

Abstract
Radiotherapy has been widely used for the clinical management of esophageal squamous cell carcinoma. However, radioresistance remains a serious concern that prevents the efficacy of esophageal squamous cell carcinoma (ESCC) radiotherapy. REV7, the structural subunit of eukaryotic DNA polymerase ζ, has multiple functions in bypassing DNA damage and modulating mitotic arrest in human cell lines. However, the expression and molecular function of REV7 in ESCC progression remains unclear. In this study, we first examined the expression of REV7 in clinical ESCC samples, and we found higher expression of REV7 in ESCC tissues compared to matched adjacent or normal tissues. Knockdown of REV7 resulted in decreased colony formation and increased apoptosis in irradiated Eca‐109 and TE‐1 cells coupled with decreased tumor weight in a xenograft nude mouse model postirradiation. Conversely, overexpression of REV7 resulted in radioresistance in vitro and in vivo. Moreover, silencing of REV7 induced increased reactive oxygen species levels postirradiation. Proteomic analysis of REV7‐interacting proteins revealed that REV7 interacted with peroxiredoxin 2 (PRDX2), a well‐known antioxidant protein. Existence of REV7‐PRDX2 complex and its augmentation postirradiation were further validated by immunoprecipitation and immunofluorescence assays. REV7 knockdown significantly disrupted the presence of nuclear PRDX2 postirradiation, which resulted in oxidative stress. REV7‐PRDX2 complex also assembled onto DNA double‐strand breaks, whereas REV7 knockdown evidently increased double‐strand breaks that were unmerged by PRDX2. Taken together, the present study sheds light on REV7‐modulated radiosensitivity through interacting with PRDX2, which provides a novel target for ESCC radiotherapy.
Keywords: DNA double‐strand breaks, PRDX2, radioresistance, reactive oxygen species, REV7
1. INTRODUCTION
Esophageal cancer is one of the most common malignant tumors worldwide,1 and mainly involves 2 pathological patterns: adenocarcinoma and squamous carcinoma. In China, over 90% of esophageal cancers are squamous cell carcinomas,2 with 5‐year overall survival (OS) rates after diagnosis of 20%‐40%.3, 4 Surgical resection and radiotherapy are the 2 main treatments for early‐stage esophageal squamous cell carcinoma (ESCC), while radiotherapy provides postoperational local tumor control. Moreover, radiotherapy is typically recommended for inoperable ESCC and for palliative treatment of some late‐phase ESCC. Maximizing survival benefits is an ongoing issue for clinicians. Radiotherapy dose and fractionation strategies have been applied in clinical trials, with some progress demonstrated: for instance, high‐dose (>50.4 Gy) radiotherapy tends to improve local control (LC) more than low‐dose (≤50.4 Gy), but improvement in OS has not been ascertained.5, 6 Modification of conventional radiotherapy including late‐course accelerated hyperfractionated radiotherapy results in some improvement of LC and OS;7, 8 however, the prognosis of ESCC remains unfavorable.
Radioresistance limits the efficacy of radiotherapy, which can be attributed to intrinsic genetic phenotype, microenvironment alteration or acquired resistance during fractionated radiotherapy. P53 mutation or dysfunction is a signature of ESCC,9, 10 leading to aggressive phenotypes11 and enhanced insensitivity to radiation.12 High expression of REV3, PRDX613, 14 enables ESCC to tolerate gene toxic or oxidative stress. It has been established that an altered cancer microenvironment also results in radioresistance: for example, HIF augmentation enables cancer cells to tolerate hypoxia and to induce aggressive malignant phenotypes.15 Upregulated GSH/thioredoxins/peroxiredoxins counteract excessive ROS production of cancer to maintain cell survival.16 Moreover, multiple DNA damage pathways are activated in response to radiation and emerging evidence of gene suppression has demonstrated a tumor‐specific improvement in clinical trials; for instance, CHK1 and WEE1 inhibitors are lethal in P53‐mutant cancers postirradiation,17, 18 PARP inhibition kills in BRCA1/2‐mutant cancers19 and ATM‐defective cancers are susceptible to DNA‐PK or ATR inhibition.20, 21 The feasibility of using targeted gene inhibitors in radiotherapy awaits more convincing proof. The profile of radioresistance remains quite obscure in ESCC, and clinical evidence and trials for radiosensitization of ESCC remain limited. Therefore, defining more novel mechanisms of radioresistance is vital in seeking better prognoses for ESCC.
Radiation‐induced genomic DNA damage requires emergent repair and re‐synthesis mechanisms, such as homologous recombination (HR) and nonhomologous end joining (NHEJ), to maintain chromosome homeostasis. As an indispensable complement to these processes, translesion DNA synthesis (TLS), classified as a type of post–replication repair, is able to function in emergent and severe DNA lesions. TLS mainly consists of processes that bring DNA polymerases to DNA damage sites, enabling the damaged DNA strands to extend and replicate at the cost of high‐rate error‐prone base repair.22, 23 Polymerase zeta (Polζ) mainly consists of REV7 and REV3, possessing high efficiency in extending DNA from a lesion site24, 25 while displaying lower fidelity in template replication.26, 27 Current studies have shown that REV7 interacts with multiple genes to implement its TLS function, as structurally REV7 matches the middle region of Rev3,28 which increases the catalytic activity of Pol ζ.29 REV7 also maps to an additional REV7‐binding domain in the C‐terminal region of Rev1.30 In addition, REV7 participates in the DNA repair pathway choice through modulation of DNA end‐resection31, 32 to promote NHEJ repair33 or cooperating with REV1 or REV3 to promote HR repair.34 Upregulation of REV7 has been reported in many human cancers, including glioblastoma, and nasopharyngeal, breast and colon cancer,35, 36, 37, 38 and is related to poor prognoses in late‐stage ovarian, colon cancer and B‐cell lymphoma treated with rituximab.37, 39, 40 REV7 knockdown (KD) is associated with radiosensitization35 or chemosensitization38, 39 of human cancer cells through overburdened DNA lesions. However, the role of REV7 in the radioresistance of ESCC is still largely unknown. Herein, we report that REV7 contributes to the radioresistance of ESCC, which is associated with recruitment of peroxiredoxin 2 (PRDX2).
2. MATERIALS AND METHODS
2.1. Tissue sample
A total of 102 ESCC samples and 52 matched adjacent normal esophagus tissues were collected from 102 patients. A total of 21 normal esophageal tissues were obtained from surgical resections of trauma patients. These tissues were obtained postoperatively between 2012 and 2015 from the Gastrointestinal Center, Jiangyin People's Hospital, Medical School of University of Southeast of China (Jiangyin, China), as reported previously.41 The tumor and normal tissues were diagnosed as carcinomas on the basis of pathological evidence: the histological features of the specimens were evaluated by 2 senior pathologists. All patients provided signed, informed consent for their tissue to be used for scientific research. Ethical approval for this study was obtained from Changzhou Cancer Hospital Affiliated to Soochow University. All experiments were performed in accordance with the relevant guidelines and regulations of Soochow University.
2.2. Cell culture and irradiation
Human ESCC cell lines Eca109 and TE‐1 were purchased from Shanghai Institute of Cell Research (Shanghai, China). All cells were cultured in DMEM supplemented with 10% FBS (Biological Industries, Kibbutz Beit‐Haemek, Israel) at 37°C in 5% CO2. The cells were exposed to a single dose of X‐rays using a linear accelerator (RadSource, Suwanee, GA, USA) at a dose rate of 1.15 Gy/min and 160 kV X‐ray energy. Xenografted tumors in nude mice were exposed to a linear accelerator (Varian, Palo Alto, CA, USA) and irradiated at a dose rate of 4 Gy/min and 6 MeV X‐ray energy.
2.3. Plasmid transfection
The RNAi targeting REV7 (shREV7), pcDNA3.1‐REV7, PRDX2 (shPRDX2) and their respective controls were chemically synthesized or purchased from GenePharma (Shanghai, China). Cells were seeded in 6‐well plates and subjected to transfection the next day with a mixture of plasmids and ExFect Transfection Reagent (Vazyme, Nanjing, China) according to the manufacturer's instructions. Detailed information regarding shREV7, shNC and shPRDX2 is as follows:
shREV7:(5′‐GGAGCTGAATCAGTATATCCA‐3′); shNC:(5′‐GTTCTCCGAACGTGTCACGT‐3′); shPRDX2:(5′‐TCCTCTTTATCATCGATGGCAACTCGAGTTGCCATCGATGATAAAGAGGTTTTTTC‐3′).
2.4. Tumor xenograft models
Forty male BALB/c nude mice (age, 6 weeks; gender, male; weight, 20 g) were obtained by SLAC Laboratory Animal Company (Shanghai, China) and were fed in a pathogen‐free animal facility at 24°C with access to distilled food and water. Mice were randomly assigned to groups (5 mice/group); 3 × 106 plasmid transfected cells were injected subcutaneously into the right posterior limb of nude mice, and the tumor volume was then measured. The tumors were measured using vernier calipers on weeks 1, 2, 3, 4, 5, 6, 7 and 8. Xenografted mice were irradiated at 7 weeks then killed at 8 weeks. The final volume of tumor tissues was measured and determined using the formula: Tumor volume (V): V (mm3) = length (mm) × width2(mm2)/2. Investigators were blinded to the treatment groups. All animal experiments were conducted with the approval of the Soochow University Institutional Committee for Animal Research and in conformity with national guidelines for the care and use of laboratory animals.
2.5. MTT assay
Planted cells (5000/well in 96‐well plate) were washed with PBS twice then incubated with MTT solution at a concentration of 5 mg/mL for 3 hours at 37°C. DMSO was added to dissolve the formazan crystals after the medium was aspirated. Finally, the absorbance at 570 nm (reference wavelength: 630 nm) was measured by Bio‐Tek (Suwanee, GA, USA).
2.6. DCFH assay and NAPDH assay
In the DCFH‐DA assay, a DCFH fluorescence probe (Beyotime, Nantong, China) was diluted into a concentration of 10 μL/10 mL using DMEM without FBS. In dark conditions, PBS washed cells were subjected to incubation with a diluted DCFH fluorescence probe (1 mL/well) for 30 minutes in a cell incubator without light, then digested with trypsin and resuspended in DMEM containing FBS for neutralizing trypsin. Cells were washed with PBS twice before detection by flow cytometric analysis at emission wavelengths of 519 nm. In the NAPDH assay, NADPH content was detected using an NADP/NADPH Kit (Biovision, Milpitas, CA, USA) according to the manufacturer's instructions.
2.7. Colony formation assay
Cells (200, 500, 1000, 2000 or 4000/well) were separately seeded into 6‐well plates then exposed to 0, 2, 4, 6 or 8 Gy X‐ray irradiation subsequent to plating. After 15 days, the cells were stained with crystal violet. The colonies consisting of more than 40 cells were counted. The survival fraction was calculated as the fraction of colonies divided by that of the control group. The cell survival curves were then fitted using single hit multi‐target radiobiological models.
2.8. Immunohistochemistry
All tissues were fixed in 10% formalin, embedded in paraffin, and cut into 5‐μm sections. The sections were deparaffinized in xylene and rehydrated in graded concentrations of ethyl alcohol. Antigen retrieval was performed by heating the sections in a microwave oven for 10 minutes with .01 mol/L citrate buffer (pH 6.0). Endogenous peroxidases and nonspecific reactions were blocked with 3% hydrogen peroxide and 5% BSA for 30 minutes, respectively. The sections were then incubated with an anti–REV7 antibody (Abcam, Cambridge, UK) at a dilution of 1:100 overnight. Labeling for REV7 was detected using biotinylated secondary antibodies, visualized with diaminobenzidine substrate (Sigma‐Aldrich, St Louis, MO, USA), and counterstained with hematoxylin (Sigma‐Aldrich). All steps were performed at room temperature. As a negative control, PBS was used in the absence of the primary antibody, confirming the specificity of this antibody. Immunostained slides were evaluated by 2 independent observers using light microscopy in a blinded manner. Immunohistochemistry (IHC) images were taken using an Olympus microscope. aScores were evaluated in a range of 0‐3 points at intervals of .2 points. Counts of IHC staining (Ki67 and CD31) were performed in images at a magnification of ×20, and cases were regarded as positive only if both observers independently defined them as such.
2.9. Flow cytometric analysis of cell apoptosis
An annexin V/7‐aminoactinomycin D (7‐AAD) Apoptosis Kit (BD Biosciences, Franklin Lakes, NJ, USA) was used to evaluate apoptosis. The prepared cells were stained with Annexin V/7‐AAD for 30 minutes. The results were analyzed using a FACSCalibur System with ModFit's LT software (Becton Dickinson, Franklin Lakes, NJ, USA).
2.10. Immunofluorescence
Cells plated on glass slides in 6‐well plates were fixed in 4% paraformaldehyde for 10 minutes, and then 1% Triton X‐100 (Sigma‐Aldrich) was added to increase membrane permeability for 10 minutes. Next, 5% BSA was added to block nonspecific protein binding sites for 1 hour. The glass slides were incubated with anti–REV7 mAb (Abcam), anti–γH2AX mAb (Abcam) and/or anti–PRDX2 mAb (Abnova, Taipei, Taiwan) at a dilution of 1:200 overnight in a humidified chamber. The secondary antibody for REV7 and γH2AX was Cy3‐labeled goat anti–rabbit IgG (Beyotime), and the secondary antibody for PRDX2 was AlexaFluor 488‐labeled goat anti–mouse IgG (Beyotime). Both antibodies were used at a dilution of 1:200 for 1 hour, and the corresponding red or green fluorescence was detected at emission wavelengths of 519 or 570 nm by laser scanning confocal microscope (PerkinElmer, Waltham, MA, USA), respectively.
2.11. Immunoprecipitation
Cells were cultured in 10‐cm dishes, harvested at 90% cell density with 500 μL RIPA lysis buffer with PMSF per dish, incubated for 20 minutes on ice, and centrifuged at 13 000 g for 5 minutes. Primary antibody was added at 20 μg/mL into the centrifuged protein solution, and the dishes were incubated overnight with gentle rocking. Resuspended Protein A + G agarose (Beyotime) was added into the solution at 40 μL/mL, and the cells were incubated with gentle rocking at 4°C for 3 hours and then centrifuged at 1000 g for 5 minutes. The precipitate was resuspended and repeatedly washed with RIPA lysis buffer at 1.0 mL/assay 6 times. A volume of 40 μL SDS loading buffer (1×) was added to detach the immunoprecipitated proteins. As a negative control, rabbit IgG for REV7 (Abcam) or mice IgG (Beyotime) for PRDX2 (Abnova) was used at 20 μg/mL in the absence of the primary antibody, confirming the specificity of this antibody.
2.12. Western blotting
The proteins in the lysates were resuspended using SDS‐PAGE electrophoresis and transferred to a nitrocellulose membrane, which was then blocked with PBS/Tween‐20 containing 5% nonfat milk. The membrane was incubated with antibodies against REV7 (Abcam), PRDX2 (Abnova), GAPDH (Beyotime), Lamin B1 (Santa Cruz, CA, USA), Bcl‐2 and BAX (Cell Signaling Technology, Danvers, MA, USA). The protein‐bound antibodies were detected using an enhanced chemiluminescence (ECL) stable peroxide solution (Beyotime). All protein bands were visualized using a FluoroChem MI imaging system (AlphaInnotech, Santa Clara, CA, USA) at room temperature.
2.13. Statistical analysis
The data are expressed as the mean ± SEM from at least 3 independent experiments. Differences among samples were analyzed with one‐way ANOVA. P values of <.05 were considered statistically significant.
3. RESULTS
3.1. REV7 is overexpressed in esophageal squamous cell carcinoma clinical samples
REV7 has been reported to be overexpressed in many cancer cells35, 36, 37, 38 and REV7 overexpression is associated with resistance to ionizing radiation35 or chemotherapy.38, 39 To determine the expression of REV7 in ESCC, IHC analysis was performed on 102 ESCC tissue samples, 52 tumor adjacent tissues and 21 normal esophageal mucosa tissues of ESCC patients. As shown in Figure 1A,B, REV7 staining was stronger in ESCC tissues (2.2 ± .15) than in the tumor‐adjacent (1.4 ± .11) or normal (.8 ± .17) tissues. The expression of REV7 was pronounced in the nucleus of cancer cells. Thus, higher expression of REV7 in ESCC may be a hallmark of this malignancy.
Figure 1.
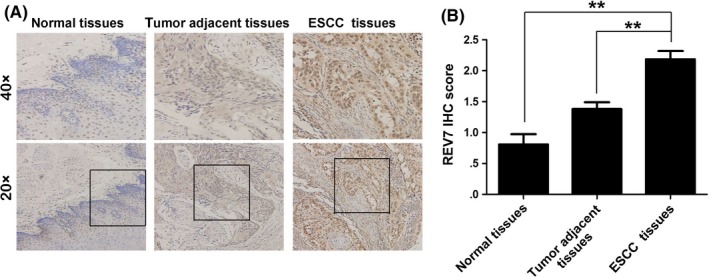
Higher expression of REV7 in esophageal squamous cell carcinoma (ESCC) samples. A, Representative immunohistochemistry (IHC) staining of REV7 expression in ESCC tissue, tumor‐adjacent tissue and normal esophageal tissue specimens (magnification 20× or 40×). B, Bar plot representing the IHC staining score of REV7 in ESCC tissues (n = 102), tumor‐adjacent tissues (n = 52) and normal esophageal tissues (n = 21). **P < .01
3.2. REV7 protects esophageal squamous cell carcinoma cells against irradiation‐induced apoptosis in vitro
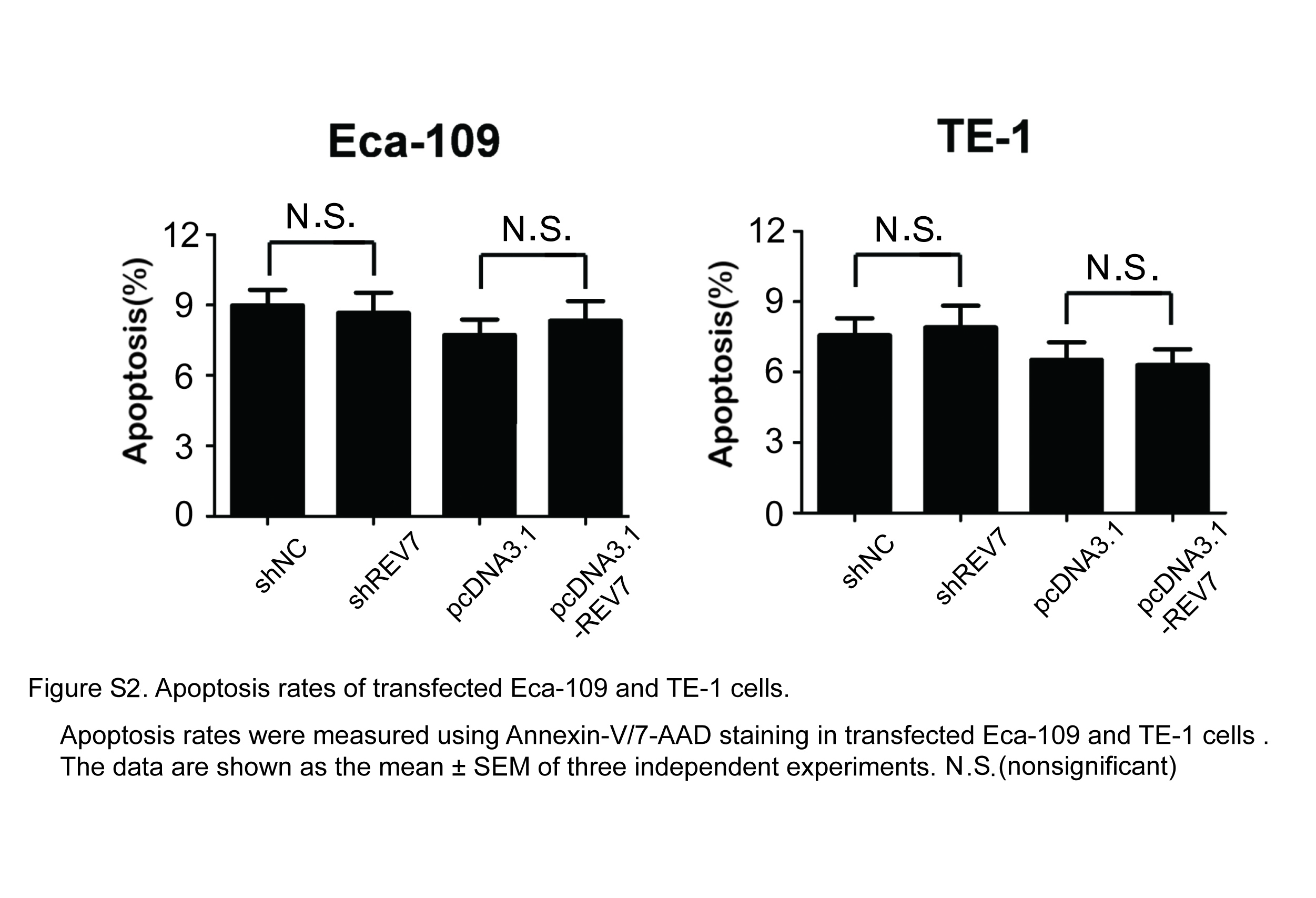
To determine whether REV7 is associated with radiosensitivity in ESCC cells, we performed knockdown and overexpression of REV7 in Eca109 and TE‐1 cell lines (Figure 2A). We first confirmed that REV7 knockdown (KD) or overexpression negatively impacted cell viability and migration ability (Figure S1). Next we noted that REV7 KD cells had a significant reduction in colony forming ability (SER = 1.38 for Eca109 cells, SER = 1.15 for TE‐1 cells) postirradiation (Figure 2B). In contrast, REV7‐overexpressing cells retained more colony formation ability than their corresponding control group (SER = .83 for Eca109 cells and SER = .87 for TE‐1 cells; Figure 2B). Because no significant transfection toxicity was observed on apoptosis (Figure S2), we further assayed the apoptotic rate in REV7‐overexpressing and REV7 KD cells in response to 4 Gy X‐ray irradiation. As shown in Figure 2C, a significant reduction in the number of apoptotic cells (P < .01 in Eca109, P < .05 in TE‐1 cells) was coupled with an increase in Bax and a decrease in the Bcl‐2 IHC level in REV7‐overexpressing cells (Figure 2D). In contrast, REV7 KD cells showed significantly elevated apoptosis in parallel with elevated Bax and decreased Bcl‐2 level (Figure 2C,D). These results indicate that REV7 overexpression protects ESCC cells against irradiation‐induced apoptosis, while REV7 KD sensitizes ESCC cells to irradiation.
Figure 2.
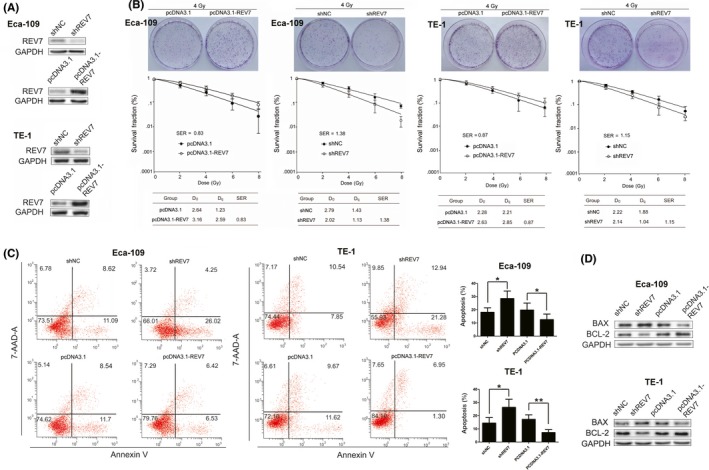
REV7 confers radioresistance on esophageal squamous cell carcinoma cells. A, Western blotting analysis of REV7 expression in Eca‐109 and TE‐1 cells transfected with shREV7, shNC, pcDNA3.1 or pcDNA3.1‐REV7. GAPDH served as an internal control. B, Colony formation of REV7‐overexpressing or REV7‐knockdown Eca‐109 and TE‐1 cells. Clonogenic survival curves were generated for Eca‐109 and TE‐1 cells that were stably transfected with the indicated vectors and were then exposed to 2, 4, 6 or 8 Gy X‐ray irradiation. The D0, Dq and sensitization enhancement ratio (SER) value of the corresponding groups is shown. The survival curve was derived from a multi‐target single‐hit model: SF = 1‐1‐exp(‐D/D0)n. D0 was defined as the dose that gave an average of 1 hit per target. Dq represents the repair of nonlethal injury, a higher Dq value means a higher dose is required to cause the death of cells. The SER was measured according to the multi‐target single‐hit model. C, Apoptosis rates were measured using Annexin‐V/7‐AAD staining in Eca‐109 and TE‐1 cells treated with the indicated vectors at 48 h after 4 Gy X‐ray irradiation. The data are shown as the mean ± SEM of 3 independent experiments. *P < .05, **P < .01. D, Western blotting analysis of Bcl‐2 and Bax expression for indicated group
3.3. REV7 confers esophageal squamous cell carcinoma resistance to irradiation in vivo
To test whether REV7 confers ESCC radioresistance in vivo, nude mice bearing tumor cell xenografts were used. Transfected Eca‐109 cells were subcutaneously injected into nude mice. As shown in Figure 3A,B, mice were exposed to 6 MV X‐rays at a dose of 20 Gy (dose rate 4 Gy/min) following confirmation of ESCC engraftment for 7 weeks. The xenograft tumors were harvested at the 8th week. As shown by Figure 3C,D, REV7 KD resulted in a significant decrease in tumor weight compared to the control group, whereas REV7 overexpression resulted in an increase in these parameters compared to the pcDNA3.1 group. IHC analysis showed that REV7 KD promoted apoptosis due to increased Bax and decreased Bcl‐2 expression (Figure 3E,F). Moreover, assessments of cell proliferation (Ki67 marker) and microvessel density (CD31 marker) showed no significant change between the indicated groups (Figure S3). Hence, the radiosensitivity modulated by REV7 seems to be independent of tumor proliferation and the microvessel formation process. Taken together, these findings indicate that REV7 contributes to resistance to irradiation through activation of apoptosis in vivo.
Figure 3.
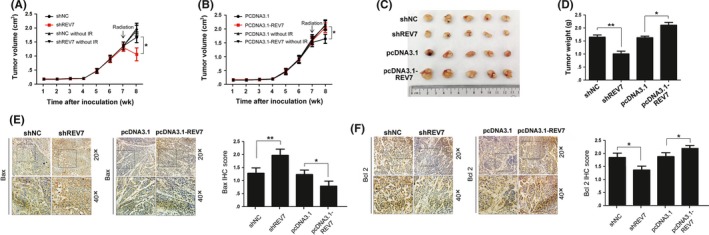
REV7 modulates the radioresistance of esophageal squamous cell carcinoma (ESCC) tumor xenografts. A‐B, Growth curve of Eca109 in subcutaneous tumor xenografts. C‐D, Representative images of the volume and weight of tumors originating from ESCC tumor xenografts in nude mice at 8 wk (1 wk postirradiation). The tumor weights are presented as the mean ± SEM of 3 independent experiments. **P < .01, *P < .05. E‐F, Representative immunohistochemistry (IHC) staining of Bax and Bcl2 in the indicated tumor xenografts. Scores or counts of IHC were evaluated by 2 independent observers in a blinded manner and are presented as the mean ± SEM of 3 independent experiments. **P < .01, *P < .05
3.4. REV7 protects esophageal squamous cell carcinoma cells against irradiation‐induced oxidative stress
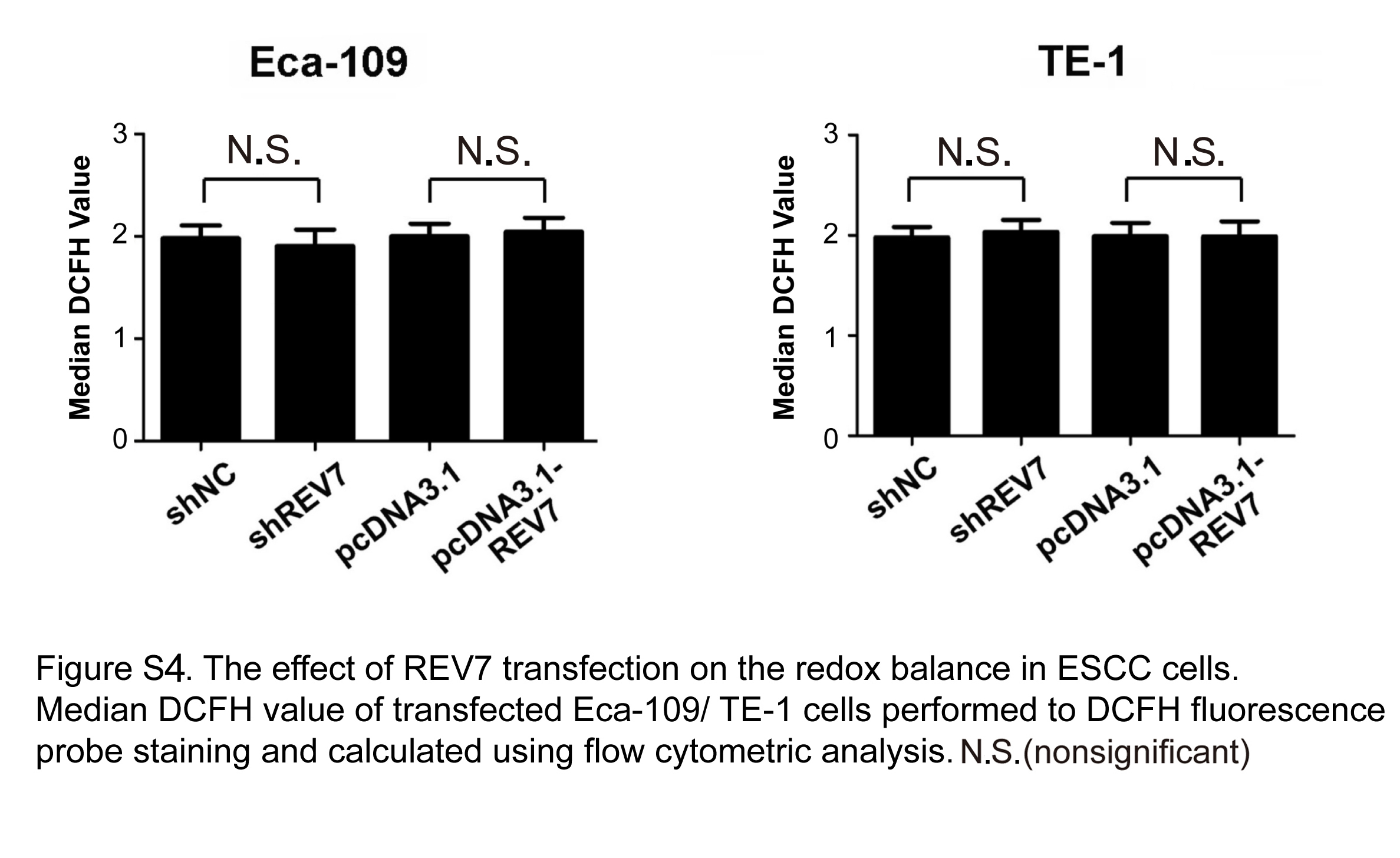
Ionizing radiation induces excess reactive oxygen species (ROS) production, leading to cell death through oxidative damaging effects to cell organelles and DNA structure.42 To test whether REV7 is involved in the modulation of ROS postirradiation, a ROS assay was performed and no significant ROS fluctuation was observed in purely transfected ESCC cells (Figure S4). However, ROS production was dramatically increased in irradiated REV7 KD cells (13.3 ± 2.1 vs 7.7 ± 1.8 of Eca109, 12.7 ± 1.9 vs 7.5 ± 1.8 of TE‐1 in Figure 4A). Meanwhile, REV7 KD reduced more than 30% of cellular NADPH in irradiated ESCC cells (Figure 4B). In contrast, REV7‐overexpression cells possessed decreased ROS levels (3.5 ± .4 vs 6.4 ± .7 of Eca109, 4.2 ± .3 vs 7.6 ± .9 of TE‐1 in Figure 4A), accompanied by 20%‐30% NADPH augmentation (Figure 4B). These results indicate that REV7 modulates the oxidation‐reduction balance postirradiation.
Figure 4.
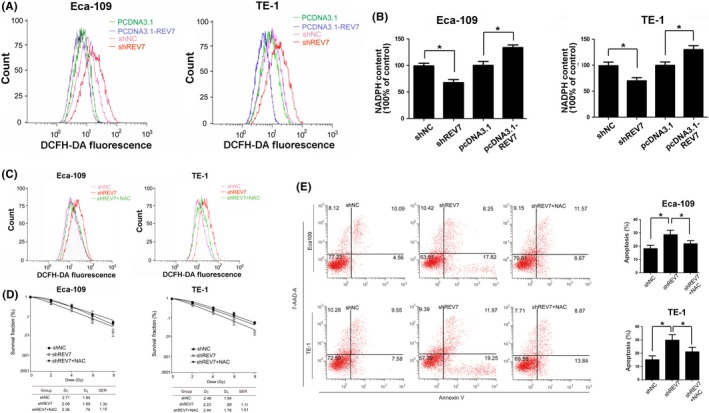
REV7 maintains the redox balance in irradiated esophageal squamous cell carcinoma cells. A, Representative curves of the reactive oxygen species (ROS) level, as indicated by DCFH fluorescence probe staining, were obtained using flow cytometric analysis of Eca109/TE‐1 cells 2 h after 4 Gy of X‐ray irradiation. B, Corresponding relative NADPH content was assayed by NADPH kit, presented as the mean ± SEM of 3 independent experiments. **P < .01,*P < .05. C, Representative curves of ROS level in REV7 KD and REV7 KD + NAC Eca109 cells. D, Colony formation of REV7 KD and REV7 KD + NAC Eca109 cells. E, Apoptosis rates were measured using Annexin‐V/7‐AAD staining in REV7 KD and REV7 KD + NAC Eca‐109 cells at 48 h after 4 Gy X‐ray irradiation. The data are shown as the mean ± SEM of 3 independent experiments. *P < .05
Furthermore, to test whether this oxidation disturbance in irradiated REV7 KD cells is responsible for radiosensitization, N‐acetyl cysteine (NAC) was applied to neutralize ROS. We found that pretreatment of 2 mmol/L NAC substantially restored the ROS level of irradiated REV7 KD cells to shNC (Figure 4C). Following that, as shown by Figure 4D,E, 2 mmol/L NAC decreased the apoptosis and increased colony formation in REV7 KD cells, which partially reversed the radiosensitization induced by REV7 KD. These results support that REV7 KD‐induced radiosensitization of ESCC cells is impacted by increased ROS level.
3.5. REV7 interacts with PRDX2 to modulate reactive oxygen species in esophageal squamous cell carcinoma cells
Interestingly, nuclear REV7 can modulate whole‐cell ROS levels, which implies that there may exist an interplay between REV7 and other antioxidant proteins. To that end, cell components immunoprecipitated by anti–REV7 antibody from whole‐cell lysates from Eca‐109 cells were subjected to proteomic analysis. A total of 141 proteins were identified in an untreated sample, and 140 proteins were identified in an irradiated sample. As a negative control, rabbit IgG was used, and 98 proteins were identified in the IgG sample. We recruited proteins that existed in both the untreated and the irradiated sample, while we rejected proteins that were present in the IgG sample. Twelve proteins meeting these criteria are shown in Figure 5A, of which only 7 proteins have a higher rank of containment in irradiated samples than untreated samples. These proteins include HNRH1, PRDX2, HNRPK, S10A9, DDX3X, IGHG1 and SPB12. Among them, PRDX2 was further investigated in greater detail for its well‐known antioxidant ability, as PRDX2 is abundantly expressed in cytoplasm, nucleus43 even in chromosomes44, and PRDX2 eliminates intracellular ROS through thioredoxin system.45 Next, immunofluorescence (IF) assays were used to visualize the co‐localization of REV7 and PRDX2 in ESCC cells. As shown in Figure 5B, co‐localization of PRDX2 and REV7 was observed in the nucleus of ESCC cells. Immunoprecipitation assays further validated the binding between PRDX2 and REV7 (Figure 5C). The proteins immunoprecipitated by beads coupled to anti–REV7 antibody from Eca‐109 cell lysate were purified, and then western blotting assays were performed using IgG as a negative control. As shown in Figure 5C, REV7 was able to bind PRDX2 (21.4 kDa). The REV7 protein (24.0 kDa) could also be detected in PRDX2‐immunoprecipitated proteins. The above results indicate that REV7 interacts with PRDX2 in ESCC cells.
Figure 5.
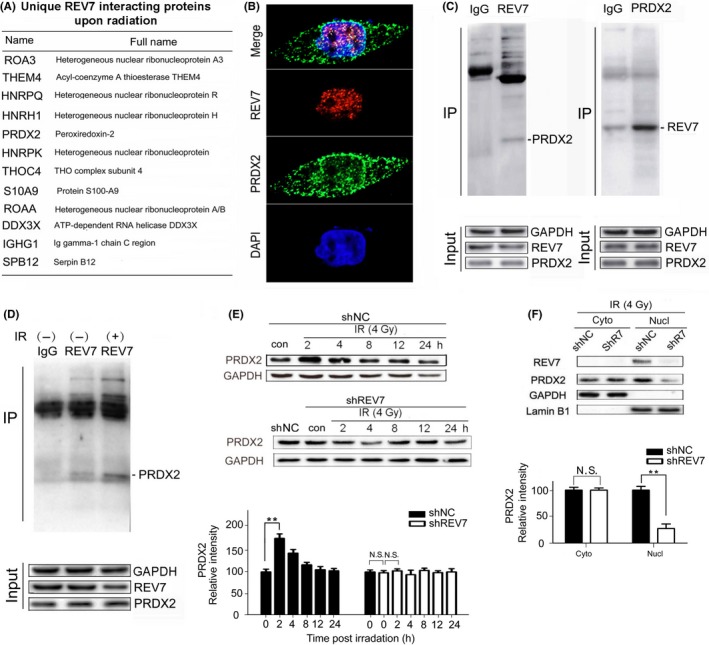
REV7 interacts with PRDX2 in the nucleus of esophageal squamous cell carcinoma cells. A, List of unique proteins that increased in percentage based on proteomic analysis at 2 h after 4 Gy X‐ray irradiation. B, Immunofluorescent staining of REV7 and PRDX2 in Eca‐109 cells (coexistence of REV7 and PRDX2 are indicated by the arrows). C, Confirmation of PRDX2/REV7 protein co‐immunoprecipitation by beads containing REV7/PRDX2 antibody using whole cell lysate from Eca‐109 cells, as revealed by western blot analysis. D, Immunoprecipitation assay by beads containing REV7 antibody in whole cell lysate from Eca‐109 cell with or without irradiation (2 h after 4 Gy X‐ray), as revealed by western blot analysis. E, Western blotting analysis of PRDX2 expression of shNC/shREV7 group postirradiation. Relative intensity of PRDX2 normalized to GAPDH is presented as the mean ± SEM of 3 independent experiments. **P < .01, N.S. nonsignificant. F, Cytosolic and nuclear fractions isolated from shNC/shREV7 Eca109 cells were assayed by western blotting for PRDX2. Relative intensity of PRDX2 normalized to GAPDH is presented as the mean ± SEM of 3 independent experiments. **P < .01, N.S. nonsignificant
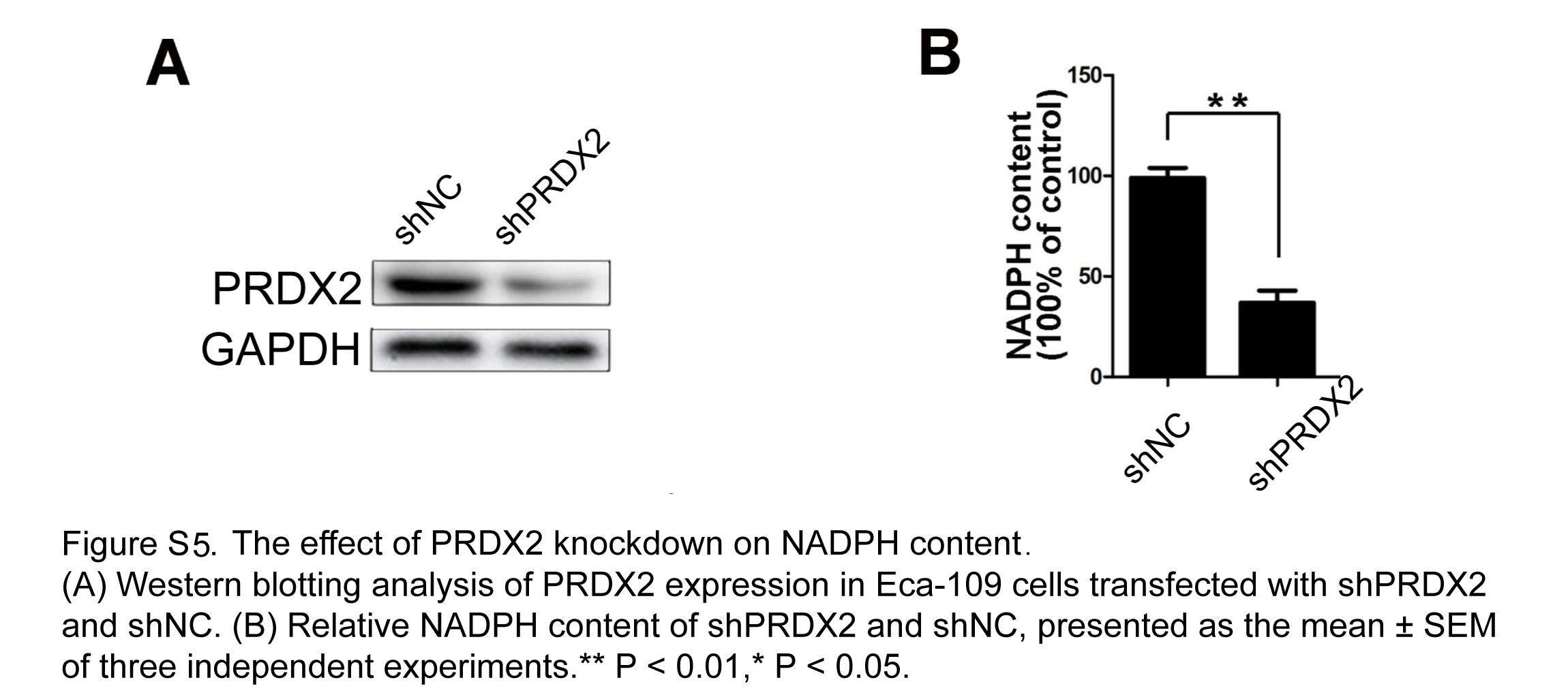
As exposure of 4 Gy X‐Ray was confirmed to efficiently increase REV7‐PRDX2 binding (Figure 5D), we wondered whether the binding increases for ROS modulation. Initially, we found an increase in the total PRDX2 postirradiation in shNC cells (Figure 5E), which dramatically peaked at 2 hours then gradually returned to baseline. In contrast, no significant change of total PRDX2 was observed in REV7 KD cells (Figure 5E). Furthermore, we extracted nuclear proteins from the cytoplasm and we found that REV7 KD reduced approximately 70% of the nuclear PRDX2 content of shNC cells, with no significant change in cytoplasm (Figure 5F); thus, the nuclear PRDX2 increase depends on REV7 presence. In NADPH assays, silencing PRDX2 exhausted NAPDH (more than 55% of shNC cells; Figure S5); thus, REV7 KD contributes to oxidative stress postirradiation through suppression of nuclear PRDX2.
3.6. REV7 interacts with PRDX2 for double‐strand break repair in esophageal squamous cell carcinoma cells
In nonirradiated cells, REV7 and PRDX2 initially distributed dispersedly and proportionally bound with each other (Figure 6A). Upon irradiation, we observed that REV7 co‐localized with nuclear PRDX2 in an assembled manner highly specifically (Figure 6A,B), whereas REV7 KD efficiently eliminated the indicated binding (Figure 6A,B). Because radiation‐induced double‐strand breaks (DSB) are considered lethal to tumor cells and are closely linked with the assembly of REV7,33 we hypothesized that PRDX2‐REV7 complex may be associated with DSB repair. As shown in Figure 6C, the majority of nuclear PRDX2 localization resembles DSB (marked by γH2AX) postirradiation, peaking at 2 hours then gradually returning to baseline at 24 hours following radiation, whereas REV7 KD significantly preserved more DSB (more than twice that of shNC cells), which was retained at 24 hours post–radiation (Figure 6C,D). Moreover, REV7 KD significantly increased the presence of DSB that were unmerged with nuclear PRDX2 postirradiation (Figure 6C,D), suggesting that PRDX2 is recruited by DSB through REV7. All these results indicated that the REV7‐PRDX2 axis is a process in DSB repair through which REV7 adapts PRDX2 to DSB sites in case of randomized radiation attack.
Figure 6.
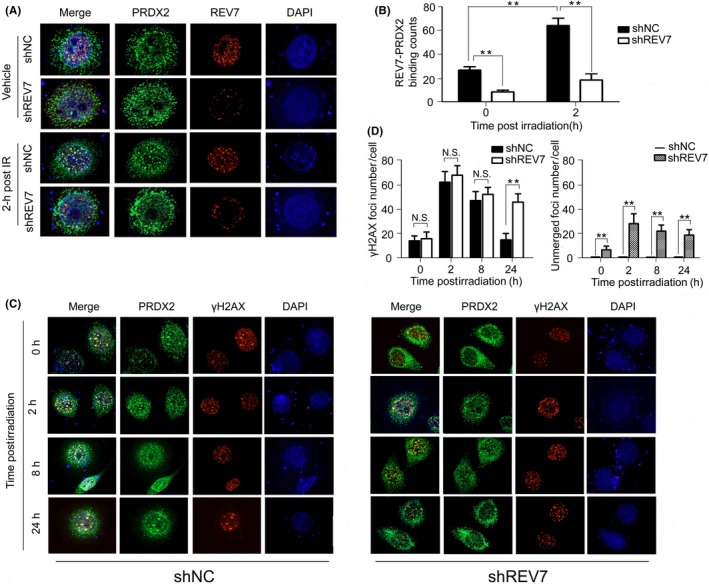
REV7‐PRDX2 complex is recruited onto double‐strand breaks (DSB) postirradiation. A‐B, Immunofluorescent staining of REV7 and PRDX2 in shNC/shREV7 Eca‐109 cells with or without irradiation (2 h after 4 Gy X‐ray). Bindings of REV7‐PRDX2 complex are observed by 2 independent observers in a blinded manner and are regarded as positive only if both observers counted independently and are presented as the mean ± SEM of 3 independent experiments. **P < .01. C‐D, Immunofluorescent staining of PRDX2 and γH2AX in shNC/shREV7 Eca‐109 cells with radiation (0, 2, 8 and 24 h after 4 Gy X‐ray). γH2AX foci merged or unmerged with nuclear PRDX2 were observed and calculated as indicated above. N.S., nonsignificant. ** P < .01, N.S. nonsignificant
4. DISCUSSION
In this study, we initially identified upregulation of REV7 in ESCC tissues. To confirm the function of this hallmark, REV7 KD was performed and found to sensitize ESCC to radiation through activation of apoptosis in ESCC cell lines and tumor xenografts, whereas REV7 overexpression went the opposite way. These results are consistent with accumulating evidence on the resistance of REV7 to irradiation or DNA‐damaging agents;35, 38, 39 however, the focus is the DNA repair process. REV7 functions range from TLS to NHEJ/HR repair. To launch TLS, REV7 together with REV3 extends the DNA from the inserted deoxynucleotides with aid from the DNA polymerase iota, which incorporates deoxynucleotides opposite DNA lesions.24 To implement DSB repair, recruitment of REV7 is achieved through the H2AX‐53BP1 chromatin pathway,33 controlling DNA repair by inhibiting 5′ end resection at the DSB31, 32 to promote NHEJ repair,32, 33 or cooperating with REV1 or REV3 to promote HR repair.34 Our work showed REV7 as immediately assembled onto DSB postirradiation (Figure 6A,C) while REV7 KD preserved elevated DSB (Figure 6C), which is associated with blockage of the indicated DNA repair processes.
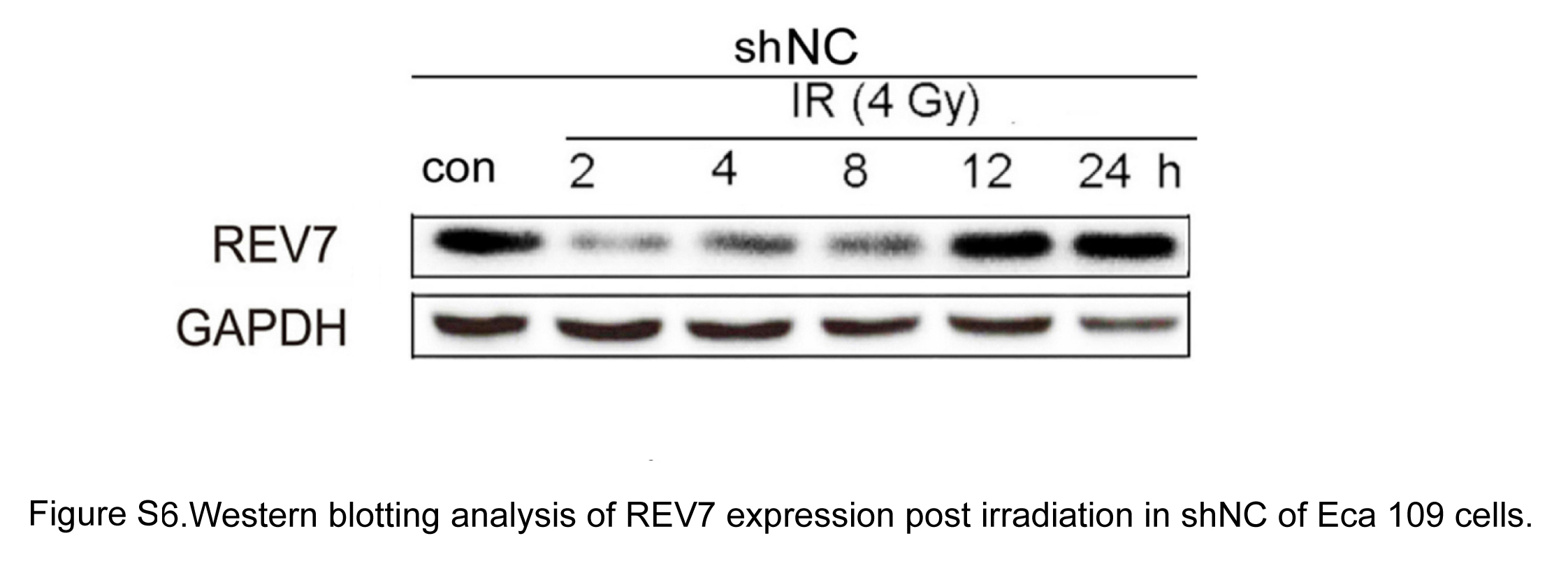
It is interesting that REV7 decreased at 2‐hours postirradiation (Figure S6), which is consistent with REV7 degradation post–UV treatment.46 Under irradiation conditions, REV7 intensely participates in both TLS and HR/NHEJ pathways.24, 33, 34 Studies have shown that a few proteins decrease, including helicases function in balancing TLS and HR repair at blocked replication forks or choice of post–replication repair pathway.47 It is conceivable that decreased REV7 is associated with DNA repair pathway transition, a safeguard mechanism to avoid TLS inducing excessive genetic mutagenesis. It has been reported that the error‐free or error‐prone manner of TLS depends on the DNA lesion species and polymerases involved,48 and replication‐blocking DSB leads to a prominent increase in TLS and an inhibition of HR through DNA lesion proximity.49 Therefore, accompanied by restoration of DNA lesions, overloaded REV7 tends to be switched then degraded. The mechanisms whereby REV7 is processed to transition and how REV7 is eliminated deserve further investigation.
Exposure to irradiation increases DNA lesions, dysfunction of organelles and intracellular free radicals.42 In the ROS assay, we found that REV7 KD increased ROS postirradiation while overexpression of REV7 decreased ROS (Figure 4A). In seeking answers for this, REV7 interacting proteins were identified by proteomic analysis and we regarded PRDX2 protein as a qualified candidate on the basis of its anti–oxidation function and wide distribution. Physiologically, PRDX2 possesses robust reductive ability through providing reductive electrons via thiol oxidation for the NADPH‐dependent thioredoxin (Trx)/Trx‐reductase system.45 Consistently, upregulated PRDX2 protected cells from H2O2‐induced oxidative stress,50 whereas PRDX2 knockdown was found to increase ROS and to impair cell cycle progression and proliferation.51 It has been well established that PRDX2 functions as a radio‐protector as PRDX2 expression increases immediately in rat skin after UV radiation52 and is highly inducible by therapeutic radiation in cancer cells,53 correspondingly null of PRDX2 enhances oxidized DNA damage in vivo or vitro.54 In our work, knockdown of PRDX2 induced over 50% NADPH loss (Figure S5) and REV7 KD suppressed nuclear PRDX2 augmentation (Figure 5E,F), hence contributing to oxidative stress. Moreover, increasing evidence suggests that PRDX2 is strongly connected with DNA integrity as PRDX2 is structurally enriched at chromosomes in mouse oocytes for chromosome organization and spindle assembly,44 and PRDX2 KD potently impaired DNA while other PRDX could not.43 Our work demonstrated that irradiation induces increased REV7‐PRDX2 complex (Figure 5D) and tends to promote its assembly onto DSB (Figure 6A), which implies a novel connection of PRDX2 and DNA lesions. That oxidative stress induces oxidized DNA damage55 has been well elucidated; however, there is a lack of proof regarding how DNA damage sites protect themselves from randomized oxidative attack. In this study, the existence of REV7‐PRDX2 complex seems to adapt PRDX2 to become a locally reductive microenvironment to guarantee the implementation of DNA repair. REV7‐PRDX2 complex acts to bridge DNA repair and anti–oxidation processes, which provides a novel notion of REV7‐modulated genomic homeostasis, and silencing REV7 showed promising potential in enhancing radiosensitization of ESCC.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest to this work.
Supporting information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gu C, Luo J, Lu X, et al. REV7 confers radioresistance of esophagus squamous cell carcinoma by recruiting PRDX2. Cancer Sci. 2019;110:962–972. 10.1111/cas.13946
Gu, Luo and Lu contributed equally to this study.
Contributor Information
Xifa Zhou, Email: zhouxifacz@sina.com.
Shuyu Zhang, Email: zhang.shuyu@hotmail.com.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Chung CS, Lee YC, Wu MS. Prevention strategies for esophageal cancer: perspectives of the East versus West. Best Pract Res Clin Gastroenterol. 2015;29:869‐883. [DOI] [PubMed] [Google Scholar]
- 3. Cunningham D, Allum WH, Stenning SP, et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N Engl J Med. 2006;355:11‐20. [DOI] [PubMed] [Google Scholar]
- 4. van Hagen P, Hulshof MC, Van Lanschot JJB, et al. Preoperative chemo‐radiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366:2074‐2084. [DOI] [PubMed] [Google Scholar]
- 5. Suh YG, Lee IJ, Koom WS, et al. High‐dose versus standard‐dose radiotherapy with concurrent chemotherapy in stages II–III esophageal cancer. Jpn J Clin Oncol. 2014;44:534‐540. [DOI] [PubMed] [Google Scholar]
- 6. He L, Allen PK, Potter A, et al. Re‐evaluating the optimal radiation dose for definitive chemoradiotherapy for esophageal squamous cell carcinoma. J Thorac Oncol. 2014;9:1398‐1405. [DOI] [PubMed] [Google Scholar]
- 7. Zhang YW, Chen L, Bai Y, Zheng X. Long‐term outcomes of late course accelerated hyper‐fractionated radiotherapy for localized esophageal carcinoma in Mainland China: a meta‐analysis. Dis Esophagus. 2011;24:495‐501. [DOI] [PubMed] [Google Scholar]
- 8. Wang JH, Lu XJ, Zhou J, Wang F. A randomized controlled trial of conventional fraction and late course accelerated hyperfraction three‐dimensional conformal radiotherapy for esophageal cancer. Cell Biochem Biophys. 2012;62:107‐112.9. [DOI] [PubMed] [Google Scholar]
- 9. Barnas C, Martel‐Planche G, Furukawa Y, Hollstein M, Montesano R, Hainaut P. Inactivation of the p53 protein in cell lines derived from human esophageal cancers. Int J Cancer. 1997;71:79‐87. [DOI] [PubMed] [Google Scholar]
- 10. Hollstein MC, Metcalf RA, Welsh JA, Montesano R, Harris CC. Frequent mutation of the P53 gene in human esophageal cancer. Proc Natl Acad Sci USA. 1990;87(24):9958‐9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim MP, Lozano G. Mutant p53 partners in crime. Cell Death Differ. 2018;25:161‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang N, Wang Y, Guo S, et al. Mutant TP53 G245C and R273H promote cellular malignancy in esophageal squamous cell carcinoma. BMC Cell Biol. 2018;19:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu X, Zou S, Zhou J, et al. REV3L, the catalytic subunit of DNA polymerase ζ, is involved in the progression and chemoresistance of esophageal squamous cell carcinoma. Oncol Rep. 2016;35:1664‐1670. [DOI] [PubMed] [Google Scholar]
- 14. He Y, Xu W, Xiao Y, et al. Overexpression of peroxiredoxin 6 (PRDX6) promotes the aggressive phenotypes of esophageal squamous cell carcinoma. J Cancer. 2018;9:3939‐3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393‐410. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y, Martin SG. Redox proteins and radiotherapy. Clin Oncol. 2014;26:289‐300. [DOI] [PubMed] [Google Scholar]
- 17. Bridges KA, Hirai H, Buser CA, et al. MK‐1775, a novel Wee1 kinase inhibitor, radiosensitizes p53‐defective human tumor cells. Clin Cancer Res. 2011;17:5638‐5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitchell JB, Choudhuri R, Fabre K, et al. In vitro and in vivo radiation sensitization of human tumor cells by a novel checkpoint kinase inhibitor, AZD7762. Clin Cancer Res. 2010;16:2076‐2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Biddlestone‐Thorpe L, Sajjad M, Rosenberg E, et al. ATM kinase inhibition preferentially sensitizes p53‐mutant glioma to ionizing radiation. Clin Cancer Res. 2013;19:3189‐3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Riabinska A, Daheim M, Herter‐Sprie GS, et al. Therapeutic targeting of a robust non‐oncogene addiction to PRKDC in ATM‐defective tumors. Sci Transl Med. 2013;5:189ra78. [DOI] [PubMed] [Google Scholar]
- 21. Reaper PM, Maccormick S, Charlton PA, et al. Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol. 2011;7:428‐430. [DOI] [PubMed] [Google Scholar]
- 22. Friedberg EC, Wagner R, Radman M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science. 2002;296(5573):1627‐1630. [DOI] [PubMed] [Google Scholar]
- 23. Lehmann AR. Translesion synthesis in mammalian cells. Exp Cell Res. 2006;312:2673‐2676. [DOI] [PubMed] [Google Scholar]
- 24. Johnson RE, Washington MT, Haracska L, Prakash S, Prakash L. Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature. 2000;406:1015‐1019. [DOI] [PubMed] [Google Scholar]
- 25. Haracska L, Prakash S, Prakash L. Yeast DNA polymerase zeta is an efficient extender of primer ends opposite from 7,8‐dihydro‐8‐Oxoguanine and O6‐methylguanine. Mol Cell Biol. 2003;23:1453‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakamoto AN, Stone JE, Kissling GE, McCulloch SD, Pavlov YI, Kunkel TA. Mutator alleles of yeast DNA polymerase zeta. DNA Repair (Amst). 2007;6:1829‐1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harfe BD, Jinks‐Robertson S. DNA polymerase zeta introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol Cell. 2000;6:1491‐1499. [DOI] [PubMed] [Google Scholar]
- 28. Hara K, Hashimoto H, Murakumo Y, et al. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem. 2010;285:12299‐12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nelson JR, Lawrence CW, Hinkle DC. Thymine‐thymine dimer bypass by yeast dna polymerase zeta. Science. 1996;272:1646‐1649. [DOI] [PubMed] [Google Scholar]
- 30. Kikuchi S, Hara K, Shimizu T, et al. Structural basis of recruitment of DNA polymerase ζ by interaction between REV1 and REV7 proteins. J Biol Chem. 2012;287:33847‐33852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simonetta M, de Krijger I, Serrat J, et al. H4K20me2 distinguishes pre‐replicative from post‐replicative chromatin to appropriately direct DNA repair pathway choice by 53BP1‐RIF1‐MAD2L2. Cell Cycle. 2018;17:124‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boersma V, Moatti N, Segura‐Bayona S, et al. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015;7553:537‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu G, Chapman JR, Brandsma I, et al. REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature. 2015;521:541‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma S, Hicks JK, Chute CL, et al. REV1 and polymerase ζ facilitate homologous recombination repair. Nucleic Acids Res. 2012;40:682‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao J, Cao S, Wang H, et al. Mitotic arrest deficient protein MAD2B is overexpressed in human glioma, with depletion enhancing sensitivity to ionizing radiation. J Clin Neurosci. 2011;18:827‐833. [DOI] [PubMed] [Google Scholar]
- 36. Yuan B, Gabrielson E. Increased expression of mitotic checkpoint genes in breast cancer cells with chromosomal instability. Clin Cancer Res. 2006;12:405‐410. [DOI] [PubMed] [Google Scholar]
- 37. Rimkus C, Janssen KP, Rosenberg R, Holzmann B, Siewert JR, Janssen KP. Expression of the mitotic checkpoint gene MAD2L2 has prognostic significance in colon cancer. Int J Cancer. 2007;120:207‐211. [DOI] [PubMed] [Google Scholar]
- 38. Cheung HW, Chun AC, Wang Q, et al. Inactivation of human MAD2B in nasopharyngeal carcinoma cells leads to chemosensitization to DNA‐damaging agents. Cancer Res. 2006;66:4357‐4367. [DOI] [PubMed] [Google Scholar]
- 39. Niimi K, Murakumo Y, Watanabe N, et al. Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci. 2014;105:545‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Okina S, Yanagisawa N, Yokoyama M, et al. High expression of REV7 is an independent prognostic indicator in patients with diffuse large B‐cell lymphoma treated with rituximab. Int J Hematol. 2015;102:662‐669. [DOI] [PubMed] [Google Scholar]
- 41. Luo J, Zhou X, Ge X, et al. Upregulation of Ying Yang 1 (YY1) suppresses esophageal squamous cell carcinoma development through heme oxygenase‐1. Cancer Sci. 2013;104:1544‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dayal R, Singh A, Pandey A, Mishra KP. Reactive oxygen species as mediator of tumor radiosensitivity. J Cancer Res Ther. 2014;10:811‐818. [DOI] [PubMed] [Google Scholar]
- 43. Lee KW, Lee DJ, Lee JY, Kang DH, Kwon J, Kang SW. Peroxiredoxin II restrains DNA damage‐induced death in cancer cells by positively regulating JNK‐dependent DNA repair. J Biol Chem. 2011;286:8394‐8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jeon HJ, Park YS, Cho DH, et al. Peroxiredoxins are required for spindle assembly, chromosome organization, and polarization in mouse oocytes. Biochem Biophys Res Commun. 2017;489:193‐199. [DOI] [PubMed] [Google Scholar]
- 45. Chae HZ, Oubrahim H, Park JW, Rhee SG, Chock PB. Protein glutathionylation in the regulation of peroxiredoxins: a family of thiol‐specific peroxidases that function as antioxidants, molecular chaperones, and signal modulators. Antioxid Redox Signal. 2012;16:506‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhat A, Qin Z, Wang G, et al. REV7, the regulatory subunit of Polζ, undergoes UV‐induced and Cul4‐dependent degradation. FEBS J. 2017;284:1790‐1803. [DOI] [PubMed] [Google Scholar]
- 47. Sommers JA, Suhasini AN, Brosh RM Jr. Protein degradation pathways regulate the functions of helicases in the DNA damage response and maintenance of genomic stability. Biomolecules. 2015;5:590‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Henrikus SS, van Oijen AM, Robinson A. Specialised DNA polymerases in Escherichia coli: roles within multiple pathways. Curr Genet. 2018;64:1189‐1196. [DOI] [PubMed] [Google Scholar]
- 49. Chrabaszcz É, Laureti L, Pagès V, et al. DNA lesions proximity modulates damage tolerance pathways in Escherichia coli. Nucleic Acids Res. 2018;46:4004‐4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stresing V, Baltziskueta E, Rubio N, et al. Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene. 2013;32:724‐735. [DOI] [PubMed] [Google Scholar]
- 51. Avitabile D, Ranieri D, Nicolussi A, et al. Peroxiredoxin 2 nuclear levels are regulated by circadian clock synchronization in human keratinocytes. Int J Biochem Cell Biol. 2014;53:24‐34. [DOI] [PubMed] [Google Scholar]
- 52. Lee SC, Chae HZ, Lee JE, et al. Peroxiredoxin is ubiquitously expressed in rat skin: isotype‐specific expression in the epidermis and hair follicle. J Invest Dermatol. 2000;115:1108‐1114. [DOI] [PubMed] [Google Scholar]
- 53. Smith‐Pearson PS, Kooshki M, Spitz DR, et al. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H(2)O(2). Free Radic Biol Med. 2008;45:1178‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kwon TH, Han YH, Hong SG, et al. Reactive oxygen species mediated DNA damage is essential for abnormal erythropoiesis in peroxiredoxin II(‐/‐) mice. Biochem Biophys Res Comm. 2012;424:189‐195. [DOI] [PubMed] [Google Scholar]
- 55. Kryston TB, Georgiev AB, Pissis P, Georgakilas AG. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res. 2011;711:193‐201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials