

Abstract
Introduction
Gabapentin is currently used ‘off-label’ in children and adolescents with chronic neuropathic pain, and reliable evidence of its effects and optimal dosing are lacking.
Objectives
The GABA-1 trial aims to compare the efficacy and safety of gabapentin liquid formulation relative to tramadol and to explore the pharmacokinetics of both drugs in the treatment of chronic, neuropathic or mixed pain in the paediatric population.
Methods and analysis
The trial is a multicentre, double-blind, double-dummy, randomised, active-controlled, non-inferiority trial. Participants aged from 3 months to <18 years of age with moderate to severe (≥4/10 in age-appropriate pain scales) chronic neuropathic or mixed pain will be recruited in 14 clinical sites in eight European countries. A total of 94 subjects will be randomised to receive gabapentin and tramadol placebo or tramadol and gabapentin placebo throughout 16–19 weeks (including 3 weeks of titration [optimisation period], 12 weeks of treatment at a stable dose [maintenance period] and 1–4 weeks of tapering [discontinuation period]). The primary objective is to assess the efficacy of gabapentin relative to tramadol for the treatment of moderate to severe chronic neuropathic or mixed pain by comparing the difference in average pain scores (assessed by age-appropriate pain scales) between intervention arms after 15 weeks of treatment. Secondary objectives include the assessment of the safety, quality of life and global satisfaction with treatment and the description of the pharmacokinetic–pharmacodynamic relationship of gabapentin liquid formulation and tramadol oral drops to validate the recommended paediatric doses. Only rescue pain medication by paracetamol and/or ibuprofen is allowed during the trial.
Ethics and dissemination
Ethic approval was obtained in the eight participating countries. Results will be submitted for publication in a peer-reviewed journal and presented at one or more scientific conferences.
Trial registration numbers
2014-004851-30 and NCT02722603.
Trial status
Ongoing research study, currently recruiting.
Keywords: neuropathic pain, mixed pain, gabapentin, tramadol, randomised controlled trial, paediatrics
Strengths and limitations of this study.
This is the first clinical trial evaluating the efficacy of gabapentin for the management of chronic neuropathic or mixed pain in children and adolescents.
The trial is a multicentre, double-blind (participants and caregivers), double-dummy, randomised, active-controlled, non-inferiority trial ensuring rigorous study design to assess drug effects.
A newly developed oral liquid formulation of gabapentin will be used, and the dosing regimen of gabapentin has been optimised using pharmacokinetic–pharmacodynamic modelling to bridge adult data to children.
In contrast to standard efficacy trials in pain, data arising from this trial will be used in conjunction with modelling and simulation concepts to confirm the dose rationale for children from 3 months to 3 years of age.
As the rarity of the disease is a challenge to recruitment, the participation of a large number of European centres (n=14) with paediatric pain expertise was deemed necessary.
Introduction
Background
Neuropathic pain (NP) is caused by a lesion or disease of the somatosensory nervous system, but its physiopathology is frequently complex.1 In adults, prevalence of chronic pain with neuropathic features is estimated at 3.3%–8.2%, and prognosis for recovery from NP is often poor.2 In children, there is growing evidence that prevalence is much lower, and that prognosis of NP in childhood differs from that seen when neural lesions occur in adulthood.3 This may be due to the higher plasticity of the young nervous system that leads to a better restitution of function and lower incidence of pain than adults.4 5 Moreover, conditions with which NP is associated differ between children and adults.3 6 Diabetic neuropathy (DNP) and postherpetic neuralgia (PHN) commonly related with NP in adults are rare in children. Established causes of NP in children include post-traumatic or postsurgery nerve injury, phantom limb pain, cancer and chemotherapy, genetic/metabolic diseases (eg, Fabry disease) and some chronic conditions and infections like HIV/AIDS that may cause nerve damage. Complex regional pain syndrome (type I and II) is a condition that occurs in both children and adults, and although largely debated, it is generally considered to comprise NP features.7
Current guidelines for assessment and diagnosis of NP are designed for adults and thus may be less applicable in children.1 8 Since there is no specific biomarker, clinical laboratory or functional test for NP, diagnosis is made merely on the basis of clinical symptoms. Hence, pain history is the mainstay of diagnosis and includes: evaluation of intensity; quality (sensory descriptors); temporal aspects of pain (frequency, spontaneous/paroxysmal or continuous, aggravating and relieving factors); and response to previous treatment (pharmacological or non-pharmacological). Physical examination should attempt to verify and locate the lesion of the somatosensory system and document associated neurological signs, but this is often subject to the well-described challenges of pain assessment in young children. Sensory abnormalities are also more difficult to elicit in infants and young children. Several tests can be indicated such as quantitative sensory testing and electroneuromyography but are rarely performed in routine medical practice except to rule out other underlying pathology.3 Finally, assessment of pain-related disability is important, and quality of sleep, mood and role functioning should be standard assessments for chronic pain conditions including NP.
In adults, drug therapy in NP is often unsatisfactory as the specific underlying mechanism is rarely well understood.9 10 To date, in the absence of age-specific trials, guidelines for the management of NP in children have been developed exclusively based on data from clinical trials in adults.11 The analgesics that are most frequently prescribed for NP in children include tricyclic antidepressants (amitryptiline and nortriptyline) and gabapentinoids (gabapentin and pregabalin).3 12 Opioids like tramadol have shown to be efficacious on NP in adults, but their use is often limited by their unfavourable benefit/risk balance in chronic conditions.9 10 13 14
Rationale
In adults, gabapentin has been shown to be effective in reducing neuropathic and mixed pain associated with DNP, PHN, multiple sclerosis and cancer-related pain and is marketed for the treatment of peripheral NP.9 15
In paediatric patients, reliable evidence of efficacy is lacking, and only case series reporting on the efficacy of gabapentin in treating chronic pain (always with a neuropathic component) are available.16–18 Although paediatric pain specialists have extensive experience with gabapentin off-label use for children, the potential benefits of gabapentin in treating chronic mixed and NP were never demonstrated in this population. Consequently, the need for an adequately designed clinical efficacy/safety trial has been acknowledged as a means to ensure supporting evidence for current empirical clinical practice. In addition, this requirement has been specifically prioritised by the WHO Guideline Development Committee on the pharmacological treatment of persisting pain in children with medical illnesses.19 An additional constraint has been the way gabapentin is used off-label, as there is currently no marketed liquid formulation of gabapentin adequate for administration in infants and young children in Europe.
In this context, the Gabapentine in Paediatric Pain (GAPP) project is a European-funded project that comprises a full paediatric development programme for gabapentin in the treatment of chronic neuropathic or mixed pain in children.20 The development strategy, requirements and regulatory deliverables have been outlined in a paediatric investigation plan (PIP), which has agreed with and approved by the European Medicines Agency’s (EMA) Paediatric Committee.21 The PIP includes: (1) the development of a liquid oral gabapentin formulation; (2) the evaluation of gabapentin safety in juvenile animal toxicity studies (PRE-GABA); (3) two clinical trials to evaluate the efficacy and safety of gabapentin as monotherapy (GABA-1) and as adjuvant therapy (GABA-2); and (4) a modelling bridging study (GABA-3) to specifically address the paucity of pharmacokinetic (PK) data in children and enhance the dose rationale for the paediatric population.20 The study protocol presented in this paper concerns the GABA-1 trial.
Objectives
Primary objective
The primary objective of the GABA-1 trial is to assess the efficacy of gabapentin relative to tramadol for the treatment of moderate to severe chronic neuropathic or mixed pain in children from 3 months to less than 18 years of age by comparing the difference in average pain scores between intervention arms at the end of the treatment period.
Secondary objectives
To assess the effect of gabapentin relative to tramadol on quality of life (physical, emotional, social and school functioning) and global satisfaction with treatment.
To assess the safety of gabapentin relative to tramadol for the treatment of chronic neuropathic or mixed pain in children 3 months to less than 18 years of age.
To characterise the population pharmacokinetic–pharmacodynamic (PKPD) relationship of gabapentin liquid formulation and provide confirmation of the recommended paediatric dose.
Additional exploratory objectives of the trial include the following:
To describe the metabolomic profile following gabapentin and tramadol pain management treatments.
To explore genetic polymorphisms and their impact on PKs and pharmacodynamics (PDs) of both gabapentin and tramadol.
To assess the population PKs of tramadol and its PKPD relationship in the paediatric population.
Methods and analysis
GABA-1 trial design and setting
This GABA-1 trial is designed as a randomised, double-blind, double-dummy, active-controlled, parallel group, multicentre, non-inferiority trial. Tramadol was considered to be an adequate comparator to gabapentin in GABA-1 study because: (1) its efficacy in the management of NP has been shown in adults14 and opioids’ efficacy data from adults can be extrapolated to children,22 and (2) it is currently the only weak opioid marketed for children throughout Europe. The choice to conduct a non-inferiority trial was based on the fact that gabapentin is not thought to be necessarily more effective than tramadol for the treatment of neuropathic or mixed chronic pain in children; however, gabapentin may be better tolerated than tramadol.
Seven countries involving 14 academic paediatric hospital centres will be recruiting eligible children and adolescents (France: five centres; Italy: two centres; the Netherlands: two centres and Albania, Germany, Greece, Poland, UK with one centre each). In all of these hospitals, recruitment of potentially eligible patients will take place in specialised paediatric pain management units and in neurology, oncology, surgery and anaesthesia departments through local study promotion by the investigators. Patient enrolment begun in July 2018 (seven centres open to recruitment at that date) and is expected to end in 2019. Written informed consent will be obtained from parents and/or legal guardians as well as assent from all eligible patients prior to trial participation in accordance with applicable national laws and guidelines for enrolment of children into clinical research. Informed consent and assent to take a blood sample for pharmacogenomics and metabolomic studies will be obtained separately. Informed consent and assent forms and age-tailored informative booklets are provided in online supplementary appendix 1.
bmjopen-2018-023296supp001.pdf (13.4MB, pdf)
A complete list of participating sites can be found on the ClinicalTrials.gov site (https://clinicaltrials.gov/ct2/show/NCT02722603).
Eligibility criteria
Children and adolescents satisfying the following inclusion criteria will be considered for participation to the GABA-1 trial:
Male or female, aged 3 months to less than 18 years at the screening visit.
- Subjects that meet the diagnostic criteria for neuropathic or mixed pain that is:
- NP: ‘Pain arising as a direct consequence of a lesion or disease affecting the somatosensory system’. Subjects may present central or peripheral neuropathic pain
- Mixed pain: ‘Pain that has both a neuropathic and a nociceptive component’.
Since no validated tools exist to assess the presence of NP in children, diagnosis will be based on medical history (underlying disease, quality and temporal aspects of pain, plausible neurological distribution and response to previous treatment) clinical examination (positive and negative sensory criteria) and results of diagnostic tests.
Four criteria from the grading system for definition of NP will be considered2 23:
Pain distribution neuroanatomically plausible.
History that suggests relevant lesion or disease that affects the peripheral or central somatosensory system and with a temporal link between the lesion and disease causing the pain.
Clinical examination with demonstration of neurological signs (negative or positive sensory signs with or without motor or autonomic signs) confined to innervation territory of the lesioned nervous structure.
Diagnostic test confirming lesion or disease explaining NP: surgical or radiological confirmation of lesion (eg, MRI or CT confirming nerve compression), laboratory confirmation of disease (eg, metabolic disorder and multiple sclerosis (MS)), nerve biopsy confirmation of neuropathy and so on.
Patients under 3 years of age must meet at least one out of the four criteria and patients of 3 years of age and above must meet at least two out of the four criteria. To improve the diagnosis of neuropathic or mixed pain in children of less than 3 years of age, a list of genetic, metabolic and neurodegenerative diseases that are specifically associated with occurrence of NP is also provided (online supplementary appendix 2).
bmjopen-2018-023296supp002.pdf (4.7KB, pdf)
For patients affected by CRPS type I: as diagnosis of this condition may not satisfy two out of four criteria of the Treede classification, the ‘Budapest’ diagnostic criteria as recommended for this condition by the International Association for the Study of Pain (IASP) will be used.24 25
Subjects that present with chronic pain defined as a recurrent or continuous pain persisting more than 3 months.26 Duration of pain will be determined from the date of the first painful experience.
- Subjects that present with at least moderate pain as defined by an average pain intensity of ≥4/10 assessed during the 3-day screening period (ie, baseline assessment period, where pain intensity is assessed two times daily during 3 days; at least 5 out of the 6 pain intensity assessments must be available). Pain will be assessed using the following recommended pain scales according to age at the screening visit27:
- Face, Legs, Activity, Cry, Consolability (FLACC) Scale in children aged 3 months to less than 3 years.
- Faces Pain Scale – Revised (FPS-R) for children aged 3 years to less than 8 years; children in this age group that cannot perform self-assessment of pain intensity will use the FLACC Scale.
- Pain Numeric Rating Scale (NRS-11) for children aged 8 years to less than 18 years.
Stable underlying disease and treatment.
In presence of malignant diseases, subjects in clinical remission and/or no expected changes in their therapeutic protocol during participation to the present study.
Informed consent by parent(s) and/or legal guardian according to each country legal requirement.
Assent by the patient, where applicable, according to each country legal requirement.
Exclusion criteria include the following: pain duration ≥5 years; current use of gabapentin or tramadol or history of failure to respond to adequate treatment by gabapentin or tramadol/opioids for NP; history of epileptic condition except febrile seizure disorder; sleeping apnoea syndrome of any origin or subjects with history of severe respiratory impairment; sickle cell disease; significant cognitive impairment; current, controlled or uncontrolled, comorbid psychiatric diagnosis that can impair pain diagnosis and assessment such as severe depressive conditions or psychosis; history of suicidal ideation or behaviour; history of substance abuse in particular opioids; use of prohibited concomitant medication (detailed list is provided in online supplementary appendix 3); current oral corticosteroid treatment or corticosteroid infiltrations to treat pain caused by infiltration or compression of neural structures, for example, peripheral nerves or spinal cord; subjects born prematurely at ≤36 weeks’ gestational age if recruited during the first year of age; body mass index for age and gender of <5th percentile or >95th percentile glomerular filtration rate <90 mL/min/1.73 m2 (Schwarz equation); significant hepatic impairment or aspartate transaminase or alanine transaminase enzymes three times the upper limit of the age-specific reference range; known allergy, hypersensitivity or clinically significant intolerance to gabapentin or tramadol or any component found in the study drugs; subjects with fructose intolerance, diabetes, glucose-galactose malabsorption or lactase-isomaltase deficiency; clinically relevant abnormal ECG at the screening; surgery scheduled or in recovery from surgery occurring within 3 months of baseline assessment; female subjects who are pregnant or currently lactating; subjects who failed screening or were previously enrolled in this study; and participation in another clinical interventional trial.
bmjopen-2018-023296supp003.pdf (6.4KB, pdf)
Intervention arms
The gabapentin oral liquid solution (syrup, 75 mg/mL, 200 mL bottle) and both dummy placebos of gabapentin (syrup) and tramadol (oral drops) will be provided by Dompé Farmaceutici S.p.A (Milan, Italy). The tramadol oral liquid solution (oral drops, 100 mg/mL, 10 mL bottle) is marketed by EG S.p.A (Milan, Italy). Patients will be administered two investigational medicinal products (IMPs) (active product and placebo), orally, three times daily. To facilitate IMPs intake and increase compliance, the dosing intervals will be flexible including a dose in the morning (before school), a dose in the afternoon (after school) and one in the evening (bed time). A period of at least 4 hours should be observed between IMPs doses. Patients will be instructed only to use the provided devices (syringes) for the administration of the IMPs throughout the treatment period.
Selection of doses and dosing regimen
The doses to be used in this paediatric population were selected based on the results of a dose-optimisation approach using population PKPD modelling and simulation to bridge adult data to children. A brief description of the approach is summarised below for both IMPs.
Gabapentin
Assuming that the exposure–analgesic response relationship can be considered comparable between adults and children, a PK analysis was performed to evaluate the dosing requirements to ensure drug concentrations equivalent to those observed and found to be therapeutically efficacious and safe in adults.28 29 Reported adult values show that gabapentin efficacy in NP is reached with doses between 900 mg/day and 3600 mg/day.30 Based on this range and taking into account interindividual variability in the pharmacokinetics of gabapentin, the median value obtained for area under the concentration versus time curve at steady state (area under the concentration-time curve [AUC0–8h]) in adults was selected as target exposure for the paediatric population. The gabapentin population PK model proposed by Ouellet et al 31 was adapted to account for the mechanisms of gabapentin disposition accounting for the role of known demographic and clinical covariates. A range of doses and dosing regimens were then evaluated using clinical trial simulations to ensure comparable gabapentin exposure across the population, irrespective of differences in age or body weight. A dosing regimen based on weight bands was selected taking into account the proposed dosing frequency (three times daily) and rounding rules.
Tramadol
Using published tramadol PK and PKPD models, which have been developed to describe acute pain control in children,32 we have attempted to characterise the pharmacokinetics across the overall paediatric population (3 months to <18 years of age) and identify suitable titration steps for tramadol. Assuming that the exposure–response relationships of tramadol do not differ significantly in acute and chronic pain conditions, the publication by Garrido et al 33 was used to derive exposure values associated with the analgesic effect in the paediatric population. Similarly, comparable exposure–response relationships were assumed for toxicity, which has been previously shown to occur at varying blood concentrations but all above 30.0 mg/L (range: 30–134 mg/L).34 The population PK model published by Bressolle et al 35 was selected for the evaluation of a range of doses and dosing regimens that mimic those previously described for gabapentin. For all scenarios, tramadol was administered according to a three times daily dosing regimen so that dosing intervals could be aligned with the regimen used for gabapentin.
Prohibited and authorised concomitant medications
The concomitant use of drugs that may interfere with the assessment of efficacy, safety or tolerability of both IMPs is prohibited during the entire study period and need to be discontinued before start of enrolment into the trial. The detailed list of drugs is given in online supplementary appendix 3.
All other concomitant pharmacological and non-pharmacological medications implemented during the study must be reported by the patients in the patient’s diary and documented in the electronic case report form (eCRF). Although antacids containing aluminium and magnesium are not prohibited, they may interfere with gabapentin PK profile. For this reason, intake of gabapentin should occur at least 2 hours before or after taking antacids.
Non-pharmacological interventions for the management of NP will be continued if initiated before inclusion to the protocol but should not be initiated during study period.
Rescue medications
Unrestricted use of paracetamol and/or ibuprofen will be used as rescue therapy if the patient experiences pain (FLACC, FPS-R or NRS-11 pain scale >4/10) at any time during study period. These medications will be used at standard therapeutic doses: paracetamol: 15 mg/kg, with a maximum of 1 g (oral or rectal), every 4–6 hours as necessary; maximum total daily dose is 60 mg/kg with a maximum of 4 g, divided into four doses; ibuprofen: 5–10 mg/kg (oral or rectal) every 6–8 hours, oral doses can be taken with or after food; maximum total daily dose is 30 mg/kg/day divided into 3–4 doses. The investigator is the person responsible to instruct the parents/participants on the use of paracetamol and/or ibuprofen as rescue medication and those instructions are to be reported in the patients’ diary. Use of comedication (dose and duration) must be carefully monitored in the patient diary.
Corticosteroids can be continued as part of the specific oncological treatment. However, systemic corticosteroids administered for pain caused by infiltration or compression of neural structures, for example, peripheral nerves or spinal cord, are not allowed.
Trial procedures
Figure 1 summarises the design of the trial and visits described in detail below.
Figure 1.
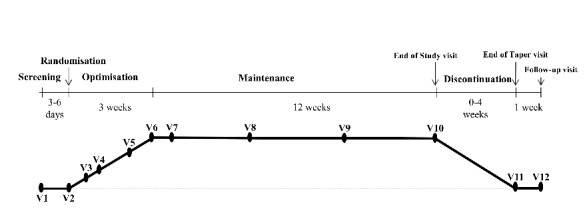
Design of the trial.
Screening
On the first day of participation, following consent, screening assessment of patients for participation will be performed. The screening period will be of maximum 7 days to allow for all screening results to be obtained and validated, in particular the assessment of the subject’s baseline pain intensity (mean over three consecutive days) prior to randomisation.
A wash-out period may be required if the subject is on an analgesic medication that could interfere with the baseline pain intensity assessment. The wash-out period must be at least five times the half-life of the medication. However, study wash-out period will be limited to a maximum of 3 days. Subjects under medications that require a wash-out period of more than 3 days will be excluded unless these medications have been discontinued as part of usual medical care before inclusion to the protocol. During wash-out and screening period, rescue treatment will be allowed (ibuprofen or/and paracetamol).
Randomisation
Random sequence generation
A computer random number generator (R+ script, blockrand package) will be used to select random permuted blocks of randomly varying sizes for the generation of the two intervention arms. Patients will be individually randomised to receive either gabapentin 75 mg/mL syrup and a tramadol placebo, three times/day (experimental group) or tramadol oral drops 100 mg/mL and a gabapentin placebo, three times/day for 15 weeks (control group) in an equal allocation ratio. Randomisation will be stratified according to three age groups: 3 months–<3 years; 3–<8 years and 8–<18 years. In addition, eligible patients will also be randomised for PK sampling purposes. Indeed, PK blood samples will be collected at one of three visits after the start of treatment namely, 14 days±2 (V5), 21 days±2 (V6) or 105 days±3 (V10). All lists of randomisation will be created centrally at Erasmus Medical Centre, Rotterdam, the Netherlands.
Allocation concealment
Central web-based allocation will be used to conceal allocation. The treatment allocation scheme has already been created and has been integrated in the trial’s eCRF to ensure concealment of treatment allocation. Each patient will be given a unique study number on trial screening and then a unique randomisation number on randomisation. Patients will be randomly assigned when all eligibility criteria are entered and validated in the eCRF. Only the locally authorised investigators responsible for recruitment will be able to randomise patients. Patients’ eligibility and randomisation will be confirmed by email sent from the eCRF server to the principal site investigator.
Treatment period
Optimisation period
Dosing of both IMPs will initiate at a starting dose and will be titrated to response up to a maximum dose, according to a predefined matrix. Dosing for gabapentin is defined according to two weight groups from a starting dose of 5 to 7 mg/kg/day to a maximum dose of 45 to 63 mg/kg/day (table 1). By contrast, dosing of tramadol will be performed according to the following dosing schedule: starting dose (V2)=1 mg/kg/day; day 3±1 after start of IMPs (V3)=2 mg/kg/day; day 5±2 (V4)=3 mg/kg/day; day 14±2 (V5)=5 mg/kg/day; day 21±2 (V6)=8 mg/kg/day. Maximum allowed daily dose of gabapentin and tramadol will be 3600 mg and 400 mg, respectively.
Table 1.
Dose optimisation schedule of gabapentin
| Weight group | V2 | V3 | V4 | V5 | V6 |
| ≤15 kg | 7 mg/kg/day | 14 mg/kg/day | 21 mg/kg/day | 42 mg/kg/day | 63 mg/kg/day |
| >15 kg | 5 mg/kg/day | 10 mg/kg/day | 15 mg/kg/day | 30 mg/kg/day | 45 mg/kg/day |
IMPs, investigational medicinal products; V2, start of IMPs; V3, day 3±1 after start of IMPs; V4, day 5±2 after start of IMPs; V5, day 14±2 after start of IMPs; and V6, day 21±2 after start of IMPs.
Titration to response will be flexibly optimised in order to maximise the potential benefits while minimising risk of AEs. There will be a maximum of five possible dose adjustments during the 3-weeks optimisation period. A dose will be indicated as optimal if the subject has reached a pain intensity of less than 4/10 in all pain assessments in the last 48 hours or the maximum tolerable dose. In that case, doses will not be further increased. If necessary, the investigator will be allowed to lower the subject’s dose level once (to the previous dose level) at any point during optimisation. This dose reduction may be followed by subsequent dose increases. Only one dose reduction will be allowed during optimisation period. Subjects who are unable to tolerate the IMPs after one dose reduction will be discontinued. When a dose adjustment is required, subjects will be asked to adjust the dosing of both IMPs (active and placebo IMPs). No dose adjustments can be made after V6 (day 21 after start of IMPs).
Maintenance period
Both IMPs will be maintained at the dose reached at the end of the titration phase for a total of 12 weeks. Dose adjustments will not be possible during maintenance period even if the maximum dose has not been reached during optimisation (last possible adjustment at V6).
Discontinuation period
All subjects who will have completed the study or who will have been withdrawn earlier must be tapered off from the IMPs to prevent withdrawal effects. At the visit of early termination or end of study (EOS visit; V10), subjects will be dispensed a blinded taper dose, and a tapering schedule will be determined based on the subject’s dose administered in the dose maintenance period (table 2).
Table 2.
Tapering schedule of investigational medicinal products
| Last maintenance dose | Days* 1–7 | Days* 8–14 | Days* 15–21 | Days* 22–28 |
| V6 dose level | V5 dose | V4 dose | V3 dose | V2 dose |
| V5 dose level | V4 dose | V3 dose | V2 dose | – |
| V4 dose level | V3 dose | V2 dose | – | – |
| V3 dose level | V2 dose | – | – | – |
| V2 dose level | – | – | – | – |
*Days after the end of study visit (V10).
During the taper period (if applicable), site staff will contact subjects every week to ensure that they are complying with the taper schedule and to remind subjects to start the next week of dose taper.
Parents and/or participants will be given instructions on how to keep a daily patient dairy to record the following items: daily pain score, study drug intake changes, comedication intake, including rescue medications and any adverse events.
Overall, eight visits (V1, V2, V5, V6, V8, V9, V10 and V11) will be performed at the study site during the study period. Remaining visits (V3, V4, V7 and follow-up) will be conducted by phone. Supplementary visits at site can be performed at the discretion of the investigator during both optimisation and maintenance periods.
Outcome measures
Primary endpoint
Average pain score at the end of the treatment period (average of two measures each day for 3 days before EOS visit, V10) as assessed by age-appropriate pain scales:
FLACC Scale (observational assessment scale) in children aged less than 3 years.36
FPS-R (self-assessment scale) for children aged 3 years to less than 8 years; children in this age group that cannot perform self-assessment of pain intensity will use the FLACC Scale.37 38
Pain NRS-11 (self-assessment scale) for children aged 8 years to less than 18 years.27
Secondary efficacy endpoints
Percentage of responders to treatment defined as subjects with a reduction of 30% from baseline or equal to 3/10 of pain intensity assessed by appropriate scale (FLACC, FPS-R and NRS-11) at the end of the study.
Daily pain intensity assessed by age appropriate scale (FLACC, FPS-R or NRS-11) during dose optimisation (V3–V6).
Observational assessment using the NRS-11 completed by parents and investigator (or caregiver) at each visit.
Self-assessment of pain for children ≥8 years of age using the FPS-R pain scale at each visit.
Extent of pain evaluated as the number of painful areas using the pain charts at screening visit (V1), randomisation (V2) and EOS visit (V10).
Number of episodes of breakthrough pain (>4/10 pain score and use of rescue medications) during treatment period.
Number of rescue interventions required during treatment period.
Number of pain-free (<4/10 average pain score without the use of rescue medications) days during treatment period.
Number of participant dropouts due to lack of efficacy.
The total cumulative weight normalised dose of each rescue drug.
Quality of life, physical, emotional, social and school functioning and quality of sleep as assessed on the Paediatric Quality of Life Inventory (PedsQL) Generic Core Scales (by parent and patient)39 assessed at randomisation (V2) and EOS visit (V10).
Acceptability of treatment (Five-Point Facial Hedonic Scale, by patient) at EOS visit (V10).
Global satisfaction with treatment (NRS-11, by parent and patient) at EOS visit (V10).
Clinical Global Impression of Change (Clinical Global Impression – Severity scale (CGI-S) and Clinical Global Impression – Improvement scale (CGI-I); by investigator) at randomisation (V2) for CGI-S and V6 and EOS visit (V10) for CGI-I.40
Patient/parent Global Impression of Change (by parent and patient) at V6 and at EOS visit (V10).41
Assessment of blinding: guess of the subject’s treatment group (by investigator, parents and subject if at adequate maturity level) at V10.
Secondary PK outcomes
Primary (Apparent Oral Clearance (CL/F), Apparent Volume of Distribution (Vd/F), apparent first-order absoption rate constant (Ka)) and secondary (AUC, maximum concentrations (Cmax), time to maximum concentration (Tmax), concentration at steady state (Css) and minimum concentration (Cmin)) PK parameters for gabapentin and tramadol.
Systemic exposure to investigational products during maintenance period, as assessed by predicted steady-state concentrations.
Secondary safety endpoints
Incidence of adverse events at all visits (V1–V12).
Percentage of subjects discontinuing the trial due to treatment-emergent adverse events.
Aggressive behaviour in children aged >6 years using the Retrospective-Modified Overt Aggression Scale at V2, V6 and EOS visit (V10).42
Suicidal ideation/behaviour in subjects aged 6 years and older using the Columbia – Suicide Severity Rating Scale scores before IMP (screening V1), V6 and at the EOS visit (V10) and end of taper visit (V11).43
Secondary exploratory endpoints
Metabolomic profile at screening (V1) and at EOS visit (V10), in responders and non-responders.
PK or PD outcomes based on genetic variation.
All scales that will be implemented in the GABA-1 trial are validated and recommended for use in infants, children and adolescents. All age-appropriate versions and the validated translations of the different scales are considered. For observational assessments, raters (including parent or caregiver and the investigator or study centre designee) observing the subject’s behaviour should be consistent throughout the study. Parents (or principal caregiver) and patients will be trained to adequately use these scales at the screening visit, and they will also be provided with an individual patient diary that includes instructions for use. Patients and/or parents (caregiver) will be instructed to measure their pain level twice a day for assessment of primary outcome, if possible always at the same time of the day, for example, in the morning and in evening, before going to bed. Outside these periods, they will be instructed to provide daily pain level at their convenience but preferably at the same time of the day and before drug administration. Outcomes will be assessed according to the schedule of trial assessments presented in table 3.
Table 3.
Schedule of trial assessments
| Period | Screening | Randomisation | Optimisation (titration) |
Maintenance | End of study (EOS) |
Taper* | End of taper | Follow-up | |||||
| Visit | V1 | V2 | V3† | V4† | V5 | V6 | V7† | V8 | V9 | V10/ET | V11 | V12† | |
| Day (D)/week (W) | D 1 W1 |
D3±1 W1 |
D 5±2 W1 |
D14±2 W2 |
D21±2 W3 |
D28±1 W4 |
D49±3 W7 |
D77±3 W11 |
D105±3 W15 |
D105−133±3 W15-19 |
D112−140±3 W16-20 |
D119−147±3 W17-21 |
|
| Enrolment | |||||||||||||
| Informed consent | X | ||||||||||||
| Inclusion/exclusion criteria | X | X | |||||||||||
| Wash-out instructions | X | ||||||||||||
| Demographics | X | ||||||||||||
| Randomisation | X | ||||||||||||
| Interventions | |||||||||||||
| Gabapentin+tramadol placebo |

|
||||||||||||
| Tramadol+gabapentin placebo |

|
||||||||||||
| Assessments | |||||||||||||
| Medical/medication history | X | ||||||||||||
| Concomitant/rescue medication | X | X | X | X | X | X | X | X | X | X | X | X | |
| Physical examination | X | X | X | X | X | X | |||||||
| Vital signs | X | X | X | X | X | X | X | X | |||||
| Weight/height/BMI | X | X | |||||||||||
| Clinical laboratory test‡ | X | X | |||||||||||
| Serum (s)/urine hCG (u)§ | X (s) | X (u) | X (u) |
X (u) |
|||||||||
| ECG | X | X | |||||||||||
| Blood sampling for pharmacokinetics | X¶ | X¶ | X¶ | ||||||||||
| Investigator dose assessment | X | X | X | X | X | ||||||||
| Pain intensity score (FLACC, FPS-R, NRS-11) | X | X | X | X | X | X | X | X | X | X | X | X | |
| Observational assessment of pain intensity (NRS-11)** | X | X | X | X | X | X | X | X | X** | ||||
| Self-assessment of pain for children ≥8 years (FPS-R) | X | X | X | X | X | X | X | X | X | X | X | X | |
| Pain charts | X | X | X | ||||||||||
| Patient diary assessment | X | X | X | X | X | X | X | ||||||
| Global satisfaction with treatment (NRS-11; parent, subject) | X | ||||||||||||
| CGI-S (investigator) | X | ||||||||||||
| CGI-I (investigator) | X | X | |||||||||||
| PGIC (parent and subject) | X | X | |||||||||||
| PedsQL | X | X | |||||||||||
| C-SSRS | X | X | X | X | |||||||||
| UMSS (investigator and parent)†† | X | X | X | X | X | ||||||||
| Assessment of blinding (investigator, parent and subject‡‡) | X | ||||||||||||
| Five-point Facial Hedonic Scale | X | ||||||||||||
| Adverse events collection | X | X | X | X | X | X | X | X | X | X | X | X | |
| R-MOAS | X | X | X | ||||||||||
| Renal ultrasound scan (only for <3 years of age) | X | X | |||||||||||
| Investigational product dispensed | X | X | X | X | X | ||||||||
| Taper phone call | X | ||||||||||||
| Exploratory assessments (to be performed only if specific consent is provided) | |||||||||||||
| Blood sampling for metabolomics | X | X | |||||||||||
| Blood sampling for pharmacogenomics | X | ||||||||||||
*During taper period (if applicable), site staff will contact subjects every week to ensure that they are complying with the taper schedule and to remind subjects to start the next week of dose taper.
†Visits V3, V4, V7 and V12 are phone call visits. V7 will not be performed if the patient has reached optimal dose of IMPs prior to the last visit of the dose optimisation period (V6).
‡Standard clinical haematology and biochemistry, including cystatin-C and serum creatinine.
§If a blood sampling is required for the visit, only a serum beta human chorionic gonadotropin (HCG) test will be performed.
¶PK sampling will be performed according to a sampling matrix at either D14 or D21 or EOS visit. Patient allocation will be defined according to randomisation scheme. In total, four samples will be collected, one before dosing and at three Udifferent time windows postdosing (0–2 hours; 2–4 hours and 4–6 hours). Details of the sampling procedures will be described in the study procedures.
**This assessment will be performed only by parent(s) of children above 3 years of age.
††University Michigan Sedation Scale (UMSS). This scale will assist parents at home to evaluate the level of sedation of their child during the 3-week titration period and the first week of the maintenance period.
‡‡Assessment will be performed by both parent(s) and investigator. Subjects will be asked to guess the treatment arm if at an adequate level of maturity.
BMI, body mass index; C-SSRS, Columbia – Suicide Severity Rating Scale; ET, early termination; FLACC, Face, Legs, Activity, Cry, Consolability; FPS-R, Faces Pain Scale – Revised; IMPs, investigational medicinal products; NRS-11, Numeric Rating Scale; PGIC, Patient /parent Global Impression of Change; PK, pharmacokinetic; R-MOAS, Retrospective-Modified Overt Aggression Scale.
Blinding
Patients, parents and caregivers (all those assessing outcomes) will be blinded to intervention assignment. Gabapentin or tramadol and respective placebos will be indistinguishable in appearance as to maintain the study blind. Also, labelling will not allow identification of the actual treatment. During the trial, blinding will be broken by the investigator in case of predefined emergency purposes, where knowledge of the blinded treatment could influence further patient care. Also, the sponsor’s safety contact will unblind case safety reports, as per regulatory requirements. Otherwise, study blinding will only be broken upon completion of statistical analysis.
Sample size and rationale for non-inferiority
A sample size of 94 patients was planned on the basis of an expected average baseline pain score of 7 (SD=2),44 45 an expected average end of treatment pain scores of 6 in the tramadol and 5 in the gabapentin group,46 47 a one-sided alpha level of 2.5%, a power of 80%, a non-inferiority margin (Δ) of 0.75 and a dropout rate of 10%. This non-inferiority margin of 0.75 reflects published findings of clinically important differences in pain scores of 1 point among children with baseline pain of ≤5 points and of 2 points among children with baseline pain of ≥6 points on 10-point scales.48 We chose the mean of these two results, a 1.5 point difference, as a clinically important difference for our population. In keeping with the literature, this clinically significant difference in pain scores was halved to produce a Δ of 0.75 for the margin of non-inferiority, which is taken as the maximum size of the effect considered clinically irrelevant.49 As a one-way analysis of covariance (ANCOVA) accounting for baseline pain scores and other covariates will be used for assessment of the primary endpoint, this estimated sample size has been adjusted for a design factor of 1-ρ2, where ρ is an estimate of the correlation between baseline and EOS pain scores.50 Based on previous estimates, this correlation was assumed to be 0.79.37
If the null hypothesis is rejected and non-inferiority of gabapentin is demonstrated, an analysis of the superiority of gabapentin on tramadol will be conducted using these data. Taking the same parameters as those cited for the non-inferiority hypothesis, the estimated power for the superiority study will exceed 95% with a sample size of 94.
A minimum number of subjects per age group have been defined in agreement with regulators: (1) at least 12 subjects aged 3 months to less than 3 years; (2) at least 30 subjects aged 3 years to less than 8 years and (3) at least 40 subjects aged 8 years to less than 18 years. No maximum number of patients per age group is specified.
Data collection
Electronic Data Capture system for GABA-1 trial is provided by Advice Pharma (Milan, Italy), a company specialised in the computer management of clinical trials and includes the eCRF and data management services. On completion of each visit, a study coordinator specifically designed and trained for the study will record all requested data on a standardised eCRF. The investigator will be responsible for the accuracy of the data entered in the eCRF. Source documents and electronic health record data will be available to ensure the quality and integrity of all the data recorded in the eCRF. The eCRF will be available for review to designated sponsor representatives at each scheduled monitoring/audit visit.
Also, processing and storage conditions of biological specimens for PK, pharmacogenomics and metabolomics analysis are specified in the ‘Study manual’ prepared specifically for the GABA-1 trial. All samples of PK, pharmacogenomics and metabolomics will be shipped together from each participating clinical site to the Department of Pharmacology of the School of Pharmacy UCL, London, UK, at the end of the trial.
Adverse events
Safety aspects of the study will be closely monitored by site investigators, by the sponsor’s medical expert and by the independent Data Safety Monitoring Committee (DSMC), which receives blinded, and on request, unblinded data for its judgement. In addition, specific adverse events related to the use of gabapentin or tramadol will be closely monitoring during the entire study period.
Potential risks related to gabapentin
Gabapentin has very few drug interactions but can produce dose-dependent dizziness and sedation, which are reduced by starting with lower dosages and titrating cautiously.51 Other common side effects of gabapentin that have been described both in adults and children include: somnolence, visual disturbances, ataxia and gait disturbance, asthenia, headache and peripheral oedema. Most may occur during the first few weeks of administration but resolve over time. Dizziness and visual disturbances may cause additional risk of trauma.
Also, gabapentin use in paediatric patients with epilepsy was associated with the occurrence of central nervous system-related adverse events.52–54 The most significant of these can be classified into the following categories: (1) emotional lability (primarily behavioural problems), (2) hostility, including aggressive behaviours, (3) thought disorder, including concentration problems and change in school performance and (4) hyperkinesia (primarily restlessness and hyperactivity). Most of these events were mild to moderate in intensity. As an antiepileptic drug, gabapentin has the potential for increased risk of suicidal thought or behaviour.
A common side effect of gabapentin use in adults and children with epileptic conditions was urinary incontinence. Also, presence of hydroureter and hydronephrosis has been observed in rats taking high doses of gabapentin (unpublished data from the pre-GABA study, D6.1 ‘DRF Study Report’, submitted to the European Commission on 10 November 2014, available on request). For these reason, a renal ultrasound exam will be performed for the youngest subjects (<3 years of age) at the beginning and the end of the study period.
Potential risks related to tramadol
In both children and adults, use of tramadol has been associated with occurrence of dry mouth, nausea, constipation, somnolence and dizziness.55 Most recently, tramadol has been associated with respiratory depression in relation to polymorphisms in the CYP2D6 activity.56 To avoid occurrence of any respiratory adverse events in the GAPP-1 trial, it was decided that: (1) the study population would not include patients who recently underwent tonsillectomy or with history of sleeping apnoea syndrome or severe respiratory impairment; (2) tramadol will not be taken with other central nervous system or respiratory depressants; and (3) occurrence of excessive sleepiness, sedation and breathing abnormalities will be closely monitored during the trial to allow prompt discontinuation of the treatment if necessary. Parents will be instructed to use the University of Michigan Sedation Scale.57 If the level of sedation is ≥2, the next drug dose will not be administered and parents are to immediately contact the investigator/research site.
Of note, it is anticipated that slow titration will reduce the impact of ultrarapid metabolism.58 Such an unlikely event might then represent a potential concern after the second or third step of titration (2 or 3 mg/kg/day). In addition, to avoid any errors of administration of tramadol, site investigators will instruct and insure that parents have fully understood tramadol administration modalities before starting the treatment.
Rare cases of tramadol dependence have been described mainly in individuals with previous history of substance abuse.59 Therefore, subjects with previous history of drug abuse, in particular opioids, will be excluded from the study. Also, careful monitoring of drug accountability based on rigorous accounting of the pharmaceutical units provided by the hospital pharmacy and returned at each visit by the participant will be implemented. To avoid the occurrence of potential opioid-like withdrawal symptoms, tramadol doses will be tapered progressively over a period of 1–4 weeks according to the level of the maintenance dose.
Removal or withdrawal of trial participants
A subject may withdraw from the study at any time and for any reason without prejudice to its future medical care by the physician or the institution. Subjects may be withdrawn from study drug treatment at any time at the discretion of the investigator for safety, behaviour, non-compliance with study procedures or administrative reasons. The reason for withdrawal must be recorded (medical record and eCRF). Reasons include but are not limited to the following: adverse event; protocol violation; consent withdrawal, lost to follow-up; lack of efficacy (efficacy is defined as pain intensity of less than 4/10 in all pain assessments in the last 48 hours during the maintenance period); continuous use of rescue medication (defined as use of the maximum daily dose of paracetamol and/or ibuprofen for 12 days in a period of 15 continuous days during the maintenance period); response criteria not met (optimisation period only); and pregnancy.
Any interruption in treatment must be recorded. If IMP is discontinued, regardless of the reason, subjects will be asked to return any unused IMP and return to the study centre for final safety evaluations. Also, at least three documented attempts (including one written communication) will be made to contact any subject lost to follow-up at any time point prior to the last scheduled contact (office visit or telephone contact).
No patient who has been randomised and withdraws from the study for any reason will be replaced.
Statistical analysis
Analysis of efficacy variables
According to the CONSORT 2010 Statement for Non-Inferiority trials, endpoints will be analysed for the per-protocol (PP) and the intend-to-treat (ITT) populations.60 In our trial, the ITT population will consist of all randomised patients who received at least one dose of IMPs, based on their randomisation arm, regardless of the treatment actually received (modified ITT). The PP population will be based on the treatment actually received. All patients excluded from the analysis will be described. No interim analysis is planned.
The primary analysis of efficacy will be conducted using ANCOVA with baseline average pain score as a covariate. Other covariates will include centre, treatment, age group and any baseline characteristics (weight, height, gender, pain diagnosis, pain severity, underlying disease, duration of pain and use of non-pharmacological treatment approaches) found to differ significantly between the two groups in univariate analysis. A second ANCOVA model including a treatment-centre interaction term will be used to assess consistency across sites. The secondary efficacy endpoint of proxy pain as measured by a caregiver using a numeric ranking scale will also be assessed using ANCOVA with treatment group and baseline average proxy pain score as covariates. Non-inferiority will be assessed based on the two-sided CI approach.
Intergroup differences with regards to other secondary efficacy endpoints will be assessed by χ2 tests for categorical variables and two independent sample t-tests or Wilcoxon-Mann-Whitney tests for continuous variables.
Analysis of safety variables
The safety population will consist of all patients who received any IMP and will be based on the treatment actually received. Secondary safety endpoints including scores for quality of life, acceptability of treatment, quality of sleep, and the suicide assessment protocol will be assessed using ANCOVA with treatment group and baseline scores as covariates. Intergroup differences in the mean number of adverse events will be assessed using two independent sample t-tests or Wilcoxon-Mann-Whitney tests.
PK and PKPD analysis
The population PK analysis of gabapentin and tramadol will provide the time course and variability of drug concentrations in plasma and a covariate analysis will also be performed to explore the influence of relevant demographic factors. Non-linear mixed effects modelling will be performed using NONMEM (Icon Development Solution, Maryland, USA) and covariate search will be based on a two-step approach including forward selection and backward elimination of each covariate.61 The likelihood ratio test will be used for comparison of hierarchical models and goodness of fit will be assessed by graphical methods. All data manipulation, including graphical and statistical summaries will be implemented in R+ software.62
In the PKPD analysis, secondary PK parameters describing systemic exposure (AUC, Cmax and trough concentration (Cτ) will be derived and used subsequently to assess potential correlations with measures of efficacy. The latter include the pain scores assessed using age-appropriate pains scales. Initially, linear and non-linear regression techniques will be used to establish the statistical significance of the correlation between model-predicted estimates of drug exposure and measures of efficacy, as determined by linear interpolation of the area under the effect curve during the course of treatment, the maximum change from baseline at the start of treatment (ΔEmax). The analysis will also take into account treatment and other demographic and clinical factors as potential covariates on the intercept and slope of the correlations.
Missing data
Efforts will be made to reduce the rate of missing data; however, if that occurs, multiple imputation methods will be implemented. A mixed model repeated measures approach will be conducted as a sensitivity analysis of the primary outcome.
Statistical analyses will be performed using the SAS V.9.3 software package for PC.
Patient and public involvement
The GAPP Patient Advisory Board (PAB) managed by the patient organisation Childhood Cancer International (the Netherlands) and composed of patient and family representatives, psychologists and researchers, ensured that patient and family views were clearly accounted for in the development of the study protocol and all patient and parents informative and consent/assent materials. A representative of the organisation assisted in all project general assemblies where trial design and outcome measures were presented and discussed. Another representative was part of the GAPP Ethics Board that reviewed and advised on the study protocol from an ethical perspective. The PAB also provided input on the content and design of all materials, print and video and provided substantial revisions in an iterative review process together with the GAPP Communications Team. The PAB then set up focus groups with children and adolescents meeting many of the GABA-1 inclusion criteria in the Netherlands and Italy to provide direct patient input on these materials. Focus group results were synthesised in a final report and included in the final revision of these materials.
A layman report will be prepared and translated in different languages (covering all participating countries) at the end of GABA-1 trial with the aim to inform the participants and their parents on the results of the trial, as well as the general public.
Monitoring
The sponsor will perform a pre-study qualification visit (PSQV) to verify that all clinical sites fulfilled requirements for the participation to the GABA-1 trial with regards to their capacity to recruit eligible patients but also from a human and technical resources point of view. Taking into consideration all available information/documents already collected by the sponsor, specifically mandated Clinical Research Assistants (CRAs) checked with the sites the compliance of technical tools and facilities (eg, centrifuge, fridge, freezer, surgery, pharmacy and laboratory) and other requirements. Once the PSQV is successfully concluded and after arrival of the IMPs at site and completion of all regulatory procedures, the site initiation visit will be performed by the sponsor. Thereafter, all sites will be monitored periodically for quality and regulatory compliance to the GABA-1 study protocol, International Conference for Harmonisation - Good Clinical Practice (ICH-GCP) and regulatory requirements. All source documents will be reviewed with regards to completion of the eCRF, and IMPs accountability will be closely audited. The frequency is set at 5 weeks after first inclusion in the site, and then on average every 3 months, and in any case on sponsor’s request. The interval between monitoring visits may be shorter than stated above, depending on recruitment rate, quality issues, trial site compliance or other site-specific issues.
Ethics and dissemination
A body with consultative functions, the GAPP Ethics Board, formed by independent experts and patient representatives external to the project, with the necessary expertise and knowledge to assist and advise the Project Scientific Committee on all ethical aspects of the conduct of the trial. Also, ethics approval for the conduct of the GABA-1 trial was requested in each individual country according to national procedures. All participants will undergo a process of informed consent and will be aware that trial participation is strictly voluntary and that they have the right to withdraw at any time. An independent DSMC with extensive experience in either paediatric pain management or clinical study methodology and biostatistics will regularly review trial data and make recommendations on the continuation, modification and termination of the trial to ensure participants’ safety. The Sponsor and Patient Advisory Board will guarantee subjects’ rights and protection against any commercial considerations and conflicts of interest. Insurance has been contracted by the sponsor to ensure post-trial care and compensation to those who suffer harm from trial participation.
The results of the GABA-1 trial will be presented at relevant scientific conferences and meetings and submitted to a peer-reviewed medical journal. The results will also be included in the regulatory file for the request of a Pediatric-Use Marketing Authorization for the new liquid formulation of gabapentin at the EMA.
Discussion
Chronic pain is estimated to affect at least 15% of children with underlying conditions and is often poorly recognised and treated.63 To date, opioids, non-steroid anti-inflammatory drugs, antidepressants and anticonvulsants are among the most commonly used medications to treat chronic pain, but very few of these are formulated or authorised for paediatric use. Gabapentin is a drug proven to be efficacious and safe in adults with NP and in children with epilepsy. However, only case series have reported on the efficacy of gabapentin in treating chronic pain with a neuropathic component in children and an adequate formulation for administration in the youngest is currently missing in Europe. Nevertheless, paediatric pain specialists have extensive and mostly reassuring experience with gabapentin ‘off-label’ use in children.
The GABA-1 trial represents a balance between methodological excellence and the reality of chronic pain management in paediatrics. It was designed to provide reliable evidence for the use of a new liquid formulation of gabapentin in children presenting with moderate to severe chronic neuropathic or mixed pain. There is a notable paucity of clinical trials in children presenting with chronic pain, and the GABA-1 trial will be the first one in the field of paediatric neuropathic or mixed pain.11 In fact, the conduct of such trials faces numerous methodological and practical challenges even in adults.64 Some of those, more specific in paediatrics, include: the absence of validated diagnostic criteria for NP, the choice of an adequate comparator, the complexity of the blinding procedure, the rarity of the condition and that of previous data to use for sample size calculations and the optimisation of dosing schemas and titration. We addressed these challenges by defining a core scientific steering committee that reviewed available literature and interacted thoroughly with partners and field experts. Hence, the trial will provide high-level evidence for the management of chronic neuropathic and mixed pain in infants, children and adolescents through the implementation of randomisation procedures consistent with Cochrane recommendations and the use of double-blind and standardised patient follow-up and data collection.
We also used an innovative approach taking into account PKPD concepts to determine dose rationale for IMPs, gabapentin and tramadol in children. Moreover, trial design, titration and maintenance periods have been defined using optimal design methods to ensure that all collected data will be highly informative. Most trials in pain research are empirical and do not involve a rational, evidence-based selection of the evaluated dosing regimens. Indeed, little or no focus is given to exposure levels, and PK data are often not collected in efficacy studies, ignoring PK variability as a potential source of variation in pain response anald adverse events. Finally, the international and multicentre aspect of this study will allow to obtain widely generalisable data.
Supplementary Material
Acknowledgments
The authors would like to thank all collaborators, study investigators and patient representatives (Mrs Patty Brouwer and Mrs Anouk Nijenhuis) involved in the context of the FP7 GAPP Project (GA n. 602962) and under the umbrella of TEDDY – European Network of Excellence for Paediatric Clinical Research for their contributions. Authors would like to acknowledge the input of the Ethics Board members, Dr Helen Sammons, Dr Annagrazia Altavilla, Mrs Anouk Nijenhuis and coordinator Mrs Viviana Giannuzzi. The authors would like to thank the CVBF, Bari, Italy, and the PARTNERS platform, Paris, France, for preparing and coordinating regulatory submissions in the different European countries as well as exchanges among partners. The authors would like to thank Mr Francois Luc from the Clinical Investigations Center of the Robert Debré Hospital, Paris, France, for assisting on preparation and layout of figures and tables.
Footnotes
Patient consent for publication: Not required.
Contributors: FK, LM, RL, SNdW, TGdL, ODP, MF, DT, AC and CA were involved in conception and trial design. All members of the Consortium participated were involved in the discussions on trial design and feasibility. FK, ELR, MF, DB, MF and ODP were involved in drafting of the article and its critical revision for important intellectual content. All the authors were involved in the final approval of the article.
Funding: The trial is part of the European GAPP project that has received funding from the European Union Seventh Framework Programme for research, technological development and demonstration under Grant Agreement No 602041.
Disclaimer: The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: DB and MF are employees and LM is a former employee of the PHARM srl, sponsor of the GABA-1 trial. Remaining authors have no conflicts of interest to declare.
Provenance and peer review: Not commissioned; externally peer reviewed.
Collaborators: Albania: Dr Donjeta Bali, Dr Alketa Hoxha and Professor Ermira Kola; Estonia: Dr Inga Talvik; France: Dr Juliette Andrieu-Galien, Dr Celine Greco, Professor Daniel Annequin, Dr Justine Avez-Couturier, Dr Romy Blanchet, Priscilla Boizeau, Dr Brigitte Charron, Professor Isabelle Desguerre, Dr Sophie Dugue, Dr Elisabeth Fournier-Charrière, Professor Evelyne Jacqz-Aigrain, Dr Cecile Mareau, Dr Ivana Milovanovic, Dr Barbara Tourniaire, Dr Silvia Pontone and Dr Chantal Wood; Germany: Dr Antje Neubert, Regina Trollmann and Dr Stefan Wimmer; Greece: Dr Eleana Garini and Dr Panagoula Mammi; Italy: Dr Marcello Allegretti, Dr Ornella Bellagamba, Proffesor Franca Benini, Dr Donato Bonifazi, Dr Daniela Caprino, Dr Sabrina Congedi, Dr Francesco Craig, Dr Sandro Dallorso, Dr Antuan Divisic, Dr Mariagrazia Felisi, Dr Marco Gentile, Dr Andrea De Giacomo, Dr Luca Manfredini, Dr Emilia Matera, Professor Lucia Margari, Dr Alessandro Mazza, Dr Virgilio Pace, Dr Chiara Di Pede, Dr Maria Giuseppina Petruzzelli, Dr Pieradelchi Ruffini, Dr Luigina Tagliavacca and Dr Maria Traverso; Poland: Dr Anna Szumowska; the Netherlands: Dr Maarten O Mensink, Dr Joost M van Rosmalen, Tjitske van der Zanden and Patty Brouwer; UK: Mr Paul Healy, Dr Daniel Howcutt and Helen Neary. All the collaborators were involved in the context of the FP7 GAPP Project (GA n. 602962) and under the umbrella of TEDDY – European Network of Excellence for Paediatric Clinical Research.
Contributor Information
Collaborators: Donjeta Bali, Alketa Hoxha, Ermira Kola, Inga Talvik, Juliette Andrieu-Galien, Celine Greco, Daniel Annequin, Justine Avez-couturier, Romy Blanchet, Priscilla Boizeau, Brigitte Charron, Isabelle Desguerre, Sophie Dugue, Elisabeth Fournier-charrière, Evelyne Jacqz-Aigrain, Cecile Mareau, Ivana Milovanovic, Barbara Tourniaire, Silvia Pontone, Chantal Wood, Antje Neubert, Regina Trollmann, Stefan Wimmer, Eleana Garini, Panagoula Mammi, Marcello Allegretti, Ornella Bellagamba, Franca Benini, Donato Bonifazi, Daniela Caprino, Sabrina Congedi, Francesco Craig, Sandro Dallorso, Antuan Divisic, Mariagrazia Felisi, Marco Gentile, Andrea De Giacomo, Luca Manfredini, Emilia Matera, Lucia Margari, Alessandro Mazza, Virgilio Pace, Chiara Di Pede, Maria Giuseppina Petruzzelli, Pieradelchi Ruffini, Luigina Tagliavacca, Maria Traverso, Anna Szumowska, Maarten O Mensink, Joost M Van Rosmalen, Tjitske Van Der zanden, Patty Brouwer, Paul Healy, Daniel Howcutt, and Helen Neary
References
- 1. Jensen TS, Baron R, Haanpää M, et al. . A new definition of neuropathic pain. Pain 2011;152:2204–5. 10.1016/j.pain.2011.06.017 [DOI] [PubMed] [Google Scholar]
- 2. Haanpää M, Attal N, Backonja M, et al. . NeuPSIG guidelines on neuropathic pain assessment. Pain 2011;152:14–27. 10.1016/j.pain.2010.07.031 [DOI] [PubMed] [Google Scholar]
- 3. Howard RF, Wiener S, Walker SM. Neuropathic pain in children. Arch Dis Child 2014;99:84–9. 10.1136/archdischild-2013-304208 [DOI] [PubMed] [Google Scholar]
- 4. Stanton-Hicks M. Plasticity of complex regional pain syndrome (CRPS) in children. Pain Med 2010;11:1216–23. 10.1111/j.1526-4637.2010.00910.x [DOI] [PubMed] [Google Scholar]
- 5. Fitzgerald M, McKelvey R. Nerve injury and neuropathic pain - A question of age. Exp Neurol 2016;275:296–302. 10.1016/j.expneurol.2015.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walco GA, Dworkin RH, Krane EJ, et al. . Neuropathic pain in children: special considerations. Mayo Clin Proc 2010;85:S33–41. 10.4065/mcp.2009.0647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Naleschinski D, Baron R. Complex regional pain syndrome type I: neuropathic or not? Curr Pain Headache Rep 2010;14:196–202. 10.1007/s11916-010-0115-9 [DOI] [PubMed] [Google Scholar]
- 8. Bouhassira D, Attal N. Diagnosis and assessment of neuropathic pain: the saga of clinical tools. Pain 2011;152:S74–83. 10.1016/j.pain.2010.11.027 [DOI] [PubMed] [Google Scholar]
- 9. Finnerup NB, Attal N, Haroutounian S, et al. . Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 2015;14:162–73. 10.1016/S1474-4422(14)70251-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dworkin RH, O’Connor AB, Audette J, et al. . Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc 2010;85:S3–14. 10.4065/mcp.2009.0649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boulkedid R, Abdou AY, Desselas E, et al. . The research gap in chronic paediatric pain: a systematic review of randomised controlled trials. Eur J Pain 2018;22:261–71. 10.1002/ejp.1137 [DOI] [PubMed] [Google Scholar]
- 12. Fournier-Charrière E, Marec-Berard P, Schmitt C, et al. . [Management of neuropathic pain in children: guidelines for good clinical practice]. Arch Pediatr 2011;18:905–13. 10.1016/j.arcped.2011.05.016 [DOI] [PubMed] [Google Scholar]
- 13. Eisenberg E, McNicol E, Carr DB. Opioids for neuropathic pain. The Cochrane database of systematic reviews 2006;3:CD006146. [DOI] [PubMed] [Google Scholar]
- 14. Duhmke RM, Cornblath DD, Hollingshead JR. Tramadol for neuropathic pain. The Cochrane database of systematic reviews 2004;2:CD003726. [DOI] [PubMed] [Google Scholar]
- 15. Moore RA, Wiffen PJ, Derry S, et al. . Gabapentin for chronic neuropathic pain and fibromyalgia in adults. The Cochrane database of systematic reviews 2011;3:CD007938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Behm MO, Kearns GL. Treatment of pain with gabapentin in a neonate. Pediatrics 2001;108:482–4. 10.1542/peds.108.2.482 [DOI] [PubMed] [Google Scholar]
- 17. McGraw T, Stacey BR. Gabapentin for treatment of neuropathic pain in a 12-year-old girl. Clin J Pain 1998;14:354–6. 10.1097/00002508-199812000-00014 [DOI] [PubMed] [Google Scholar]
- 18. Rusy LM, Troshynski TJ, Weisman SJ. Gabapentin in phantom limb pain management in children and young adults: report of seven cases. J Pain Symptom Manage 2001;21:78–82. 10.1016/S0885-3924(00)00243-8 [DOI] [PubMed] [Google Scholar]
- 19. World Health Organization (WHO). Persisting pain in children package: WHO guidelines on the pharmacological treatment of persisting pain in children with medical illnesses, 2012. [PubMed] [Google Scholar]
- 20. GAPP. Gabapentin in pediatric pain. https://www.pediatricpain.eu/ (Accessed 18 February 2018).
- 21. PDCO Opinion P/0250/2015. EMA decision of 30 October 2015 on the acceptance of a modification of an agreed paediatric investigation plan for gabapentin (EMEA-001310-PIP01-12-M02). http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/pips/EMEA-001310-PIP01-12-M02/pip_001009.jsp&mid=WC0b01ac058001d129 (Accessed 18 Feb 2018).
- 22. Berde CB, Walco GA, Krane EJ, et al. . Pediatric analgesic clinical trial designs, measures, and extrapolation: report of an FDA scientific workshop. Pediatrics 2012;129:354–64. 10.1542/peds.2010-3591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Treede RD, Jensen TS, Campbell JN, et al. . Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70:1630–5. 10.1212/01.wnl.0000282763.29778.59 [DOI] [PubMed] [Google Scholar]
- 24. Harden RN, Bruehl S, Perez RS, et al. . Validation of proposed diagnostic criteria (the “Budapest Criteria”) for Complex Regional Pain Syndrome. Pain 2010;150:268–74. 10.1016/j.pain.2010.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harden RN, Bruehl S, Stanton-Hicks M, et al. . Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007;8:326–31. 10.1111/j.1526-4637.2006.00169.x [DOI] [PubMed] [Google Scholar]
- 26. Merskey H, Bogduk N. International association for the study of pain classification of chronic pain: descriptions of chronic pain syndromes and definitions of pain terms: (SeattleIASP Press), 1994. [Google Scholar]
- 27. McGrath PJ, Walco GA, Turk DC, et al. . Core outcome domains and measures for pediatric acute and chronic/recurrent pain clinical trials: PedIMMPACT recommendations. J Pain 2008;9:771–83. 10.1016/j.jpain.2008.04.007 [DOI] [PubMed] [Google Scholar]
- 28. Bellanti F, Della Pasqua O. Modelling and simulation as research tools in paediatric drug development. Eur J Clin Pharmacol 2011;67:75–86. 10.1007/s00228-010-0974-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bellanti F, Di Iorio VL, Danhof M, et al. . Sampling optimization in pharmacokinetic bridging studies: example of the use of deferiprone in children with β-thalassemia. J Clin Pharmacol 2016;56:1094–103. 10.1002/jcph.708 [DOI] [PubMed] [Google Scholar]
- 30. Lal R, Sukbuntherng J, Luo W, et al. . Clinical pharmacokinetics of gabapentin after administration of gabapentin enacarbil extended-release tablets in patients with varying degrees of renal function using data from an open-label, single-dose pharmacokinetic study. Clin Ther 2012;34:201–13. 10.1016/j.clinthera.2011.12.004 [DOI] [PubMed] [Google Scholar]
- 31. Ouellet D, Bockbrader HN, Wesche DL, et al. . Population pharmacokinetics of gabapentin in infants and children. Epilepsy Res 2001;47:229–41. 10.1016/S0920-1211(01)00311-4 [DOI] [PubMed] [Google Scholar]
- 32. Payne KA, Roelofse JA, Shipton EA. Pharmacokinetics of oral tramadol drops for postoperative pain relief in children aged 4 to 7 years--a pilot study. Anesth Prog 2002;49:109–12. [PMC free article] [PubMed] [Google Scholar]
- 33. Garrido MJ, Habre W, Rombout F, et al. . Population pharmacokinetic/pharmacodynamic modelling of the analgesic effects of tramadol in pediatrics. Pharm Res 2006;23:2014–23. 10.1007/s11095-006-9049-7 [DOI] [PubMed] [Google Scholar]
- 34. Clarot F, Goullé JP, Vaz E, et al. . Fatal overdoses of tramadol: is benzodiazepine a risk factor of lethality? Forensic Sci Int 2003;134:57–61. 10.1016/S0379-0738(03)00100-2 [DOI] [PubMed] [Google Scholar]
- 35. Bressolle F, Rochette A, Khier S, et al. . Population pharmacokinetics of the two enantiomers of tramadol and O-demethyl tramadol after surgery in children. Br J Anaesth 2009;102:390–9. 10.1093/bja/aen405 [DOI] [PubMed] [Google Scholar]
- 36. Merkel SI, Voepel-Lewis T, Shayevitz JR, et al. . The FLACC: a behavioral scale for scoring postoperative pain in young children. Pediatr Nurs 1997;23:293–7. [PubMed] [Google Scholar]
- 37. Bieri D, Reeve RA, Champion GD, et al. . The faces pain scale for the self-assessment of the severity of pain experienced by children: development, initial validation, and preliminary investigation for ratio scale properties. Pain 1990;41:139–50. 10.1016/0304-3959(90)90018-9 [DOI] [PubMed] [Google Scholar]
- 38. Hicks CL, von Baeyer CL, Spafford PA, et al. . The faces pain scale-revised: toward a common metric in pediatric pain measurement. Pain 2001;93:173–83. 10.1016/S0304-3959(01)00314-1 [DOI] [PubMed] [Google Scholar]
- 39. pedsql. The PedsQL Measurement Model is a modular approach to measuring health-related quality of life (HRQOL) in healthy children and adolescents and those with acute and chronic health conditions. http://www.pedsql.org/.
- 40. Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry 2007;4:28–37. [PMC free article] [PubMed] [Google Scholar]
- 41. Hurst H, Bolton J. Assessing the clinical significance of change scores recorded on subjective outcome measures. J Manipulative Physiol Ther 2004;27:26–35. 10.1016/j.jmpt.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 42. American Academy of Pediatrics. Addressing mental health concerns in primary care: a clinician’s toolkit: American Academy of Pediatrics, 2010. [Google Scholar]
- 43. Oquendo MA, Halberstam B, Mann JJ. Risk factors for suicidal behavior: utility and limitations of research instruments Standardized evaluation in clinical practice. 1st edn, 2003:103–30. [Google Scholar]
- 44. Manworren RC, Hynan LS. Clinical validation of FLACC: preverbal patient pain scale. Pediatr Nurs 2003;29:140–6. [PubMed] [Google Scholar]
- 45. Sutters KA, Miaskowski C, Holdridge-Zeuner D, et al. . A randomized clinical trial of the efficacy of scheduled dosing of acetaminophen and hydrocodone for the management of postoperative pain in children after tonsillectomy. Clin J Pain 2010;26:95–103. 10.1097/AJP.0b013e3181b85f98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Butkovic D, Toljan S, Mihovilovic-Novak B. Experience with gabapentin for neuropathic pain in adolescents: report of five cases. Paediatr Anaesth 2006;16:325–9. 10.1111/j.1460-9592.2005.01687.x [DOI] [PubMed] [Google Scholar]
- 47. Rose JB, Finkel JC, Arquedas-Mohs A, et al. . Oral tramadol for the treatment of pain of 7-30 days’ duration in children. Anesth Analg 2003;96:78–81. 10.1213/00000539-200301000-00016 [DOI] [PubMed] [Google Scholar]
- 48. Voepel-Lewis T, Burke CN, Jeffreys N, et al. . Do 0-10 numeric rating scores translate into clinically meaningful pain measures for children? Anesth Analg 2011;112:415–21. 10.1213/ANE.0b013e318203f495 [DOI] [PubMed] [Google Scholar]
- 49. Hida E, Tango T. Three-arm noninferiority trials with a prespecified margin for inference of the difference in the proportions of binary endpoints. J Biopharm Stat 2013;23:774–89. 10.1080/10543406.2013.789893 [DOI] [PubMed] [Google Scholar]
- 50. Borm GF, Fransen J, Lemmens WA. A simple sample size formula for analysis of covariance in randomized clinical trials. J Clin Epidemiol 2007;60:1234–8. 10.1016/j.jclinepi.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 51. Neurontin SPC. Neurontin and Associated names. INN- Gabapentin. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Neurontin_30/WC500009308.pdf (Accessed on 18 Feb 2018).
- 52. Appleton R, Fichtner K, LaMoreaux L, et al. . Gabapentin as add-on therapy in children with refractory partial seizures: a 12-week, multicentre, double-blind, placebo-controlled study. Gabapentin Paediatric Study Group. Epilepsia 1999;40:1147–54. 10.1111/j.1528-1157.1999.tb00833.x [DOI] [PubMed] [Google Scholar]
- 53. Korn-Merker E, Borusiak P, Boenigk HE. Gabapentin in childhood epilepsy: a prospective evaluation of efficacy and safety. Epilepsy Res 2000;38:27–32. 10.1016/S0920-1211(99)00063-7 [DOI] [PubMed] [Google Scholar]
- 54. Trudeau V, Myers S, LaMoreaux L, et al. . Gabapentin in naive childhood absence epilepsy: results from two double-blind, placebo-controlled, multicenter studies. J Child Neurol 1996;11:470–5. 10.1177/088307389601100611 [DOI] [PubMed] [Google Scholar]
- 55. Tramadol drops SPC. Summary of product characteristics. https://db.cbg-meb.nl/mri/spc/nlh-0598-001.pdf (Accessed on 18 Feb 2018).
- 56. Orliaguet G, Hamza J, Couloigner V, et al. . A case of respiratory depression in a child with ultrarapid CYP2D6 metabolism after tramadol. Pediatrics 2015;135:e753–5. 10.1542/peds.2014-2673 [DOI] [PubMed] [Google Scholar]
- 57. Malviya S, Voepel-Lewis T, Tait AR, et al. . Depth of sedation in children undergoing computed tomography: validity and reliability of the University of Michigan Sedation Scale (UMSS). Br J Anaesth 2002;88:241–5. 10.1093/bja/88.2.241 [DOI] [PubMed] [Google Scholar]
- 58. Healy P, Della Pasqua O. Impact of metabolic polymorphism on the exposure to tramadol and its active metabolite in children. 2017. www.page-meeting.org/?abstract=7358
- 59. Adams EH, Breiner S, Cicero TJ, et al. . A comparison of the abuse liability of tramadol, NSAIDs, and hydrocodone in patients with chronic pain. J Pain Symptom Manage 2006;31:465–76. 10.1016/j.jpainsymman.2005.10.006 [DOI] [PubMed] [Google Scholar]
- 60. Piaggio G, Elbourne DR, Pocock SJ, et al. . Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA 2012;308:2594–604. 10.1001/jama.2012.87802 [DOI] [PubMed] [Google Scholar]
- 61. EMEA. Committee for Medicinal Products for Human Use (CHMP). Guideline on reporting the results of population pharmacokinetic analyses, CHMP/EWP/185990/06, 21 June 2007. 2008. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf (Accessed 18 Feb 2018).
- 62. Core Team R. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing., 2014. [Google Scholar]
- 63. King S, Chambers CT, Huguet A, et al. . The epidemiology of chronic pain in children and adolescents revisited: a systematic review. Pain 2011;152:2729–38. 10.1016/j.pain.2011.07.016 [DOI] [PubMed] [Google Scholar]
- 64. Dworkin RH, Turk DC, Peirce-Sandner S, et al. . Research design considerations for confirmatory chronic pain clinical trials: IMMPACT recommendations. Pain 2010;149:177–93. 10.1016/j.pain.2010.02.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2018-023296supp001.pdf (13.4MB, pdf)
bmjopen-2018-023296supp002.pdf (4.7KB, pdf)
bmjopen-2018-023296supp003.pdf (6.4KB, pdf)