations when the benefit of immediate availability outweighs the risk of less comprehensive data than normally required. In the USA, the FDA has a programme similar to the EMA’s conditional approval. The FDA’s accelerated approval programme enables early approval based on surrogate endpoints and is subject to re-evaluation following confirmatory trials.5 6 The FDA has three other expedited designations or pathways to address unmet medical need for serious conditions: fast-track, breakthrough therapy and priority review.7 8 Both regulatory agencies provide a designation for orphan drugs.
Recent reviews have focused on products submitted through specific FDA and EMA pathways using evidence from clinical studies that are not typical RCTs.9–19 Hatswell et al provided the most recent and comprehensive systematic review of products that were submitted to regulatory agencies in absence of RCT evidence, irrespective of licensing pathway and covering both the FDA and EMA regulatory agencies15; however, gaps in knowledge remain regarding the design of those non-RCT studies, how treatment effect was established when the non-RCT evidence was used and how non-RCT designs differed by pathway and approval status.
We were interested in updating the review by Hatswell et al and learning more about non-randomised trials using comparisons with external controls that are submitted to the EMA and FDA. Specifically, we aimed to identify submissions involving non-randomised clinical trials, estimate the proportion of submissions that used explicitly defined external controls and characterise external control study designs. This systematic review also focuses on the regulatory pathways adhered to in these submissions, the designation of products, effect size estimation and the relevant conditions of approval (or reasons for non-approvals).
Methods
We wanted to learn more about non-RCT study designs with external controls that are used in regulatory submissions. We focused our study on haematological malignancies, conditions related to stem cell transplantation, other (non-malignant) haematological conditions and rare metabolic diseases (table 1), based on research showing that submissions in these therapeutic areas are among the most common to involve non-randomised evidence.9 12 15 20 We conducted a systematic search of EMA and FDA regulatory submissions spanning the years 2005–2017, focusing on regulatory pathways and designations that most commonly involve submissions based on non-randomised evidence.15 18
Table 1.
Eligibility criteria
| Item | Criterion for inclusion |
| Evidence base | Primary source(s) of evidence came from uncontrolled trial(s), with or without an explicit comparison against external control(s). |
| Indication | The indication was within one of the following broad therapeutic areas:
|
| Year | Approval by FDA or EMA in or after 2005. |
EMA, European Medicines Agency; FDA, Food and Drug Administration.
In August 2017, we searched the EMA’s database of European public assessment reports (EPAR) for human medicines with conditional approval, exceptional circumstances or orphan medicine designation. We searched all products, including those listed as authorised, withdrawn, suspended and refused.
We conducted a parallel search of the Drugs@FDA database in August 2017, using lists of products licensed with the following designations: orphan drug, accelerated approvals, breakthrough therapy, fast-track and priority approval. The lists available at that time were current to December 2016. From this search, we retrieved the relevant Center for Drug Evaluation and Research (CDER) summary reviews.
Eligibility criteria (table 1) were applied to each identified product with specific indication, based on an initial assessment of the summary reviews (EPAR or CDER summary review), by two independent reviewers (reviewer team: EEK, KM, TJJL). Additional information was retrieved from the medical and statistical summaries, labels and approval letters if needed. On completion of these reviews, we crosschecked the FDA and EMA lists. If a product (with specific indication) was identified for only one regulatory body, we searched the other’s database to determine its approval status and supporting evidence base. The Drugs@FDA database is limited to approved products only; therefore, we also searched for press releases to determine if the submission was rejected by or withdrawn from the FDA.
For each product and each included indication for that product, one researcher extracted and a second researcher validated the following data from the EPAR (for EMA products) and the CDER summary review, medical and statistical summaries (for FDA products) (extractors and validators: EEK, KM, SG, TL): product name and indication; details of approval status as of October 2017; regulatory pathway and designation; and study design(s) of primary evidence and comparator group submitted as supporting evidence (sample size, data source(s), primary endpoint and associated effect estimate(s), survival-related effect estimate(s), choice of control group(s), methodology used for comparison).
We classed the evidence into three categories based on the choice and design of the external controls3: (A) used individual patient-level data (IPD) to balance intervention and external control groups (IPD-based external controls); (B) explicitly defined the external group (descriptions of one or more studies (alone or meta-analysed) were included in the summary, or citation(s) were provided to such studies), but did not attempt to adjust for differences between intervention and control groups (aggregate-level external controls); and (C) no explicit mention of a control group was provided in the summary; the comparison appeared to be based on general medical knowledge (undefined external controls). In some situations, it was evident that the manufacturer had not provided evidence of type A or B but the regulatory review committee sought these data and summarised them in their review. These submissions were assigned to category C, but annotated to acknowledge that regulatory reviewers had identified other types of controls.
The main unit of analysis was an ‘indication-specific product’. This counted each drug product, allowing products that have two or more different indications to be counted for each indication. Data were summarised using counts, proportions, means, medians and ranges. Analysis was conducted in Microsoft Excel and R V.3.4.1.
Patient and public involvement
This study was a systematic review of regulatory submission documents and did not involve patients or the public in any study phase.
Results
Products and supporting evidence
We identified and reviewed submissions for a total of 574 indication-specific products, representing all submissions that received conditional approval, exceptional circumstances, or orphan medicine designation from the EMA, or orphan drug status, accelerated approval, breakthrough therapy, fast-track, or priority approval from the FDA. Forty-three indication-specific products submitted to either the FDA (n=41) or the EMA (n=34) met our inclusion criteria (figure 1), that is, they were approved based on non-randomised evidence, reviewed in or after 2005 by at least one agency and sought approval for an indication for a haematological cancer, condition related to stem cell transplantation, other (non-malignant) haematological condition or rare metabolic disease. These 43 indication-specific products consisted of 40 unique products, three of which were submitted for regulatory approval for two indications each.
Figure 1.

Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) diagram. EMA, European Medicines Agency; FDA, Food and Drug Administration; RCT, randomised controlled trial.
Within the framework of our inclusion criteria, the most common indication-specific products seeking regulatory approval were for treatment of haematological cancers (figure 2). Twenty-five of the 26 haematological cancer indication-specific products targeted diseases that had relapsed, become refractory to treatment or intolerant to treatment (ie, unable to tolerate alternate therapies because of adverse effects); the remaining one was an expanded indication for a newly diagnosed biomarker-defined paediatric population. Additional details are presented in online supplementary table 1.
Figure 2.

Proportions of disease targets of included submissions. ADA-SCID, adenosine deaminase deficiency-severe combined immunodeficiency; aHUS, atypical haemolytic uraemic syndrome; ASCT, autologous stem cell transplant; HSCT, haematopoietic stem cell transplant; sALCL, systemic anaplastic large cell lymphoma.
bmjopen-2018-024895supp001.pdf (612.7KB, pdf)
In total, there were 96 unique studies presented as primary or supporting evidence of clinical efficacy for the 43 indication-specific products. This represented a mean of 2.2 studies per indication-specific product (median 2 studies, range 1–6 studies). Studies were most commonly designed as single-arm studies (67%; n=64), typically in phase II development (60%; n=58 studies) (figure 3). Two studies were designed as RCTs; however, neither captured randomised data in the relevant indication. One of these studies was in a different indication than that being considered for regulatory approval, and the other only presented interim data from the intervention arm. Half of the studies had a sample size of less than 60 subjects (p25, p75: 24, 103) (online supplementary table 2).
Figure 3.
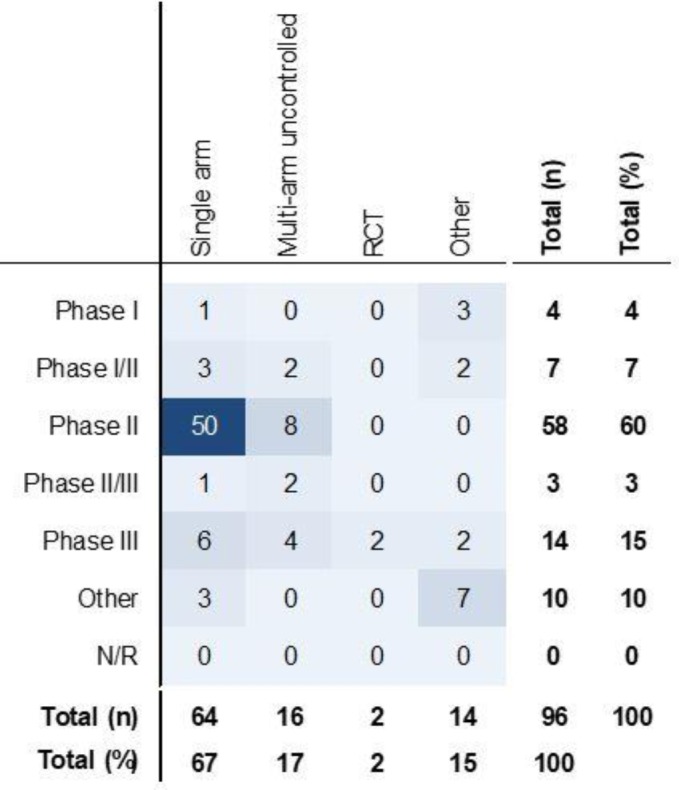
Study design features of included studies (total n=96). ‘Other’ study designs included: switchover studies (n=1), extensions or subset analyses from previous studies (n=3), dose escalation studies (n=4), pilot studies (n=1), compassionate use (n=2), retrospective studies (n=2), or consensus recommendations and other literature (n=1). N/R, not reported; RCT, randomised controlled trial.
The majority of indication-specific products included in our review were submitted to EMA and/or FDA in the second half of the study period (2012–2017; 71%), compared with the first half (2005–2011; 29%). Between 2005 and 2011, there were, on average, 1.4 submissions per year to the EMA and 1.9 submissions per year to the FDA. This compares with an average of 4.0 submissions per year to the EMA between 2012 and 2017 and 4.3 submissions per year to the FDA.
Evidence from external control(s) was explicitly provided for 16/43 (37%) of the indication-specific products: 4 (9%) involved individual-level data collected from external controls matched to participants in the interventional trials, and 12 (28%) used aggregate-level controls. For the remaining 27/43 (63%) indications, no supporting studies of outcomes in external control groups were described. The four indication-specific products with studies presenting individual-level data used a variety of methodological approaches (table 2). Among the 12 indication-specific products with studies involving aggregate-level external controls, one was resubmitted using a matched approach after an initial negative recommendation by the Committee for Medicinal Products for Human Use (CHMP), and presented the analysis as grounds for re-examination. Despite the additional data, the CHMP’s recommendation remained negative, as the data submitted were still considered by the committee to be insufficient to establish the benefits of the indication. Of the remaining 11 indication-specific products involving aggregate-level external control data, six were based on non-systematic literature reviews, one cited one retrospective study and four included detailed descriptions of data from specific retrospective studies (registry or medical chart review) but without further analysis to match the controls with the patients in the interventional study (online supplementary table 3).
Table 2.
Summary of external controls for submissions involving individual patient-level data
| Product | Source(s) | Year(s) of data capture | n | Key methods |
| Recombinant antithrombin* | Retrospective cohort at US and European sites | From 1997 | 35 | Controls selected using eligibility criteria that matched clinical trial eligibility criteria. |
| Modified T cells† | European Bone Marrow Transplant database | 2000–2013 | 853 | Pair-matched analysis, matched on four key prognostic factors using a 1:4 matching ratio. |
| Blinatumomab | Six national study groups and five large treatment centres | >1990 | 1139 | Several different matching approaches were applied to three different cohort definitions: two propensity score approaches using inverse probability of treatment weights (IPTW), and a weighted stratified analysis using established prognostic factors. |
| Blinatumomab | Systematic literature review (24 studies) | NR | 2622 | Model-based meta-analysis. |
| Defibrotide sodium | Retrospective chart review from the same sites as uncontrolled interventional trial | NR | 32 | Propensity score stratification. Strict exclusion for historical controls and a comparison of baseline characteristics. |
| Defibrotide sodium | Center for International Bone and Marrow Transplant Research | 2008–2011 | Arm 1=41 Arm 2=55 |
Observational study involving defibrotide sodium (arm 1) and standard of care (arm 2). Supporting study. |
*Included in the Food and Drug Administration (FDA) submission only.
†Modified with a retroviral vector encoding for a truncated form of the human low-affinity nerve growth factor receptor and the herpes simplex I virus thymidine kinase.
NR, not reported.
Effect size
Very few relative effect estimates were available. HRs comparing overall survival were reported for only 2 of the 43 indication-specific products (5%): HRs relative to standard of care were: 0.54 (95% CI 0.39 to 0.73) for blinatumomab for the treatment of acute lymphocytic leukaemia, and 0.39 (95% CI 0.26 to 0.60) for pralatrexate for the treatment of peripheral T cell lymphoma (table 3).
Table 3.
Effect size for oncology products reporting overall survival
| Intervention and indication | Approval status | Intervention | Control | Median OS (months) | Intervention versus control |
| Median OS (months) | Exposure | HR | |||
| Blinatumomab for ALL | FDA, EMA | 6.1 | Standard of care | 3.3 | 0.536 |
| Clofarabine for ALL | FDA, EMA | 4.1 | No further intervention | 2.2 | NR |
| Gemtuzumab ozogamicin for AML | FDA only (EMA refused) | 4.8 | NR | NR | NR |
| Pralatrexate for PTCL | FDA only (EMA refused) | 14.5 | Standard of care | 6.1–10* | 0.39† |
| Imatinib for ALL | FDA, EMA | NR | Chemotherapy±HSCT | NR | NR |
| Belinostat for PTCL | FDA (EMA not submitted) | 7.9 | NR | NR | NR |
| Ofatumumab for CLL | FDA, EMA | 15.4 | NR | NR | NR |
| Idelalisib for NHL | FDA, EMA | 20 | NR | NR | NR |
*Multiple estimates were provided: 6.1 months and 8.7 months estimated from two different US registries, and 10 months estimated from one European Union (EU) registry.
†Multiple estimates were provided. In the initial submission, no estimated HRs were provided; an updated HR was provided by the manufacturer based on a matched analysis that was conducted after an initial negative recommendation by the Committee for Medicinal Products for Human Use (CHMP); this updated HR estimate was 0.39 (95% CI 0.26 to 0.60).
ALL, acute lymphocytic leukaemia; AML, acute myeloid leukaemia; CLL, chronic lymphocytic leukaemia; EMA, European Medicines Agency; FDA, Food and Drug Administration; HSCT, haematopoietic stem cell transplant; NHL, non-Hodgkin’s lymphoma; NR, not reported; OS, overall survival; PTCL, peripheral T cell lymphoma.
Objective response rate (ORR) was the most commonly reported endpoint in the interventional oncology trials. There were 22 estimates of ORR for 16 indication-specific products; across these 16 indication-specific product submissions, only one estimate was provided for external controls. The single external control estimate was presented as a range (ORR=51%–76%), which was compared against an ORR of 87% for the new product based on a non-randomised interventional trial. Across the 16 indication-specific products, ORRs in the intervention arms varied considerably, with response rates exceeding 90% in some indications yet only approximately 25% in others (figure 4). The intraindication variability was lower than interindication. For example, the ORRs reported to support products with indications for relapsed/refractory peripheral T cell lymphoma and relapsed/refractory multiple myeloma ranged from 23% to 33%. All products were approved by at least one agency, which reflects the unmet need caused by the exceptionally poor outcomes in absence of the new investigational agents.
Figure 4.

Objective response rate in haematological oncology products, by indication. Colour coding reflects approval status: dark green=approved by both agencies; light green=approved by Food and Drug Administration (FDA) only; ‘−2’ indicates more than one estimate was available (second estimate). CLL, chronic lymphocytic leukaemia; CTCL, cutaneous T cell lymphoma; MCL, mantle cell lymphoma; MM, multiple myeloma; NHL, non-Hodgkin’s lymphoma; ORR, objective response rate; PTCL, peripheral T cell lymphoma; sALCL, systemic anaplastic large cell lymphoma; WM, Waldenström macroglobulinaemia. Note: Lenalidomide and pomalidomide were approved by European Medicines Agency (EMA) using randomised controlled trial (RCT) evidence.
Additional details are provided in online supplementary table 4.
Approvals and pathways
Of the 34 submissions to the EMA based on non-randomised evidence, 27 (79%) were approved (figure 5). Eight of these approvals (24% of submissions) were granted as conditional approvals, of which four were conditional on completing an RCT, and four were conditional on conducting subsequent non-randomised trials because postapproval RCTs were not considered feasible or necessary for full marketing authorisation. Six of the approvals (18% of submissions) were granted approval under exceptional circumstances. RCTs for these indications were considered infeasible due to the rarity of these conditions. In one submission, contravention of medical ethics was also provided as the rationale for using non-randomised evidence.
Figure 5.

Summary of approval status, and regulatory pathways and designations. Left: EMA submissions (n=34). Right: FDA submissions (n=41). EMA, European Medicines Agency; FDA, Food and Drug Administration.
The FDA approved 40 of the 41 submissions (98%), including all seven indications refused by the EMA. Accelerated approvals were granted to 23 (56%) of the 41 indications reviewed by the FDA (figure 5). Nearly 90% (n=20) of the accelerated approvals required postapproval RCTs to confirm efficacy, suggesting that RCTs were deemed feasible yet deferred. For seven of these products, however, the confirmatory RCT was to be conducted in a first-line population, whereas the accelerated approval was for a relapse/refractory setting.
There were 14 of 43 (33%) indications that received neither accelerated approval from the FDA, nor conditional approval or exceptional circumstances designation from the EMA. Among these 14, ten were approved by at least one agency (seven by the EMA and nine by the FDA). Three were expanded indications for haematological cancer products, two were new formulations of existing agents, five were biologics for a bleeding disorder (n=3) or a rare metabolic disease (n=2) and one was for a rare metabolic disease which was granted breakthrough and priority review designations by the FDA but was not reviewed by the EMA. All 14 products, except for one new formulation of an existing product, received an orphan designation. Additional data are available in online supplementary table 5.
Non-approvals and divergent opinions
Few details were available on the single FDA non-approval (1/41, 2%), which was one of the four submissions that incorporated matched historical control data.
Five indication-specific products (5/34, 15%) were refused by the EMA and applications relating to two indication-specific products (2/34, 5%) were withdrawn (table 4). For six of these seven indication-specific products, the reason for the non-approval/withdrawal was attributed to inadequate study design and lack of unbiased evidence from patients receiving comparator treatments. External controls had been explicitly defined but not matched to subjects in the interventional trial (n=5), or were undefined (n=1). A relative effect estimate was available for two indication-specific products; however, these effect sizes did not substantially differ from effect sizes among approved indications. The seventh indication could not be authorised, despite having a positive risk-benefit profile, because it was similar to another orphan product that had market exclusivity (table 4). Although all indication-specific product submissions providing matched historical control data were approved by EMA, two approvals were only granted after re-examination using new data.
Table 4.
Rationale for EMA non-approvals
| Product | Reason for non-approval/withdrawal | Divergent positions |
| Refused | ||
| Pralatrexate for PTCL | Concerns about the external validity due to the non-randomised study design. Historical data not considered to be acceptable, even when matching algorithm applied to support grounds for re-examination. | The product should have been considered for conditional approval. |
| Romidepsin for PTCL | Lack of comparator group in trial made evaluation of clinical benefit impossible. | The product should have been considered for conditional approval. |
| Gemtuzumab ozogamicin for AML | RCT was considered feasible in this population and did not accept the grounds for which non-randomised data were provided. | NR |
| Human heterologous liver cells for urea cycle disorders | Concerns regarding the risk of bias in the clinical trial (specifically: selection bias) and in the historical controls. The effect size was not considered convincing enough, even for a conditional approval. | The benefit/risk balance was seen as positive for an approval under exceptional circumstances. |
| Taliglucerase alfa for type 1 Gaucher disease | Similarity with existing marketed agent. | NR |
| Withdrawn | ||
| Omacetaxine mepesuccinate for CML | Use of single-arm trials did not demonstrate ‘dramatic activity’, and historical data were not considered reliable. | NR |
| Vorinostat for CTCL | Safety and efficacy could not be adequately assessed due to the lack of comparator group. | NR |
AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; CTCL, cutaneous T cell lymphoma; EMA, European Medicines Agency; NR, not reported; PTCL, peripheral T cell lymphoma; RCT, randomised controlled trial.
Discussion
We systematically reviewed non-randomised trials using comparisons with external controls that were submitted as the main evidence base in applications for regulatory approval for indications in haematological malignancies, conditions related to stem cell transplantation, other (non-malignant) haematological conditions and rare metabolic diseases. These were identified from a list of all products with conditional approval, exceptional circumstances, or orphan medicine designation from the EMA, or orphan drug status, accelerated approval, breakthrough therapy, fast-track, or priority approval by the FDA. We found that the majority of submissions were based on clinical efficacy data from single-arm phase II trials, and lacked an explicitly defined group of controls. The majority of indications went through an approval process for products targeting patients with high unmet need (ie, for whom no or only very limited treatment options exist, as assessed by the regulatory agency). For an additional small fraction of submissions, non-randomised evidence was used as RCTs were deemed infeasible, most often due to the rarity of the disease.
When comparing the time periods from 2005 to 2011 with that from 2012 to 2017, we found a noticeable increase in the number of submissions of interest. A similar observation was made by Beaver et al 5 in their review of accelerated approvals for indications in malignant haematology and oncology drugs by the FDA. The authors also noted that the majority of accelerated approvals (72%) were based on evidence from single-arm trials. The number of approval processes based on non-RCT evidence is thus increasing also in the broader context of oncology indications. However, even though the use of non-RCT studies may bring treatments to patients earlier, it can lead to difficulties for physicians and payers when they assess the evidence. DeLoughery and Prasad6 highlighted a number of consequences to the increase in the approval of therapies based on single-arm, uncontrolled data, specifically in oncology, where surrogate markers, such as response rate, are increasingly being used (according to their review). They argue that it is difficult for physicians and payers to gauge the efficacy of an approved therapy based on limited non-randomised evidence, no comparisons to pre-existing therapies and a finding that when larger studies are undertaken, the efficacy can oftentimes appear reduced.
We did not identify any specific characteristics that were clearly associated with approval status, and decisions reached by the FDA and EMA were often divergent. These findings illustrate the individualised multifactor decision-making process for regulatory submissions. In non-randomised studies, the questions required to address any gaps in available evidence are variable and difficult to quantify; thus, it must be left to the expert judgement of the review committees who may also request that special advisory groups (SAG) determine which questions need to be addressed. Documentation issued by the EMA and FDA stops short of providing explicit guidance on the use of non-randomised evidence. EMA guidance on clinical trials in small populations states that the use of lower quality evidence (vs small amounts of high-quality evidence) will be evaluated on a case-by-case basis.21 FDA guidance for industry states that historically controlled data would be persuasive if there was a ‘large difference’ between new data and historical experience, without specifying the desired magnitude of effect.7 While some researchers have detected a statistical difference in the distribution of effect sizes between EMA approvals that require further testing in RCTs versus approvals requiring no further testing, no clear threshold has been detected. To our knowledge, no empirical data have been used to compare effect sizes between approvals and non-approvals.13
Both the FDA and EMA provide individual guidance to manufacturers throughout drug development. Enhanced communication is available for products with specific designation/pathways, such as the FDA’s fast-track and breakthrough designations and EMA’s Priority Medicines (PRIME) programme.7 22 Other adaptive pathways in Europe extend early discussions to include patient advocacy groups and health technology assessment bodies.23 In previous research that evaluated EMA approvals of oncology products through conditional approval versus full marketing authorisation, products receiving conditional approval had more frequent referrals to SAGs, providing expert recommendation on scientific and technical matters.14 In our review, we similarly saw several examples where the inclusion of historical control data was based on regulators’ recommendations either prior to or during the review process.
There were few submissions involving individual-level data (4 indications (9%) (3 (9%) EMA submissions; 3 (7%) FDA submissions)). The use of matching, stratification or reweighting in the three most recent submissions involving IPD is consistent with literature on observational study design24 25 and with recent guidance on using IPD to estimate treatment effect in observational studies.26 In the absence of head-to-head evidence, and given that new interventions are typically limited to investigational settings, this form of adjusted comparison with external control groups represents the best approach to producing an unbiased estimate of relative effect; however, they were surprisingly uncommon. This may be due to the fact that an optimal study design may not always be possible, as data availability for external controls can be limited, so that the study design must be chosen within the options available.
Two-thirds (n=27 out of 41) of FDA submissions and just over half of EMA submissions included in this study (n=18 out of 34) would appear to involve undefined external controls, and 100% of these FDA submissions and 95% of these EMA submissions were approved. Among our three predefined types of external controls, these represent the lowest quality of evidence carrying the greatest risk of bias in estimating effect size. Although the external controls were not described in the regulatory bodies’ summary reports of the evidence presented by manufacturers, it is possible that further clinical context was provided in the actual manufacturers’ submissions, or requested by the agencies but not described in the summary report: For example, the values used as null and alternative hypotheses in the clinical trial study design. Furthermore, we observed some cases when manufacturers did not provide external control data but the regulators themselves conducted a review of expected outcomes among external controls. Thus, decisions were made in reference to the clinical context current at the time of decision-making, but that context was not always made explicit in the summary documents.
Our review findings corroborated a previously identified trend towards a relatively higher likelihood of approval by the FDA relative to the EMA.15 Using semistructured interviews with regulators, Tafuri et al 27 revealed that both agencies are aware that they make different decisions based on the same data. In the interviews, regulators attributed this to several factors: (1) a greater willingness by the FDA to approve drugs based on phase II single-arm trials and based on promising effects rather than demonstrated clinical benefit; (2) the FDA’s ability to independently review trials’ raw data files versus EMA’s reliance on FDA’s findings; (3) an assumption of cultural differences with US regulators more willing to guarantee quick access to new anticancer treatments; and (4) the organisational structure of the review committees such that the USA review is done by committees specialised into specific therapeutic areas, whereas the European Union system is based on committee members who do not necessarily hold the specific expertise (although they do consult with medical experts).
Our review was conducted using a systematic and transparent approach, and involved in-depth extractions and validations of the submission summaries. Despite this, our study was subject to several limitations. First, we were limited to regulators’ summary documents, rather than the manufacturers’ original submissions. As a result, some details on external control groups may have been omitted, and information relating to communication between regulators and manufacturers was sparse. Knowledge of the ongoing communication between manufacturer and regulatory agencies would help contextualise the decision to submit based on non-randomised evidence. Second, the FDA does not publicly provide information on non-approvals. However, we did identify several of these FDA non-approvals by searching for press releases for all EMA submissions that were not in the FDA database; although we may have missed some submissions that were refused by the FDA and not submitted to the EMA. Third, our search, while systematic in nature, was not an exhaustive search of all submissions to FDA and EMA. We limited our scope to products with prespecified designations, with prespecified indications and reviewed between 2005 and 2017 by at least one agency. In cross-referencing our list of included products with those identified by Hatswell et al, who reviewed every single submission to the EMA and FDA, we were confident that our strategy yielded a reasonably comprehensive set of products within the prespecified indications of interest and the time frame of our review. Furthermore, we did not conduct a formal inter-reviewer reliability assessment during the study selection process, and we are therefore not able to quantify the level of agreement of the two reviewers on the inclusion or exclusion of the reviewed submissions in this study. Finally, by limiting our search to select designations and a subset of indications of interest, we found a high overall approval rate (98% of submissions were approved by one or both agencies); however, this is not intended to generalise to all submissions based on non-randomised evidence. Our evidence base was limited to a selected set of indications, and submissions were identified based on the product having been granted one of several designations that are more likely to be associated with non-randomised evidence. Furthermore, our search was not designed to identify situations where manufacturers considered seeking approval based on non-randomised evidence but instead elected to conduct an RCT, based on initial discussions with regulators.
This current review builds on what is currently known regarding the use of uncontrolled trials in regulatory submissions, and accelerated pathways for approval. We have brought other research more up-to-date, and we characterised the use of external control groups, which had not been addressed previously. Several researchers previously focused on features of EMA submissions for orphan medicines (2000–2013),19 and medicines receiving conditional approval or accelerated assessment (2006–2016).16 17 Others have researched FDA approvals of oncology orphan drugs (up to 2010),18 20 oncology drugs approved without an RCT (1973–2006),9 drugs granted accelerated approval (1992–2017; 2009–2013)10 and characteristics of postapproval studies approved with limited evidence (2005–2012).11 Hatswell et al 15 included both FDA and EMA decisions in their review of products submitted without RCT evidence, with the primary objective being to enumerate and compare approvals by these two regulatory bodies. Though not a specific objective of their study, these studies discussed the lack of apparent threshold regarding what constituted a sufficiently large effect size to merit approval in absence of an RCT, which is consistent with our findings that encompassed an additional 17 new indications approved after the end of their study period.
Conclusion
There has been a large increase in submissions to EMA and FDA used non-randomised study designs involving comparisons with external controls in recent years, with most submissions approved in the diseases we reviewed, often conditional to further confirmatory evidence from postapproval studies. Based on this selection of 43 indications that had pathways/designations relating to expedited review or orphan drug status, it is evident that when a product’s benefit outweighs the limitations of the evidence, regulators may accept evidence from uncontrolled trials, with or without explicit comparisons to external controls, as the primary evidence base for regulatory approval. The approvals are usually provisional, such that a commitment by the submitting entity to provide further evidence is required. With the rise of personalised medicine, which can address urgent unmet need, yet is associated with smaller biomarker-defined target populations, this trend of relying on non-randomised evidence in initial regulatory review may continue to grow.
Supplementary Material
Footnotes
Contributors: SG, AT, KM, TJJL, EEK, ARL and NF contributed to the conception and design of the study, and the interpretation of data. SG, KM, EEK and TJJL performed the systematic review, synthesised the data and wrote the manuscript. SG, AT, KM, TJJL, EEK, ARL and NF critically reviewed and approved all versions of the manuscript.
Funding: This study was sponsored by Takeda Pharmaceuticals International.
Competing interests: AT is an employee of Takeda. EEK, TJJL and KM are employees of ICON, a CRO that was paid by Takeda to perform this study and to draft this manuscript. SG was an employee and is a consultant for ICON. ARL is a consultant for ICON. NF has received funding for research, travel or consultation from Sanofi Aventis, Takeda, Allegan, Biopharma, Ipsen, Servier, Pfizer and Novo Nordisk.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data available.
Patient consent for publication: Not required.
References
- 1. Gassman AL, Nguyen CP, Joffe HV. FDA Regulation of Prescription Drugs. N Engl J Med 2017;376:674–82. 10.1056/NEJMra1602972 [DOI] [PubMed] [Google Scholar]
- 2. Nardini C. The ethics of clinical trials. Ecancermedicalscience 2014;8:387 10.3332/ecancer.2014.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline: Choice of control group and related issues in clinical trials E10, 2000. [Google Scholar]
- 4. European Medicines Agency (EMA). Guideline on procedures for the granting of a marketing authorisation under exceptional circumstances, 2005. [Google Scholar]
- 5. Beaver JA, Howie LJ, Pelosof L, et al. . A 25-Year Experience of US Food and Drug Administration Accelerated Approval of Malignant Hematology and Oncology Drugs and Biologics: A Review. JAMA Oncol 2018;4:849 10.1001/jamaoncol.2017.5618 [DOI] [PubMed] [Google Scholar]
- 6. DeLoughery EP, Prasad V. The US Food and Drug Administration’s use of regular approval for cancer drugs based on single-arm studies: implications for subsequent evidence generation. Ann Oncol 2018;29:527–9. 10.1093/annonc/mdy008 [DOI] [PubMed] [Google Scholar]
- 7. US Food and Drug Administration (FDA). Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics, 2014. [Google Scholar]
- 8. Darrow JJ, Avorn J, Kesselheim AS. The FDA Breakthrough-Drug Designation - Four Years of Experience. N Engl J Med 2018;378:1444–53. 10.1056/NEJMhpr1713338 [DOI] [PubMed] [Google Scholar]
- 9. Tsimberidou AM, Braiteh F, Stewart DJ, et al. . Ultimate fate of oncology drugs approved by the us food and drug administration without a randomized Trial. J Clin Oncol 2009;27:6243–50. 10.1200/JCO.2009.23.6018 [DOI] [PubMed] [Google Scholar]
- 10. Naci H, Smalley KR, Kesselheim AS. Characteristics of Preapproval and Postapproval Studies for Drugs Granted Accelerated Approval by the US Food and Drug Administration. JAMA 2017;318:626–36. 10.1001/jama.2017.9415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pease AM, Krumholz HM, Downing NS, et al. . Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: systematic review. BMJ 2017;357:j1680 10.1136/bmj.j1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morant AV, Vestergaard HT. European Marketing Authorizations Granted Based on a Single Pivotal Clinical Trial: The Rule or the Exception? Clin Pharmacol Ther 2018;104:169–77. 10.1002/cpt.900 [DOI] [PubMed] [Google Scholar]
- 13. Djulbegovic B, Glasziou P, Klocksieben FA, et al. . Larger effect sizes in nonrandomized studies are associated with higher rates of EMA licensing approval. J Clin Epidemiol 2018;98:24–32. 10.1016/j.jclinepi.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 14. Hoekman J, Boon WP, Bouvy JC, et al. . Use of the conditional marketing authorization pathway for oncology medicines in Europe. Clin Pharmacol Ther 2015;98:534–41. 10.1002/cpt.174 [DOI] [PubMed] [Google Scholar]
- 15. Hatswell AJ, Baio G, Berlin JA, et al. . Regulatory approval of pharmaceuticals without a randomised controlled study: analysis of EMA and FDA approvals 1999-2014. BMJ Open 2016;6:e011666 10.1136/bmjopen-2016-011666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martinalbo J, Bowen D, Camarero J, et al. . Early market access of cancer drugs in the EU. Ann Oncol 2016;27:96–105. 10.1093/annonc/mdv506 [DOI] [PubMed] [Google Scholar]
- 17. Banzi R, Gerardi C, Bertele' V, et al. . Approvals of drugs with uncertain benefit-risk profiles in Europe. Eur J Intern Med 2015;26:572–84. 10.1016/j.ejim.2015.08.008 [DOI] [PubMed] [Google Scholar]
- 18. Kesselheim AS, Myers JA, Avorn J. Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA 2011;305:2320–6. 10.1001/jama.2011.769 [DOI] [PubMed] [Google Scholar]
- 19. Hofer MP, Hedman H, Mavris M, et al. . Marketing authorisation of orphan medicines in Europe from 2000 to 2013. Drug Discov Today 2018;23:424–33. 10.1016/j.drudis.2017.10.012 [DOI] [PubMed] [Google Scholar]
- 20. Cheng MM, Ramsey SD, Devine EB, et al. . Systematic review of comparative effectiveness data for oncology orphan drugs. Am J Manag Care 2012;18:47–62. [PubMed] [Google Scholar]
- 21. European Medicines Agency (EMA). Guideline on clinical trials in small populations, 2006. [Google Scholar]
- 22. European Medicines Agency (EMA). Launch of PRIME – Paving the way for promising medicines for patients, 2016, 2016. [Google Scholar]
- 23. Vella Bonanno P, Ermisch M, Godman B, et al. . Adaptive Pathways: Possible Next Steps for Payers in Preparation for Their Potential Implementation. Front Pharmacol 2017;8:497 10.3389/fphar.2017.00497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eichler HG, Bloechl-Daum B, Bauer P, et al. . “Threshold-crossing”: A Useful Way to Establish the Counterfactual in Clinical Trials? Clin Pharmacol Ther 2016;100:699–712. 10.1002/cpt.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Austin PC. An Introduction to Propensity Score Methods for Reducing the Effects of Confounding in Observational Studies. Multivariate Behav Res 2011;46:399–424. 10.1080/00273171.2011.568786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Faria R, Alava MH, manca A, et al. . NICE DSU Technical Support Document 17: Theuse of observational data to inform estimates of treatment effectiveness in technology appraisal: Methods for comparative individual patient data, 2015. [Google Scholar]
- 27. Tafuri G, Stolk P, Trotta F, et al. . How do the EMA and FDA decide which anticancer drugs make it to the market? A comparative qualitative study on decision makers’ views. Ann Oncol 2014;25:265–9. 10.1093/annonc/mdt512 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2018-024895supp001.pdf (612.7KB, pdf)