

Abstract
Recent clinical trials of non‐small cell lung cancer with immune checkpoint inhibitors revealed that patients with epidermal growth factor receptor (EGFR) mutations had more unfavorable outcomes compared with those with wild‐type EGFR. However, the underlying mechanism for the link between EGFR mutations and immune resistance remains unclear. We performed T cell receptor (TCR) repertoire analysis of resected lung adenocarcinoma tissues with and without EGFR mutations to investigate the characteristics of TCR repertoires. We collected a total of 39 paired (normal and tumor) lung tissue samples (20 had EGFR mutations) and conducted TCR repertoire analysis as well as whole‐exome sequencing (WES) and transcriptome analysis. The TCR diversity index in EGFR‐mutant tumors was significantly higher than that in EGFR‐wild‐type tumors (median [range] 552 [162‐1,135] vs 230 [30‐764]; P < .01), suggesting higher T cell clonal expansion in EGFR‐wild‐type tumors than in EGFR‐mutant tumors. In WES, EGFR‐mutant tumors showed lower numbers of non‐synonymous mutations and predicted neoantigens than EGFR‐wild‐type tumors (P < .01, P = .03, respectively). The number of non‐synonymous mutations revealed a positive correlation with the sum of frequencies of the TCRβ clonotypes of 1% or higher in tumors (r = .52, P = .04). The present study demonstrates significant differences in TCR repertoires and the number of predicted neoantigens between EGFR‐mutant and wild‐type lung tumors. Our findings provide important information for understanding the molecular mechanism behind EGFR‐mutant patients showing unfavorable responses to immune checkpoint inhibitors.
Keywords: EGFR mutation, lung adenocarcinoma, neoantigen, non‐synonymous mutation, T cell receptor repertoire
Abbreviations
- HLA
human leukocyte antigen;
- ICI
immune checkpoint inhibitors;
- NSCLC
non‐small cell lung cancer;
- PD‐1
programmed death‐1;
- PD‐L1
programmed death‐1 ligand‐1;
- RNA
ribonucleic acid;
- TCR
T cell receptor;
- WES
whole‐exome sequencing
1. INTRODUCTION
Lung cancer is the most common cause of cancer‐related death worldwide.1 Despite advances in treatment modalities, such as combination chemotherapy and molecular‐targeted therapy, over the past few decades the survival benefit has been restricted to a subset of patients with advanced diseases. New treatment modalities are urgently needed to target and eliminate invading tumor cells.
Recently, therapeutic antibodies that block the programmed death‐1 (PD‐1)/programmed death‐ligand 1 (PD‐L1) pathway have induced robust and durable clinical responses in patients with various cancers, including advanced non‐small cell lung cancer (NSCLC).2, 3 However, clinical benefits have been observed only in a small subset of patients, with response rates of approximately 20%‐40% for advanced NSCLC.2, 3, 4, 5 In particular, retrospective analyses of clinical trials with PD‐1/PD‐L1 blockade, in which patients with NSCLC harboring epidermal growth factor receptor (EGFR) mutations were enrolled in clinical trials, demonstrated that patients with EGFR‐mutant tumors responded more poorly to immune checkpoint inhibitors (ICI) compared to those with wild‐type EGFR.6, 7, 8 However, the molecular mechanism underlying the lower response rates to ICI remains unclear. Thus, elucidating the mechanism that cause the differences in the clinical responses to ICI between EGFR‐mutation‐positive and EGFR‐mutation‐negative groups is important to further improve outcomes and to optimize the use of these agents in lung cancer patients.
The progress in analyzing T cell receptor (TCR) repertoires in cancer tissues made it possible to evaluate the diversity of T cell clonotypes and the extent of clonal T cell expansion, and to characterize neoantigen‐specific TCR.9 Detailed information on the tumor microenvironment may serve as a predictive marker for immunomodulatory therapies and may also be useful for development of new treatment strategies, including personalized T cell‐mediated cancer immunotherapy and neoantigen vaccine therapy.10, 11, 12, 13, 14 TCR repertoire analyses could be used to monitor the dynamics of T cell clonality and the individual tumor‐reactive T cell clones in cancer patients treated with ICI.15, 16 Profiling the immune repertoire by quantifying the TCR composition in tumor tissues enables assessment of T cell diversity and immune‐related characteristics. Therefore, in the present study, we aimed to investigate the differences in immune‐related conditions in EGFR‐mutant/wild‐type tumors using TCR sequencing along with whole‐exome sequencing (WES) analysis.
Here, we report distinct characteristics of TCR repertoire patterns in lung adenocarcinomas with and without EGFR mutations and demonstrate the association between the diversity of TCR repertoires and the numbers of non‐synonymous mutations in tumors. Our results should contribute to a better understanding of the molecular mechanism behind EGFR‐mutant patients having shown an unfavorable response to ICI.
2. MATERIALS AND METHODS
2.1. Patients
All subjects in the present study received curative surgery between 2014 and 2017 at the Tohoku University Hospital. A total of 39 patients were enrolled based on the following inclusion criteria: (i) pathologically diagnosed with lung adenocarcinoma; (ii) pathological stage was I to III; (iii) frozen tumor and normal paired tissue samples were available; and (iv) written informed consent was obtained. This study was approved by the Institutional Review Board of Tohoku University (Sendai, Japan: approval no. 2013‐1‐592) and the University of Chicago (Chicago, IL, USA: approval no. 13‐0797).
2.2. Tissue samples
Tumor and adjacent normal lung tissue samples were obtained from the resected tissues. These resected tissue samples were immediately soaked in RNA later Stabilization Solution (Thermo Fisher Scientific, Waltham, MA, USA) and stored at −80°C until extraction of nucleic acids. All the samples were histologically confirmed as adenocarcinoma by pathologists.
2.3. T cell receptor sequencing
We performed TCR sequencing as described previously.17, 18 Briefly, we synthesized complementary DNAs (cDNAs) with the common 5′‐RACE adapter sequence, and amplified TCR β‐chain (TCRβ) by PCR. We added Illumina index sequences with barcodes using the Nextera XT Index Kit v2 (Illumina, San Diego, CA, USA), which allows barcode tagging and pooling of multiple samples. Subsequently, the prepared library was sequenced using the MiSeq Reagent Kit v3 (600‐cycle) on the MiSeq System (Illumina). We calculated the diversity index (inverse Simpson's index) to quantify the clonality of the TCRβ repertoires, as previously described.19
2.4. Whole‐exome sequencing and data analysis
DNA and RNA were isolated using the standard procedures and the AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA, USA). Whole‐exome libraries were prepared from genomic DNA using the SureSelect XT Human All Exon V6 kit (Agilent Technologies, Santa Clara, CA, USA) and analyzed as previously described.18, 20 Human leukocyte antigen (HLA) class I genotypes were determined using the WES data of normal tissues and the OptiType algorithm.21 Neoantigens were predicted for each non‐synonymous variant by defining all 8‐mer to 11‐mer peptides resulting from the mutation and determining the predicted binding affinity to HLA‐A, B and C of <500 nM, using NetMHCv3.4 and NetMHCpanv2.8 software.11, 22, 23, 24
2.5. TCGA dataset analysis
We obtained the TCGA dataset from a previous publication.25 Among these, 467 patients from the TCGA lung adenocarcinoma cohort had information of non‐synonymous mutations as well as the EGFR status.
2.6. Gene expression analysis
Gene expression quantitative PCR in tumor tissues was performed using the TaqMan gene expression assays (Thermo Fisher Scientific) on a ViiA 7 Real‐Time PCR System (Thermo Fisher Scientific). mRNA expression levels of PD‐1 (assay ID, Hs01550088_m1), PD‐L1 (CD274; assay ID, Hs01125301_m1), CD8 (assay ID, Hs002335520_m1) and FOXP3 (assay ID, Hs01085834_m1) were evaluated and normalized to GAPDH expression (assay ID, Hs02758991_g1).
2.7. Statistical methods
Student's t test and Fisher's exact test were performed for comparison between tumors with and without EGFR mutations. The Mann‐Whitney U test was used for comparison of the numbers of non‐synonymous mutations, the TCRβ diversity index, the proportions of expanded TCRβ clonotypes, and the numbers of predicted neoantigens between tumors with and without EGFR mutations. Multiple logistic regression models were applied to assess the association between the EGFR mutation status and the binary measures of patient characteristics, including the diversity index. Pearson correlation (r) was used to analyze the association between the proportion of TCRβ clones and non‐synonymous mutations. Statistical analysis was carried out using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA, USA). A P‐value of <.05 was considered statistically significant.
3. RESULTS
3.1. Patients
The characteristics of patients are summarized in Table 1. The median age of these patients was 68 years old (range: 41‐85) and 25 patients (64%) were women. Nineteen patients (49%) were never‐smokers, and pathological stages after curative lung resection were I for 9 patients (23%), II for 16 patients (41%) and III for 14 patients (36%). Tumors from 20 patients (51%) had EGFR mutations (an exon 19 deletion in 9 patients and an L858R mutation in 11 patients); 80% of these patients were women and 70% of them were never‐smokers.
Table 1.
Patient characteristics
| Characteristics | Total (n = 39) | EGFR status | P‐value | |
|---|---|---|---|---|
| Mutant (n = 20) | Wild‐type (n = 19) | |||
| Age (median, years) (range) | 68 (41‐79) | 68 (41‐85) | 66 (56‐85) | .31 |
| Sex | ||||
| Male | 14 (36%) | 4 (20%) | 10 (53%) | <.05 |
| Female | 25 (64%) | 16 (80%) | 9 (47%) | |
| Smoking | ||||
| Current/extent | 20 (51%) | 6 (30%) | 14 (74%) | .01 |
| Never | 19 (49%) | 14 (70%) | 5 (26%) | |
| Pathological stage (pStage) | ||||
| Stage I | 9 (23%) | 6 (30%) | 3 (16%) | .74 |
| Stage II | 16 (41%) | 5 (25%) | 11 (58%) | |
| Stage III | 14 (36%) | 9 (45%) | 5 (26%) | |
EGFR, epidermal growth factor receptor.
The significant P‐values are shown in bold (P < .05).
3.2. T cell receptor repertoire analysis
To elucidate whether an EGFR mutation status affects the diversity of TCR repertoires, we performed next‐generation sequencing‐based TCRβ repertoire analysis and calculated the diversity index of 39 lung adenocarcinoma samples. In the TCRβ sequencing, we obtained a total of 804 134 ± 358 995 sequence reads (average ± SD) mapped to the V, J and C segments, and identified 55 343 ± 32 756 unique CDR3 clonotypes (Table S1). Notably, tumors with EGFR mutations had a higher TCRβ diversity index than those without EGFR mutations (median [range] 552 [162‐1135] vs 230 [30‐764]; P < .01; Figure 1A, Table S1). Multivariate logistic regression analyses were undertaken to examine whether the correlation between the diversity index and the EGFR status would be influenced by differences in patient characteristics such as age, sex, smoking and pathological stage. We found that the TCRβ diversity index (High: greater than median value) and smoking status (non‐smoker) were independently associated with EGFR mutation status (P = .04 and .02, respectively; Table 2).
Figure 1.
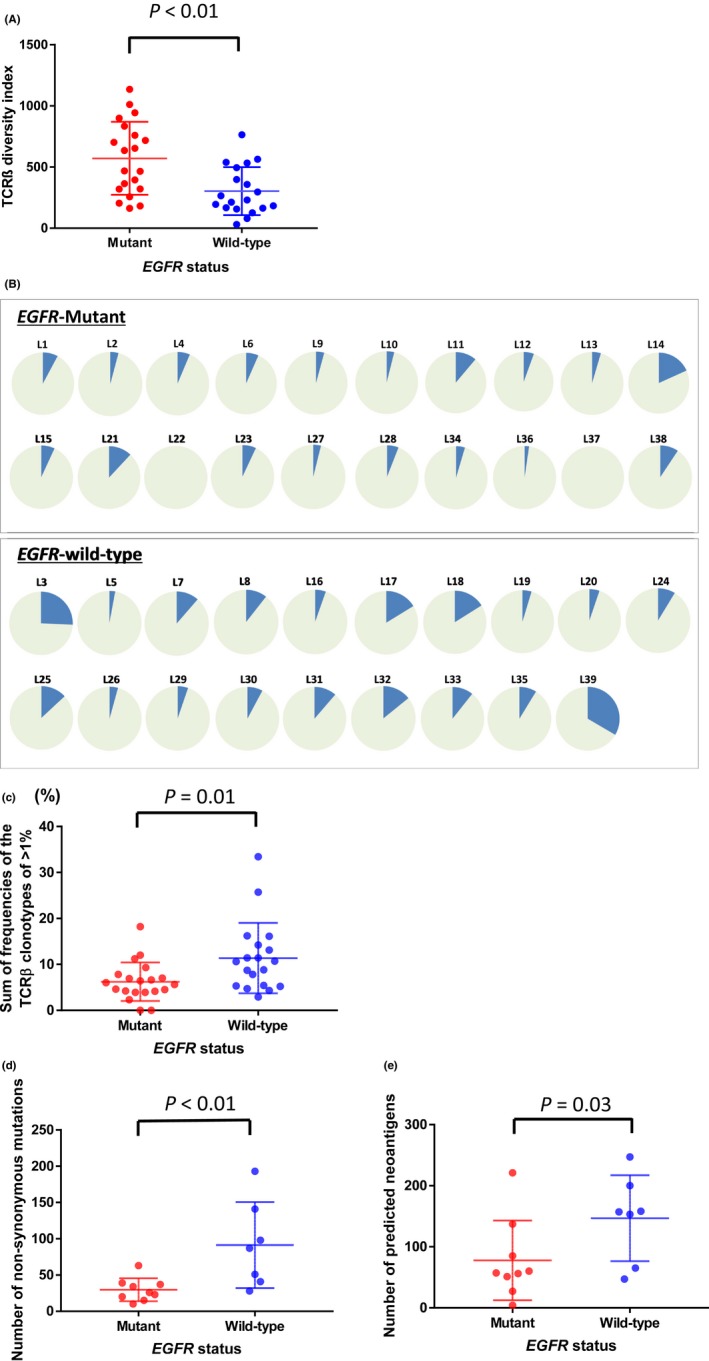
TCRβ diversity index (A) and the sum of frequencies of the TCRβ clonotype of 1% or higher EGFR‐mutant and wild‐type tumors (N = 39) (B, C). A, The EGFR‐mutant tumors had a higher TCRβ diversity index than wild‐type tumors (median [range] 552 [162‐1135] vs 230 [30‐764]; P < .01). B, The blue portion of each pie chart shows the cumulative sum of frequencies of the TCRβ clonotype of 1% or higher in EGFR‐mutant and wild‐type tumors. C, The proportions of TCRβ clones in tumors with wild‐type EGFR were significantly higher than those in tumors with EGFR mutations (median [range]: 5.8% [0%‐18.2%] vs 10.6% [2.9%‐33.4%]; P = .01). Number of non‐synonymous mutations (D) and predicted neoantigens (E) in EGFR‐mutant and wild‐type tumors (N = 16). D, Number of non‐synonymous mutations in EGFR‐mutant and wild‐type tumors. The number of non‐synonymous mutations was significantly lower in EGFR‐mutant than wild‐type tumors (median [range]: 26 [10‐63] vs 87 [28‐193]; P < .01). E, Number of predicted neoantigens in EGFR‐mutant and wild‐type tumors. EGFR‐mutant tumors had a significantly lower number of predicted neoantigens than wild‐type tumors (median: 57 [4‐221] vs 157 [47‐247]; P = .03). P‐values were calculated to test the difference between the EGFR‐mutant and wild‐type groups using the unpaired t test. EGFR, epidermal growth factor receptor; TCR, T cell receptor
Table 2.
Multivariate logistic regression analyses of variables related to EGFR mutation status
| Variable | Category | Regression coefficient | Standard error | Odds ratio (95% CI) | P‐value |
|---|---|---|---|---|---|
| Diversity index in tumor | High (>364a) | 1.91 | .93 | 6.76 (1.09‐41.80) | .04 |
| Age | 65 or more | .74 | .89 | 2.10 (.37‐11.98) | .40 |
| Sex | Female | 1.11 | .88 | 3.04 (.54‐17.22) | .20 |
| Smoking | Non‐smoker | 2.15 | .93 | 8.57 (1.38‐53.32) | .02 |
| pStage | llb/III | −.29 | .86 | .75 (.14‐4.07) | .74 |
| Constant | −2.68 | 1.04 | .07 | .01 |
CI, Confidence interval.
Median value.
The significant P‐values are shown in bold (P < .05).
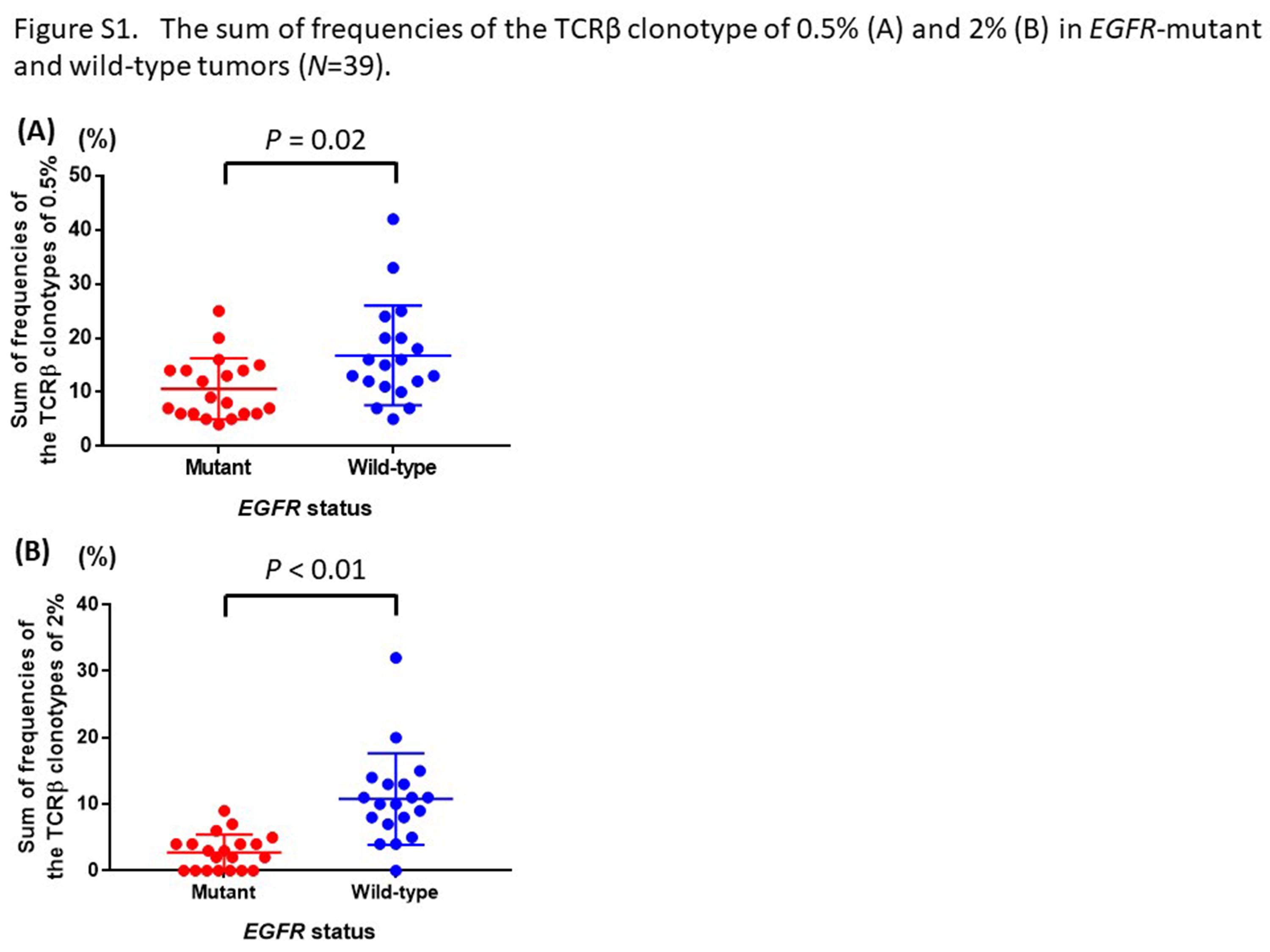
To further examine differences in the proportions of the expanded T cell clones in the 2 groups with and without EGFR mutations, we compared the sum of frequencies of the TCRβ clonotypes of 1% or higher in the 2 groups (Figure 1B, Table S1). The sum of frequencies of the TCRβ clonotypes of 1% or higher in tumors with wild‐type EGFR were significantly higher than those in tumors with EGFR mutations (median [range]: 5.8% [0‐18.2%] vs 10.6% [2.9‐33.4%]; P = .01; Figure 1C). The results showed the same tendencies when we used the sum of frequencies of the TCRβ clonotypes of .5% and 2% as cut‐off values (P = .02, P < .01, respectively; Figure S1)
3.3. Comparison of the numbers of somatic non‐synonymous mutations/predicted neoantigens between epidermal growth factor receptor‐mutant and wild‐type tumors
To assess a relationship between the EGFR status and the numbers of somatic non‐synonymous mutations, we compared the numbers in tumors with and without EGFR mutations. WES analysis was performed for 16 randomly selected cases (Del19/L858R/wild type were 5/4/7, respectively) from the 39 lung adenocarcinomas. We identified a total of 906 somatic non‐synonymous mutations (10‐193 per individual patients; Table S2). The number of non‐synonymous mutations was significantly lower in EGFR‐mutant tumors than in EGFR‐wild‐type tumors (median [range]: 26 [10‐63] vs 87 [28‐193]; P < .01; Figure 1D). To verify this result, we examined the data of lung adenocarcinomas in the TCGA dataset and found similar results supporting that the mutational burden was lower in the EGFR‐mutant group than the EGFR‐wild‐type group (median [range]: 48 [16‐241] vs 191 [0‐1277], P < .01; Figure S2).
To further investigate the relation between the numbers of predicted neoantigens and the EGFR status, we performed in silico neoantigen prediction for non‐synonymous mutations in the 16 lung adenocarcinomas in which we conducted WES. We predicted the binding affinity of peptides including an amino‐acid substitution to individual HLA‐A, B and C molecules that were estimated from the WES data of normal DNAs. We obtained neoantigen epitope candidate sequences, which were filtered with the binding affinity to the HLA molecules of 500 nM or lower, and identified a total of 469 neoantigen candidates (4‐247 neoantigens in individual patients; Table S2). Subsequently, we compared the number of candidate peptides in the 2 groups with and without EGFR mutations and found that tumors with EGFR mutations had significantly lower numbers of predicted neoantigens than those without EGFR mutations (median: 57 [4‐221] vs. 157 [47‐247]; P = .03; Figure 1E).
3.4. Correlation between the proportion of expanded T cell receptor β clones and the number of non‐synonymous mutations
To analyze the relationship between the clonal expansions of T cells in tumor microenvironment and the numbers of non‐synonymous mutations, we compared the sum of frequencies of the TCRβ clonotypes of 1% or higher with the number of non‐synonymous mutations in the 2 groups with and without EGFR mutations (Figure 2). The numbers of non‐synonymous mutations were significantly correlated with the clonal T cell expansions in the tumor tissues (r = .52, P = .04).
Figure 2.
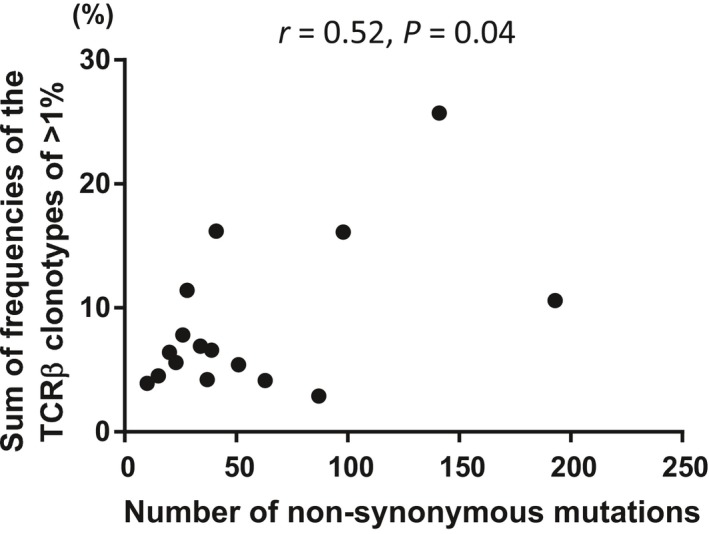
Correlation analysis of the number of non‐synonymous mutations and the sum of frequencies of the TCRβ clonotype of 1% or higher. The number of non‐synonymous mutations was significantly correlated with the sum of frequencies of the TCRβ clonotype of 1% or higher. TCR, T cell receptor
3.5. Immune‐related gene expression analysis
Because PD‐1/PD‐L1 expression is important to immune responses in tumors, including the efficacy to ICI, we assessed PD‐1/PD‐L1 expression levels in tumors with and without EGFR mutations. We measured mRNA expression levels of PD‐1 and PD‐L1 in 39 lung adenocarcinomas and compared the expression levels between the 2 groups. We observed no significant differences in PD‐1 and PD‐L1 mRNA expression levels between the EGFR‐mutant and wild‐type groups (P = .09, P = .25, respectively). We also observed no significant differences in the CD8/FOXP3 ratio between the 2 groups (P = .38). However, we found the tendency of a higher PD‐1/CD8 expression ratio in EGFR‐wild‐type tumors compared to EGFR‐mutant tumors (median: .10 [.02‐.48] vs .13 [.01‐2.35]; P = .24), implying that EGFR‐wild‐type tumors might have a more immune‐active microenvironment.
4. DISCUSSION
The underlying biology for lower clinical response rates of ICI in lung adenocarcinomas having EGFR mutations is not well understood. Hence, identification of predictive biomarkers for responses to ICI is critical for lung cancer patients with EGFR mutations. We analyzed immune‐related microenvironment in tumors with and without EGFR mutations using TCRβ repertoire analysis and WES. Our study is the first to characterize distinct TCR repertoire patterns between 2 groups of lung adenocarcinoma with and without EGFR mutations; we also clarified the association between the diversity of TCR repertoires and the mutational load in tumors. Our findings may evoke further understanding of the molecular mechanism through which EGFR‐mutant patients show poor clinical responses to ICI.
In this study, we demonstrated that the sum of frequencies of the TCRβ clonotypes of 1% or higher in tumors with wild‐type EGFR were significantly higher than those in tumors with EGFR mutations (Figure 1C). Clonal T cell expansion in the tumor microenvironment is essential for the effective anti‐tumor immune response. Recent studies suggested that responses to ICI were associated with the clonal expansion of tumor‐infiltrating T lymphocytes.25, 26 Therefore, our findings implied that the low clonal T cell expansion in tumors with EGFR mutations might be a critical factor related to the unfavorable response to ICI. Furthermore, TCR sequencing might be applicable for the treatment selection in patients with EGFR mutations by evaluating the proportions of TCRβ clones in the tumor.
This is the first study showing significant differences in the numbers of neoantigens between lung adenocarcinomas with and without EGFR mutations; we demonstrated that tumors with EGFR mutations had lower numbers of non‐synonymous mutations and predicted neoantigens than those without EGFR mutations. Previous studies have reported that the efficacy of antibody targeting PD‐1/PD‐L1 is associated with the tumor mutation burden (TMB) and the numbers of predicted neoantigens in NSCLC.27, 28, 29 Hence, we assume that the lower TMB may partly explain the lower efficacy of lung cancer with an EGFR mutation to ICI.30
In addition, patients’ characteristic may affect the efficacy of ICI. EGFR mutations are found in female patients with no history of tobacco smoking.31, 32 Our study population also showed more women than men in the EGFR‐mutated group. Interestingly, the pooled analysis showed that a higher benefit of ICI treatment was observed in men than women, regardless of histological type.33 Furthermore, several studies report that tobacco smoking leads to a higher mutation burden in human cancers.27, 34, 35
PD‐L1 expression levels may affect the clinical benefit of ICI. In the combined analysis of 15 reported studies, EGFR‐mutated tumors showed low PD‐L1 expression in tumors30 and recent studies demonstrated that PD‐L1 expression levels were associated with EGFR mutation status.36, 37 However, in this study, PD‐1/PD‐L1 expression levels were not significantly different between the 2 groups with and without EGFR mutations. Accumulated data now indicates that the PD‐L1 status alone is not a useful biomarker for the prediction of the efficacy of ICI.38, 39, 40
In conclusion, the present study offers novel evidence that lung adenocarcinoma with EGFR mutations have a higher TCRβ diversity index and a lower number of neoantigens compared with tumors without EGFR mutations, and could explain impaired responses to ICI. Furthermore, TCR repertoire analysis might provide useful information for identification of good responders for immunotherapy in EGFR‐mutant NSCLC.
CONFLICT OF INTEREST
Y. N. is a stock holder and a scientific advisor of OncoTherapy Science. K. K. and S.‐K. L. are scientific advisors of Cancer Precision Medicine. No potential conflicts of interest were disclosed by the other authors.
Supporting information
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
We thank Drs Rui Yamaguchi, Seiya Imoto and Satoru Miyano at The University of Tokyo for developing the algorithm for TCR repertoire analysis and helpful support in data management. The super‐computing resource (http://sc.hgc.jp/shirokane.html) was provided by the Human Genome Center, Institute of Medical Science, The University of Tokyo. We also gratefully acknowledge Hirotsugu Notsuda, Teruyuki Sato, Ryota Saito, Mitsu Takahashi and Masako Honda for the collection of patients’ samples.
Miyauchi E, Matsuda T, Kiyotani K, et al. Significant differences in T cell receptor repertoires in lung adenocarcinomas with and without epidermal growth factor receptor mutations. Cancer Sci. 2019;110:867–874. 10.1111/cas.13919
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;373:123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;373:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carbone DP, Reck M, Paz‐Ares L, et al. First‐line nivolumab in stage IV or recurrent non‐small‐cell lung cancer. N Engl J Med. 2017;376:2415‐2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 6. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD‐1 pathway blockade in non‐small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22:4585‐4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR‐mutated non‐small cell lung cancer‐a meta‐analysis. J Thorac Oncol. 2017;12:403‐407. [DOI] [PubMed] [Google Scholar]
- 8. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet. 2016;387:1540‐1550. [DOI] [PubMed] [Google Scholar]
- 9. Correia‐Neves M, Waltzinger C, Mathis D, et al. The shaping of the T cell repertoire. Immunity. 2001;14:21‐32. [DOI] [PubMed] [Google Scholar]
- 10. Saini SK, Rekers N, Hadrup SR. Novel tools to assist neoepitope targeting in personalized cancer immunotherapy. Ann Oncol. 2017;28(Suppl 12):xii3‐xii10. [DOI] [PubMed] [Google Scholar]
- 11. Kiyotani K, Chan HT, Nakamura Y. Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Sci. 2018;109:542‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kato T, Matsuda T, Ikeda Y, et al. Effective screening of T cells recognizing neoantigens and construction of T cell receptor‐engineered T cells. Oncotarget. 2018;9:11009‐11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsuda T, Leisegang M, Park JH, et al. Induction of neoantigen‐specific cytotoxic T cells and construction of T cell receptor‐engineered T cells for ovarian cancer. Clin Cancer Res. 2018;24:5357‐5367. [DOI] [PubMed] [Google Scholar]
- 14. Gubin MM, Artyomov MN, Mardis ER, et al. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125:3413‐3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santa‐Maria CA, Kato T, Park JH, et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Expert Rev Anticancer Ther. 2014;14:129‐141.24467217 [Google Scholar]
- 16. Haymaker CL, Kim D, Uemura M, et al. Metastatic melanoma patient had a complete response with clonal expansion after whole brain radiation and PD‐1 blockade. Cancer Immunol Res. 2017;5:100‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fang H, Yamaguchi R, Liu X, et al. Quantitative T cell repertoire analysis by deep cDNA sequencing of T cell receptor α and β chains using next‐generation sequencing (NGS). Oncoimmunology. 2015;3:e968467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choudhury NJ, Kiyotani K, Yap KL, et al. Low T cell receptor diversity, high somatic mutation burden, and high neoantigen load as predictors of clinical outcome in muscle‐invasive bladder cancer. Eur Urol Focus. 2016;2:445‐452. [DOI] [PubMed] [Google Scholar]
- 19. Venturi V, Kedzierska K, Turner SJ, et al. Methods for comparing the diversity of samples of the T cell receptor repertoire. J Immunol Methods. 2007;321:182‐195. [DOI] [PubMed] [Google Scholar]
- 20. Kiyotani K, Park JH, Inoue H, et al. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017;6:e1278330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szolek A, Schubert B, Mohr C, et al. OptiType: precision HLA typing from next‐generation sequencing data. Bioinformatics. 2014;30:3310‐3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24:1397‐1398. [DOI] [PubMed] [Google Scholar]
- 23. Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA‐A and ‐B locus protein of known sequence. PLoS ONE. 2007;2:e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rizvi H, Sanchez‐Vega F, La K, et al. Molecular determinants of response to anti‐programmed cell death (PD)‐1 and anti‐programmed death‐ligand 1 (PD‐L1) blockade in patients with non‐small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol. 2018;36:633‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dong ZY, Zhang JT, Liu SY, et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD‐1 blockade in non‐small cell lung cancer. Oncoimmunology. 2017;6:e1356145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomizawa Y, Iijima H, Sunaga N, et al. Clinicopathologic significance of the mutations of the epidermal growth factor receptor gene in patients with non‐small cell lung cancer. Clin Cancer Res. 2005;11:6816‐6822. [DOI] [PubMed] [Google Scholar]
- 32. Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339‐346. [DOI] [PubMed] [Google Scholar]
- 33. Conforti F, Pala L, Bagnardi V, et al. Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta‐analysis. Lancet Oncol. 2018;19:737‐746. [DOI] [PubMed] [Google Scholar]
- 34. Pfeifer GP, Denissenko MF, Olivier M, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking‐associated cancers. Oncogene. 2002;21:7435‐7451. [DOI] [PubMed] [Google Scholar]
- 35. Alexandrov LB, Ju YS, Haase K, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang M, Li G, Wang Y, et al. PD‐L1 expression in lung cancer and its correlation with driver mutations: a meta‐analysis. Sci Rep. 2017;7:10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lan B, Ma C, Zhang C, et al. Association between PD‐L1 expression and driver gene status in non‐small‐cell lung cancer: a meta‐analysis. Oncotarget. 2018;9:7684‐7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kerr KM, Tsao MS, Nicholson AG, et al. Programmed Death‐Ligand 1 Immunohistochemistry in Lung Cancer: in what state is this art? J Thorac Oncol. 2015;10:985‐989. [DOI] [PubMed] [Google Scholar]
- 39. Takada K, Toyokawa G, Shoji F, et al. The significance of the PD‐L1 expression in non‐small‐cell lung cancer: trenchant double swords as predictive and prognostic markers. Clin Lung Cancer. 2018;19:120‐129. [DOI] [PubMed] [Google Scholar]
- 40. Mathew M, Safyan RA, Shu CA. PD‐L1 as a biomarker in NSCLC: challenges and future directions. Ann Transl Med. 2017;5:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials