

Abstract
Aflibercept targets vascular endothelial growth factor. The present study involved assessing the efficacy, safety and pharmacokinetics of aflibercept plus 5‐fluorouracil/levofolinate/irinotecan (FOLFIRI) as a second‐line treatment for metastatic colorectal cancer (mCRC) in Japanese patients. Aflibercept (4 mg/kg) plus FOLFIRI was administered every 2 weeks in 62 patients with mCRC until disease progression, unacceptable toxicity or patient withdrawal. Tumors were imaged every 6 weeks. The primary endpoint was objective response rate (ORR); secondary endpoints were progression‐free survival, overall survival, safety, and pharmacokinetics of aflibercept, irinotecan and 5‐fluorouracil. A total of 60 patients were evaluated for ORR; 50 had received prior bevacizumab. The ORR was 8.3% (95% confidence interval [CI]: 1.3%‐15.3%), and the disease control rate (DCR) was 80.0% (69.9%‐90.1%). The median progression‐free survival was 5.42 months (4.14‐6.70 months) and the median overall survival was 15.59 months (11.20‐19.81 months). No treatment‐related deaths were observed, and no significant drug‐drug interactions were found. The most common treatment‐emergent adverse events were neutropenia and decreased appetite. Free aflibercept had a mean maximum concentration (coefficient of variation) of 73.2 μg/mL (15%), clearance of 0.805 L/d (22%) and volume of distribution of 6.2 L (18%); aflibercept bound with vascular endothelial growth factor had a clearance of 0.162 L/d (9%) (N = 62). Aflibercept did not significantly affect the pharmacokinetics of irinotecan or 5‐fluorouracil: The clearance was 11.1 L/h/m2 (28%) for irinotecan and, at steady state, 72.6 L/h/m2 (56%) for 5‐fluorouracil (N = 10). Adding aflibercept to FOLFIRI was shown to be beneficial and well‐tolerated in Japanese patients with mCRC. ClinicalTrials.gov Identifier: NCT01882868.
Keywords: aflibercept, angiogenesis inhibitors, colorectal neoplasms, neoplasm metastasis, vascular endothelial growth factor A
1. INTRODUCTION
Colorectal cancer is the third most commonly occurring cancer worldwide and the second leading cause of cancer‐related deaths.1 In Japan, it is the most commonly occurring cancer in both sexes combined.2, 3 The 5‐year relative survival rate for metastatic colorectal cancer (mCRC) is approximately 12%‐13%.4 Thus, it is important to develop new, more effective therapies.
Vascular endothelial growth factor (VEGF) is overexpressed in primary colon tumors that have metastasized compared to those that have not.5 Furthermore, high VEGF expression predicts poor relapse‐free and overall survival of individuals with colorectal cancer.6 For this reason, VEGF has in recent years become a target for anti–cancer therapies, in combination with standard chemotherapy regimens folinic acid/5‐fluorouracil (5‐FU)/oxaliplatin (FOLFOX), FOLFOX/irinotecan (FOLFOXIRI) or 5‐FU/levofolinate/irinotecan (FOLFIRI). One anti–VEGF agent, bevacizumab, specifically blocks VEGF‐A and, in combination with FOLFOX,7 FOLFOXIRI8, 9 or FOLFIRI,10 has been shown to increase survival of patients with mCRC.
Aflibercept, also known as VEGF‐trap or ziv‐aflibercept, is a relatively new anti–VEGF agent.11 It is a recombinant fusion protein containing portions of the extracellular domains of human VEGF receptors 1 and 2. Unlike bevacizumab, which binds only to VEGF‐A, aflibercept binds to VEGF‐A, VEGF‐B and placental growth factor, thus blocking their downstream activity.
Several clinical trials have demonstrated the relative safety and efficacy of aflibercept plus FOLFIRI. The largest trial to date was an international randomized double‐blind phase III study conducted outside of Japan (VELOUR study [NCT00561470]),12 which consisted of 1226 patients with mCRC who had previously received oxaliplatin. Aflibercept plus FOLFIRI significantly improved both overall survival (OS; 13.50 vs 12.06 months, P = .0032) and progression‐free survival (PFS; 6.90 vs 4.67 months, P = .0001) compared to placebo plus FOLFIRI. Likewise, in a recently published randomized Phase III study of patients from the Asia‐Pacific region, aflibercept plus FOLFIRI improved both OS (14.59 vs 11.93 months, hazard ratio: .794) and PFS (6.93 vs 5.59 months, hazard ratio: .629) compared to placebo plus FOLFIRI (NCT01661270).13 A phase I dose‐escalation study of aflibercept plus FOLFIRI (NCT00921661)14 was conducted in Japanese patients with mCRC. No dose‐limiting toxicities or major safety issues were observed for 4 mg/kg aflibercept, the standard dose.
The objectives of the current phase II study were to assess the efficacy, safety and pharmacokinetics (PK) of aflibercept plus FOLFIRI as a second‐line treatment for mCRC in Japanese patients.
2. METHODS
2.1. Patients
Inclusion criteria were: histologically or cytologically proven adenocarcinoma of the colon or rectum; measurable disease, per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1; inoperable metastatic disease; 1 prior chemotherapeutic, oxaliplatin‐containing regimen for metastatic disease, during which or within 6 months after completion of which the disease progressed or patients relapsed; Eastern Cooperative Oncology Group performance status of 0 or 1; adequate organ function; and signed, dated informed consent. Exclusion criteria included active infectious disease, gastrointestinal ulcer, bleeding, urine protein‐creatinine ratio >1, and uridine‐5‐diphospho‐glucuronosyltransferase 1A1 (UGT1A1) genotype of *6/*6, *28/*28 or *6/*28.
2.2. Study design
This was a prospective, multicenter, open‐label, single‐arm study (Figure 1). All patients received the following treatment regimen once every 2 weeks: aflibercept (4 mg/kg) over 1‐2 hours by intravenous (i.v.) infusion; then levofolinate (200 mg/m2) over 120 ± 20 minutes, plus irinotecan (180 mg/m2) over 90 ± 15 minutes, simultaneously by i.v. infusion; then 5‐FU (400 mg/m2) as a bolus over 2‐4 minutes; and then 5‐FU (2400 mg/m2) over 41‐46 hours by continuous i.v. infusion. Treatment was given until disease progression, unacceptable toxicity or patient withdrawal.
Figure 1.
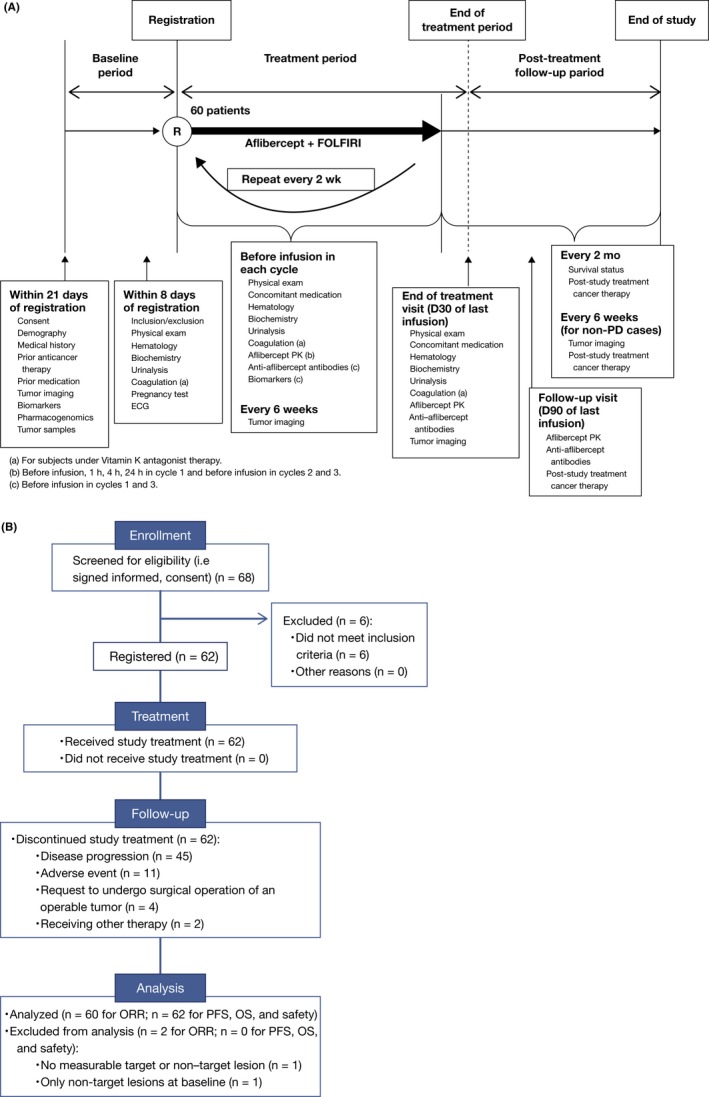
A, Study design. This prospective, multicenter, open‐label, single‐arm study consisted of a baseline period, a treatment period and a post–treatment follow‐up period. During the follow‐up period, tumors were imaged every 6 ± 1 wk until disease progression. (a) For subjects under Vitamin K antagonist therapy. (b) Before infusion, 1, 4 and 24 h in cycle 1 and before infusion in cycles 2 and 3. (c) Before infusion in cycles 1 and 3. B, Flow chart of patient participation
The primary endpoint was objective response rate (ORR). Secondary endpoints were PFS, OS, safety and PK.
2.3. Efficacy assessments
Tumors were imaged every 6 ± 1 weeks and at the end‐of‐treatment visit 30 ± 3 days after the last study treatment. In the post–treatment follow‐up period, tumors were imaged every 6 ± 1 weeks until disease progression. Survival status was determined every 2 months. ORR was defined as the percentage of patients with either a complete response (CR) or partial response (PR) to study treatment, determined based on tumor assessment by an independent radiological review committee (IRRC) using RECIST version 1.1 criteria. PFS was defined as the time interval from the first study treatment administration to either the first observation of radiologically documented disease progression, determined based on tumor assessment by the IRRC, or death due to any cause, whichever came first. OS was defined as the time interval from the first study treatment administration to death due to any cause.
In an exploratory analysis, ORR, PFS and OS were compared in patients with mutated KRAS (exon 2) vs those with wild‐type KRAS and in patients with left‐sided primary tumors (descending colon, sigmoid colon, rectosigmoid colon and/or rectum) vs those with right‐sided primary tumors (caecum, ascending colon and/or transverse colon).
2.4. Safety assessments
Safety assessments were performed as shown in Figure 1 and included physical examination, evaluation of laboratory data and assessment of adverse events. Laboratory safety tests were performed at baseline, at every visit before treatment administration and at the end‐of‐treatment visit, and included hematology, biochemistry, urinalysis, coagulation and any other tests as clinically indicated. Laboratory abnormalities were recorded as adverse events only if they led to study treatment discontinuation or modification (eg, dose reduction, cycle delay or omission) and/or were serious (ie, were life‐threatening and/or resulted in hospitalization, disability and death). Adverse events assessed included treatment‐emergent adverse events (TEAE), serious adverse events and death.
For immunogenicity evaluation, blood samples were collected before aflibercept infusion in treatment cycles 1 and 3, at 30 ± 3 days and 90 ± 7 days after the last aflibercept infusion, and in cases of infusion‐related allergic reaction (Grade ≥ 2) or proteinuria (>3.5 g/24 hours or of renal origin associated with hematuria). The presence of anti–aflibercept antibodies was evaluated in serum using a validated non–quantitative titer‐based bridging immunoassay. If the result was positive, then the presence of aflibercept‐neutralizing antibodies was evaluated using a non–quantitative competitive ligand‐binding assay.
2.5. Population pharmacokinetics
A population PK approach was used to estimate individual PK parameters for free and VEGF‐bound aflibercept in all 62 patients. Blood samples were obtained during treatment cycle 1: pre‐treatment, before the end of infusion (EOI) of aflibercept (1 hour), and 3, 23 and 335 hours (Day 14) after the EOI of aflibercept. Blood samples were also obtained pre‐dose of every odd‐numbered cycle, and 30 and 90 days after the last administration of aflibercept.
Plasma concentrations were measured by validated enzyme‐linked immunosorbent assays. Concentrations of VEGF‐bound aflibercept were expressed as the free aflibercept equivalent by multiplying by 0.717, the ratio of the molecular weights of free and VEGF‐bound aflibercept. The lower limits of quantification (LLOQ) were 15.6 and (adjusted) 31.5 ng/mL, respectively.
The PK parameters were maximum concentration (C max), area under the curve over the dosing interval (AUC0‐14 day), total body clearance (CL) and volume of distribution at steady state (V ss) for free aflibercept, and CL for VEGF‐bound aflibercept.
2.6. Non–compartmental pharmacokinetics
Pharmacokinetics parameters were calculated for irinotecan, its active metabolite SN‐38, and 5‐FU in the first 10 patients by non–compartmental analysis (PKDMS Version 2 running with WinNonLin Professional, Version 5.2.1, PharSight, Raleigh‐Durham, NC, USA). Blood samples for irinotecan and SN‐38 were obtained before aflibercept infusion, just before EOI of irinotecan (1.5 hours), and 2, 4.5 and 23 hours after the start of irinotecan infusion during cycle 1. Blood samples for 5‐FU were obtained before the start of aflibercept infusion and 2.5, 21 and 45 hours after the start of 5‐FU infusion during cycle 1. Concentrations were measured using validated electrospray liquid chromatography tandem mass spectrometry for irinotecan, SN‐38 and 5‐FU (LLOQ: 10.0, 1.0 and 5.0 ng/mL, respectively).
The PK parameters for irinotecan and SN‐38 were C max, time to reach C max(t max), AUC until last quantifiable time point (AUClast), AUC extrapolated to infinity (AUC), terminal elimination half‐life (t 1/2z) and metabolic ratio based on molecular weight (R met). For irinotecan, CL and V ss were also estimated. For 5‐FU, steady‐state concentration during constant‐rate infusion (C ss) and clearance at steady state (CLss) were estimated.
2.7. Statistical analysis
This study aimed to estimate the ORR in Japanese patients with mCRC at a certain precision. In prior studies of patients with mCRC treated in the second line with FOLFIRI or FOLFIRI plus aflibercept, ORR ranged from approximately 10% to 20%.10, 11, 12, 13, 14, 15, 16, 17 If the ORR observed in this study was in the same range, then 60 patients would provide precision (range of 95% CI) from 0.16 to 0.20.
The best objective response was summarized with descriptive statistics. ORR with its associated 95% CI was calculated using normal approximation based on the best objective response judged by the IRRC.
The median PFS, median OS, and their associated probability of survival at each time point and 95% CI were estimated using the Kaplan‐Meier method. The time points were every 3 months, for a total of 15 months for PFS and 24 months for OS.
For each laboratory parameter, a patient was considered evaluable if ≥1 measurement was available on treatment. Laboratory toxicities were graded from 1 (least severe) to 5 per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03 and were summarized as “all grades” or “Grade ≥3.” For patients with multiple occurrences of a particular laboratory parameter during the study treatment period, the maximum (worst) grade was used.
Treatment‐emergent adverse events were adverse events reported between the first study treatment infusion and 30 days after the last one. TEAE were summarized with respect to frequency and intensity/severity, as graded by the worst NCI CTCAE version 4.03 criteria. All TEAE were coded using the Medical Dictionary for Regulatory Activities version 18.0.
2.8. Ethical considerations
The study was conducted in accordance with the principles of the Declaration of Helsinki and in compliance with all international and Japanese laws, regulations and guidelines. All aspects of the study were approved by the independent ethics committee and institutional review board. Patients were fully informed of the study and provided written consent. The study was registered with ClinicalTrials.gov with the identifier NCT01882868.
3. RESULTS
This study started when the first patient was enrolled in July 2013 and closed on the day of database lock in August 2015. Sixty‐two patients from 19 clinical sites in Japan were enrolled. Two patients were not evaluable for ORR: one had neither a target nor a non–target lesion, and one had only non–target lesions at baseline. The demographics and disease characteristics of the 60 evaluable patients appear in Table 1. Fifty evaluable patients (83.3%) had received prior bevacizumab.
Table 1.
Patient and disease characteristics at baseline
| Characteristic | Value (N = 60) |
|---|---|
| Age in years | |
| Mean (SD) | 61.6 (9.8) |
| Median | 62.5 |
| Min:Max | 39:78 |
| Age group in years (n [%]) | |
| <65 | 33 (55.0) |
| ≥65 but <75 | 24 (40.0) |
| ≥75 | 3 (5.0) |
| Sex (n [%]) | |
| Male | 34 (56.7) |
| Female | 26 (43.3) |
| Race (n [%]) | |
| Asian/Oriental | 60 (100) |
| Other | 0 (0) |
| ECOG performance status (n [%]) | |
| 0 | 40 (66.7) |
| 1 | 20 (33.3) |
| Body weight in kg | |
| Mean (SD) | 59.67 (10.48) |
| Median | 57.70 |
| Min:Max | 39.8:86.8 |
| Body surface area in m2 | |
| Mean (SD) | 1.626 (0.167) |
| Median | 1.613 |
| Min:Max | 1.29:1.99 |
| Prior hypertension (n [%]) | |
| Yes | 31 (51.7) |
| No | 29 (48.3) |
| UGT1A genotype (n [%])a | |
| Wild‐type | |
| *1/*1 | 20(60.6) |
| Heterozygous | |
| *1/*28 | 4 (12.1) |
| *1/*6 | 9 (27.3) |
| Complex heterozygous | |
| *6/*28 | 0 |
| Homozygous | |
| *28/*28 | 0 |
| *6/*6 | 0 |
| Primary tumor location (n [%]) | |
| Left (descending colon, sigmoid, rectosigmoid, rectum) | 45 (75.0) |
| Right (caecum, ascending colon, transverse colon) | 15 (25.0) |
| Histology type (n [%]) | |
| Adenocarcinoma | 60 (100) |
| Histopathology | |
| Well or moderately differentiated | 55 (91.7) |
| Undifferentiated or poorly differentiated | 2 (3.3) |
| Unknown | 3 (5.0) |
| Number of metastatic organs | |
| >1 | 36 (60.0) |
| 1 | 24 (40.0) |
| Liver metastasis only (n [%]) | |
| Yes | 9 (15.0) |
| No | 51 (85.0) |
| Prior bevacizumab (n [%]) | |
| Yes | 50 (83.3) |
| No | 10 (16.7) |
| Prior adjuvant therapy (n [%]) | |
| Yes | 8 (13.3) |
| No | 52 (86.7) |
| Time from 1st diagnosis to 1st study treatment administration (mo) | |
| Mean (SD) | 18.79 (14.74) |
| Median | 14.93 |
| Min:Max | 3.7:89.6 |
ECOG, Eastern Cooperative Oncology Group; UGT1A1, uridine‐5‐diphospho‐glucuronosyltransferase 1A1.
N = 33.
As of the final database lock, all patients had discontinued study treatment (Figure 1). The median number of treatment cycles received per patient was 8.0 (range: 1‐31), and the median duration of study treatment exposure was 21.8 weeks (range: 2‐73 weeks). The median relative dose intensities were 72% for aflibercept, 65% for irinotecan and 71% for 5‐FU.
3.1. Efficacy
Five of the 60 evaluable patients had PR and none had CR, resulting in an ORR of 8.3% (95% CI: 1.3%‐15.3%). In addition, 43 (71.7%) had stable disease, for an overall DCR of 80.0% (95% CI: 69.9%‐90.1%; see also Figure 2).
Figure 2.

Waterfall plot showing best objective response in 60 patients. CI, confidence interval; DCR, disease control rate; ORR, objective response rate; OS, overall survival; PD, progressive disease; PFS, progression free survival; PR, partial response; SD, stable disease
The other efficacy endpoints were evaluated for all 62 patients. The median PFS was 5.42 months (95% CI: 4.14‐6.70; Figure 3). As of the last tumor assessment, 47 (75.8%) patients had documented disease progression, 11 (17.7%) had died without disease progression and 4 (6.5%) had no disease progression. The median OS was 15.59 months (range: 11.20‐19.81 months; Figure 4). As of the final cutoff date, 21 (33.9%) patients had survived. The 2‐year survival rate was 28.2% (95% CI: 14.8%‐41.6%).
Figure 3.
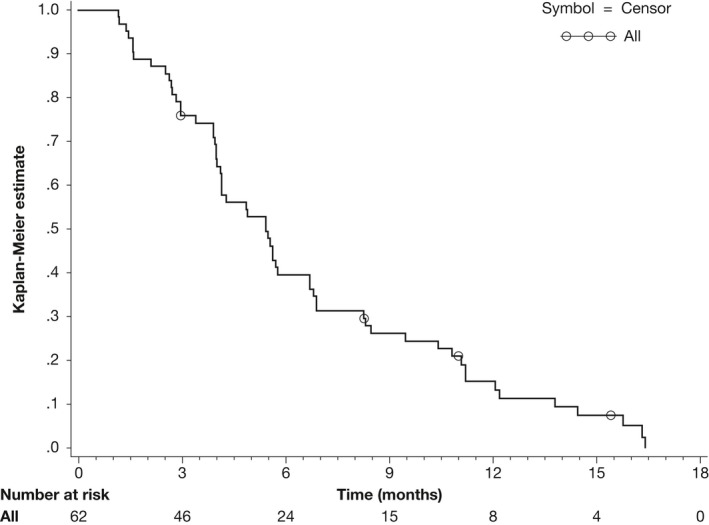
Kaplan‐Meier curve of progression free survival
Figure 4.
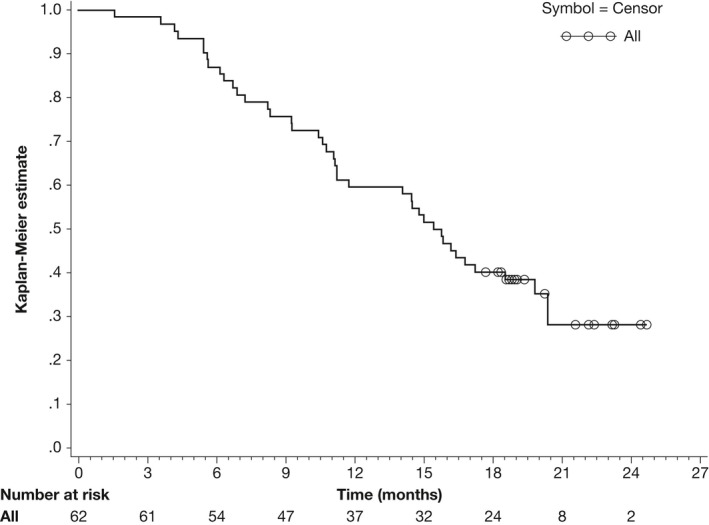
Kaplan‐Meier curve of overall survival
Of the 62 patients, 26 had mutant KRAS, 29 had wild‐type KRAS and 7 had unknown KRAS status. Forty‐seven patients had a left‐sided primary tumor and 15 had a right‐sided tumor. The ORR, PFS and OS based on KRAS status and primary tumor location are shown in Table S1 and Figures S1 and S2.
After discontinuing the study treatment, 51 (82.3%) patients received ≥1 further anti–cancer therapy, including 39 (62.9%) who received further biologics/small molecules (cetuximab: 9 [14.5%]; bevacizumab: 9 [14.5%]; regorafenib: 15 [24.2%]; panitumumab: 10 [16.1%]).
3.2. Safety
Hematological abnormality occurred in most patients: leukopenia (87.1% of patients), neutropenia (85.5%), anemia (82.3%) and thrombocytopenia (62.9%) (Table 2). Of all clinical laboratory abnormalities assessed, an abnormal creatinine level was the most common, affecting 60 patients (96.8%). Proteinuria occurred in 51 patients (82.3%); of these cases, 22 (43.1%) occurred in treatment cycle 1.
Table 2.
Clinical laboratory abnormalities
| Clinical laboratory tests | Abnormalities, n (%) | |
|---|---|---|
| All grades | Grade ≥3 | |
| Hematological tests (N = 62) | ||
| Leukopenia | 54 (87.1) | 19 (30.6) |
| Neutropenia | 53 (85.5) | 39 (62.9) |
| Anemia | 51 (82.3) | 1 (1.6) |
| Thrombocytopenia | 39 (62.9) | 0 |
| Liver and renal tests (N = 62) | ||
| ALT | 35 (56.5) | 3 (4.8) |
| AST | 38 (61.3) | 4 (6.5) |
| Alkaline phosphatase | 45 (72.6) | 3 (4.8) |
| Total bilirubin | 20 (32.3) | 2 (3.2) |
| Creatinine | 60 (96.8) | 1 (1.6) |
| Urine tests (N = 62) | ||
| Proteinuria | 51 (82.3) | 13 (21.0) |
| UPCR | ||
| ≤1a | 37 (59.7) | – |
| >1, ≤2 | 10 (16.1) | – |
| >2, ≤3 | 5 (8.1) | – |
| >3 | 10 (16.1) | – |
| Dipstick RBC | ||
| + | 16 (25.8) | – |
| ++ | 9 (14.5) | – |
| +++ | 7 (11.3) | – |
| ++++ | 1 (1.6) | – |
| Metabolism tests (N = 62) | ||
| Hypoglycemia | 1 (1.6) | 0 |
| Hyperglycemia | 49 (79.0) | 1 (1.6) |
| Hypoalbuminemia | 51 (82.3) | 2 (3.2) |
| Electrolyte tests (N = 62) | ||
| Hypocalcemia | 11 (17.7) | 0 |
| Hypercalcemia | 5 (8.1) | 0 |
| Hypokalemia | 20 (32.3) | 1 (1.6) |
| Hyperkalemia | 17 (27.4) | 0 |
| Hyponatremia | 25 (40.3) | 2 (3.2) |
| Hypernatremia | 5 (8.1) | 0 |
| Hypophosphatemia | 21 (33.9) | 4 (6.5) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; RBC, red blood cells; UPCR, urine protein‐creatinine ratio.
Includes UPCR in the normal range of 0 to <0.15.
All patients had ≥1 TEAE. Fifty‐six patients (90.3%) had Grade 3 or 4 TEAE; the most common TEAE were neutropenia, hypertension, diarrhea and decreased appetite (Table 3). Furthermore, 20 patients had ≥1 serious adverse event. Forty‐one (66.1%) patients died due to disease progression, all >30 days after the last study treatment administration. No patients died as a result of treatment.
Table 3.
Most commonly reported treatment‐emergent adverse events
| Primary system organ class | n (%) | |
|---|---|---|
| All grades | Grade ≥3 | |
| Preferred term | ||
| Any class | 62 (100) | 56 (90.3) |
| Blood and lymphatic system disorders | 46 (74.2) | 38 (61.3) |
| Neutropenia | 46 (74.2) | 38 (61.3) |
| Metabolism and nutrition disorders | 47 (75.8) | 10 (16.1) |
| Decreased appetite | 46 (74.2) | 8 (12.9) |
| Nervous system disorders | 18 (29.0) | 0 |
| Headache | 7 (11.3) | 0 |
| Vascular disorders | 33 (53.2) | 17 (27.4) |
| Hypertension | 29 (46.8) | 17 (27.4) |
| Respiratory, thoracic, and mediastinal disorders | 45 (72.6) | 2 (3.2) |
| Epistaxis | 25 (40.3) | 0 |
| Dysphonia | 18 (29.0) | 0 |
| Cough | 7 (11.3) | 0 |
| Hiccups | 7 (11.3) | 0 |
| Gastrointestinal disorders | 56 (90.3) | 18 (29.0) |
| Diarrhea | 42 (67.7) | 12 (19.4) |
| Nausea | 36 (58.1) | 2 (3.2) |
| Stomatitis | 29 (46.8) | 5 (8.1) |
| Vomiting | 17 (27.4) | 0 |
| Constipation | 10 (16.1) | 0 |
| Abdominal pain | 9 (14.5) | 0 |
| Skin and subcutaneous tissue disorders | 44 (71.0) | 1 (1.6) |
| Alopecia | 30 (48.4) | 0 |
| Palmar‐plantar erythrodysesthesia syndrome | 8 (12.9) | 0 |
| Rash | 7 (11.3) | 0 |
| Renal and urinary disorders | 19 (30.6) | 6 (9.7) |
| Proteinuria | 19 (30.6) | 6 (9.7) |
| General disorders and administration site conditions | 44 (71.0) | 4 (6.5) |
| Fatigue | 39 (62.9) | 3 (4.8) |
| Pyrexia | 13 (21.0) | 0 |
One patient was positive for anti–aflibercept antibodies at baseline (and positive in the neutralizing antibody assay); however, all subsequent samples from this patient were negative.
3.3. Pharmacokinetics
Population PK data (free and VEGF‐bound aflibercept) and non–compartmental PK data (irinotecan, SN‐38 and 5‐FU) appear in Table 4. Plasma concentrations for all 5 analytes are shown in Figure 5.
Table 4.
Summary of PK parameters in treatment cycle 1
| PK parametersa | Mean ± SD (CV%) |
|---|---|
| Free aflibercept | |
| C max, μg/mL | 73.2 ± 10.7 (15%) |
| AUC0‐14 d, μg·day/mL | 247 ± 41 (17%) |
| Clearance, L/d | 0.805 ± 0.178 (22%) |
| V ss, L | 6.20 ± 1.11 (18%) |
| VEGF‐bound aflibercept | |
| Clearance, L/d | 0.162 ± 0.014 (9%) |
| Irinotecan | |
| C max, ng/mL | 2220 ± 528 (24%) |
| AUC, ng·h/mL | 17 700 ± 6400 (36%) |
| t 1/2z, h | 5.19 ± 0.74 (14%) |
| Clearance, L/h/m2 | 11.1 ± 3.2 (28%) |
| V ss, L/m2 | 55.7 ± 16.0 (29%) |
| SN‐38b | |
| C max, ng/mL | 32.2 ± 11.4 (36%) |
| AUC, ng·h/mL | 341 ± 72 (21%) |
| t 1/2z, h | 10.3 ± 3.1 (30%) |
| 5‐FU | |
| CLss, L/h/m2 | 72.6 ± 40.4 (56%) |
AUC0‐14 d, area under the concentration vs time curve 0‐14 d post start of infusion; CLss, clearance at steady state; C max, maximum plasma concentration observed; CV%, coefficient of variation; 5‐FU, 5‐fluorouracil; PK, pharmacokinetics; SD, standard deviation; t 1/2z, terminal elimination half‐life; V ss, volume of distribution at steady state.
N = 62 for free and vascular endothelial growth factor‐bound aflibercept. N = 10 for irinotecan, SN‐38 and 5‐FU, for which a non–compartmental PK analysis was performed after a 1‐h infusion of aflibercept (4 mg/kg).
Represented 3% of irinotecan exposure.
Figure 5.
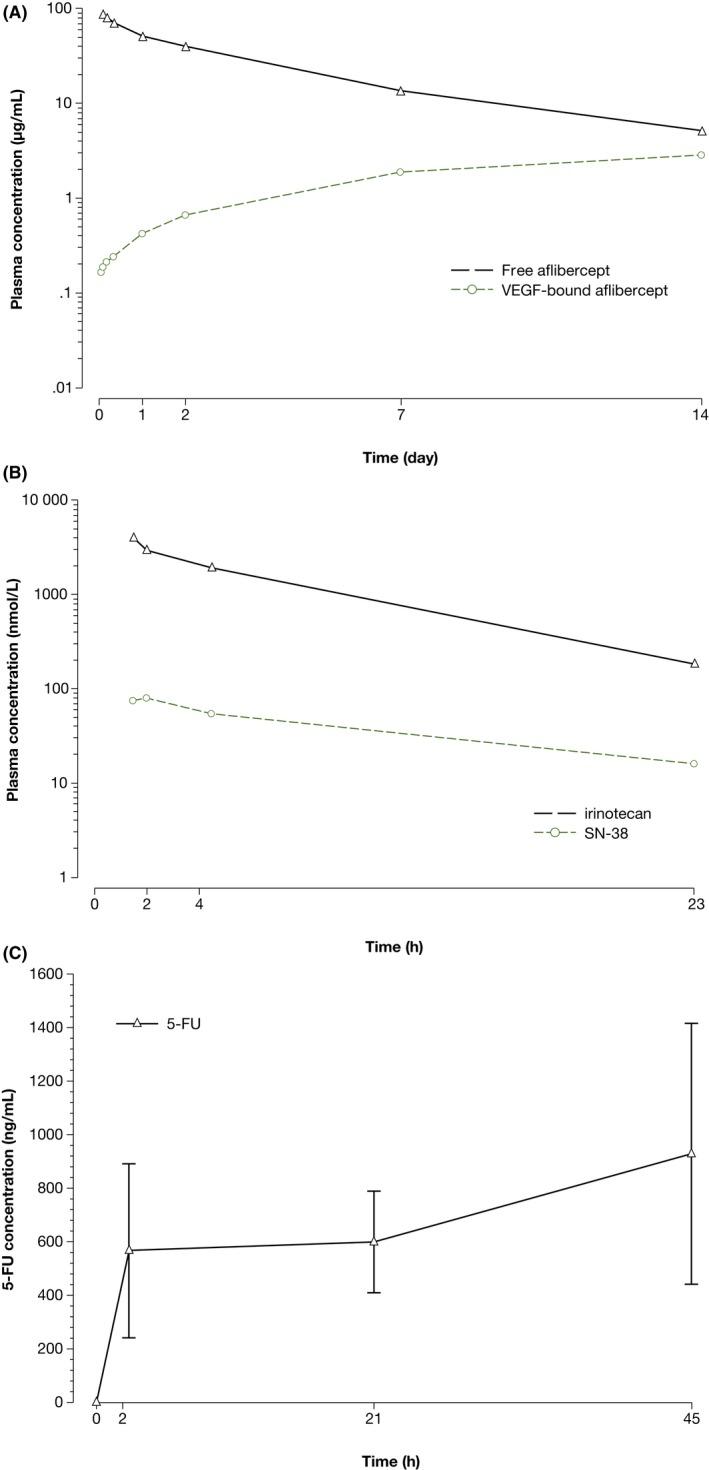
Plasma concentrations of study drugs over time in treatment cycle 1. Blood samples were collected from 10 patients before treatment cycle 1 and at various time points throughout cycle 1. Plasma concentrations of (A) free and vascular endothelial growth factor (VEGF)‐bound aflibercept were determined using validated enzyme‐linked immunosorbent assays. Plasma concentrations of (B) irinotecan, SN‐38 and (C) 5‐fluorouracil (5‐FU) were determined using validated electrospray liquid chromatography tandem mass spectrometry
4. DISCUSSION
This study examined the efficacy, safety and PK of aflibercept plus FOLFIRI in Japanese patients with mCRC that was refractory or intolerant to a first‐line oxaliplatin‐containing regimen. The primary endpoint, ORR by the IRRC, was 8.3% (95% CI: 1.3%‐15.3%).
The ORR was lower than that in the VELOUR study (19.8%; 95% CI: 16.4%‐23.2%). Possible reasons for this discrepancy are differences between the study populations and the limited size of our study. Consistent with a lower ORR, the PFS in the current study was slightly shorter than that in the VELOUR study: 5.42 months (95% CI: 4.14‐6.70) vs 6.90 months (6.51‐7.20) overall.12 Based on these findings and the DCR of 80.0% (95% CI: 69.9%‐90.1%), adding aflibercept to FOLFIRI yielded results consistent with those of the VELOUR study. The ORR and PFS were also comparable to those in the Asia‐Pacific study13 (ORR = 8.3% [95% CI: 1.3%‐15.3%] vs 18% [13.3%‐23.5%] and PFS = 5.42 months [4.14‐6.70] vs 6.93 months [6.045‐7.655]).
The median OS of 15.59 months (95% CI: 11.20‐19.81) was similar to the median OS in both the VELOUR study12 (13.50 months; 95% CI: 12.52‐14.95) and the Asia‐Pacific study13 (14.59 months; 95% CI: 13.18‐16.46), with overlapping 95% CI. Neither the KRAS oncogene nor primary tumor sidedness significantly affected ORR, PFS or OS.
The safety profile was as expected of an anti–VEGF agent and was comparable to that observed in the VELOUR study. Together, the TEAE represent an enhancement of the toxicity profile associated with usage of FOLFIRI. The absence of a sample positive for anti–aflibercept antibodies post–treatment suggests that IV administration of 4 mg/kg aflibercept confers no immunogenicity in patients with mCRC.
The PK values of free aflibercept were comparable to those in previous studies for Chinese patients.18 Adding aflibercept to FOLFIRI did not significantly affect the PK of irinotecan, SN‐38 or 5‐fluorouracil: the PK values of these FOLFIRI components were comparable between the current study, in which aflibercept was added, and published studies in which it was not. For example, the clearance of irinotecan in the current study was similar to the values in the Gupta et al19 study and in the Satoh et al20 study. The AUC for irinotecan and SN‐38 overlaps19 or is slightly high.20 The clearance of 5‐FU in our study had a relatively large coefficient of variance (56%), consistent with studies showing that 5‐FU plasma clearance is widely variable among patients (reviewed by Lee et al21). In previous population pharmacokinetic analyses, no clinically relevant drug‐drug interactions between aflibercept and irinotecan or fluorouracil were found.22 Likewise, no significant drug‐drug interactions were found in the current study.
A limitation of this study is its small sample size compared to that of the VELOUR study. In addition, unlike the VELOUR study, the current study had no control arm, so ability to compare the ORR results in the 2 studies is limited. Studies containing larger numbers of Japanese patients are needed to corroborate our findings.
In conclusion, adding aflibercept to FOLFIRI was shown to be beneficial and well‐tolerated in Japanese patients with mCRC.
CONFLICT OF INTEREST
TH received honoraria from Taiho, Chugai, Takeda, Yakult and Merck Serono, a consulting fee from NanoCarrier, and research funding from MSD, Ono, Sanofi, Daiichi Sankyo, Sumitomo Dainippon Pharma, Taiho, Teijin and NanoCarrier. NS received honoraria from Chugai and Eli Lilly, and research funding from Chugai, Eli Lilly, Dainippon Sumitomo, Taiho, MSD, Ono, Dai‐ichi Sankyo and Sanofi. TU received honoraria from Merck Serono, Taiho, Chugai and Takeda, and research funding from Sanofi. KY received honoraria from Chugai, Takeda, Yakult, Daiichi‐sankyo, Merck Serono, Bristol, Bayer, Eli Lilly and Taiho, and research funding from BMS and Sanofi. HF received research funding from Sanofi. ST received honoraria from Asahikasei, and research funding from Merck Serono, Ono and Sanofi. YK received honoraria from BMS and Sanofi, and research funding from Eli‐Lilly, BMS and Sanofi. TE received honoraria from Eli Lilly, and research funding from Boehringer, Daiichi‐Sankyo, Dainippon Sumitomo, Eli Lilly, Merck Serono, MSD, Novartis, Ono and Taiho, and a scholarship from Ono. EO received honoraria from Bayer, Chugai, Eli Lilly, Merck Serono, Taiho, Takeda and Yakult. TY received honoraria from Chugai, Eli Lilly, Merck Serono, Sanofi and Taiho, and research funding from Boehringer Ingelheim, Chugai, Dainippon Sumitomo, GlaxoSmithKline, MSD, Novartis and Sanofi. The affiliated medical institutions of physician authors received study funding from Sanofi. TS, YS, SZL and CB are employees of Sanofi. Funding for this research was provided by Sanofi. Because Sanofi is the company that initiated the clinical trial, the company employees were involved in planning the study and analyzing the data.
Supporting information
ACKNOWLEDGMENTS
The authors thank all patient participants. This study was funded by Sanofi and medically supervised by Ichinosuke Hyodo (Tsukuba University) and Hiromichi Suzuki (Musashino Tokushukai Hospital). Independent monitoring of safety and efficacy was provided by the Efficacy and Safety Evaluation Committee (Hiroyuki Uetake, Tokyo Medical and Dental University; Yoshihiko Tomita, Niigata University; and Ichiei Narita, Niigata University). Writing assistance was provided by Michelle L. Jones, PhD, and funded by Sanofi.
Denda T, Sakai D, Hamaguchi T, et al. Phase II trial of aflibercept with FOLFIRI as a second‐line treatment for Japanese patients with metastatic colorectal cancer. Cancer Sci. 2019;110:1032–1043. 10.1111/cas.13943
REFERENCES
- 1. The Global Cancer Observatory, International Agency for Research on Cancer. September 2018. http://gco.iarc.fr/today/data/factsheets/cancers/10_8_9-Colorectum-fact-sheet.pdf. Accessed October 18, 2018.
- 2. The Global Cancer Observatory, International Agency for Research on Cancer. September 2018. http://gco.iarc.fr/today/data/factsheets/populations/392-japan-fact-sheets.pdf. Accessed October 18, 2018.
- 3. Center for Cancer Control and Information Services, National Cancer Center Japan. Projected Cancer Statistics, 2018. https://ganjoho.jp/en/public/statistics/short_pred.html. Accessed October 23, 2018.
- 4. American Cancer Society, Inc. What are the survival rates for colorectal cancer, by stage? http://www.cancer.org/cancer/colonandrectumcancer/detailedguide/colorectal-cancer-survival-rates. Accessed October 18, 2018.
- 5. Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995;55:3964‐3968. [PubMed] [Google Scholar]
- 6. Des Guetz G, Uzzan B, Nicolas P, et al. Microvessel density and VEGF expression are prognostic factors in colorectal cancer. Meta‐analysis of the literature. Br J Cancer. 2006;94:1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giantonio BJ, Catalano PJ, Meropol NJ, et al. Bevacizumab in combination with oxaliplatin, fluorouracil, and leucovorin (FOLFOX4) for previously treated metastatic colorectal cancer: results from the Eastern Cooperative Oncology Group Study E3200. J Clin Oncol. 2007;25:1539‐1544. [DOI] [PubMed] [Google Scholar]
- 8. Loupakis F, Cremolini C, Masi G, et al. Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer. N Engl J Med. 2014;371:1609‐1618. [DOI] [PubMed] [Google Scholar]
- 9. Cremolini C, Loupakis F, Antoniotti C, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first‐line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open‐label, phase 3 TRIBE study. Lancet Oncol. 2015;16:1306‐1315. [DOI] [PubMed] [Google Scholar]
- 10. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335‐2342. [DOI] [PubMed] [Google Scholar]
- 11. Regeneron . Our history. https://www.regeneron.com/history. Accessed September 18, 2017.
- 12. Van Cutsem E, Tabernero J, Lakomy R, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin‐based regimen. J Clin Oncol. 2012;30:3499‐3506. [DOI] [PubMed] [Google Scholar]
- 13. Li J, Xu R, Qin S, et al. Aflibercept plus FOLFIRI in Asian patients with pretreated metastatic colorectal cancer: a randomized phase III study. Future Oncol. 2018;14:2031‐2044. [DOI] [PubMed] [Google Scholar]
- 14. Yoshino T, Yamazaki K, Yamaguchi K, et al. A phase I study of intravenous aflibercept with FOLFIRI in Japanese patients with previously treated metastatic colorectal cancer. Invest New Drugs. 2013;31:910‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muro K, Boku N, Shimada Y, et al. Irinotecan plus S‐1 (IRIS) versus fluorouracil and folinic acid plus irinotecan (FOLFIRI) as second‐line chemotherapy for metastatic colorectal cancer: a randomised phase 2/3 non‐inferiority study (FIRIS study). Lancet Oncol. 2010;11:853‐860. [DOI] [PubMed] [Google Scholar]
- 16. Sobrero AF, Peeters M, Price TJ, et al. Final results from study 181: randomized phase III study of FOLFIRI with or without panitumumab (pmab) for the treatment of second‐line metastatic colorectal cancer (mCRC). J Clin Oncol. 2012;28(4_Suppl):387. [Google Scholar]
- 17. Tournigand C, André T, Achille E, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229‐237. [DOI] [PubMed] [Google Scholar]
- 18. Xu J, Li Y, Sun X, et al. A phase I and pharmacokinetic study of afilbercept with FOLFIRI: comparison of Chinese and Caucasian populations. Invest New Drugs. 2017;35:463‐470. [DOI] [PubMed] [Google Scholar]
- 19. Gupta E, Mick R, Ramirez J, et al. Pharmacokinetic and pharmacodynamic evaluation of the topoisomerase inhibitor irinotecan in cancer patients. J Clin Oncol. 1997;15:1502‐1510. [DOI] [PubMed] [Google Scholar]
- 20. Satoh T, Yasui H, Muro K, et al. Pharmacokinetic assessment of irinotecan, SN‐38, and SN‐38‐glucuronide: a substudy of the FIRIS study. Anticancer Res. 2013;33:3845‐3854. [PubMed] [Google Scholar]
- 21. Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5‐fluorouracil. Cancer Chemother Pharmacol. 2016;78:447‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sanofi‐Aventis . Highlights of prescribing information, ZALTRAP® (ziv‐aflibercept); 2012. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125418s000lbl.pdf. Accessed October 19, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials