

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy associated with an extremely poor prognosis. Chemotherapy, such as gemcitabine (GEM), is the only treatment for PDAC patients who are not suitable for radical surgical treatment; however, its anti‐tumor efficacy is limited. In this study, we investigated the host immune system response in murine PDAC models undergoing GEM treatment. We found that PDAC tumor tissues were infiltrated with a substantial number of Gr‐1+ myeloid cells and had relatively small numbers of CD4+ and CD8+ cells. In addition, there were increased numbers of myeloid cells expressing CD11b+ and Gr‐1+ in peripheral blood. When mice with PDAC tumors in the intraperitoneal cavity or liver were treated with GEM, numbers of myeloid cells in tumor tissues and in peripheral blood decreased. In contrast, numbers of CD4+ or CD8+ cells increased. In peripheral blood, the numbers of CD8+ cells expressing interferon‐gamma (IFN‐γ) were higher in GEM‐treated mice than in untreated mice. In addition, GEM treatment in combination with myeloid cell depletion further prolonged the survival of PDAC mice. The gene expression profile of peripheral blood in myeloid cell‐depleted PDAC mice treated with GEM showed biological processes related to anti‐cancer immunity, such as natural killer cell‐mediated cytotoxicity, type I IFN signaling, and co‐stimulatory signaling for T cell activation. Thus, in PDAC murine models, GEM treatment was associated with an immune response consistent with an anti‐cancer effect, and depletion of myeloid‐lineage cells played an important role in enhancing anti‐cancer immunity associated with GEM treatment.
Keywords: anti‐cancer immunity, chemotherapy, Gr‐1, myeloid‐lineage cells, pancreatic cancer
Abbreviations
- 7‐AAD
7‐aminoactinomycin D
- APC
allophycocyanin
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- FITC
fluorescein isothiocyanate
- GEM
gemcitabine
- IFN‐γ
interferon‐gamma
- P/S
penicillin and streptomycin
- PBS
phosphate‐buffered saline
- PDAC
pancreatic ductal adenocarcinoma
- PE
phycoerythrin
- RPMI
Roswell Park Memorial Institute
- TICs
tumor‐infiltrating inflammatory cells
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a lethal malignancy with a mortality rate that is almost the same as the morbidity rate.1 A clinical reason for the extremely poor prognosis of PDAC is the difficulty of diagnosis in the early stages,2 when complete remission following surgical removal is possible.3 Invasion of PDAC into adjacent vessels or neurons4, 5 also contributes to the extremely poor prognosis of PDAC patients. In addition, patients on chemotherapeutic anti‐cancer agents6 fail to achieve complete remission and do not show a marked improvement in prognosis. Therefore, the pathology of PDAC needs to be better understood so that novel diagnostics and therapeutics that improve the prognosis can be developed.
The host immune system plays a major role in protecting the host from external invading pathogens and internal neoplasms. However, the immune system is composed of numerous mediators that have pro‐ or anti‐cancer effects.7 We previously reported that PDAC was associated with immune responses both in local tumor tissues and peripheral blood cells.8 However, natural immune responses in PDAC patients are inadequate in combatting cancer. This is evidenced by the extremely poor prognosis of PDAC patients even after chemotherapy treatment, which usually fails to yield a completely curative response.
In this study, we established intraperitoneal‐dissemination and liver‐metastasis PDAC mouse models. We observed Gr‐1‐positive myeloid cells in local PDAC tissue and peripheral blood. In intraperitoneal‐dissemination and liver‐metastasis murine models of PDAC, gemcitabine (GEM) treatment significantly prolonged survival times. In addition, treatment was associated with decreased numbers of myeloid cells and increased numbers of CD4+ and CD8+ T cells. Furthermore, GEM treatment combined with anti‐Gr‐1 antibody treatment further improved the prognosis, as indicated by the gene expression profiles of peripheral blood cells, which revealed the expression of genes associated with anti‐cancer immune responses.
2. MATERIALS AND METHODS
2.1. Cell line
The PDAC cell line, PAN02 (NCI‐Frederick, Frederick, MD, USA), was cultured and expanded in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Carlsbad, CA, USA) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) and 100 μg/mL penicillin and streptomycin (P/S; Life Technologies). The cells were cultured at 37°C in an incubator with 5% CO2.
2.2. Murine PDAC models and treatments
C57BL/6J mice (Charles River Laboratories, Yokohama, Japan) were injected with 1 × 106 PAN02 cells suspended in 200 μL phosphate‐buffered saline (PBS) (Wako Pure Chemical Industries, Osaka, Japan), intraperitoneally or via subcapsule into the spleen under phenobarbital anesthesia to establish intraperitoneal‐dissemination and liver‐metastasis PDAC‐tumor mouse models, respectively. The chemotherapy to treat the murine PDAC tumors was a dose of 50 mg/kg GEM (Sigma‐Aldrich, St. Louis, MO, USA) injected intravenously via the tail vein. To evaluate the immune response in peripheral blood and observe tumors and tumor‐infiltrating inflammatory cells (TICs), a single dose of GEM was administered. To determine the effect on survival, GEM was administered weekly or twice a week for up to 5 weeks in the intraperitoneal‐dissemination PDAC models, and for up to 8 weeks in the liver‐metastasis PDAC models. When antibody therapy was used in conjunction with GEM, a single dose of 200 μg anti‐Gr‐1 antibody (clone: RB6‐8C5; BD Biosciences, Franklin Lakes, NJ, USA) was injected intraperitoneally. To determine the effect on survival, anti‐Gr‐1 was administered twice a week for up to 5 weeks in the intraperitoneal‐dissemination PDAC models, and for up to 8 weeks in the liver‐metastasis PDAC models. The experimental plan for the use of mice was approved by our institutional review board.
2.3. Isolation of TICs
Pancreatic ductal adenocarcinoma tissue samples were obtained and cut into small pieces using scissors, placed in 100 μL PBS containing heparin sodium (1000 U/mL; Mochida, Tokyo, Japan), homogenized, and digested with 300 U/mL collagenase Type I and 2000 U/mL DNase I (Sigma‐Aldrich) in PBS in gentleMACS™ C Tubes, using a gentleMACS™ Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). Then, the dissociated cells were collected by centrifugation. The cells were treated with ACK Lysing Buffer (Life Technologies) to lyse erythrocytes. The cell suspension was sequentially filtered through 100 and 40‐μm cell strainers (BD Biosciences) and then centrifuged. The cells were suspended in 5 mL 40% Percoll™ PLUS (GE Healthcare, Uppsala, Sweden), which was then gently transferred over 3 mL Histopaque®‐1083 (Sigma‐Aldrich). The tube was then centrifuged to collect inflammatory cells in the layer between the Percoll™ and Histopaque® layers.
2.4. Isolation of white blood cells (WBCs)
To isolate WBCs, withdrawn blood from mice under anesthesia was transferred to a heparin sodium vacutainer (Venoject II, VP‐H100K; Terumo, Tokyo, Japan) pre‐filled with PBS. The cell suspension was filtered through a 40‐μm strainer and centrifuged. The pellet was resuspended in ACK buffer to lyse the red blood cells and then culture medium was added to neutralize the buffer. After centrifugation, the cells were collected and used for the experiments.
2.5. Flow cytometry
Isolated WBCs and TICs from mice were suspended in PBS supplemented with 2% bovine serum albumin (Sigma‐Aldrich). The cells were incubated with fluorescein isothiocyanate (FITC)‐conjugated anti‐CD4 (clone: RM4‐5), allophycocyanin (APC)‐conjugated anti‐CD8a (clone: 53‐6.7), FITC‐conjugated anti‐CD11b (clone: M1/70) (BD Pharmingen, San Jose, CA, USA), and phycoerythrin (PE)‐conjugated anti‐Gr‐1 (clone: RB6‐8C5; Miltenyi Biotec) antibodies for 15 minutes at 4°C. The samples were processed using a BD Accuri™ C6 Cytometer (BD Biosciences). The data were analyzed using FlowJo™ software (v.10.4.1; Tree Star Inc, Ashland, OR, USA).
2.6. DNA microarray analysis
RNA was isolated from blood samples using a Mouse RiboPure™‐Blood RNA Isolation Kit (Life Technologies) and then amplified and labeled with Cy3 using a Quick Amp Labeling Kit (Agilent Technologies, Santa Clara, CA, USA) as per the manufacturers’ protocols. The cRNA was hybridized on a Whole Mouse Genome 4 × 44K Array and readings were performed using a G2505B DNA microarray scanner (Agilent Technologies). The software BRB‐ArrayTools v.4.6.0 (http://linus.nci.nih.gov/BRB-ArrayTools.html) was used for gene expression analysis. Using this software, quantile normalization was applied and 27 144 genes passed the preliminary filtering criteria. Furthermore, gene set enrichment analysis was performed using class comparison by Gene Sets in the BRB tool software, which allowed analysis of gene sets showing differential expression among classes. The gene sets are pre‐included as a module of BRB‐ArrayTools, using publicly available Gene Ontology tools, lymphoid signatures or BioCarta Pathways domains. The Bioconductor GO package, in combination with SOURCE annotation, was also used for the analysis. The resulting gene sets passed either the LS/KS permutation test or the Efron‐Tibshirani test (P < 0.005).
2.7. Quantitative real‐time PCR (qRT‐PCR)
The qRT‐PCR was performed as previously described9 with modifications: the reaction was conducted with the cDNA mixed with qPCR MasterMix Plus® (Eurogentec, Seraing, Belgium) and the following hydrolysed Taqman® Gene Expression Assay probes: Adam8, Amica1, Trem1, Trem3, Bnip3 l, Bpgm, Cln3, Fbxo9, Fech, Hemgn, Hp, Mmp8, Mmp9. Relative expression levels were calculated with Gapdh as a reference gene using the 2−∆∆Ct method.
2.8. Apoptosis detection assay
CD8+ TICs were sorted by FACS ARIA II® and activated/expanded for 7 days with RPMI 1640 media supplemented with 10% FBS, 1% antibiotic–antimycotic solution (Gibco, Life Technologies, Carlsbad, CA, USA), 100 units/mL of murine IL‐2 (PeproTech, Rocky Hill, NJ, USA) and Anti‐Biotin MACSiBead particles loaded with CD3ε‐ and CD28‐Biotin (Miltenyi Biotec). The CD8+ TICs were co‐cultured with PAN02 at a ratio of 13:1 for 20 hours in a low‐grade attachment Falcon™ Round‐Bottom Polypropylene Tube (Thermo Fisher Scientific, Waltham, MA, USA). The FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen) was used for the detection of dead and early/late apoptosis PAN02 cells, the measurements were performed with a BD Accuri™ C6 Cytometer. Apoptotic cells were identified by FACS as FITC‐Annexin V + 7‐AADneg, the dead cells by FITC‐Annexin V + 7‐AAD+. The FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen) was also used for the evaluation in vitro of the chemotoxic effect of GEM over PAN02 cells.
2.9. Caspase‐3 activity assay
Caspase‐3 activity was assessed using a colorimetric CaspACE™ Assay System (Promega, Madison, WI, USA) in accordance with the manufacturer's protocol. Briefly, PAN02 cells were cultured in culture media with 300 μg/mL GEM and either the pan‐caspase inhibitor Z‐VAD‐FMK (Promega) or PBS (negative control) for 16 hours. After harvesting, centrifuging and washing the cells with PBS, the cells obtained were lysed. The lysates were incubated with labeled Asp‐Glu‐Val‐Asp‐p‐nitroanilide (DEVD‐pNA) substrate, and then absorbance at a wavelength of 405 nm was measured.
2.10. Arginase assay
White blood cells from PDAC mice and control mice were stained with FITC‐conjugated anti‐CD11b and PE‐conjugated anti‐Gr‐1 antibodies and then analyzed with a FACS ARIA II® cytometer (BD Biosciences) to sort CD11b+Gr‐1+ cells. The collected cells were used for colorimetric quantification of arginase activity using a QuantiChrom™ Arginase Assay Kit (BioAssay Systems, Hayward, CA, USA) as per the manufacturer's protocol. Briefly, the cells were lysed and centrifuged, and the collected supernatants were incubated with a chromogen that forms a colored complex with urea. The emitted color was read at an optical density of 430 nm using a Tecan Sunrise™ microplate reader (Tecan Group Ltd., Männedorf, Switzerland) and the arginase activity of each sample was calculated.
2.11. Immunohistochemical analysis
Immunohistochemistry was performed as described previously,10 with slight modifications. Briefly, tumor tissue samples were obtained from murine PDAC models, preserved with IHC Zinc Fixative® (BD Pharmingen), embedded in paraffin, sectioned at 2 μm, and stained with H&E and azan. For immunohistochemical analysis, tumor tissue samples were fixed and sliced as described above, embedded in OCT compound (Sakura Finetek Japan Co., Ltd. Tokyo, Japan), frozen, and then sectioned at 7 μm. The sections were incubated with rat anti‐CD4 (clone: RM4‐5), anti‐CD8a (clone: 53‐6.7), and anti‐Gr‐1 (clone: RB6‐8C5) (BD Pharmingen), anti‐CD279 (PD‐1; clone: 29F.1A12, BioLegend, San Diego, CA, USA) and anti‐CD274 (PD‐L1; clone: MIH6, LifeSpan BioSciences, Seattle, WA, USA) primary antibodies, and then incubated with the reagent anti‐rat Histofine Simple Stain Mouse MAX POR (Nichirei Corporation, Tokyo, Japan) for 45 minutes. Staining was obtained after incubation with diaminobenzidine substrate solution (Dako ChemMate EnVision Kit/HRP (DAB)®) (Dako, Kyoto, Japan), the sections were then counterstained with Myer's hematoxylin.
2.12. Interferon‐gamma (IFN‐γ) secretion assay
CD8+ cells were isolated from TICs obtained from murine PDAC models using anti‐CD8a magnetic beads (clone: 53‐6.7; Miltenyi Biotec) in accordance with the manufacturer's protocol (CD8a+ T Cell Isolation Kit; Miltenyi Biotec). The isolated CD8+ T cells were stimulated for 8 days with interleukin‐2 (PeproTech) at a concentration of 100 units/mL in RPMI 1640 media supplemented with 10% FBS. A mouse IFN‐γ Secretion Assay and Detection Kit (Miltenyi Biotec) was used as per the manufacturer's protocol for detection and staining of IFN‐γ secreting CD8+ T cells, followed by flow cytometry analysis.
2.13. Chromium‐51 release assay
PAN02 cells (5 × 104 cells) were suspended in a U‐bottomed 96‐well tissue plate (BD Biosciences) with 100 μL culture medium for 16 hours. Then, 10 μL chromium‐51 [51Cr] radionuclide (37 MBq/mL; PerkinElmer, Waltham, MA, USA) was added to each well and incubated for 1 hour. After preparing the cell culture, supernatant containing 51Cr that was not taken up by the cells was carefully removed and the cultured cells were washed twice with 200 μL medium. CD8+ cells (1 × 106) from splenocytes of liver‐metastasis PDAC mice, treated (n = 3) or not treated (n = 3) with GEM, were co‐cultured for 16 hours with the labeled cells in 100 μL RPMI 1640 supplemented with 10% FBS. Then, the supernatant was recovered, placed in a polystyrene vial and analyzed using a Gamma Counter (AccuFlex gamma ARC‐8001®; Hitachi Aloka Medical, Mitaka, Japan).
2.14. Statistical analysis
IBM SPSS Statistics (IBM Corp., Armonk, NY, USA) was used for statistical analyses. The Kaplan–Meier estimator was used to measure the fraction of survival in mice, and the log‐rank test was then performed to compare the survival distributions. The Mann‐Whitney U test was used to compare the data from the IFN‐γ secretion assay; the Student's t‐test was used for the Arginase assay. Statistical significance was determined to be P < 0.05.
3. RESULTS
3.1. Murine PDAC models and immune responses
We established murine intraperitoneal‐dissemination PDAC models and murine liver‐metastasis PDAC models by intraperitoneal injection of PAN02 cells and subcapsule insertion in the spleen under laparotomy, respectively (Figure 1). In the intraperitoneal‐dissemination PDAC mice, relatively small tumor nodules were disseminated in the intraperitoneal cavity (Figure 1B). The liver‐metastasis PDAC mice had multi‐foci tumors in the liver. Microscopic observation of these tumors showed spindle‐like pancreatic cancer cells (Figure 1C). Azan staining of the tumor tissues showed intense fibrosis formation, similar to the characteristics of human PDAC tissues (Figure 1B,C).
Figure 1.
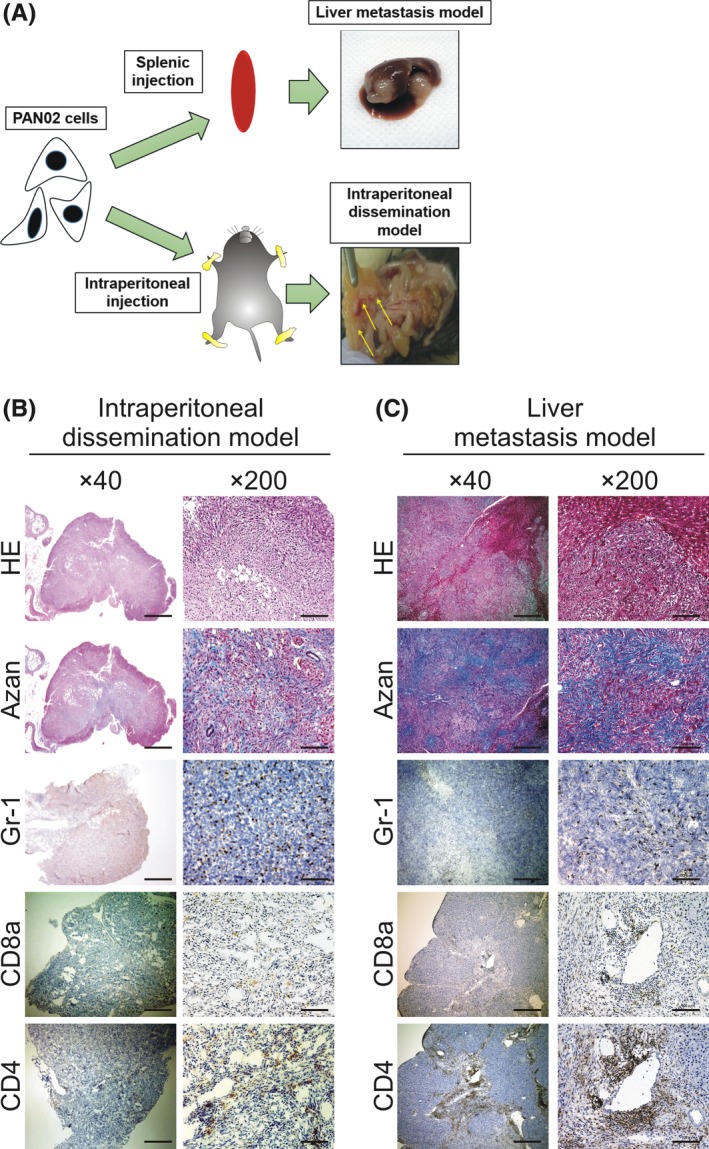
Pancreatic ductal adenocarcinoma (PDAC) murine models and histochemical characteristics of tumors. A, A scheme of PDAC models is depicted. PAN02 cells were injected intrasplenically to induce PDAC liver metastasis or into the peritoneal cavity to establish PDAC intraperitoneal‐dissemination models. B,C, Representative histochemical sections showing H&E, azan and DAB staining of inflammatory cells (Gr‐1+, CD8a+ and CD4+ cells) in tumors. B, Tumors obtained from intraperitoneal‐dissemination mice (day 28). C, Tumors obtained from liver‐metastasis mice (day 38). Bars: 500 μm for ×40 magnification and 100 μm for ×200 magnification
Since we previously observed that human PDAC tumors are associated with the host immune response in tumor tissues, we next used immunohistochemistry to analyze PDAC tumor tissues for the presence of inflammatory cells. The tumor tissues obtained from both the intraperitoneal‐dissemination and liver‐metastasis mouse models had substantial numbers of scattered Gr‐1+ myeloid cells in the tumor tissues, and some infiltrated CD4+ and CD8+ cells (Figure 1B,C).
3.2. GEM treatment and associated anti‐cancer immune response in murine PDAC models
We next assessed whether the chemotherapeutic reagent for PDAC treatment, GEM, had therapeutic efficacy for treating murine pancreatic cancer. We confirmed that GEM treatment induced exposure of annexin V on the plasma membrane of PAN02 pancreatic cancer cells in vitro (Figure 2A). We also observed that caspase‐3 activity was higher in PAN02 cells treated with GEM than in non‐treated cells (Figure 2B), implying that GEM is directly cytocidal to PAN02 cells. Next, we found that GEM treatment significantly prolonged the survival of intraperitoneal‐dissemination PDAC mice and liver‐metastasis PDAC mice compared to untreated mice in vivo (Figure 2C). Thus, GEM treatment was therapeutically beneficial for treating murine pancreatic cancer.
Figure 2.
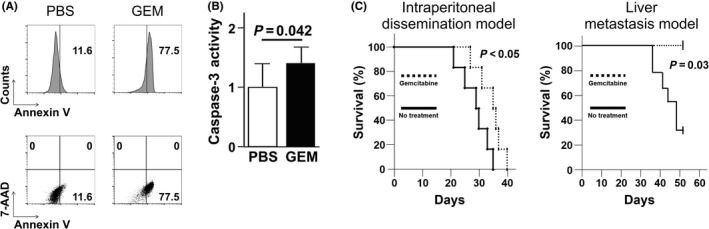
Gemcitabine (GEM)‐induced apoptosis of PAN02 cells in vitro and therapeutic effect of GEM on survival in pancreatic ductal adenocarcinoma (PDAC) models. A, PAN02 cells were incubated in culture media with GEM (300 μg/mL) or without GEM for 48 h; then, apoptotic cells were identified as FITC‐annexin V positive and 7‐AAD negative by FACS. Representative FACS histograms/scatterplots are presented. B, Quantification of caspase‐3 activity of PAN02 cells cultured in culture media for 16 h with or without GEM in the presence of the inhibitor Z‐VAD‐FMK. The cleaving activity was determined by colorimetric assay; the Student's t‐test was performed to obtain the P‐value. C, Cumulative survival curves of intraperitoneal‐dissemination and liver‐metastasis PDAC mice (n = 6) involving twice a week injection of GEM. PBS was injected in the control group that received no treatment. The log‐rank test was performed to obtain the P‐value
We next assessed how GEM affected PDAC‐associated immune responses. We observed that CD11b+Gr‐1+ myeloid cell numbers were higher in the peripheral blood of intraperitoneal‐dissemination and liver‐metastasis PDAC mice than in wild‐type mice without tumors (Figure 3). We also found that the CD11b+Gr‐1+ cells isolated from peripheral blood of intraperitoneal‐dissemination PDAC mice had high arginase activity compared to that of wild‐type mice (Figure S1), indicating that the myeloid populations in these mice included immunosuppressive cells. When the intraperitoneal‐dissemination PDAC mice and liver‐metastasis PDAC mice were treated with GEM, the proportion of CD11b+Gr‐1+ myeloid cells in peripheral blood decreased (Figure 3B,C). In contrast, the proportion of CD4+ cells and CD8+ cells increased (Figure 3B,C).
Figure 3.
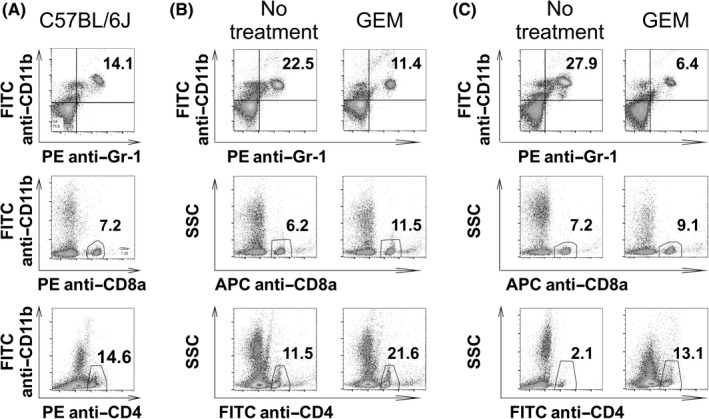
Peripheral blood analysis of intraperitoneal‐dissemination and liver‐metastasis pancreatic ductal adenocarcinoma (PDAC) mice treated with gemcitabine (GEM). Intraperitoneal‐dissemination and liver‐metastasis PDAC mice were treated with a single dose of GEM 2 d prior to the retrieval of peripheral blood cells and FACS analysis. Mice in the no‐treatment group were injected with PBS alone. A, Wild‐type C57BL/6J mice without tumors. B, Intraperitoneal‐dissemination PDAC mice with or without GEM treatment. C, Liver‐metastasis PDAC mice with or without GEM treatment. Representative FACS scatterplots are shown
Consistently, immunohistochemical staining of PDAC tumor tissues showed that Gr‐1+ cell infiltration decreased, while CD4+ and CD8+ cell numbers increased in both mouse models treated with GEM (Figure 4A,B). As for immune‐checkpoint molecules, PD‐1 and PD‐L1,11 we observed GEM treatment induced PD‐1‐expressing, and PD‐L1‐expressing cells in the tumor tissues of both intraperitoneal‐dissemination and liver‐metastasis PDAC mice (Figure S2). Flow cytometry analysis of isolated TICs also showed that the proportion of Gr‐1+ cells decreased, whereas the proportions of CD4+ and CD8+ cells increased (Figure 4C,D).
Figure 4.
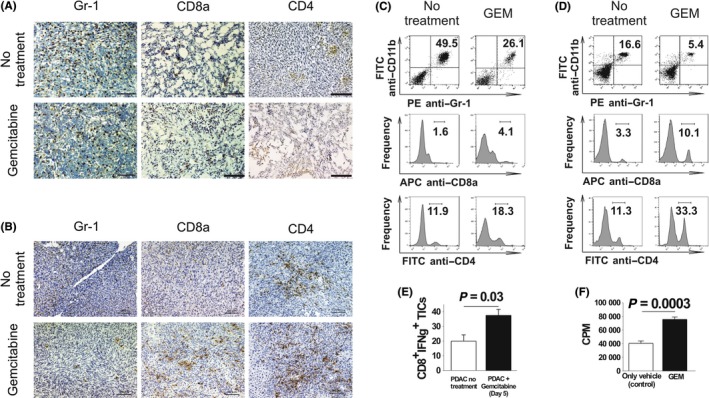
Effect of gemcitabine (GEM) on tumor‐infiltrating inflammatory cells (TICs) in pancreatic ductal adenocarcinoma (PDAC) models. Intraperitoneal‐dissemination PDAC mice and liver‐metastasis PDAC mice received a single dose of GEM on days 26 and 36, respectively. Two days later, tumor tissues were obtained and used for immunohistochemical analysis and isolation of TICs. A, B, Tumors immunohistochemically analyzed for Gr‐1+, CD8a+ and CD4+ inflammatory cells. Magnification: ×100. Scale bars: 100 μm. A, Intraperitoneal‐dissemination PDAC mice. B, Liver‐metastasis PDAC mice. C, D, FACS analysis of TICs isolated from tumors from intraperitoneal‐dissemination and liver‐metastasis PDAC mice treated with or without GEM. C, TICs isolated from intraperitoneal‐dissemination PDAC mice. D, TICs isolated from liver‐metastasis PDAC mice. One representative scatterplot or histogram in each group is presented. E, TICs were isolated from GEM‐treated (n = 4) or untreated (n = 4) liver‐metastasis PDAC mice, CD8+ cells were selectively sorted from the TICs and stimulated for 8 d with interleukin‐2. Then, IFN‐γ secretion assay was performed. F, The cytotoxicity of CD8+ cells isolated from splenocytes of liver‐metastasis PDAC mice treated (n = 3) or not treated (n = 3) with GEM was assessed by quantifying the amount of [51Cr] released from PAN02 target cells; the Student's t‐test was performed to obtain the P‐value
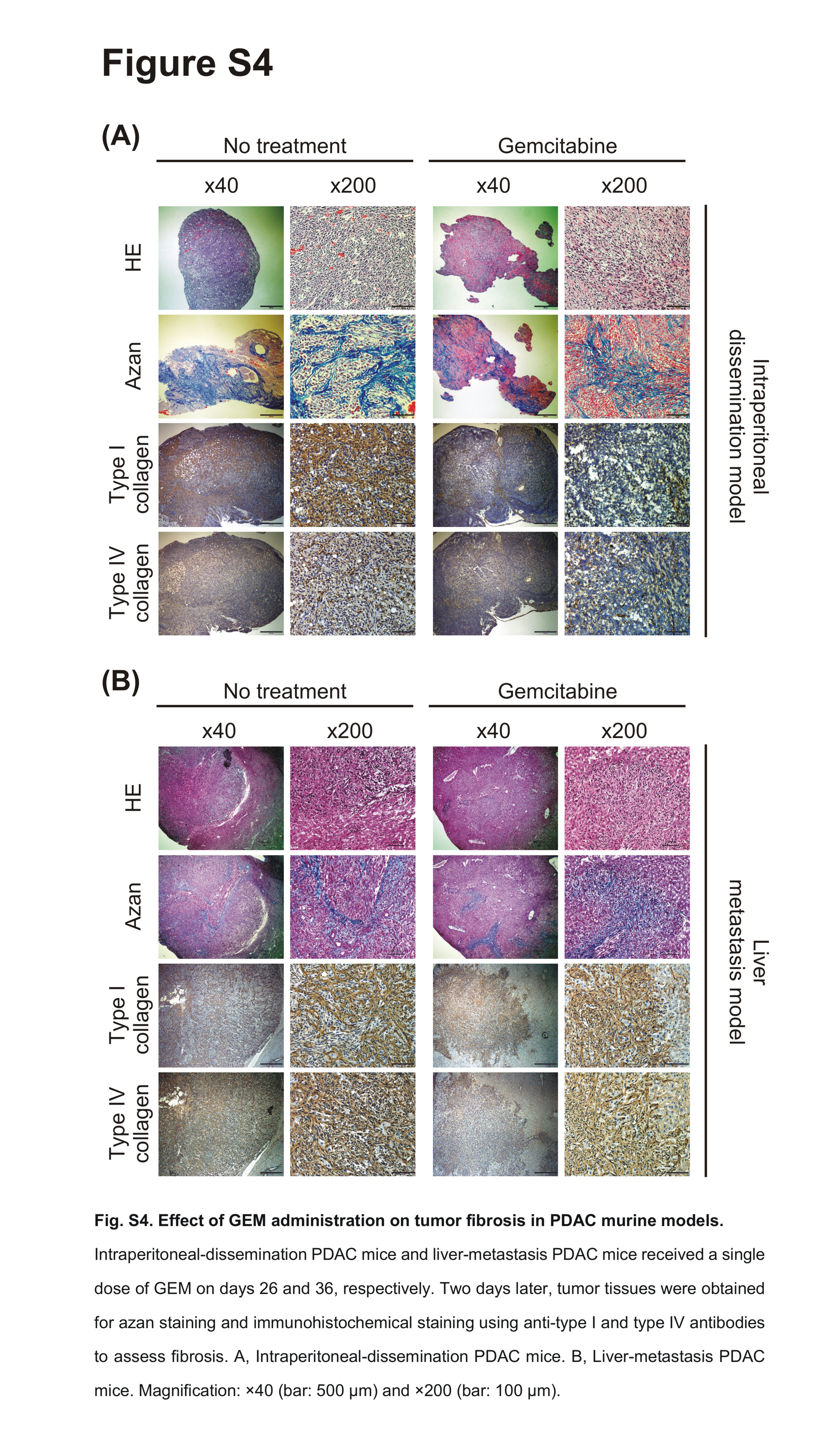
We also assessed the function of CD8+ cells isolated from peripheral blood and from tumor tissue of PDAC mice treated with GEM. We observed that the number of IFN‐γ‐secreting CD8+ TICs was increased in PDAC mice treated with GEM (Figure 4E). Furthermore, a [51Cr]‐release assay showed that the cytocidal capability of peripheral blood CD8+ cells against PAN02 cells was enhanced (Figure 4F). We also observed that CD8+ TICs obtained from liver‐metastasis PDAC mice treated with GEM induced significantly an increase in number of apoptotic and dead PAN02 cells, compared to CD8+ TICs from liver‐metastasis PDAC mice without any treatments (Figure S3). Taken together, these results show that GEM treatment of PDAC mice induced anti‐cancer immunity by decreasing numbers of suppressive myeloid cells and enhancing the cytocidal capability of CD8+ cells. Furthermore, immunohistochemical staining of PDAC tumor tissues from GEM‐treated mice showed reductions in the amounts of type I and IV collagen compared to that in untreated mice (Figure S4).
3.3. Anti‐Gr‐1 antibody treatment augmented the therapeutic effect of GEM in PDAC murine models
The above data imply that myeloid lineage cells in peripheral blood, as well as tumor tissues in PDAC murine models, are associated with tumor formation, and that GEM treatment decreases the proportion of myeloid cells in peripheral blood and tumor tissues. We next assessed whether further depletion of myeloid cells using an anti‐Gr‐1 antibody in combination with GEM treatment would augment the anti‐cancer effect. We observed that GEM treatment and anti‐Gr‐1 antibody treatment significantly prolonged survival in intraperitoneal‐dissemination PDAC mice which underwent GEM treatment twice a week (Figure 5A). When we administered GEM into the liver‐metastasis PDAC mice weekly, not twice a week, survival prolongation effect by GEM treatment alone was not obvious. However, when we administered GEM weekly together with anti‐Gr‐1 antibody treatment, we also observed this combined treatment relatively prolonged survival in liver‐metastasis PDAC mice that received GEM weekly (Figure 5B). In both intraperitoneal‐disseminated and liver‐metastasis PDAC mice treated with a combination of anti‐Gr‐1 antibody therapy and GEM, the proportion of CD11b+Gr‐1+ myeloid cells in peripheral blood was even lower than in peripheral blood isolated from mice that received GEM treatment only (Figure 5C,D). In addition, numbers of CD4+ cells were also further increased when anti‐Gr‐1 antibody therapy was combined with GEM treatment. In contrast, numbers of CD8+ cells remained unchanged in mice treated with only GEM, and with combination GEM and anti‐Gr‐1 antibody treatment.
Figure 5.
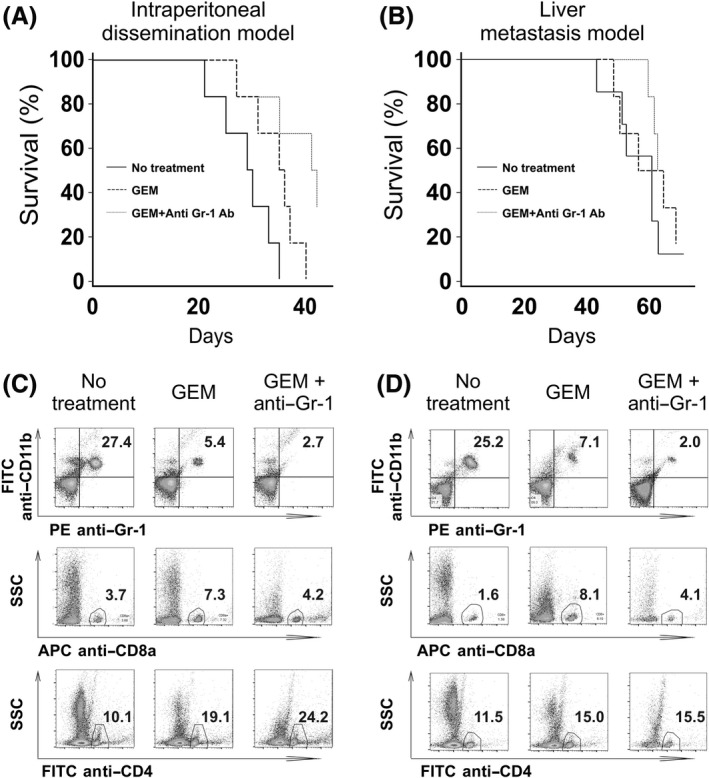
Survival after combination treatment with gemcitabine (GEM) and anti‐Gr‐1 antibody in pancreatic ductal adenocarcinoma (PDAC)‐model mice and peripheral blood cell analysis. A, B, Cumulative survival curves of intraperitoneal‐dissemination and liver‐metastasis PDAC mice that received: (a) no treatment (only PBS; n = 6 or 7), (b) twice a week (intraperitoneal‐dissemination model) or weekly (liver‐metastasis model) injection of GEM (n = 6), and (c) twice a week (intraperitoneal‐dissemination model) or weekly (liver‐metastasis model) injection of GEM in combination with anti‐Gr‐1 administration (n = 6). A, Intraperitoneal‐dissemination PDAC mice. B, Liver‐metastasis PDAC mice. The log‐rank test was performed to obtain P‐values: the P‐values for intraperitoneal‐dissemination PDAC mice were: no treatment/GEM = 0.041, no treatment/GEM + Anti‐Gr‐1 = 0.012, and GEM/GEM + Anti‐Gr‐1 = 0.046. The P‐values for liver‐metastasis PDAC mice were: no treatment/GEM = 0.663, no treatment/GEM + Anti‐Gr‐1 = 0.190, and GEM/GEM + Anti‐Gr‐1 = 0.730. C, D, Analysis of peripheral blood cells. PDAC mice were treated with GEM plus anti‐Gr‐1 antibody (GEM + Anti‐Gr‐1), GEM alone, or no treatment. Two days later, peripheral blood cells were obtained and used for FACS analysis. C, Intraperitoneal‐dissemination PDAC mice. D, Liver‐metastasis PDAC mice. Representative scatter plots of myeloid‐ and lymphoid‐derived cells are displayed
Using a cDNA microarray, we next analyzed the gene‐expression profiles of peripheral blood cells in liver‐metastasis PDAC mice treated with GEM alone or in combination with the anti‐Gr‐1 antibody. Except in one case, unsupervised clustering analysis discriminated between the mice depending on the treatment (Figure 6A). We also analyzed the expression of genes related to biological processes for neutrophils and anti‐cancer cytotoxic immune responses. Anti‐Gr‐1 treatment suppressed expression of neutrophil‐related‐gene sets (Figure 6B). To confirm the suppressed expression of neutrophil‐related gene sets demonstrated by microarray gene expression analysis, we have randomly selected the genes, Adam8, Amica1, Trem1, Trem3, Bnip3 l, Bpgm, Cln3, Fbxo9, Fech, Hemgn, Hp, Mmp8, Mmp9 from heat‐map panels, “Neutrophil extravasation” and “Blood_Module‐2.2_Neutrophils”, and examined the expression by quantitative RT‐PCR (Figure S5). Consistent with the gene expression data by microarray data, the genes’ expression was confirmed to be suppressed by anti‐Gr‐1 antibody treatment. In line with our findings that PDAC mice treated with anti‐Gr‐1 antibody and GEM had prolonged survival, expression of gene sets related to anti‐cancer immune responses, such as natural killer cell‐mediated cytotoxicity directed against tumor cell targets (GeneSet GO:0002420), the type I IFN signaling pathway (GeneSet GO:0060337), and The Co‐Stimulatory Signal During T‐cell Activation (GeneSet BioCarta Pathway: m_ctla4Pathway), was enhanced in mice treated with GEM and anti‐Gr‐1 antibody (Figure 6B).
Figure 6.
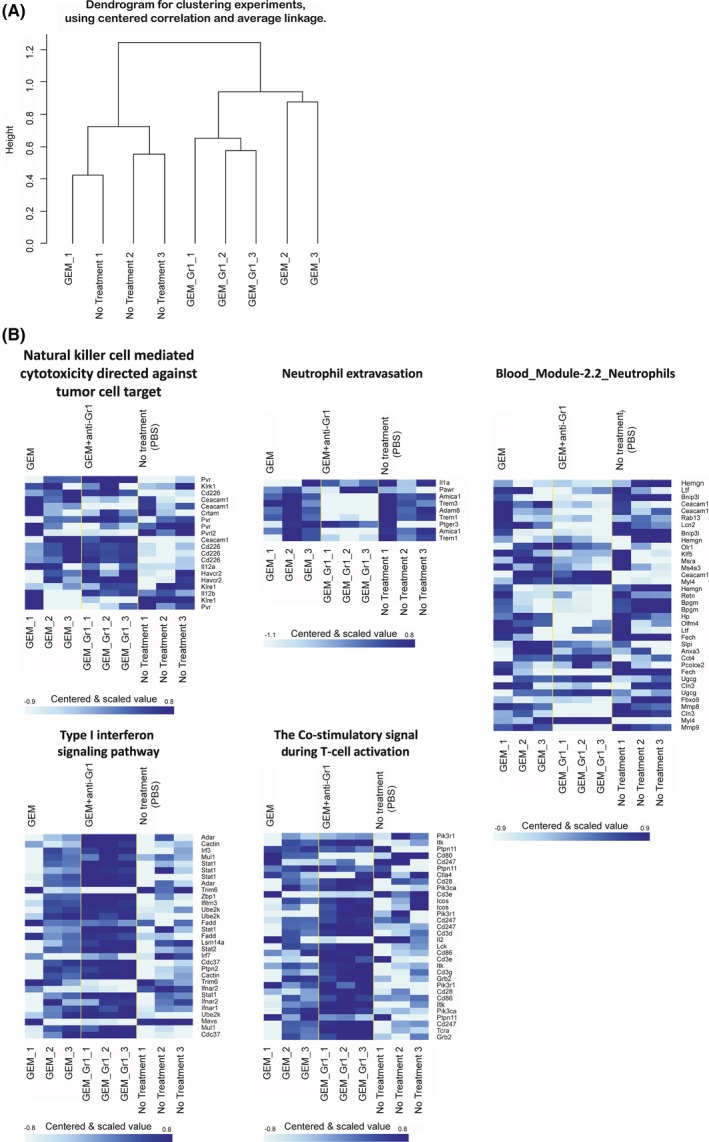
DNA microarray analysis of whole peripheral blood cells from liver‐metastasis pancreatic ductal adenocarcinoma (PDAC) mice following treatment. Liver‐metastasis PDAC mice underwent the following treatments on day 36: gemcitabine (GEM) in combination with anti‐Gr‐1 antibody (n = 3), GEM (n = 3), and no treatment (n = 3). Two days later, total peripheral blood was withdrawn and total RNA was isolated for cDNA microarray analysis. A, Dendrogram for post‐analysis sample clustering (27 144 genes which passed filter). B, Gene set class comparison analysis of biological processes related to neutrophils and anti‐cancer cytotoxic immune responses that passed the LS/KS permutation test or the Efron‐Tibshirani's test with significance threshold (P < 0.005)
4. DISCUSSION
In this study, we used murine intraperitoneal‐dissemination and liver‐metastasis PDAC models to analyze both natural host immune responses and GEM chemotherapy‐associated immune responses. We confirmed that PDAC tissues from both murine models showed substantial infiltration of inflammatory cells, including myeloid cells expressing genes related to immune‐suppressor cells, and CD4+ and CD8+ T lymphocytes. GEM induced apoptosis in PAN02 pancreatic cancer cells. Host inflammatory responses to PDAC tumors were also affected by GEM treatment. Specifically, there was a decrease in the numbers of Gr‐1+ infiltrating inflammatory cells, and an increase in the numbers of T cells with an enhanced cytotoxic CD8+ T cell response. Anti‐Gr‐1 antibody treatment in combination with GEM prolonged the survival of PDAC mice. The gene‐expression profile of peripheral blood cells showed that anti‐cancer immune responses were prominent in PDAC mice treated with GEM and anti‐Gr‐1 antibody.
Cancer‐associated inflammation has been intensively investigated. Such inflammation has an important role in disease progression/regression, and its effect on prognoses has been studied in various cancers.12, 13 Inflammatory cells consist of various subtypes and lineages, including myeloid and lymphoid cells.14, 15, 16 Consistent with the importance of inflammatory cells in cancer, the findings of the current study suggest that myeloid cells are cancer‐prone immune‐mediating cells that are found in both peripheral blood and TICs. The population of myeloid‐lineage cells originating from bone marrow is highly heterogeneous,17 and it has been proven that immune‐suppressive myeloid cells play an important role in inhibiting anti‐cancer immunity.18 Thus, further detailed investigations of myeloid‐lineage cells should be performed to understand their role in the context of PDAC tumor development.
A major goal of the current study was to examine chemotherapy‐associated alterations in host immune responses to tumor‐associated inflammation. GEM is a deoxycytidine nucleoside analog,19 and an anti‐metabolite that is cytotoxic to cancer cells.20 It would be of value to discover a chemotherapeutic agent with a mechanism of action that has a direct cytocidal effect on cancer cells and also influences host inflammatory responses to cancer tissues.21 In our intraperitoneal‐dissemination and liver‐metastasis PDAC mice models, GEM treatment decreased numbers of myeloid cells and increased CD4+ and CD8+ cells. In other cancer mice model, GEM was proven to be suppressive to myeloid‐lineage cells, especially, myeloid‐derived suppressor cells.22 Additionally, in BALB/c mice system, GEM depleted B‐lymphocytes; in contrast, tumor‐specific CD4+ and CD8+ cells were augmented.23 Our current findings of immunomodulatory effects by GEM in C57BL/6J PDAC mice models are consistent with these previous reports. Detailed relations of myeloid‐lineage suppression and augmentation of CD4+ and CD8+ cells by GEM treatment remains to be disclosed; however, decrease of suppressor cells in myeloid‐lineage cells is considered to be a mechanism since they inhibit tumor‐reactive T cell response,24 by production of arginase, inducible NO synthase, etc.25 Thus, gemcitabine's therapeutic effect is not only dependent on DNA synthesis halt, but also on immune‐modulation with anti‐cancer effects. Despite of these intriguing findings, cancer tissues affected by chemotherapeutic agents should be further investigated, since further complex host immune responses are involved26 as a consequence of anti‐cancer chemotherapeutic agents’ effects on cancer tissues.
Novel anti‐cancer immune therapies such as those that target immune checkpoints27 have emerged as treatments for cancers including melanoma28 and renal cell carcinoma.29 As for PDAC, there have been no reports of effective immune therapies in clinical trials.30 The findings in the current study show that immune modulation induction of anti‐cancer immunity by depletion of myeloid‐lineage cells is a promising novel strategy for treating PDAC when used in combination with chemotherapy. However, the prolongation effect of survival by anti‐Gr‐1 antibody treatment combined with GEM was limited, especially in liver metastasis PDAC models. PD‐1 and PD‐L1 expressing cells were significantly induced in tumor tissues of both intraperitoneal‐dissemination and liver‐metastasis murine PDAC models, when they were treated with GEM. Therefore, targeting these immune checkpoints molecules together with depletion of myeloid‐lineage cells might be novel approach for PDAC immune‐chemotherapy.
In conclusion, immune responses in murine PDAC models were affected by GEM chemotherapy. Moreover, the induced anti‐cancer immunity was further augmented by targeting myeloid‐lineage cells, showing that immunomodulatory treatment can be used as an alternative for the development of novel PDAC treatments. Further studies should be performed to determine the immunopathological features of PDAC, and to identify the most effective immunomodulation therapy for PDAC.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported in part by the grant of Japan Society for the Promotion of Science and Japan Agency for Medical Research and Development.
Sakai Y, Miyazawa M, Komura T, et al. Distinct chemotherapy‐associated anti‐cancer immunity by myeloid cells inhibition in murine pancreatic cancer models. Cancer Sci. 2019;110:903–912. 10.1111/cas.13944
Yoshio Sakai and Masaki Miyazawa equally contributed to this study.
REFERENCES
- 1. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039‐1049. [DOI] [PubMed] [Google Scholar]
- 2. Al‐Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the society of abdominal radiology and the american pancreatic association. Gastroenterology. 2014;146:291‐304. e1. [DOI] [PubMed] [Google Scholar]
- 3. Buchler MW, Kleeff J, Friess H. Surgical treatment of pancreatic cancer. J Am Coll Surg. 2007;205:S81‐S86. [DOI] [PubMed] [Google Scholar]
- 4. Kayahara M, Nakagawara H, Kitagawa H, Ohta T. The nature of neural invasion by pancreatic cancer. Pancreas. 2007;35:218‐223. [DOI] [PubMed] [Google Scholar]
- 5. Kitagawa H, Tajima H, Nakagawara H, et al. The retropancreatic fusion fascia acts as a barrier against infiltration by pancreatic carcinoma. Mol Clin Oncol. 2013;1:418‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saung MT, Zheng L. Current standards of chemotherapy for pancreatic cancer. Clin Ther. 2017;39:2125‐2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fridman WH, Pages F, Sautes‐Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298‐306. [DOI] [PubMed] [Google Scholar]
- 8. Komura T, Sakai Y, Harada K, et al. Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4+ T cells with clinical impact. Cancer Sci. 2015;106:672‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nasti A, Sakai Y, Seki A, et al. The CD45(+) fraction in murine adipose tissue derived stromal cells harbors immune‐inhibitory inflammatory cells. Eur J Immunol. 2017;47:2163‐2174. [DOI] [PubMed] [Google Scholar]
- 10. Higashimoto M, Sakai Y, Takamura M, et al. Adipose tissue derived stromal stem cell therapy in murine ConA‐derived hepatitis is dependent on myeloid‐lineage and CD4+ T‐cell suppression. Eur J Immunol. 2013;43:2956‐2968. [DOI] [PubMed] [Google Scholar]
- 11. Chikuma S, Suita N, Okazaki IM, Shibayama S, Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo. Nat Immunol. 2012;13:596‐603. [DOI] [PubMed] [Google Scholar]
- 12. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137‐148. [DOI] [PubMed] [Google Scholar]
- 13. Tenenbaum T, Hasan C, Kramm CM, et al. Oncological management of pediatric cancer patients belonging to Jehovah's Witnesses: a two‐institutional experience report. Onkologie. 2004;27:131‐137. [DOI] [PubMed] [Google Scholar]
- 14. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 16. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kiss M, Van Gassen S, Movahedi K, Saeys Y, Laoui D. Myeloid cell heterogeneity in cancer: not a single cell alike. Cell Immunol. 2018;330:188‐201. [DOI] [PubMed] [Google Scholar]
- 18. Haile LA, Greten TF, Korangy F. Immune suppression: the hallmark of myeloid derived suppressor cells. Immunol Invest. 2012;41:581‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gesto DS, Cerqueira NM, Fernandes PA, Ramos MJ. Gemcitabine: a critical nucleoside for cancer therapy. Curr Med Chem. 2012;19:1076‐1087. [DOI] [PubMed] [Google Scholar]
- 20. Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3:415‐424. [DOI] [PubMed] [Google Scholar]
- 21. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. 2008;8:59‐73. [DOI] [PubMed] [Google Scholar]
- 22. Eriksson E, Wenthe J, Irenaeus S, Loskog A, Ullenhag G. Gemcitabine reduces MDSCs, tregs and TGFbeta‐1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nowak AK, Robinson BW, Lake RA. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo‐immunotherapy. Cancer Res. 2002;62:2353‐2358. [PubMed] [Google Scholar]
- 24. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand‐Rosenberg S. Myeloid‐derived suppressor cells down‐regulate L‐selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183:937‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L‐arginine metabolism in myeloid cells controls T‐lymphocyte functions. Trends Immunol. 2003;24:302‐306. [DOI] [PubMed] [Google Scholar]
- 26. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity. 2013;39:1‐10. [DOI] [PubMed] [Google Scholar]
- 27. Romero D. CheckMate 214 ‐ a winning combination? Nat Rev Clin Oncol. 2018;15:343. [DOI] [PubMed] [Google Scholar]
- 28. Bhatia S, Thompson JA. Melanoma: immune checkpoint blockade story gets better. Lancet. 2014;384:1078‐1079. [DOI] [PubMed] [Google Scholar]
- 29. Hammers HJ, Plimack ER, Infante JR, et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 study. J Clin Oncol. 2017;35:3851‐3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brunet LR, Hagemann T, Andrew G, Mudan S, Marabelle A. Have lessons from past failures brought us closer to the success of immunotherapy in metastatic pancreatic cancer? Oncoimmunology. 2016;5:e1112942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials