

Abstract
Background:
Human apolipoprotein E3 (apoE3) is an exchangeable apolipoprotein that plays a critical role in maintaining plasma cholesterol/triglyceride homeostasis. The C-terminal (CT) domain of apoE3 (residues 201–299) is composed of amphipathic α-helices C1: W210-S223, C2: V236-E266, and C3: D271-W276, which play a dominant role in mediating high-affinity lipid binding.
Objective:
The objective is to understand the accessibility of the CT domain at the sub-domain level and the mechanistic details regarding lipid-binding interaction.
Methods:
Hydrogen-deuterium exchange coupled to mass spectrometry (HDX/MS) of recombinant wild type (WT) apoE(201–299), chemical-induced unfolding monitored as changes in fluorescence polarization (FP) of labeled apoE(201–299) bearing a probe at specified sites, and lipid binding studies were carried out.
Results:
HDX/MS revealed that residues towards the C-terminal end of the domain display significantly lower %D uptake compared to those towards the center, suggesting extensive protein-protein interaction in this segment. Functional assays showed that locking apoE(201–299) in an inter-molecular disulfide-bonded state at position 209, 223, 255, or 277 significantly decreases its ability to interact with lipids, especially when tethered towards the ends; this could be restored by reduction. Unfolding studies indicate that the C-terminal end offers less resistance to unfolding compared to the central portion of the domain.
Conclusion:
Taken together, our data suggest that two dimers of CT domain are juxtaposed around helix C3 leading to apoE3 tetramerization, and that dissociation to monomeric units is a required step in lipid binding, with helix C3 likely seeking stability via lipid interaction prior to helices C1 or C2.
Keywords: apolipoprotein E3, lipid binding mechanism, Hydrogen-deuterium exchange, mass spectrometry, fluorescence polarization, C-terminal domain, lipoprotein
1. INTRODUCTION
Human apolipoprotein E3 (apoE3) is a 299 residue exchangeable apolipoprotein that has the ability to exist in lipid-free and lipoprotein-bound states. It is an anti-atherogenic protein found on triglyceride-rich lipoproteins and a subclass of HDL in the plasma, and on HDL-like particles in the brain [1–3]. ApoE3 plays a critical role in regulating plasma and brain cholesterol homeostasis [4, 5] primarily by virtue of its ability to serve as a high-affinity ligand for the low-density lipoprotein receptor (LDLr) family of proteins, which leads to cellular uptake and internalization of the lipoprotein thereby lowering plasma lipid levels. It also functions in reverse cholesterol transport in atherosclerosis, a process involving transport of cholesterol from the peripheral tissues to the liver for eventual disposal via the bile [6, 7]. The apoE4 isoform is considered a significant risk factor for developing Alzheimer’s disease [8, 9].
ApoE3 is composed of independently folded N-terminal (NT) (1–191) and C-terminal (CT) (201–299) domains that are linked by a protease-sensitive loop. The NT domain is organized as a 4-helix bundle [10, 11] and bears the LDLr binding sites, while the CT domain is responsible for subunit interaction to form dimers, tetramers and higher state oligomers in lipid free state. Although both domains have the capability to interact with lipids, the CT domain has a relatively higher binding affinity compared to the NT domain, a feature that may play a key role in regulating plasma lipid levels [4, 12, 13]. Lipid binding is an essential prerequisite for the LDLr binding interaction of apoE3; several lines of evidence indicate that the protein undergoes a dramatic conformational re-organization upon lipid interaction that allows presentation of the receptor binding sites in an optimal configuration to the ligand binding domain of the LDLr [9, 14] [15] [16–18].
Structural predictions suggest that residues 210–266 form a long class A amphipathic α-helix (characterized by clustering of positively charged residues at the polar-non polar interface and negatively charged residues at the center of the polar face), while residues 268–289 form a class G* helix (no clustering of charged residues; random distribution of negative and positive residues around the perimeter of the polar face). Class A helices are believed to promote protein-lipid interaction, while G* helices are involved in protein-protein interaction and subunit formation [19, 20]. Consistent with the prediction, analytical ultracentrifugation and size-exclusion chromatographic studies on C-terminal truncated apoE3 reveal that residues 266–299 are likely involved in tetramerization [21, 22]. NMR analysis of a monomeric mutant apoE3 [23, 24] (F257A/W264R/V269A/L279Q/V287E) reveals 3 helices: C1: (W210-S223); C2: (V236-E266); and, C3: (D271-W276) that are wrapped around the NT domain helix bundle [11]. It is not known if this represents the organization of the monomeric unit in a tetrameric apoE3. Nonetheless, it is generally acknowledged that helix-lipid interactions replace helix-helix interactions during lipid association of apoE3, and that self association of the isolated CT domain has similar characteristics as those of full length apoE3. However, the details regarding the structural basis of interaction with lipids and the mechanism of lipid interaction of apoE3 CT domain remain unclear.
In the present study, we determined the extent of subunit contact in lipid-free apoE3 CT domain by identifying the relative accessibilities to hydrogen/deuterium exchange (HDX) by mass spectrometry (MS). Further, we monitored changes in fluorescence polarization (FP) of probes placed on specified sites to evaluate the order of unfolding of the helices, which may be a predictor of the order of lipid binding. Lastly, we tested the hypothesis that the subunits dissociate to a monomeric form to facilitate lipid interaction by assessing the lipid-binding ability of the CT domain that has been ‘locked’ at specified sites along the class A and G* helices.
2. MATERIALS AND METHOD
2.1. Expression, isolation, and purification of apoE(201–299)
A pET22b expression vector encoding wild-type (WT) human apoE(201– 299) (also referred to as apoE CT domain) or apoE(201–299) bearing single Cys substitutions: A209C, S223C, E255C, and A277C, with an N-terminal hexa-His tag was used as described previously [25]. The proteins were over-expressed in E. coli, isolated and purified using a Ni-affinity matrix (Hi-Trap chelating column; GE Healthcare). Protein purity was verified by SDS-PAGE analysis using a 4–20% acrylamide gradient under reducing conditions. Western blot analysis was carried out using anti-His-tag antibodies followed by goat anti-mouse IgG (IR dye, 680RD) (LI-COR, Superior St. Lincoln, NE).
2.2. Disulfide bond formation, NEM and fluorescence labeling
Typically, the purified single Cys apoE CT variants were engaged in disulfide bond formation during the purification process. Where indicated, the samples (5 mg protein in 10 mM ammonium bicarbonate, pH 7.4) were treated with 6 M GdnHCl (Applied Biosystems, Eugene, OR), and 5-fold molar excess of dithiothreitol (DTT) for 2 h at 37°C and labeled with 25-fold excess NEM for 17 h at 37°C. In other cases, they were reduced with 2-fold excess of tris(2-carboxyethyl)phosphine hydrochloride (Sigma-Aldrich, St. Louis, MO) and treated with 5-fold excess 1,5-IAEDANS, 5-((((2-Iodoacetyl)amino)ethyl)amino)napthalene-1-sulfonic acid (IAEDANS) (Invitrogen Molecular Probes, Eugene, OR) for 2 h at 37°C. All samples were dialyzed extensively against 10 mM ammonium bicarbonate, pH 7.4. The extent of labeling was calculated using the molar extinction coefficients of apoE(201–299) (ε280 nm =16,500 M−1 cm−1) and IAEDANS (ε340 nm = 5,700 M−1cm−1).
2.3. Gel filtration chromatography
A277C/apoE(201–299) was subjected to gel filtration chromatography on a Superdex 200 10/30 G/L column by FPLC (Amersham Biosciences, Piscataway, NJ) in the disulfide bonded or DTT/NEM treated state in PBS (flow rate of 0.5 ml/min).
2.4. HDX coupled to MS
HDX was initiated by a 10-fold dilution of cold WT apoE(201–299) (0.1 mg/ml in 10 mM sodium phosphate buffer, pH 7.4) into deuterated buffer (10 mM sodium phosphate buffer, pD 7.4, 25°C). At this concentration, apoE exists as a tetramer as observed by earlier studies from other labs [26]. At various time points (30 – 7200 s), the exchange-in reaction was arrested with cold deuterated phosphate buffer, pD 2.5, and flash frozen with liquid nitrogen until further use. Following deuteration the flash frozen, quenched samples were injected into a Porosyme Immobilized Pepsin Cartridge (Applied Biosystems, Eugene, OR). The resulting peptides from peptic cleavage were separated on a nanoAcquity UPLC BEH C18 1.7 µm analytical column (Waters, Milford, MA) operating at 50 µL/s, coupled to a Synapt G1 electrospray ionization Quadrupole time-of-flight mass spectrometer (Waters, Milford, MA). The gradient used for elution was set at 40 µL/min flow rate using solvent A (LC-MS grade water, 0.1% formic acid) and solvent B (acetonitrile, 0.1% formic acid). Gradient settings were 8–40% solvent B in 6 min, 40–85% solvent B at 7 min, isocratic flow of 85% solvent B for 2 min, returning to 8% solvent B in 1 min. Back exchange was prevented by lowering the HDX workflow to ~0°C during analysis.
The mass of each peptide was identified with the data acquisition software, MassLynxTM. The centroid m/z peak of each isotopic envelope taken from each mass spectrum was used to calculate the peptide mass and thus D uptake. Percent D (% D) incorporation of each peptide was calculated as:
where Hx is the number of exchanged hydrogens in each peptide, M is the molecular mass of the undeuterated species, MD is the measured mass of the partially deuterated species, and N is the total number of amide hydrogens on each peptide.
2.5. GdnHCl-induced unfolding followed by CD, fluorescence intensity (FI) and FP measurements
Unfolding studies were carried out with unlabeled or AEDANS-labeled apoE CT variants (0.2 mg/ml for CD analysis; 0.05 – 0.1 mg/ml for fluorescence analysis (the protein is expected to be in a tetrameric state at these concentrations) in 10 mM ammonium bicarbonate, pH 7.4 as described previously [27].
Far-UV CD scans were obtained on a Jasco J-810–150S spectropolarimeter at 24 °C using a cylindrical cell (Hellma Cells, Plainview, NY), with a path length of 0.2 mm. The scans were recorded at 20 nm/min, response time of 1 s, bandwidth of 1 nm; an average of 4 scans were recorded from 260 nm-185 nm. The molar ellipticity ([θ]) in deg.cm2dmol−1 at 222 nm was obtained using:
where MRW is the mean residue weight (molecular weight divided by the number of residues), θ is the measured ellipticity in degrees at 222 nm, l is the cuvette path-length (in cm) and c is the protein concentration (in g/ml). The percent α-helix content was calculated as described previously [34].
FI measurements of AEDANS labeled apoE CT variants were carried out on a Perkin-Elmer LS55B fluorometer at 24 °C. Fluorescence emission spectra were recorded in 10 mM sodium phosphate buffer, pH 7.4, containing 150 mM NaCl (phosphate buffered saline, PBS) between 350 and 600 nm following excitation at 340 nm, at a scan speed of 50 nm/min (3 nm excitation and emission slit widths); an average of 10 scans were recorded.
FP measurements of AEDANS labeled apoE CT variants were carried out on a Perkin-Elmer LS55B fluorometer or FluoroMax-2 spectrometer (Horiba Scientific, Edison, NJ) at 24 °C. The excitation and emission wavelengths were 340 nm and 480 nm, respectively, with an integration time of 0.1 s and slit width of 5 nm. The % maximal change in the read outs at each concentration of GdnHCl was calculated as shown below for ellipticity values at 222 nm (θ):
| (1) |
where θ is the molar ellipticity at different GdnHCl concentrations, θN is that of the native protein, and θD that of the completely denatured protein prevailing under the conditions employed. Similar calculations were carried out for changes in FI or FP values at 480 nm.
2.6. Lipid binding assay
The ability of apoE(201–299) to convert DMPC (Avanti Polar Lipids, Alabaster, AL) MLV to discoidal lipoprotein complexes was determined as described previously [27–31] with some modifications. The assay was performed in a Perkin Elmer spectrophotometer equipped with a Peltier controlled (PTP-6) cell holder. DMPC MLV (100 μg) were preincubated at 23.7 °C in PBS in a cuvette followed by addition of 300 μg of WT or single Cys containing apoE(201–299). Prior to use, the protein samples were freshly re-natured following denaturation with 3 M GdnHCl (1 h at 25°C) and dialyzed against 10 mM ammonium bicarbonate, pH 7.4, 24 – 48 h. The decrease in absorbance was measured at 325 nm as a function of time. Data were normalized to the initial absorbance at 325 nm prior to addition of protein, and the T1/2 and rate constant K (reciprocal of T1/2), were determined.
2.7. Statistical analysis
Data are representive of three independent experiments. Statistical analysis was performed using Student’s t-test; P < 0.05 was considered to be statistically significant.
3. RESULTS
3.1. Solvent accessibility assessed by HDX/MS
The isolated CT domain (also referred to as apoE(201–299) without the isoform designation since this segment is common to the major isoforms) bearing the high-affinity lipid binding sites of apoE3 recapitulates the functional features associated with the intact protein in terms of its ability to facilitate protein-protein interaction and promote tetramerization and oligomerization. However, the mechanistic details of the protein-protein or protein-lipid interaction are not known at the sub-domain or segmental level.
We assessed the solvent accessibility of apoE(201–299) by HDX/MS to determine if specified segments are engaged in stronger protein-protein interactions than others. Table 1 shows the list of peptides obtained following pepsin cleavage of apoE(201–299) at pH 2.5. Of the 99 residues, 84 were identified with several overlapping peptides, leading to a robust sequence coverage of ~85%, as shown in Figure 1. From this set, 19 peptides that would cover most of the CT domain and reflect HDX/MS dynamics were selected for further analysis.
Table 1.
Peptides derived from peptic digestion of deuterated apoE(201–299).
| Peptide # | Amino Acid Position | Sequence | Predicted Ave Mass (Da) | Peptide Length | Amide Hydrogens | |
|---|---|---|---|---|---|---|
| 1 | 206 | 214 | RAQAWGERL | 1086.2 | 9 | 8 |
| 2 | 206 | 216 | RAQAWGERLRA | 1313.5 | 11 | 10 |
| 3 | 206 | 218 | RAQAWGERLRARM | 1600.8 | 13 | 12 |
| 4 | 217 | 229 | RMEEMGSRTRDRL | 1636.9 | 13 | 12 |
| 5 | 217 | 231 | RMEEMGSRTRDRLDE | 1881.1 | 15 | 14 |
| 6 | 219 | 229 | EEMGSRTRDRL | 1349.5 | 11 | 10 |
| 7 | 219 | 231 | EEMGSRTRDRLDE | 1593.7 | 13 | 12 |
| 8 | 230 | 249 | DEVKEQVAEVRAKLEEQAQQ | 2327.5 | 20 | 19 |
| 9 | 232 | 243 | VKEQVAEVRAKL | 1369.6 | 12 | 11 |
| 10 | 232 | 249 | VKEQVAEVRAKLEEQAQQ | 2083.3 | 18 | 17 |
| 11 | 250 | 254 | IRLQA | 599.7 | 5 | 4 |
| 12 | 253 | 261 | QAEAFQARL | 1033.1 | 9 | 8 |
| 13 | 253 | 263 | QAEAFQARLKS | 1248.4 | 11 | 10 |
| 14 | 262 | 270 | KSWFEPLVE | 1134.3 | 9 | 7 |
| 15 | 262 | 272 | KSWFEPLVEDM | 1380.6 | 11 | 9 |
| 16 | 265 | 270 | FEPLVE | 732.8 | 6 | 4 |
| 17 | 265 | 272 | FEPLVEDM | 979.1 | 8 | 6 |
| 18 | 271 | 279 | DMQRQWAGL | 1104.2 | 9 | 8 |
| 19 | 272 | 279 | MQRQWAGL | 989.2 | 8 | 7 |
Peptides derived from peptic digestion of deuterated apoE(201–299). The sequence, length, predicted mass and the number of exchangeable amide hydrogens of deuterated peptides are shown. The peptides shown in this Table were used for Protection Plot analysis. Average mass takes into account all major isotopes of the elements.
Figure 1.
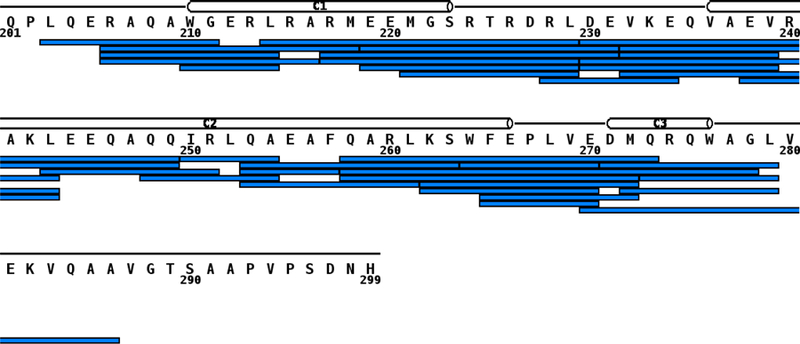
Sequence coverage of peptic digestion of apoE(201–299). On-line digestion of apoE(201–299) using pepsin at pD 2.5 produced the peptic fragment map. Each blue bar represents a peptide that was identified by MS. The secondary structure is shown as cylinders for α-helices based on NMR structure of monomeric apoE3 (PDB 2L7B) [11]. The helices are numbered C1 (W210-S223); C2 (V236-E266) and C3 (D271-W276).
The HDX data was analyzed using MSTools [32] and a protection plot generated for the peptide fragments, Figure 2. HDX/MS data are not available for the segment 280–299. In general, it can be seen that the central portion encompassing residues 230–260 has a relatively higher %D uptake (40–60%) indicative of a lower extent of protection and increased solvent accessibility compared to the N-terminal segment (201–230) and the C-terminal segment (260 to 280), both of which display a relatively lower extent of uptake (10–30%). This suggests that the terminal segments are engaged in significant protein-protein interactions. Earlier studies from other lab support this conclusion especially with regard to the C-terminal segment beyond residue 266 [21, 22].
Figure 2.
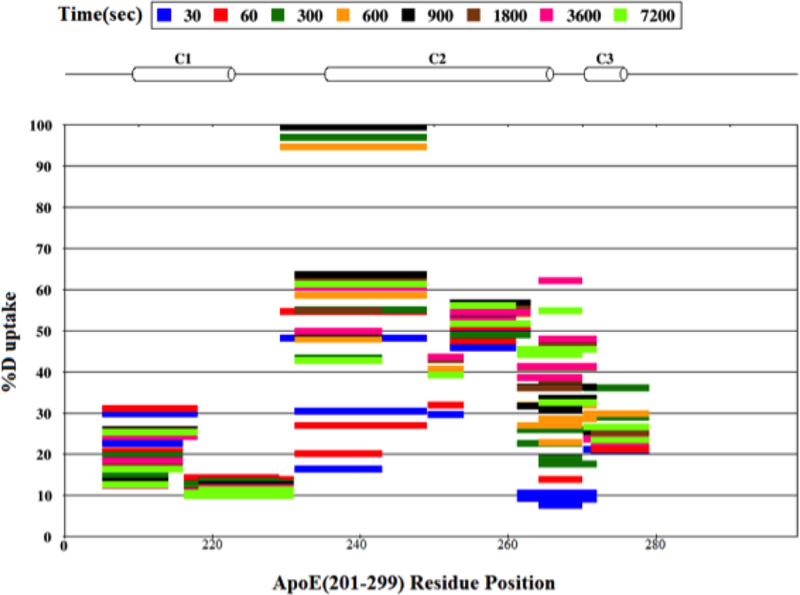
Protection plot of apoE(201–299). The %D uptake of each peptide is shown as a function of time. Peptides are color coded with respect to each time point (30, 60, 300, 600, 900, 1800, 3600 and 7200 s) and presented as horizontal bars. The horizontal cylinders represent α-helical structure (C1, C2 and C3) based on NMR structure of apoE3 (PDB ID: 2L7B).
3.2. Order of GdnHCl-induced unfolding of helices
To understand the extent of tertiary contact and to assess if there are differences in the stability within the sub domains of apoE(201–299), we monitored guanidine hydrochloride (GdnHCl)-induced changes in the order of helix unfolding. Specified segments in the CT domain were sampled by covalently labeling single substituted Cys with AEDANS. The rationale for this approach is that identifying the ease of unfolding is reflective of the ease of binding to lipids, since less structured segments tend to seek stability by lipid binding interaction [28]. The localized changes in environment and mobility were followed by monitoring changes in the fluorescence behavior of AEDANS. Specifically, we: (i) determined if the presence of AEDANS altered the secondary structure and overall fold of the CT domain by monitoring GdnHCl-induced changes in secondary structure by circular dichroism (CD) analysis; and, (ii) identified GdnHCl-induced changes in probe’s fluorescence characteristics and mobility as reflectors of change in localized structure by monitoring changes in FP values.
The location of the single Cys in the variants that were generated sampled different segments of the CT domain. Wild type (WT) apoE(201–299) (with no endogenous Cys) was used as control where relevant. The probe of choice was IAEDANS, an environmentally sensitive fluorophore that has been used to monitor protein dynamics [33]. SDS-PAGE analysis of AEDANS-labeled variants was carried out under reducing conditions Figure 3. A major band corresponding to ~13 kDa was noted in all cases. The stoichiometry of labeling for all variants was ~1.0 indicative of near complete labeling, with the exception of apoEA277C(201–299), which was ~0.5; this is likely due to lower solvent accessibility in this segment of the protein.
Figure 3.
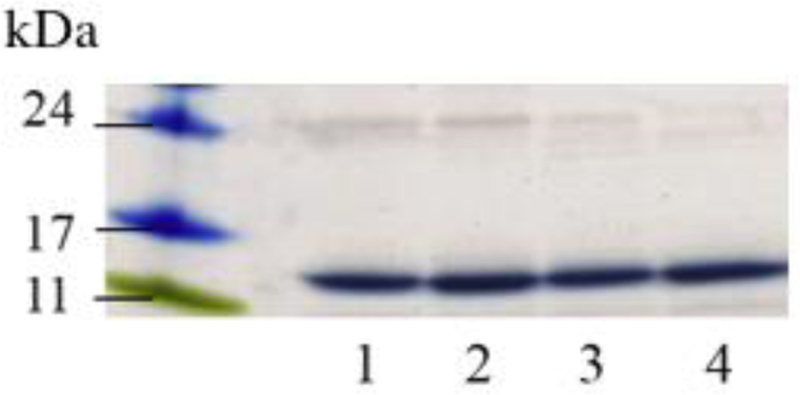
SDS-PAGE of AEDANS-labeled single Cys variants of apoE(201–299). About 10 μg of the purified, labeled variants were electrophoresed on a 4–20% acrylamide gradient SDS-PAGE gel under reducing conditions. Lane assignments following the molecular mass standards are: Lane 1, A209C; lane 2, S223C; lane 3, E255C; lane 4, A277C.
Far-UV CD analysis of AEDANS-labeled variants revealed a typical α-helical profile characterized by troughs at 208 and 222 nm (~65% α-helical content), comparable to that of WT apoE(201–299) (~60%). Unfolding studies of the AEDANS-labeled variants were carried out with increasing concentrations of GdnHCl, following treatment with TCEP to reduce any residual S-S bonds. The denaturation profiles as followed by CD were monophasic for all the labeled variants, with the mid-point of denaturation (D1/2) between 1.2 and 1.5 M GdnHCl, Figure 4A, which is comparable to that for WT apoE(201–299) CT (1.2 M). This suggests that covalent attachment of AEDANS at the various Cys substituted locations did not significantly alter the overall fold and secondary structure of apoE CT domain.
Figure 4.
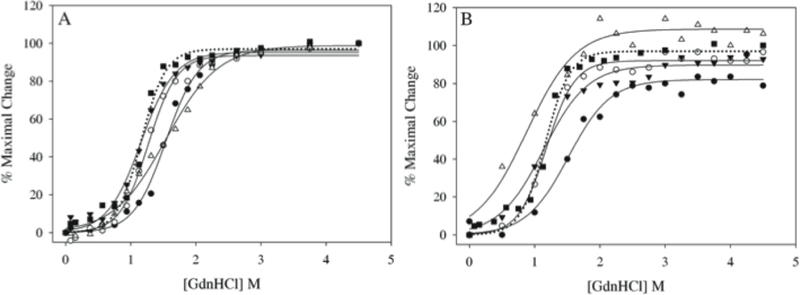
GdnHCl-induced unfolding of AEDANS-labeled single Cys variants of apoE(201–299). AEDANS-labeled apoE(201–299) variants (0.2 mg/ml) were treated with 5-fold molar excess of TCEP and increasing concentrations of GdnHCl. The unfolding process was followed as changes in: (A) molar ellipticity, and, (B) FP values. All values are plotted as % maximal change in a specific signal versus [GdnHCl]. The data were fitted in Sigma Plot using best-fit dynamic curve fitting. The designations of the different apoE(201–299) constructs are as follows: filled circle, A209C; open circle, S223C; filled triangle, E255C; open triangle, A277C; filled square, WT. In Panel B, the unfolding of WT apoE(201–299) followed as changes in molar ellipticity by CD is included (dotted line, filled square) for comparison.
With regard to GdnHCl-induced changes in the AEDANS fluorescence emission characteristics, a decrease in fluorescence emission intensity and/or a shift in λmax of 10–20 nm towards longer wavelengths were noted for all variants (data not shown), indicative of relocation of the probe to a more polar microenvironment. The decrease in emission intensity is attributed to quenching by water as the probe is exposed to the aqueous environment, while the red shift is attributed to solvent relaxation effect in the excited state.
The data on GdnHCl-induced changes in FP values revealed interesting details in probe’s mobility and dynamics, Figure 4B. In general, all variants displayed a high FP value: 0.351, 0.356, 0.364 and 0.336 for probes attached at positions 209, 223, 255 and 277, respectively. This is possibly due to their location in highly structured segments in the domain. The corresponding D1/2 and ΔGD(H2O)values were A209C: 1.56 M and 3.9 kcal/mol; S223C: 1.19 M and 4.34 kcal/mol; E255C: 1.16 M and 2.67 kcal/mol; A277C: 0.88 M and 1.40 kcal/mol. The D1/2 values for AEDANS-labeled A277C obtained by following changes in molar ellipticity was ~ 2-fold higher than that obtained by following change in FP values. Further, the D1/2 values obtained by monitoring FP values reveal the following decreasing order: A209C > E255C = S223C > A277C, suggesting a defined order of unfolding of the helices.
3.3. Dissociation to monomeric forms essential for lipid binding
We then posited that the differences in the stabilities between segments within the CT domain would correlate with their lipid binding abilities. Previous studies have suggested that upon solubilization of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) vesicles, apoE(201–299) forms discoidal lipoprotein complexes, wherein a pair of apoE molecules circumscribes a bilayer of phospholipids [34]. In such an organization, the end product still shows helix-helix interaction, though it is not known if the intermolecular interactions in the lipid-free protein are retained or altered after dissociation of the tetramer and re-association at the periphery of the particle. To address the scenario that they are retained, two monomeric units of apoE CT domain were “locked” by a single disulfide bond located either towards the ends or the central part of the single Cys constructs and its effect evaluated by monitoring the rate of lipid binding.
Although there were some variability in the extent of disulfide bond formation amongst the different constructs, likely a result of sample handling conditions during purification, previous studies from our lab [35] showed that the protein preparations following affinity purification were in a predominantly disulfide bonded state (with the exception of E255C, which had ~50% non-disulfide bonded species). In initial DMPC solubilization assays, we used the samples as such after purification, Figure 5 (curves a – d). As controls, WT apoE(201–299) (curve e) and apolipoprotein AI (apoAI) (curve f), an exchangeable apolipoprotein that is known to be very efficient in lipid binding, were employed. Addition of apoAI to DMPC multilamellar vesicles (MLV) under the conditions described caused a rapid decrease in absorbance with the time required for initial absorbance to decrease by 50% (T1/2) being 164 s. In comparison, WT apoE(201–299) reveals a T1/2 of 1,338 s (the corresponding rate constants, K, are shown in Table 2). The single Cys proteins display a range of abilities with T1/2 of > 3,000 s for A209C, 2,688 s for S223C, 1,422 s for E255C, and > 3,000 s for A277C. All (except E255C) displayed a lower ability to mediate vesicle solubilization compared to WT apoE(201–299).
Figure 5.
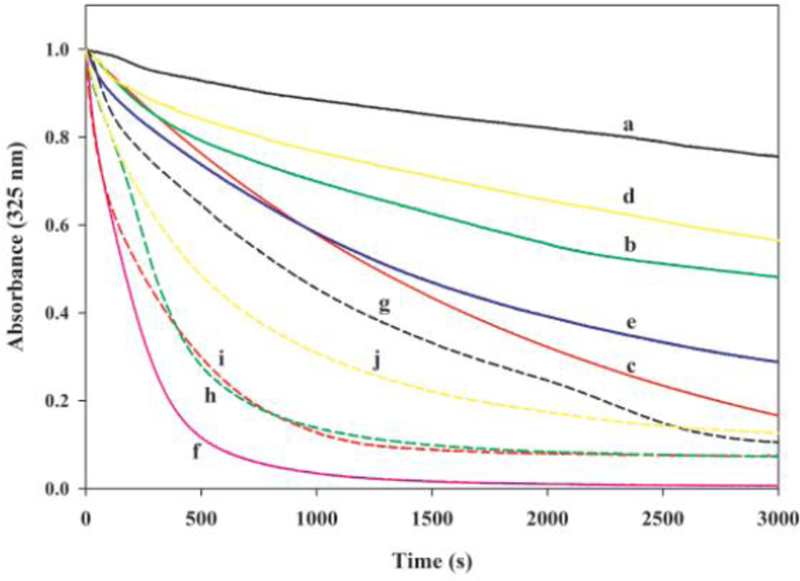
Effect of “locking” apoE CT domain at various locations on DMPC solubilization. DMPC MLV (100 μg) were treated with 300 μg of apoE(201–299) bearing single Cys at different locations in the absence (a - d) or presence (g - j) of DTT/NEM: (a) A209C; (b) S223C; (c) E255C; (d) A277C; (e) WT apoE(201–299); (f) apoAI; (g) A209C/DTT/NEM; (h) S223C/DTT/NEM; (i) E255C/DTT/NEM; and, (j) A277C/DTT/NEM.
Table 2.
Comparison of DMPC solubilization abilities of disulfide-bonded and DTT/NEM treated apoE(201–299) variants.
| apoE(201–299)1 | T1/2 (s) | K (s−1) |
|---|---|---|
| ApoAI2 | 164 | 5.98 × 10−3 |
| A209C | > 3,000 | - |
| S223C | 2,688 | 3.72 × 10−4 |
| E255C | 1,422 | 7.03 × 10−4 |
| A277C | > 3,000 | - |
| WT | 1,338 | 7.20 × 10−4 |
| A209C/DTT/NEM | 862 | 1.16 × 10−3 |
| S223C/DTT/NEM | 290 | 3.45 × 10−3 |
| E255C/DTT/NEM | 234 | 4.27 × 10−3 |
| A277C/DTT/NEM | 472 | 2.11 × 10−3 |
For convenience, only the position of substituted single Cys is shown for each apoE(201–299) variant
ApoAI was used as a control
To verify if reducing the disulfide bond can increase lipid-binding ability, the samples were treated with DTT followed by conjugation with N-ethylmaleimide (NEM), a small molecular weight sulfhydryl-specific compound, which would prevent the reformation of disulfide bonds. This treatment resulted in a significant population of non-disulfide bonded protein (~99%) as seen by SDS-PAGE. DTT/NEM treatment increased the initial rate of lipid binding of all the variants by 5 – 10 fold, Figure 5 (curves g – j). The observed T1/2 were 862 s for A209C/DTT/NEM, 290 s for S223C/DTT/NEM, 234 s for E255C/DTT/NEM and 472 s for A277C/DTT/NEM. The substantial difference in rates between disulfide-bonded and DTT/NEM-conjugated variants suggests that the dissociation of the oligomeric assembly to monomeric units is an essential step in lipid association.
One of the constructs, A277C/apoE(201–299), was then subjected to size-exclusion chromatography to determine size distribution of species in the purified preparation under native (non-denaturing) conditions as a consequence of disulfide bond formation, Figure 6, closed circles. A major peak was noted at 12.5 ml (25 min) (Fractions 23, 24, 25, 26, & 27, red), with a minor peak (Fractions 14, 15 & 16) appearing in the void volume (~8 ml). The molecular masses correspond to ~299,000 and > 500,000 Da, respectively. Western blot of each fraction (Figure 6, left inset, fractions 14–16 and 23–27) following SDS-PAGE under non-reducing conditions revealed an ~30 kDa band, indicative of the presence of disulfide-bonded states. It appears that the major peak represents disulfide-bonded states not only between monomeric units in a tetramer, but also between ~6 neighboring tetramers (1 tetramer = ~52 kDa). The same applies to the minor peak as well.
Figure 6.
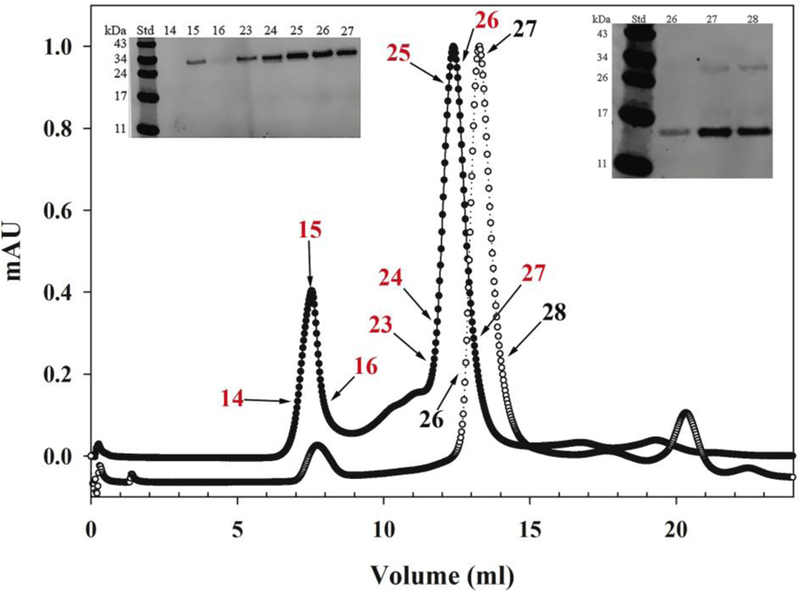
Size-exclusion chromatography of apoEA277C(201–299). Approximately 4 mg of apoEA277C(201–299) were applied on a Superdex 200 10/30 G/L column under non-reducing (filled circles) or reducing (DTT/NEM) (open circles) conditions in PBS and operated at a flow rate of 0.5 ml/min. Inset: Western blot of FPLC fractions of non-reduced (left) and reduced (right) protein. Fraction numbers correspond to those shown on chromatogram (in red for non-reducing and black for reducing conditions). Proteins of known molecular mass (bovine serum albumin (67 kDa), lactate dehydrogenase (140 kDa), catalase (232 kDa), ferritin (440 kDa), and thyroglobulin (669 kDa)) were subjected to a similar separation to estimate the approximate molecular mass of the sample.
Size-exclusion analysis of DTT/NEM treated A277C/apoE(201–299) (Figure 6, open circles) shows a major peak appearing at 13.5 ml (27 min) (Fractions 26, 27 & 28, black). Western blot of these fractions (Figure 6, right inset) following SDS-PAGE under non-reducing conditions reveals a major band at ~13 kDa, indicative of the presence of a predominantly non-disulfide bonded species. The minor peak appearing at 8 ml could not be detected in Western blot. The remaining single Cys variants were not subject to size-exclusion chromatography since we showed that the reduction and NEM treatment was efficient and complete under the conditions employed.
4. DISCUSSION
Conformational flexibility and lipid binding ability are signature features of exchangeable apolipoproteins; the flexibility allows them to adopt different types of lipid-associated structures that vary in size (5 nm to > 200 nm diameter), geometry (discoidal and spherical lipoproteins) and composition (protein, lipid and other components). Inherent in this process is the thought that less structured (therefore less stable) segments of apolipoproteins seek stability via lipid binding prior to those segments that bear strong secondary and tertiary interactions [28]. This is exemplified by observations that the NT domain of apoE3, bearing the helix bundle structure, shows a 2–3 fold higher mid-point of denaturation and a corresponding lower lipid binding affinity compared to the CT domain [15].
Peptide level HDX/MS data suggest that the C-terminal segment of the CT domain (helix C3) bears extensive protein-protein interactions and forms the primary subunit interface in a tetramer (the likely oligomeric state at the concentrations used). This interpretation is also supported by the lower accessibility to labeling by AEDANS at position 277 compared to positions located centrally. These findings are in agreement with earlier studies from our lab that suggest that the entire CT domain dimerizes, and that two dimers further dimerize via the segment bearing the G* helix (267–289) [25, 35]. Further support is derived from truncation [21, 22] and site directed mutagenesis [23, 24], which suggest that residues 267–299 are likely involved in apoE tetramerization. HDX/MS of intact apoE3 performed by others [36] cannot be directly compared with our current study since we examined the isolated CT domain without the confounding influence of the presence of the NT domain. Nevertheless, studies with intact apoE3 show that the CT domain is relatively less structured than the NT domain, an aspect also reported through biophysical analysis of isolated domains [37]. Taken together with our earlier data [25, 35], we suggest a dimer of a dimer model wherein the terminal helices from 4 subunits are juxtaposed perhaps in an intermolecular 4-helix bundle configuration, brought together by two pairs of subunits that maintain close helix-helix interactions at the central segment. Since the %D uptake was roughly twice as high in the central segment compared to the C-terminal end, we propose a refined model wherein the N-terminal end of each dimer is either splayed, Figure 7A, or oriented in an anti-parallel manner, Figure 7B. The current information does not allow us to discriminate between these two possibilities.
Figure 7.
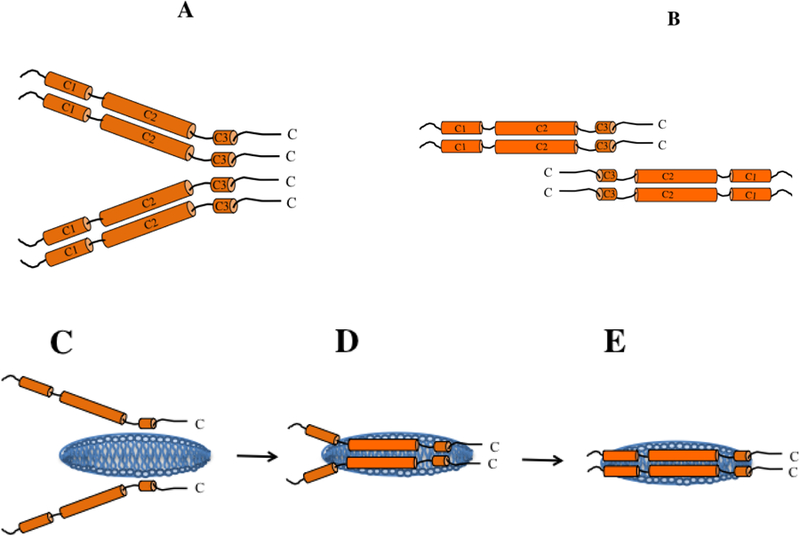
Schematic illustration of subunit organization and mode of lipid binding of apoE CT domain. Subunit organization of lipid-free apoE CT domain is depicted as a dimer of two dimers mediated by helix C3 from each subunit; each pair is aligned in a parallel (A) or antiparallel (B) orientation with respect to each other. Subunit dissociation to monomeric state allows inter-molecular helix-helix interactions to be replaced by helix-lipid interactions as shown schematically from (C) – (E). The discoidal bilayer (blue) is not pre-existing as shown; it is formed as a result of apoE interaction with phospholipid vesicles. The final discoidal particle is estimated to have 4–6 apoE molecules per particle (only two are shown for the sake of simplicity) (not to scale).
The denaturation studies monitored as changes in FP reveal further information regarding the order of unfolding of helices. FP provides a measure of the mobility of the fluorophore, which is susceptible to variation in rotation, viscosity and molecular weight [38, 39]. Implicit in this process is the concept that segments with defined secondary structure (α-helix or β-strand) would be relatively more rigid, while unstructured segments would be less rigid [40]. Thus, if a selected segment within a protein loses its secondary structure, it would be reflected as changes in FP of a probe covalently attached to that segment. The presence of AEDANS at the different locations did not significantly alter the global fold as indicated by the similar extent of denaturant-induced changes in molar ellipticity of labeled proteins. At this point, we cannot disregard the possibility that its presence may cause steric perturbations in its microenvironment and tertiary/quaternary contacts (as discussed below with regard to lipid binding). Nevertheless, a major observation was that the C-terminal end of the protein offers less resistance to GdnHCl-induced unfolding compared to the central portion, as suggested by the ease with which the probe becomes more mobile at lower GdnHCl concentrations when it is located at position 277 (compared to 223 or 255). The N-terminal segment of the CT domain appears to maintain stronger tertiary contacts than the rest of the CT domain. The D1/2 value of AEDANS-A277C obtained by following changes in molar ellipticity is ~2-fold higher than that noted by changes in FP. This suggests that loss of tertiary and quaternary contacts precedes loss of secondary structure, likely reflecting dissociation of tetramers and loss of inter-molecular helix-helix contacts prior to loss of helical structure. Although the accessibility from HDX data is lower around C3 compared to C2, based on the rationale that lower stability is a likely predictor of ease of lipid binding, the order of helix unfolding suggests that C3 likely seeks stability via lipid interaction prior to C1 or C2, Figure 7, C-E. Interestingly, although helix C3 (residues DMQRQW) is a short and relatively polar G* helix that lacks a large apolar surface, it is possible that charge interactions may be involved in initiating lipid binding with the head groups of phospholipids. Such a situation was noted in other studies: the 80’s loop (residues 79–90 with a distribution of –vely charged residues) at one end of the 4-helix bundle of the N-terminal domain of apoE3 [41], the short helix (helix 1’, residues SEQVQEELLS) at the other end of the helix bundle [42], or a short 6-residue helix (PDVEKE) in an insect apolipoprotein [43]. Further studies are needed to determine if the above order is followed in the context of the intact protein as well. It is not known if the presence of the bulky NT domain helix bundle attenuates the order of unfolding of the CT domain in intact apoE3.
In the DMPC vesicle solubilization assay, the presence of inter-molecular disulfide bonds at any of the locations tested along the polypeptide, except at position 255, significantly lowered the initial rate of lipid binding of apoE(201–299). DTT treatment alone was insufficient to determine if the lipid binding could be recovered since DTT is easily air oxidized and the proteins revert to the disulfide-bonded state (due to proximity between neighboring sulfhydryls) during the course of the vesicle solubilization assay (our unpublished observations). Therefore, it was necessary to ‘cap’ the reduced proteins with NEM to prevent reoxidation. This treatment increased the ability of all the variants to mediate vesicle solubilization. As a result, all capped variants displayed a higher efficiency of vesicle solubilization compared to WT apoE(201–299); this is likely due to steric hindrance offered by the conjugated NEM, which allows the tetrameric subunits (expected state at the concentrations used in this assay) to dissociate to monomeric subunits in a facile manner, a caveat to bear in mind. Further, the presence of a disulfide bond towards the N- or C-terminal end of apoE(201–299) (at positions 209 or 277) causes more impairment in the initial rate of binding compared to those located in the central portion. This suggests that lipid binding is initiated at one end of apoE(201–299); considering that in case of the intact protein, the NT domain bearing a highly stable and large helix bundle is located towards the N-terminal end of apoE(201–299), we propose that the lipid binding is initiated at the C-terminal end of the CT domain, Figure 7C.
5. CONCLUSION
In summary, our results suggest that the tetramer is formed by dimerization of two dimers via the C-terminal segment of apoE CT domain. Dissociation of the tetramer to a monomeric form is essential for lipid binding since disulfide bonding restricts the complete dissociation to two monomeric units. This is consistent with other studies that use a kinetic approach to show the importance of the monomeric state of apoE3 for lipid binding [44]. We propose that the lipid binding interaction is initiated by the C-terminal end of the protein, followed by the central portion of the CT domain. Further studies are needed to determine if the lipid-induced reconfiguration involves retention, alteration or re-establishment of inter-molecular interactions of the lipid-free protein.
Acknowledgements
We thank Tuyen N. Tran for his technical assistance and help with data analysis. CSF, PNN, RVH, SN and MD performed the research; RVH, SN, CSM and VN provided intellectual contribution and were involved in project design and data analysis; CSF, RVH and VN prepared the manuscript.
Footnotes
References:
- [1].Lane-Donovan C; Philips GT; Herz J More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron, 2014, 83, 771–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hottman DA; Chernick D; Cheng S; Wang S; Li L HDL and cognition in neurodegenerative disorders. Neurobiol. Dis, 2014, 72, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vitali C; Wellington CL; Calabresi L HDL and cholesterol handling in the brain. Cardiovasc. Res, 2014, 103, 405–413. [DOI] [PubMed] [Google Scholar]
- [4].Phillips MC Apolipoprotein E isoforms and lipoprotein metabolism, IUBMB Life, 2014, 66, 616–623. [DOI] [PubMed] [Google Scholar]
- [5].Huang Y; Mahley RW Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis, 2014, 72 Pt A, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Annema W; von Eckardstein A High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ. J, 2013, 77, 2432–2448. [DOI] [PubMed] [Google Scholar]
- [7].von Eckardstein A; Nofer JR; Assmann G High density lipoproteins and arteriosclerosis role of cholesterol efflux and reverse cholesterol transport. Arterioscler. Thromb. Vasc. Biol, 2001, 21, 13–27. [DOI] [PubMed] [Google Scholar]
- [8].Mahley RW; Huang Y; Weisgraber KH Detrimental effects of apolipoprotein E4: potential therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res, 2007, 4, 537–540. [DOI] [PubMed] [Google Scholar]
- [9].Mahley RW; Weisgraber KH; Huang Y Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid. Res, 2009, 50 Suppl, S183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wilson C; Wardell MR; Weisgraber KH; Mahley RW; Agard DA Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science, 1991, 252, 1817–1822. [DOI] [PubMed] [Google Scholar]
- [11].Sivashanmugam A; Wang J A unified scheme for initiation and conformational adaptation of human apolipoprotein E N-terminal domain upon lipoprotein binding and for receptor binding activity. J. Biol. Chem, 2009, 284, 14657–14666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Satio H; Lund-Katz S; Phillips MC Contributions of domain structure and lipid interaction to the functionality of exchangeable human apolipoproteins. Progress in Lipid. Res, 2004, 43, 350–380. [DOI] [PubMed] [Google Scholar]
- [13].Nguyen D; Dhanasekaran P; Phillips MC; Lund-Katz S Molecular mechanism of apolipoprotein E binding to lipoprotein particles. Biochemistry, 2009, 48, 3025–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Narayanaswami V; Maiorano JN; Dhanasekaran P; Ryan RO; Phillips MC; Lund-Katz S; Davidson WS Helix orientation of the functional domains in apolipoprotein e in discoidal high density lipoprotein particles. J. Biol. Chem, 2004, 279, 14273–14279. [DOI] [PubMed] [Google Scholar]
- [15].Narayanaswami V; Kiss RS; Weers PM The helix bundle: a reversible lipid binding motif. Comp. Biochem. Physiol. A. Mol. Integr. Physiol, 2010, 155, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Narayanaswami V; Szeto SS; Ryan RO Lipid association-induced N- and C-terminal domain reorganization in human apolipoprotein E3. J. Biol. Chem, 2001, 276, 37853–37860. [DOI] [PubMed] [Google Scholar]
- [17].Gupta V; Narayanaswami V; Budamagunta MS; Yamamato T; Voss JC; Ryan RO Lipid-induced extension of apolipoprotein E helix 4 correlates with low density lipoprotein receptor binding ability. J. Biol. Chem, 2006, 281, 39294–39299. [DOI] [PubMed] [Google Scholar]
- [18].Lu B; Morrow JA; Weisgraber KH Conformational reorganization of the four-helix bundle of human apolipoprotein E in binding to phospholipid. J. Biol. Chem, 2000, 275, 20775–20781. [DOI] [PubMed] [Google Scholar]
- [19].Segrest JP; Jones MK; De Loof H; Brouillette CG; Venkatachalapathi YV; Anantharamaiah GM The amphipathic helix in the exchangeable apolipoproteins: a review of secondary structure and function. J. Lipid. Res, 1992, 33, 141–166. [PubMed] [Google Scholar]
- [20].Segrest JP; Garber DW; Brouillette CG; Harvey SC; Anantharamaiah GM The amphipathic alpha helix: a multifunctional structural motif in plasma apolipoproteins. Adv. Protein Chem, 1994, 45, 303–369. [DOI] [PubMed] [Google Scholar]
- [21].Westerlund JA; Weisgraber KH Discrete carboxyl-terminal segments of apolipoprotein E mediate lipoprotein association and protein oligomerization. J. Biol. Chem, 1993, 268, 15745–15750. [PubMed] [Google Scholar]
- [22].Sakamoto T; Tanaka M; Vedhachalam C; Nickel M; Nguyen D; Dhanasekaran P; Phillips MC; Lund-Katz S; Saito H Contributions of the carboxyl-terminal helical segment to the self-association and lipoprotein preferences of human apolipoprotein E3 and E4 isoforms. Biochemistry, 2008, 47, 2968–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fan D; Li Q; Korando L; Jerome WG; Wang J A monomeric human apolipoprotein E carboxyl-terminal domain. Biochemistry, 2004, 43, 5055–5064. [DOI] [PubMed] [Google Scholar]
- [24].Zhang Y; Vasudevan S; Sojitrawala R; Zhao W; Cui C; Xu C; Fan D; Newhouse Y; Balestra R; Jerome WG; Weisgraber K; Li Q; Wang J A monomeric, biologically active, full-length human apolipoprotein E. Biochemistry, 2007, 46, 10722–10732. [DOI] [PubMed] [Google Scholar]
- [25].Choy N; Raussens V; Narayanaswami V Inter-molecular coiled-coil formation in human apolipoprotein E C-terminal domain. J. Mol. Biol, 2003, 334, 527–539. [DOI] [PubMed] [Google Scholar]
- [26].Yokoyama S; Kawai Y; Tajima S; Yamamoto A Behavior of human apolipoprotein E in aqueous solutions and at interfaces. J. Biol. Chem, 1985, 260, 16375–16382. [PubMed] [Google Scholar]
- [27].Tran TN; Kosaraju MG; Tamamizu-Kato S; Akintunde O; Zheng Y; Bielicki JK; Pinkerton K; Uchida K; Lee YY; Narayanaswami V Acrolein modification impairs key functional features of rat apolipoprotein E: identification of modified sites by mass spectrometry. Biochemistry, 2014, 53, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Weers PM; Abdullahi WE; Cabrera JM; Hsu TC Role of buried polar residues in helix bundle stability and lipid binding of apolipophorin III: destabilization by threonine 31. Biochemistry, 2005, 44, 8810–8816. [DOI] [PubMed] [Google Scholar]
- [29].Dettloff M; Niere M; Ryan RO; Luty R; Kay CM; Wiesner A; Weers PM Differential lipid binding of truncation mutants of Galleria mellonella apolipophorin III. Biochemistry, 2002, 41, 9688–9695. [DOI] [PubMed] [Google Scholar]
- [30].Tran TN; Kim SH; Gallo C; Amaya M; Kyees J; Narayanaswami V Biochemical and biophysical characterization of recombinant rat apolipoprotein E: similarities to human apolipoprotein E3. Arch. Biochem. Biophys, 2012, 529, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E Acrolein is a product of lipid peroxidation reaction - Formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. of Biol. Chem, 1998, 273, 16058–16066. [DOI] [PubMed] [Google Scholar]
- [32].Kavan D, Man P MSTools-Web based application for visualization and presentation of HXMS data. International Journal of Mass Spectrometry, 2011.
- [33].Vos WL; Schor M; Baumgaertner A; Tieleman DP; Hemminga MA Molecular dynamics simulations reveal that AEDANS is an inert fluorescent probe for the study of membrane proteins. Eur. Biophys. J, 2010, 39, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Raussens V; Drury J; Forte TM; Choy N; Goormaghtigh E; Ruysschaert JM; Narayanaswami V Orientation and mode of lipid-binding interaction of human apolipoprotein E C-terminal domain. Biochem. J, 2005, 387, 747–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Patel AB; Khumsupan P; Narayanaswami V Pyrene fluorescence analysis offers new insights into the conformation of the lipoprotein-binding domain of human apolipoprotein E. Biochemistry, 2010, 49, 1766–1775. [DOI] [PubMed] [Google Scholar]
- [36].Huang RY; Garai K; Frieden C; Gross ML Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein E oligomerization. Biochemistry, 2011, 50, 9273–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Aggerbeck LP; Wetterau JR; Weisgraber KH; Wu CS; Lindgren FT Human apolipoprotein E3 in aqueous solution. II. Properties of the amino- and carboxyl-terminal domains. J. Biol. Chem, 1988, 263, 6249–6258. [PubMed] [Google Scholar]
- [38].Mocz G Information content of fluorescence polarization and anisotropy. J. Fluoresc, 2006, 16, 511–524. [DOI] [PubMed] [Google Scholar]
- [39].Taraska JW; Zagotta WN Fluorescence applications in molecular neurobiology. Neuron, 2010, 66, 170–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hubbell WL; Cafiso DS; Altenbach C Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol, 2000, 7, 735–739. [DOI] [PubMed] [Google Scholar]
- [41].Segelke BW; Forstner M; Knapp M; Trakhanov SD; Parkin S; Newhouse YM; Bellamy HD; Weisgraber KH; Rupp B Conformational flexibility in the apolipoprotein E amino-terminal domain structure determined from three new crystal forms: implications for lipid binding. Protein Sci, 2000, 9, 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Redmond KA; Murphy C; Narayanaswami V; Kiss RS; Hauser P; Guigard E; Kay CM; Ryan RO Replacement of helix 1’ enhances the lipid binding activity of apoE3 N-terminal domain. FEBS J, 2006, 273, 558–567. [DOI] [PubMed] [Google Scholar]
- [43].Narayanaswami V; Wang J; Schieve D; Kay CM; Ryan RO A molecular trigger of lipid binding-induced opening of a helix bundle exchangeable apolipoprotein. Proc. Natl. Acad. Sci. USA, 1999, 96, 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Garai K; Baban B; Frieden C Dissociation of apolipoprotein E oligomers to monomer is required for high-affinity binding to phospholipid vesicles. Biochemistry, 2011, 50, 2550–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]