

Abstract
Thoracic aortic aneurysms that progress to acute aortic dissections are often fatal. Thoracic aneurysms have been managed with treatment with β-adrenergic blocking agents (β-blockers) and routine surveillance imaging, followed by surgical repair of the aneurysm when the risk of dissection exceeds the risk for repair. Thus, there is a window to initiate therapies to slow aortic enlargement and delay or ideally negate the need for surgical repair of the aneurysm to prevent a dissection. Mouse models of Marfan syndrome—a monogenic disorder predisposing to thoracic aortic disease—have been used extensively to identify such therapies. The initial fi that TGFβ (transformation growth factor-β) signaling was increased in the aortic media of a Marfan syndrome mouse model and that its inhibition via TGFβ neutralization or At1r (Ang II [angiotensin II] type I receptor) antagonism prevented aneurysm development was generally viewed as a groundbreaking discovery that could be translated into the first cure of thoracic aortic disease. However, several large randomized trials of pediatric and adult patients with Marfan syndrome have subsequently yielded no evidence that At1r antagonism by losartan slows aortic enlargement more effectively than conventional treatment with β-blockers. Subsequent studies in mouse models have begun to resolve the complex molecular pathophysiology underlying onset and progression of aortic disease and have emphasized the need to preserve TGFβ signaling to prevent aneurysm formation. This review describes critical experiments that have influenced the evolution of our understanding of thoracic aortic disease, in addition to discussing old controversies and identifying new therapeutic opportunities.
Keywords: aneurysm, aneurysm, dissecting, aorta, Marfan syndrome, therapeutics
Aortic aneurysms involving the aortic root and proximal ascending aorta are generally the result of constant biomechanical forces from pulsatile blood flow from the heart associated with structural weakening of the aortic wall caused by genetic lesions and environmental factors. The natural history of thoracic aortic aneurysms is that they progressively enlarge, and over time, this enlargement increases the risk for an acute ascending (Stanford type A) aortic dissection. An aortic dissection occurs when a tear in the inner layer of the aortic wall allows blood to fl w from the lumen into the wall of the aorta, forming a false lumen. Population-based studies have determined that up to half of individuals affected with a type A dissection die before reaching the hospital and that the mortality and morbidity remains high even after emergent surgical repair of the ascending aorta.1,2 These deaths because of dissection can be avoided if aneurysms are diagnosed and monitored and if surgical repair of aneurysmal segments of the aorta is performed to prevent type A dissections. Surgical repair of the aorta is pursued when vessel dilation reaches approximately twice the normal diameter (5.0–5.5 cm).3 The diameter of the aortic aneurysm is the major criterion for surgical repair, but this size can vary based on the underlying cause of the disease, the rate of growth of the aorta, the status of the aortic valve, symptomatology, and the presence of other cardiovascular disease. Because thoracic aortic enlargement is present in the majority of patients before an acute dissection, there is a window to initiate therapies to slow aortic enlargement and delay or ideally negate the need for surgical repair of the aneurysm to prevent a dissection.
Thoracic Aortic Disease Risk Factors and Current Therapy
Hypertension is the major risk factor for thoracic aortic disease.5–7 Other factors that heighten biomechanical forces of the pulsatile blood (eg, pregnancy, cocaine abuse, body-building weightlifting) also increase the risk for thoracic aortic disease. Genetic predisposition is the second major risk factor. Up to 20% of individuals presenting with an aneurysm or dissection have single-gene mutations that confer a high risk for disease development with or without additional systemic features.8,9 More than 15 genes have been thus far identified that harbor highly penetrant mutations predisposing to thoracic aortic disease.10 The most common congenital heart defect—a bicuspid aortic valve—increases the risk for the disease.11 Additionally, the risk for developing thoracic aortic disease also increases with age, and more men than women are affected. Interestingly, recent data indicate that diabetes mellitus protects from thoracic aortic aneurysms and dissections.12–14 The majority of families with an inherited predisposition for thoracic aortic disease have no associated syndromic features.8 One widely studied exception is Marfan syndrome (MFS), in which the risk for thoracic aortic aneurysms and dissections is associated with additional abnormalities in the eyes, heart, and musculoskeletal system. MFS results from mutations in FBN1 that interfere with fibrillin-1–dependent ECM (extracellular matrix) assembly of functional microfibrils and elastic fibers.15
Thoracic aortic aneurysms and dissection are associated with degenerative pathological changes in the medial layer of aortic wall.16 The aorta has 3 histologically distinct layers: the intima, composed of a single layer of endothelial cells attached to a basal lamina; the media, containing >50 alternating layers of elastic fibers and smooth muscle cells (SMCs) in humans; and the adventitia, made up of a loose connective tissue, fibroblasts, and vasa vasorum (Figure 1). Thoracic aortic disease is associated with genetic alterations believed to primarily impair the contractile function of the SMCs in the medial layer, thus disrupting the proper response to the hemodynamic load constantly imposed on the aortic wall. Among others, major histopathologic findings include fragmentation and loss of elastic fibers, fewer SMCs, and excessive accumulation of collagen (vascular fibrosis) and proteoglycans (Figure 1).17,18 These same histopathologic changes of the medial layer also occur with normal aging but to a less degree and later than in individuals with thoracic aortic disease.
Figure 1.
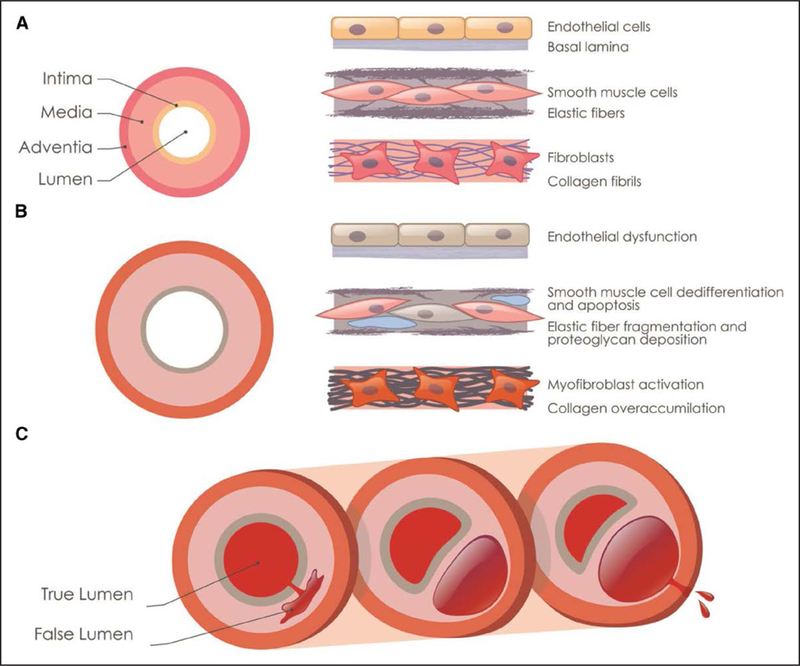
Pathology and progression of thoracic aortic aneurysms and dissections. A, Schematic illustration of the cellular and ECM (extracellular matrix) components in the 3 layers of the thoracic aorta. B, Cellular and ECM changes associated with aneurysm progression are illustrated, including endothelial dysfunction, elastin fiber fragmentation and loss, increased proteoglycan accumulation (blue), and smooth muscle cell loss (gray cell). C, Illustration of an acute aortic dissection because of a tear in intimal layer, progressing through the medial layer to form a false lumen and rupturing from the false lumen through the adventitial layer.
β-Adrenergic blockade (β-blockers) has traditionally been the treatment of choice for thoracic aortic disease.3 The effectiveness of β-blockers in preventing aortic dissection was originally demonstrated >70 years ago, when it was determined that turkeys eating sweet pea (Lathyrus odoratus) seeds, which contain the lysyl oxidase inhibitor, β-aminopropionitrile, die of acute aortic dissections.19,20 As β-blocker therapy reduces the rate of change in central aortic pressure with respect to time, treatment with the β-blocker propanol dramatically decreased deaths from dissection in β-aminopropionitrile–fed turkeys.21 Based, in part, on these data, an open-label, randomized β-blocker treatment trial was pursued in patients with MFS, which showed that propranolol decreased the rate of growth of the aortic root and reduced aortic complications.22 Additional studies interrogating β-blocker therapy to prevent aortic growth and events in patients with MFS have yielded mixed results.23–25
Losartan Controversy
Two mouse models of MFS (Fbn1mgR/mgR and Fbn1C1039/+ mice) have been widely used for ≈2 decades to identify molecular pathways associated with onset and progression of thoracic aortic disease. Fbn1mgR/mgR mice produce significantly less fibrillin-1 than wild-type animals and rapidly form thoracic aortic aneurysms that dissect and rupture within the first 6 to 8 months of postnatal life.26 Fbn1C1039/+ mice instead produce equal amounts of normal and mutant fibrillin-1 and slowly develop thoracic aortic aneurysms that rarely progress to dissection.27 The initial finding that TGFβ (transformation growth factor-β) signaling was increased in the aortic media of Fbn1C1039G/+ mice and that its inhibition via TGFβ neutralization or At1r (Ang II [angiotensin II] type I receptor) antagonism prevented aneurysm development was generally viewed as a groundbreaking discovery that could be translated into the first cure of a heritable thoracic aortic disease.27 However, several large randomized trials of pediatric and adult patients with MFS have subsequently yielded no evidence that At1r antagonism by losartan slows aortic enlargement more effectively than conventional treatment with β-blockers (Table 1).28–31 Several explanations have been invoked to reconcile this discrepancy between experimental findings and clinical trials; among others, they include differences in drug dosing and mode of administration, as well as stage of the disease and genetic variability of the cohorts studied.32,33 The presence of 2 At1rs in rodents (At1ar and At1br) and only 1 in humans (AT1R) could potentially be another factor accounting for the diverse outcome of losartan treatment in the 2 species.
Table 1.
Randomized Clinical Trials of Losartan in Marfan Syndrome*
| Trial Name | Trial Type | Drugs Tested | Dose Range | No. Enrolled | Enroll Age, y | Follow-Up, y | Primary Outcome | Results | Notes |
|---|---|---|---|---|---|---|---|---|---|
| Pediatric Heart Network28,29 | Double blind, stratified | Atenolol | 0.5–4.0 mg/kg per d | 608 | 0.5–25 | 3.0±0.1 | Rate of change in root Z score per year | Not significant (P=0.08) | FBN1 sequencing and pharmacogenomics studies pending |
| Losartan | 0.4–1.4 mg/kg per d | ||||||||
| COMPARE33 | Open label | Losartan | 50 or 100 mg/d | 233 | >18 | 3.1±0.4 | Change in absolute root diameter | P=0.014 | Significant only in individuals with FBN1 haploinsufficiency mutations |
| No additional drug | |||||||||
| Spanish31 | Double blind | Atenolol | 25–100 mg/d | 140 | 5–60 | 3.0 | Change in absolute diameter or Z score of root and ascending aorta | Not significant (P=0.193) | Ambulatory blood pressures were not different between groups |
| Losartan | 25–100 mg/d | ||||||||
| Marfan Sartan30 | Double blind | Losartan | 50 or 100 mg/d | 299 | >10 | 3.5 | Rate of change in root Z score per year | Not significant (P=0.36) | 100% sequenced and 78% had FBN1 mutations |
| Placebo | |||||||||
| Taiwanese34 | Open label | Losartan+BB | Losartan: 25–100 mg/d or 0.7 mg/kg per d to 50 mg/d | 29 | P=0.02 | ||||
| BB: 50 mg atenolol/20 mg propranolol or 1 mg/kg per d | |||||||||
| BB | Any dose <150 mg/d or <2 mg/kg/d | ||||||||
| Placebo | |||||||||
| Ghent Marfan Trial35 | Double blind, stratified | Losartan+BB | Losartan: 50 or 100 mg/d | 22 | >10 | 3 | Rate of change in absolute root diameter or Z score per year | Not significant | |
| Placebo+BB | mg/d BB: NA |
BB indicates β-blocker; COMPARE, Cozaar in Marfan Patients Reduces Aortic Enlargement; and NA, not available.
All studies included the modified Ghent criteria as an inclusion criterion.
A role for TGFβ signaling in thoracic aortic disease was further emphasized when additional mutations predisposing to thoracic aortic disease were identified that disrupt proteins involved in canonical TGFβ signaling.36–39 A paradox also emerged in the field because mutations in these genes decrease rather than increase TGFβ signaling.36–39 It should be noted that clinical evidence supporting a TGFβ-driven mechanism of aneurysm formation was largely based on staining of phosphorylated Smad proteins in end-stage diseased aortas.38,39 Below, we discuss studies using mouse models of thoracic aortic disease that have begun to resolve the complex molecular pathophysiology underlying onset and progression of arterial disease, in addition to emphasizing the need to preserve early TGFβ signaling to prevent aneurysm formation.
Initial Evidence Suggesting Excessive TGFβ Signaling Drives Thoracic Aortic Enlargement
TGFβ1, TGFβ2, and TGFβ3 (hereafter collectively referred to as TGFβ) are multifunctional signaling molecules that orchestrate a large variety of physiological processes, including SMC differentiation and ECM assembly and remodeling, in a context-specific manner.40 TGFβ signals through receptor-induced phosphorylation of Smad2/3 proteins followed by formation, nuclear translocation, and binding of activated Smad2/3:Smad4 complexes to specific DNA targets in association with transcriptional activators or repressors (Figure 2). TGFβ can also signal through non-Smad (noncanonical) pathways, including those mediated by MAPKs (mitogen-activated protein kinases) that can transduce Ang II stimuli as well; additionally, TGFβ and Ang II pathways can interact with each other to modulate vascular tone and SMC phenotype (Figure 2).4,41 Work by Habashi et al27 originally showed that either TGFβ inhibition by a neutralizing antibody or At1r blockade by losartan prevented onset of thoracic aortic disease in Fbn1C1039/+ mice. The authors interpreted these findings to indicate that aneurysm formation in MFS is the result of excessive TGFβ synthesis stimulated by heightened At1r signaling and compounded by uncontrolled activation of latent TGFβ improperly released from a fibrillin-1–deficient ECM.27 Several lines of indirect evidence supported a causal relationship between TGFβ hyperactivity and aortic aneurysm onset. First, in vitro binding assays implied that fibrillin-1 can modulate TGFβ bioavailability through interaction with LTBPs (latent TGFβ-binding proteins) that bind and sequester the inactive (C-pro-domain associated) ligand in the ECM (Figure 2).42 TGFβ hyperactivity was also reported in the diseased aortas of patients with heritable forms of thoracic aortic disease caused by mutations in genes encoding proteins not involved in modulating TGFβ bioavailability, such as fibulin-4 and the smooth muscle-specific isofoms of α-actin and myosin heavy chain.43–45 Surprisingly, TGFβ hyperactivity was also reported in the diseased aortas of patients and mice with heterozygous loss-of-function mutations in genes encoding components of canonical TGFβ signaling pathway, such as ligands, receptors, and signal transducers.38,39 Lastly, aneurysmal tissue and SMC cultures derived from patients with MFS had been shown to contain abnormally high levels of phosphorylated Smad2 due in part to TGFβ-independent epigenetic upregulation of the corresponding gene.46 Hence, the proposal has been made of grouping together these genetically and clinically distinct disease under the rubric of TGFβ signalopathies, thus implying the possible use of a common anti-TGFβ pharmacotherapy.47 It was, therefore, surprising when Holm et al48 reported that aortic disease was dramatically augmented in Fbn1C1039G/+ mice haploinsufficient for the obligatory TGFβ and BMP (bone morphogenetic protein) signal transducer Smad4.48 Nonetheless, the authors concluded that TGFβ signaling through noncanonical signaling pathways is the predominant driver of aneurysm formation in MFS because inhibition of Erk1/2 (extracellular signal–regulated kinase) or Jnk1 (Jun N-terminal kinase) mitigated arterial disease in Fbn1C1039G/+ and Fbn1C1039G/+;Smad4−/− mice, respectively.48
Figure 2.
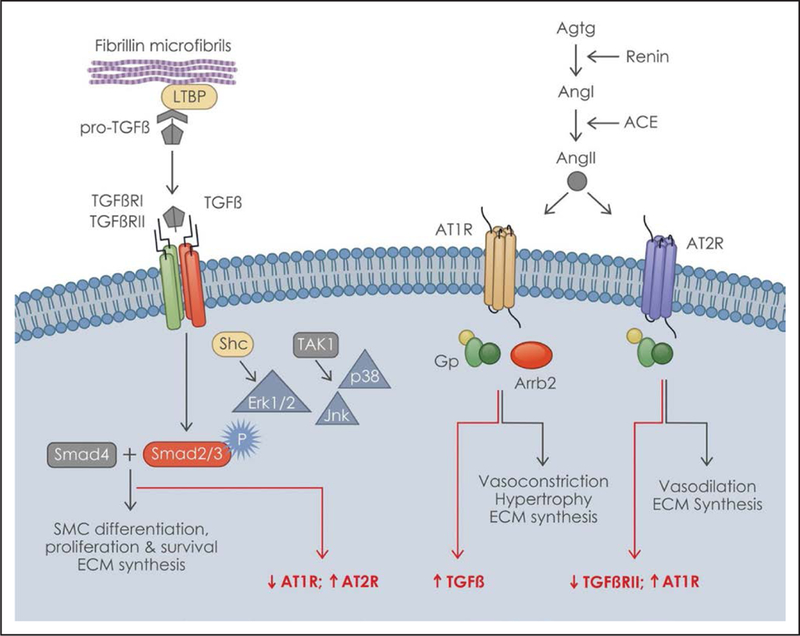
TGFβ (transformation growth factor-β) and angiotensin signaling pathways. Depicted on the (left) and (right) are the TGFβ- and Ang II (angiotensin II)-signaling pathways, respectively, and their effects on aortic homeostasis. The interaction of the latent TGFβ complex with the extracellular fibrillin-containing microfibrils is shown outside the cell, whereas the interaction between TGFβ canonical (Smad) and noncanonical (MAPKs [mitogen-activated protein kinases]) signaling pathways is shown within the cell. Some of the regulatory interactions between the TGFβ and Ang II pathways are indicated in red. Ang I indicates angiotensin I; Arrb2, beta-arrestin-2; AT1R, angiotensin II type I receptor; AT2R, angiotensin II type II receptor; ECM, extracellular matrix; Erk1/2, extracellular signal–regulated kinase; Jnk, Jun N-terminal kinase; LTBP, latent TGFβ-binding protein; Shc, Src homology and collagen family of docking proteins; SMC, smooth muscle cell TAK1, TGFβ-activated kinase; TGFβRI, TGFβ receptor type I; and TGFβRII, TGFβ receptor type II. Modified from Yu and Jeremy with permission.4 Copyright ©2018, the Authors.
Emerging Evidence of an Alternative Role of TGFβ Signaling in Thoracic Aortic Disease
More recent studies of genetically engineered mice have substantially revised the original proposal of Habashi et al27 that placed TGFβ central to the development of thoracic aortic disease. Neutralization of TGFβ signaling starting at postnatal day 16 in Fbn1mgR/mgR mice was in fact found to accelerate rather than mitigate aneurysm formation, thus leading to earlier dissection and death of these MFS mice.49 Similarly, the studies by Li et al50, Hu et al51, and Wei et al52 have independently demonstrated that genetic inhibition of TGFβ signaling via postnatal Tgfbr2 gene inactivation in SMCs of newborn Fbn1C1039G/+ mice worsened rather than mitigated thoracic aortic disease, as originally reported.27 Surprisingly, phosphorylated Smad2 and Erk1/2 levels were not increased in the dilating aortas of young Fbn1C1039G/+ mice.52 The additional finding that SMC-specific Tgfbr2 inactivation in young wild-type mice promoted aneurysm formation and dissection has demonstrated that TGFβ signaling is required for postnatal aortic growth and homeostasis.50,51 TGFβ2 is likely to be the signaling molecule required for normal development of the aorta because haploinsufficiency of this ligand causes thoracic aortic aneurysm in both humans and mice.39,53 Tgfbr2 inactivation has been casually related to perturbed SMC contractility, IGF1 (insulin-like growth factor) production by adventitial fibroblasts, and altered paracrine cross talk between the medial and outer compartments of the vessel wall.50 Of note, inhibition of mTOR (mammalian target of rapamycin) signaling eliminated the risk of dissection in mice with Tgfbr2 inactivation by restoring SMC differentiation and quiescence. By demonstrating that TGFβ signaling protects from thoracic aortic disease in MFS and wild-type mice, these genetic studies have called into question the proposed use of anti-TGFβ therapies as a common treatment of thoracic aortic aneurysms, particularly when it applies to the pediatric population.47
Differences in selectivity or efficacy of genetic versus serological TGFβ blockade were advocated to reconcile the opposite outcomes of the earlier and more recent studies of thoracic aortic disease in Fbn1C1039G/+ mice.50 However, this explanation has been refuted by the finding that either TGFβ neutralization in either the newborn Fbn1mgR/mgR mice or adult wild-type mice infused with Ang II increased aneurysm severity.49,54 Furthermore, the opposite effects of neonatal TGFβ neutralization (detrimental) and neonatal losartan treatment (beneficial) on arterial disease progression in Fbn1mgR/mgR mice disproved the original postulate that TGFβ acts as a mediator of At1r hyperactivity in promoting aneurysm formation.49 The additional finding that starting TGFβ neutralization soon after completion of vascular growth (postnatal day 45) decreased the rate of aortic enlargement and improved median survival of Fbn1mgR/mgR mice implied a pathogenic role for TGFβ signaling later in aneurysm progression.49 As it should always be the case, genetic experiments are still needed to independently corroborate the pharmacological evidence suggesting that TGFβ hyperactivity is a secondary driver of maladaptive tissue remodeling in advanced stages of thoracic aortic disease in MFS.
The proposed dual, contextual role of TGFβ signaling during thoracic aortic disease progression resembles what happens in the many types of cancer, in which TGFβ acts as tumor suppressor in the early stages of a malignancy and as a prometastatic factor at later stages of the disease.55 Recent work using non–small-cell lung carcinoma as a model system has implicated Smad2 inhibition by molecular chaperone CCT6A (chaperonin containing TCP1 subunit 6A) in switching TGFβ-stimulated gene expression to a prometastatic program.56 Similar stage-specific mechanisms may diversify TGFβ action in thoracic aortic disease. For example, prevention of aneurysmal dissection and premature death of Fbn1mgR/mgR mice lacking Ltbp3 might be accounted for by selective association of this ECM protein with the TGFβ isotype(s) responsible for detrimental signaling driving late stages of thoracic aortic disease.57 Conversely, association of Ltbp3 with a disease-preventing TGFβ (most likely TGFβ239) might be responsible for aortic disease in human and mice with Ltbp3 deficiency.57,58 Alternatively, this epistatic effect might reflect an unsuspected involvement of Ltbp3 in ECM assembly and vascular homeostasis, similar to the role Ltbp4 plays in lung elastogenesis.59 This alternative explanation is in line with the distribution of LTBP3 mutations predisposing to thoracic aortic disease in protein domains other than the one that binds pro-TGFβ dimers.58 Neonatal deletion of each of the 3 TGFβ isotypes in MFS mice before and after aneurysm formation is expected to validate this hypothetical mechanism. Similar experiments could also test the potential involvement of TGFβ receptors and transducers in signal diversification. Irrespectively, it is clear from all these studies that TGFβ hyperactivity is a secondary driver of thoracic aortic disease, largely associated with promoting and sustaining maladaptive vessel wall remodeling.
Emerging Ang II Controversies
Although it is well established that augmented Ang II signaling is a prominent determinant of both thoracic and abdominal aortic aneurysm development,4,60–62 there are also significant differences and emerging controversies about the source of increased Ang II signaling in various mouse models of aneurysms. Current evidence suggests that different mechanisms increase Ang II signaling in various genetic forms of thoracic aortic disease. For example, Ang II signaling is potentiated by upregulating angiotensin-converting enzyme levels in the aorta of mice with SMC-specific loss of fibulin-4 (Fbln4SMKO mice). In contrast, NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells)-dependent At1ar overexpression potentiates Ang II signaling in mice deficient for smooth muscle α-actin (Acta2−/− mice).63,64 It would be interesting to determine whether these 2 mechanisms reflect the distinct nature of the genetic lesion driving aneurysm formation (ECM versus contractile unit impairment). Another unresolved important issue is how angiotensin-converting enzyme augmentation translates into a narrow postnatal window of losartan sensitivity in the dilated aorta of Fbln4SMKO mice.63
The recent finding that genetic inactivation of At1ar in Fbn1C1039G/+ mice does not mitigate aneurysm formation has raised the intriguing possibility that losartan might exert its therapeutic effects independently of the targeted receptor.65 Based on losartan inactivation by NOS (NO synthase) inhibition and eNOS (endothelial NOS) protection from aneurysm formation in Fbn1C1039G/+ mice,66 Sellers at al65 have concluded that endothelial NO release may mediate this losartan off-target effect, thus implying a significant role of endothelial dysfunction in aneurysm progression. In contrast to these findings, mitigated aneurysm formation in Fbn1mgR/mgR mice with either germ line or endothelial At1ar inactivation has implied that receptor signaling is a prominent arterial disease determinant likely to perturb normal intima-to-media communication.67 At2r (angiotensin II type II receptor) role in thoracic aneurysm formation is also controversial. On one hand, Fbn1C1039G/+ mice lacking At2r have a significantly reduced response to losartan, suggesting that the beneficial effect of At1r antagonism is, in part, mediated by shunting Ang II activity through protective At2r signaling.68 On the other hand, the comparable aortic phenotypes of losartan-treated Fbn1C1039G/+ mice with or without At1ar have excluded the predicted shift toward At2r signaling in the latter group of mutant animals.65 Furthermore, thoracic aortic disease pathology was not reversed after short-term treatment of Fbn1C1039G/+ mice with the At2r agonist C21.69 Because At1ar antibodies are unavailable to monitor receptor dynamics during aneurysm formation and growth, combinatorial deletions of angiotensin receptor genes in mice treated with various receptor agonists and antagonists should eventually resolve these discrepancies.
Other Pathogenic Pathways and Potential New Therapies for Thoracic Aortic Disease
The above studies, together with an evergrowing list of treatments reported to modify thoracic aortic disease in MFS mice (Table 1), clearly indicate that arterial disease involves the gradual stratification of stress-stimulated interactions among different cell types and multiple regulatory pathways, of which At1r- and TGFβ-signaling pathways are an important subset.70,71 It follows that combinatorial drug treatments are likely to be required to more effectively slow down aneurysm progression or even prevent disease onset. Below are discussed a few examples of potential new treatments identified by studies of thoracic aortic disease in mouse models of MFS.
Inhibition of NOS, specifically Nos2, has recently emerged as an attractive therapeutic strategy against both syndromic and nonsyndromic forms of thoracic aortic disease. A combination of genetic and pharmacological approaches has causally connected thoracic aneurysm development with augmented Nos2 activity in the media of Fbn1C1039G/+ mice.66 Phenotypic similarities between these and Adamts1 (a disintegrin and metalloproteinase with thrombospondin motifs 1)-deficient mice were interpreted to suggest that a decrease of the metalloproteinase in the MFS aorta may lead to excessive accumulation of its substrates with the result of stimulating Nos2 through Akt and NFκB signaling and of promoting Mmp9 (matrix metalloproteinase-9)-driven medial degeneration through NO overactivation. In support of this disease model, elevated Nos2 and decreased Adamts1 levels were also noted in aneurysmal specimens from patients with MFS. NO involvement in MFS pathogenesis should be confirmed using the more severe mouse model of the disease (Fbn1mgR/mgR mouse) because improved survival is a more informative surrogate parameter of thoracic aortic disease severity than modifications in the rate of aortic dilation and in the degree of aortic tissue degeneration. As an illustrative example of this point, genetic disruption of IL6 (interleukin 6)-STAT3 (signal transducer and activator of transcription 3) signaling in Fbn1mgR/mgR mice was reported to decrease vascular fibrosis and Mmp9-mediated elastin degradation (2 key histopathologic evidence of arterial disease) without improving animal survival.72 Because a gain-of-function mutation in the type I cGMP (cyclic guanosine monophosphate)-activated protein kinase—an NO target in SMCs—causes thoracic aortic disease by decreasing SMC contractility,73 it would also be informative to determine whether disruption of the contractile unit is associated with dysregulated NO signaling in other mouse models of thoracic aortic disease.
Epigenetic analyses of Fbn1C1039G/+ mice has potentially expanded the therapeutic options for thoracic aortic disease in MFS and related diseases. It has recently been shown that the methyltransferase EZH2 (enhancer of zeste homolog 2) is responsible for inhibiting Smad3-binding sites to the first intron of the gene coding for SM22α—an early marker of SMC differentiation.74 Importantly, targeting the chromatin modifier with a small molecule inhibitor restored expression of the contractile protein and alleviated arterial pathology in Fbn1C1039G/+ mice. The same investigators have subsequently identified the same epigenetic pathway promoting SMC dysfunction because of pathogenic variants in 2 other genes causative for thoracic aortic disease.75 They have also connected this pathway with the assembly of an HDAC9-MALAT1-BRG1 (histone deacetylase 9-metastasis associated lung adenocarcinoma transcript 1-Brahma-related gene 1)–repressing complex on the promoters of genes encoding contractile proteins.75 Together, these studies support the possibility of using chromatin modifying factors as therapeutic targets in thoracic aortic disease resulting from various etiologies. Additionally, Fischbein et al76 have found that increased expression of miR29b—a regulator of apoptosis and ECM remodeling—contributes to aneurysm onset and progression, as evidenced by the ability of miR29b blockade to significantly reduce aneurysm development. Although promising, development of pharmacotherapies that target either of these epigenetic programs exclusively in the dilating aorta needs to first resolve the notorious problems associated with site-specific delivery of such therapeutics.
A recent preclinical study in MFS mice has called into question a class of medications currently prescribed for some patients with MFS. In spite of limited clinical data about drug efficacy and safety in MFS, calcium channel blockers are usually given to afflicted individuals who are intolerant to β-blockers under the assumption that the drug would lower stress on the aortic wall during cardiac systole. Experimental and clinical data have led to conclude that calcium channel blockers aggravate thoracic aortic disease in Fbn1C1039G/+ mice and increase aneurysmal growth and risk of dissection in patients with MFS.77 The investigators have also reported that the deleterious effect of calcium channel blockers in MFS mice involves PKCβ (protein kinase C beta)-mediated Erk1/2 activation and that an antihypertensive drug (hydralazine) that inhibits activation of this pathway prevented aneurysm formation. Independent experimental confirmation of this important finding is needed, along with thorough clinical evaluation of all causes of thoracic aortic disease that might be negatively affected by treatment with this common antihypertensive drug.
Closing Remarks and Future Perspectives
By addressing old and new controversies, the studies discussed in this review have highlighted the molecular complexity of thoracic aortic disease onset and progression and consequently the significant challenges we still face to identify suitable new treatments. Although the losartan experience has cast some doubt on using experimental mice as a reliable model of human thoracic aortic disease, several new insights into cardiovascular disease gathered from the study of MFS mice make them the best experimental model currently available.53,66,77,78 Induced pluripotent stem cells derived from patients with MFS and differentiated into vascular SMC have recently been used as an alternative model to interrogate thoracic aortic disease mechanisms.79 These studies have corroborated and extended prior in vivo findings of TGFβ’s pathological connection with MMP-driven maladaptive ECM remodeling and MAPK p38’s contribution to arterial disease independent of TGFβ.49,80 However, a major limitation of this in vitro model system is the lack of paracrine stress/regulatory stimuli from intima and adventitial cells that are involved in progression of thoracic aortic disease.50,65,67
In closing, we would like to offer a few suggestions for research directions that could be pursued to advance our understanding of thoracic aortic disease and that may eventually lead to better, evidence-based treatments. Although by necessity these suggestions reflect our own biases, we nonetheless offer them to stimulate a conversation among interested scientists and clinicians. The first and most important lesson we have learned from recent studies of thoracic aortic disease largely performed with MFS mice is that the notion of a common anti-TGFβ therapy for MFS and thoracic aortic disease in general is unwarranted, particularly in pediatric patients. In this respect, it would be productive to standardize and increase the number of surrogate markers for thoracic aortic disease progression so as to more rigorously compare data on varying treatments in different mouse models. Moreover, biomechanical analyses should be integral part of aortic disease characterization under different genetic lesions and pharmacological interventions. This suggestion stems from having recognized for many years that although patients with MFS have increased aortic stiffness particularly evident in individuals <40 years of age,81 the data have been conflicted as to whether this stiffness is beneficial or not in terms aortic growth and risk for dissection. Despite the anti-inflammatory effects of Atr2, Ang II has an established role in driving fibrosis, including aortic stiffness associated with hypertension, and these fibrotic actions are linked to activation of TGFβ signaling.82 Therefore, it is surprising that a recent follow-up study of the largest clinical trial assessing losartan versus atenolol in patients with MFS found that treatment with atenolol >3 years decreased baseline aortic root stiffness, whereas losartan did not.29,83 This study also found that patients with MFS with higher baseline stiffness measurements had the fastest rate of growth of the aortic root. Although these results favor the use of β-blockers over Atr1 inhibition to prevent aortic growth in patients with MFS, it remains unclear why an Atr1 blocker failed to block aortic stiffness. Additionally, although lower aortic root stiffness was associated with slower growth rates of the root, how this impacts risk for aortic dissection is unknown. In other words, decreasing the aortic stiffness may decrease growth rates but increase the risk for dissection or rupture because aortic fibrosis could potentially prevent these events. Biomechanical analyses in relevant mouse models should be used to address such questions.
The ultimate goal for any treatment for thoracic aortic disease is to prevent acute aortic dissections and its associated mortality and morbidity. Prophylactic aortic surgery to prevent aortic dissections is based largely on aortic dimensions >5 to 5.5 cm.84 However, almost half of individuals presenting with acute ascending aortic dissections have aortic diameters <5.5 cm.92 These findings emphasize the need to improve our understanding of the molecular pathways triggering dissections independent of aneurysm formation and identify therapeutics that block not just aortic enlargement but also acute aortic dissections. As illustrated in Table 2, the majority of preclinical trials have used Fbn1C1039/+ mice, which develop aneurysms by 12 months of age but rarely progresses to dissection. Going forward, the more severe mouse model of the disease (Fbn1mgR/mgR mice) should be used for preclinical trials because it more faithfully replicates the natural history of thoracic aneurysm progression to dissection observed in individuals diagnosed with MFS.
Table 2.
Treatment Trials in Mouse Models of Marfan Syndrome
| MFS Model | Treatment | Dilation | Histopathology | Dissection | Study |
|---|---|---|---|---|---|
| Fbn1C1039G/+ | TGFβ blockade | + | + | Habashi et al, 200627 | |
| At1r antagonism | + | + | |||
| β-blockers | 0 | 0 | |||
| ErK inhibition | + | ND | Holm et al, 201148 | ||
| At2r agonism | 0 | 0 | Verbrugghe et al, 201869 | ||
| NO inhibition | + | + | Oller et al, 201766 | ||
| EZH2 inhibition | + | + | Lino Cardenas et al, 201875 | ||
| EZH2 inhibition+AT1r antagonism | + | 0 | |||
| miR29 blockade | + | + | Merk et al, 201276 | ||
| Ca+2 channel blockade | − | − | − | Doyle et al, 201577 | |
| Statins | + | + | McLoughlin et al, 201185 | ||
| Resveratrol | + | + | Hibender et al, 201686 | ||
| MMP inhibition | + | + | Chung et al, 200887 | ||
| Moderate exercise | + | 0 | Mas-Stachurska et al, 201788 | ||
| Caspase inhibition | + | + | Enrich et al, 201589 | ||
| Fbn1MgR/MgR | At1r antagonism | + | + | Delayed | Cook et al, 201549 |
| Early TGFβ blockade | − | − | Accelerated | ||
| Late TGFβ blockade | + | + | Delayed | ||
| AT1r+late TGFβ blockade | + | + | Prevented | ||
| MMP inhibition | + | + | Delayed | Xiong et al, 200890 | |
| Indomethacin | ND | + | ND | Guo et al, 201391 |
Symbols indicate that thoracic aortic disease was mitigated (+), exacerbated (−), or unchanged (0) with the treatment. At1r indicates angiotensin II type I receptor; At2r, angiotensin II type II receptor; ERK, extracellular signal–regulated kinase; EZH2, enhancer of zeste homolog 2; MFS, Marfan syndrome; MMP, matrix metalloproteinase; ND, not determined; and TGFβ, transformation growth factor-β.
Finally, the combination of computational and experimental approaches should be more extensively applied to the study of thoracic aortic disease pathophysiology. Although a few recent reports have included proteomic and transcriptomic profiles of aortic tissues from patients with MFS and mice,67,74,93 none of them has used this information to computationally model the dynamics of rewiring networks of molecular signals during aneurysm progression to dissection. Computational methods can also be used to identify druggable nodes within disease-associated molecular networks, as exemplified by the successful identification of a new drug treatment for inflammatory bowel disease.94 Our last suggestion is, therefore, to prioritize such computational studies according to standardized clinical and experimental surrogate readouts of thoracic aortic disease progression.
Although significant progress has been made during the past 20 years in our understanding of the molecular pathogenesis of thoracic aortic aneurysm, there are still limited therapeutic options to delay disease progression and none to prevent it. Therefore, it is imperative to build on the knowledge gained from the previous studies by improving our experimental tools and clinical analyses, as well as by fostering cross-communication between the bench and bedside. Hopefully, global collaborative efforts, like the recently revived GenTAC Alliance, will help move with the standardization. In addition, patient support organizations, like the Marfan Foundation, and John Ritter Foundation, and international patient registries, like the Montalcino Aortic Consortium95–97 and BAV (bicuspid aortic valve) Consortium,11,98 are poised to approach individuals with thoracic aortic disease for further clinical trials to prevent or delay disease progression.
Highlights.
Thoracic aneurysms have been managed with treatment with β-adrenergic blocking agents (β-blockers) and routine surveillance imaging, followed by surgical repair of the aneurysm when the risk of dissection exceeds the risk for repair. Thus, there is a window to initiate therapies to slow aortic enlargement and delay or ideally negate the need for surgical repair of the aneurysm to prevent a dissection.
Mouse models of Marfan syndrome—a monogenic disorder predisposing to thoracic aortic disease—have been used extensively to identify such therapies.
Initial experiments suggested that TGFβ (transformation growth factor-β) signaling was increased in the aortic media of a Marfan syndrome mouse model and that its inhibition via TGFβ neutralization or At1r (Ang II [angiotensin II] type I receptor) antagonism prevented aneurysm development.
Subsequent studies in mouse models have begun to resolve the complex molecular pathophysiology underlying onset and progression of aortic disease and have emphasized the need to preserve TGFβ signaling to prevent aneurysm formation.
This review describes critical experiments that have influenced the evolution of our understanding of the molecular pathogenesis of this disease and discusses the controversies and identifies new therapeutic opportunities.
Acknowledgments
We thank K. Johnson for organizing the manuscript. Described studies from the laboratories of the authors were supported by National Institutes of Health grants HL62594 and P01HL110869 (D.M. Milewicz) and AR069307 and HL134605 (F. Ramirez).
Nonstandard Abbreviations and Acronyms
- Ang II
angiotensin II
- At1r
angiotensin II type I receptor
- At2r
angiotensin II type II receptor
- ECM
extracellular matrix
- eNOS
endothelial NO synthase
- LTBP
latent transforming growth factor-β–binding protein
- MAPK
mitogen-activated protein kinase
- MFS
Marfan syndrome
- NOS
NO synthase
- SMC
smooth muscle cell
- TGFβ
transforming growth factor-β
Footnotes
Disclosures
None.
Contributor Information
Dianna M. Milewicz, Division of Medical Genetics, Department of Internal Medicine, McGovern Medical School, University of Texas Health Science Center at Houston
Francesco Ramirez, Department of Pharmacological Sciences, Institute for Systems Biomedicine, Icahn School of Medicine at Mount Sinai, New York.
References
- 1.Howard DP, Banerjee A, Fairhead JF, Perkins J, Silver LE, Rothwell PM; Oxford Vascular Study. Population-based study of incidence and outcome of acute aortic dissection and premorbid risk factor control: 10-year results from the Oxford Vascular Study. Circulation 2013;127:2031–2037. 10.1161/CIRCULATIONAHA.112.000483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006;114:2611–2618. 10.1161/CIRCULATIONAHA.106.630400 [DOI] [PubMed] [Google Scholar]
- 3.Hiratzka LF, Bakris GL, Beckman JA, et al. ; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. J Am Coll Cardiol 2010;55:e27–e129. 10.1016/j.jacc.2010.02.015 [DOI] [PubMed] [Google Scholar]
- 4.Yu C, Jeremy RW. Angiotensin, transforming growth factor β and aortic dilatation in Marfan syndrome: of mice and humans. Int J Cardiol Heart Vasc 2018;18:71–80. 10.1016/j.ijcha.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagan PG, Nienaber CA, Isselbacher EM, et al. The International Registry of Acute Aortic Dissection (IRAD): new insights into an old disease. JAMA 2000;283:897–903. [DOI] [PubMed] [Google Scholar]
- 6.LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol 2011;8:103–113. 10.1038/nrcardio.2010.187 [DOI] [PubMed] [Google Scholar]
- 7.LeMaire SA, McDonald ML, Guo DC, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet 2011;43:996–1000. 10.1038/ng.934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg 1997;25:506–511. [DOI] [PubMed] [Google Scholar]
- 9.Coady MA, Davies RR, Roberts M, Goldstein LJ, Rogalski MJ, Rizzo JA, Hammond GL, Kopf GS, Elefteriades JA. Familial patterns of thoracic aortic aneurysms. Arch Surg 1999;134:361–367. [DOI] [PubMed] [Google Scholar]
- 10.Renard M, Francis C, Ghosh R, et al. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection. J Am Coll Cardiol 2018;72:605–615. 10.1016/j.jacc.2018.04.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michelena HI, Prakash SK, Della Corte A, et al. ; BAVCon Investigators. Bicuspid aortic valve: identifying knowledge gaps and rising to the challenge from the International Bicuspid Aortic Valve Consortium (BAVCon). Circulation 2014;129:2691–2704. 10.1161/CIRCULATIONAHA.113.007851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prakash SK, Pedroza C, Khalil YA, Milewicz DM. Diabetes and reduced risk for thoracic aortic aneurysms and dissections: a nationwide case-control study. J Am Heart Assoc 2012;1:jah3-e000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takagi H, Umemoto T; ALICE (All-Literature Investigation of Cardiovascular Evidence) Group. Negative association of diabetes with thoracic aortic dissection and aneurysm. Angiology 2017;68:216–224. 10.1177/0003319716647626 [DOI] [PubMed] [Google Scholar]
- 14.Tsai CL, Lin CL, Wu YY, Shieh DC, Sung FC, Kao CH. Advanced complicated diabetes mellitus is associated with a reduced risk of thoracic and abdominal aortic aneurysm rupture: a population-based cohort study. Diabetes Metab Res Rev 2015;31:190–197. 10.1002/dmrr.2585 [DOI] [PubMed] [Google Scholar]
- 15.Ramirez F, Dietz HC. Marfan syndrome: from molecular pathogenesis to clinical treatment. Curr Opin Genet Dev 2007;17:252–258. 10.1016/j.gde.2007.04.006 [DOI] [PubMed] [Google Scholar]
- 16.Carlson RG, Lillehei CW, Edwards JE. Cystic medial necrosis of the ascending aorta in relation to age and hypertension. Am J Cardiol 1970;25:411–415. [DOI] [PubMed] [Google Scholar]
- 17.Ladich E, Yahagi K, Romero ME, Virmani R. Vascular diseases: aortitis, aortic aneurysms, and vascular calcification. Cardiovasc Pathol 2016;25:432–441. 10.1016/j.carpath.2016.07.002 [DOI] [PubMed] [Google Scholar]
- 18.Cikach FS, Koch CD, Mead TJ, Galatioto J, Willard BB, Emerton KB, Eagleton MJ, Blackstone EH, Ramirez F, Roselli EE, Apte SS. Massive aggrecan and versican accumulation in thoracic aortic aneurysm and dissection. JCI Insight 2018;3:97167 10.1172/jci.insight.97167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachhuber TE, Lalich JJ, Angevine DM, Schilling ED, Strong FM. Lathyrus factor activity of β-aminopropionitrile and related compounds. Proc Soc Exp Biol Med 1955;89:294–297. [DOI] [PubMed] [Google Scholar]
- 20.Barnett BD, Bird HR, Lalich JJ, Strong FM. Toxicity of beta-amino-propionitrile for turkey poults. Proc Soc Exp Biol Med 1957;94:67–70. [PubMed] [Google Scholar]
- 21.Simpson CF, Kling JM, Palmer RF. The use of propranolol for the protection of turkeys from the development of beta-aminopropionitrile-induced aortic ruptures. Angiology 1968;19:414–418. 10.1177/000331976801900705 [DOI] [PubMed] [Google Scholar]
- 22.Shores J, Berger KR, Murphy EA, Pyeritz RE. Progression of aortic dilatation and the benefit of long-term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med 1994;330:1335–1341. 10.1056/NEJM199405123301902 [DOI] [PubMed] [Google Scholar]
- 23.Rossi-Foulkes R, Roman MJ, Rosen SE, Kramer-Fox R, Ehlers KH, O’Loughlin JE, Davis JG, Devereux RB. Phenotypic features and impact of beta blocker or calcium antagonist therapy on aortic lumen size in the Marfan syndrome. Am J Cardiol 1999;83:1364–1368. [DOI] [PubMed] [Google Scholar]
- 24.Selamet Tierney ES, Feingold B, Printz BF, Park SC, Graham D, Kleinman CS, Mahnke CB, Timchak DM, Neches WH, Gersony WM. Beta-blocker therapy does not alter the rate of aortic root dilation in pediatric patients with Marfan syndrome. J Pediatr 2007;150:77–82. 10.1016/j.jpeds.2006.09.003 [DOI] [PubMed] [Google Scholar]
- 25.Ladouceur M, Fermanian C, Lupoglazoff JM, Edouard T, Dulac Y, Acar P, Magnier S, Jondeau G. Effect of beta-blockade on ascending aortic dilatation in children with the Marfan syndrome. Am J Cardiol 2007;99:406–409. 10.1016/j.amjcard.2006.08.048 [DOI] [PubMed] [Google Scholar]
- 26.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci USA 1999;96:3819–3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006;312:117–121. 10.1126/science.1124287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groenink M, den Hartog AW, Franken R, Radonic T, de Waard V, Timmermans J, Scholte AJ, van den Berg MP, Spijkerboer AM, Marquering HA, Zwinderman AH, Mulder BJ. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 2013;34:3491–3500. 10.1093/eurheartj/eht334 [DOI] [PubMed] [Google Scholar]
- 29.Lacro RV, Dietz HC, Sleeper LA, et al. ; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med 2014;371:2061–2071. 10.1056/NEJMoa1404731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milleron O, Arnoult F, Ropers J, et al. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J 2015;36:2160–2166. 10.1093/eurheartj/ehv151 [DOI] [PubMed] [Google Scholar]
- 31.Forteza A, Evangelista A, Sánchez V, Teixidó-Turà G, Sanz P, Gutiérrez L, Gracia T, Centeno J, Rodríguez-Palomares J, Rufilanchas JJ, Cortina J, Ferreira-González I, García-Dorado D. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial. Eur Heart J 2016;37:978–985. 10.1093/eurheartj/ehv575 [DOI] [PubMed] [Google Scholar]
- 32.Mallat Z, Daugherty A. AT1 receptor antagonism to reduce aortic expansion in Marfan syndrome: lost in translation or in need of different interpretation? Arterioscler Thromb Vasc Biol 2015;35:e10–e12. 10.1161/ATVBAHA.114.305173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Franken R, den Hartog AW, Radonic T, Micha D, Maugeri A, van Dijk FS, Meijers-Heijboer HE, Timmermans J, Scholte AJ, van den Berg MP, Groenink M, Mulder BJ, Zwinderman AH, de Waard V, Pals G. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet 2015;8:383–388. 10.1161/CIRCGENETICS.114.000950 [DOI] [PubMed] [Google Scholar]
- 34.Chiu HH, Wu MH, Wang JK, Lu CW, Chiu SN, Chen CA, Lin MT, Hu FC. Losartan added to β-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc 2013;88:271–276. 10.1016/j.mayocp.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 35.Muiño-Mosquera L, De Nobele S, Devos D, Campens L, De Paepe A, De Backer J. Efficacy of losartan as add-on therapy to prevent aortic growth and ventricular dysfunction in patients with Marfan syndrome: a randomized, double-blind clinical trial. Acta Cardiol 2017;72:616–624. 10.1080/00015385.2017.1314134 [DOI] [PubMed] [Google Scholar]
- 36.Inamoto S, Kwartler CS, Lafont AL, et al. TGFBR2 mutations alter smooth muscle cell phenotype and predispose to thoracic aortic aneurysms and dissections. Cardiovasc Res 2010;88:520–529. 10.1093/cvr/cvq230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, Clarke L, Bernier F, Santos-Cortez RL, Leal SM, Bertoli-Avella AM, Shendure J, Rieder MJ, Nickerson DA, Milewicz DM; NHLBI GO Exome Sequencing Project. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res 2011;109:680–686. 10.1161/CIRCRESAHA.111.248161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011;43:121–126. [DOI] [PubMed] [Google Scholar]
- 39.Boileau C, Guo DC, Hanna N, et al. ; National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet 2012;44:916–921. 10.1038/ng.2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goumans MJ, Ten Dijke P. TGFβ signaling in control of cardiovascular function. Cold Sprin Harb Perspect Biol 2018;10:pii a022210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res 2009;19:128–139. 10.1038/cr.2008.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-protein. J Biol Chem 2003;278:2750–2757. 10.1074/jbc.M209256200 [DOI] [PubMed] [Google Scholar]
- 43.Renard M, Holm T, Veith R, et al. Altered TGFbeta signaling and cardiovascular manifestations in patients with autosomal recessive cutis laxa type I caused by fibulin-4 deficiency. Eur J Hum Genet 2010;18:895–901. 10.1038/ejhg.2010.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renard M, Callewaert B, Baetens M, et al. Novel MYH11 and ACTA2 mutations reveal a role for enhanced TGFβ signaling in FTAAD. Int J Cardiol 2013;165:314–321. 10.1016/j.ijcard.2011.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo DC, Pannu H, Tran-Fadulu V, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007;39:1488–1493. 10.1038/ng.2007.6 [DOI] [PubMed] [Google Scholar]
- 46.Gomez D, Kessler K, Michel JB, Vranckx R. Modifications of chromatin dynamics control Smad2 pathway activation in aneurysmal smooth muscle cells. Circ Res 2013;113:881–890. 10.1161/CIRCRESAHA.113.301989 [DOI] [PubMed] [Google Scholar]
- 47.Cannaerts E, van de Beek G, Verstraeten A, Van Laer L, Loeys B. TGF-β signalopathies as a paradigm for translational medicine. Eur J Med Genet 2015;58:695–703. 10.1016/j.ejmg.2015.10.010 [DOI] [PubMed] [Google Scholar]
- 48.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science 2011;332:358–361. 10.1126/science.1192149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol 2015;35:911–917. 10.1161/ATVBAHA.114.305150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest 2014;124:755–767. 10.1172/JCI69942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek DA. Postnatal deletion of the type II transforming growth factor-β receptor in smooth muscle cells causes severe aortopathy in mice. Arterioscler Thromb Vasc Biol 2015;35:2647–2656. 10.1161/ATVBAHA.115.306573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a mouse model of Marfan syndrome is not mediated by altered Transforming growth factor-β signaling. J Am Heart Assoc 2017;6:e004968 10.1161/JAHA.116.004968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 2012;44:922–927. 10.1038/ng.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Ait-Oufella H, Herbin O, Bonnin P, Ramkhelawon B, Taleb S, Huang J, Offenstadt G, Combadière C, Rénia L, Johnson JL, Tharaux PL, Tedgui A, Mallat Z. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J Clin Invest 2010;120:422–432. 10.1172/JCI38136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Massague J TGF in Cancer. Cell 2008;134:215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ying Z, Tian H, Li Y, Lian R, Li W, Wu S, Zhang HZ, Wu J, Liu L, Song J, Guan H, Cai J, Zhu X, Li J, Li M. CCT6A suppresses SMAD2 and promotes prometastatic TGF-β signaling. J Clin Invest 2017;127:1725–1740. 10.1172/JCI90439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zilberberg L, Phoon CK, Robertson I, Dabovic B, Ramirez F, Rifkin DB. Genetic analysis of the contribution of LTBP-3 to thoracic aneurysm in Marfan syndrome. Proc Natl Acad Sci USA 2015;112:14012–14017. 10.1073/pnas.1507652112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo DC, Regalado ES, Pinard A, et al. ; University of Washington Center for Mendelian Genomics. LTBP3 pathogenic variants predispose individuals to thoracic aortic aneurysms and dissections. Am J Hum Genet 2018;102:706–712. 10.1016/j.ajhg.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noda K, Dabovic B, Takagi K, et al. Latent TGF-β binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc Natl Acad Sci USA 2013;110:2852–2857. 10.1073/pnas.1215779110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A III, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest 2009;119:3637–3651. 10.1172/JCI38308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 2010;118:681–689. 10.1042/CS20090372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuang SQ, Geng L, Prakash SK, Cao JM, Guo S, Villamizar C, Kwartler CS, Peters AM, Brasier AR, Milewicz DM. Aortic remodeling after transverse aortic constriction in mice is attenuated with AT1 receptor blockade. Arterioscler Thromb Vasc Biol 2013;33:2172–2179. 10.1161/ATVBAHA.113.301624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang J, Yamashiro Y, Papke CL, Ikeda Y, Lin Y, Patel M, Inagami T, Le VP, Wagenseil JE, Yanagisawa H. Angiotensin-converting enzyme-induced activation of local angiotensin signaling is required for ascending aortic aneurysms in fibulin-4-deficient mice. Sci Transl Med 2013;5:183ra58, 1–183ra58,11. 10.1126/scitranslmed.3005025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J, Peters A, Papke CL, et al. Loss of smooth muscle α-actin leads to NF-κB-dependent increased sensitivity to angiotensin II in smooth muscle cells and aortic enlargement. Circ Res 2017;120:1903–1915. 10.1161/CIRCRESAHA.117.310563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sellers SL, Milad N, Chan R, Mielnik M, Jermilova U, Huang PL, de Crom R, Hirota JA, Hogg JC, Sandor GG, Van Breemen C, Esfandiarei M, Seidman MA, Bernatchez P. Inhibition of Marfan syndrome aortic root dilation by losartan: role of angiotensin II receptor type 1-independent activation of endothelial function. Am J Pathol 2018;188:574–585. 10.1016/j.ajpath.2017.11.006 [DOI] [PubMed] [Google Scholar]
- 66.Oller J, Méndez-Barbero N, Ruiz EJ, et al. Nitric oxide mediates aortic disease in mice deficient in the metalloprotease Adamts1 and in a mouse model of Marfan syndrome. Nat Med 2017;23:200–212. 10.1038/nm.4266 [DOI] [PubMed] [Google Scholar]
- 67.Galatioto J, Caescu CI, Hansen J, Cook JR, Miramontes I, Iyengar R, Ramirez F. Cell type-specific contributions of the angiotensin II type 1a receptor to aorta homeostasis and aneurysmal disease-brief report. Arterioscler Thromb Vasc Biol 2018;38:588–591. 10.1161/ATVBAHA.117.310609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science 2011;332:361–365. 10.1126/science.1192152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verbrugghe P, Verhoeven J, Clijsters M, Vervoort D, Schepens J, Meuris B, Herijgers P. The effect of a nonpeptide angiotensin II type 2 receptor agonist, compound 21, on aortic aneurysm growth in a mouse model of Marfan syndrome. J Cardiovasc Pharmacol 2018;71:215–222. 10.1097/FJC.0000000000000560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: insights from pre-disposing genes and mouse models. Annu Rev Med 2017;68:51–67. 10.1146/annurev-med-100415-022956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramirez F, Caescu C, Wondimu E, Galatioto J. Marfan syndrome; A connective tissue disease at the crossroads of mechanotransduction, TGFβ signaling and cell stemness. Matrix Biol 2018;71–72:82–89. 10.1016/j.matbio.2017.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ju X, Ijaz T, Sun H, Lejeune W, Vargas G, Shilagard T, Recinos A III, Milewicz DM, Brasier AR, Tilton RG. IL-6 regulates extracellular matrix remodeling associated with aortic dilation in a fibrillin-1 hypomorphic mgR/mgR mouse model of severe Marfan syndrome. J Am Heart Assoc [DOI] [PMC free article] [PubMed]
- 73.Guo DC, Regalado E, Casteel DE, et al. ; GenTAC Registry Consortium; National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet 2013;93:398–404. 10.1016/j.ajhg.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lino Cardenas CL, Kessinger CW, Cheng Y, et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun 2018;9:1009 10.1038/s41467-018-03394-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lino Cardenas CL, Kessinger CW, MacDonald C, Jassar AS, Isselbacher EM, Jaffer FA, Lindsay ME. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight 2018;3:97493 10.1172/jci.insight.97493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, Spin JM, Alvira CM, Robbins RC, Fischbein MP. miR-29b participates in early aneurysm development in Marfan syndrome. Circ Res 2012;110:312–324. 10.1161/CIRCRESAHA.111.253740 [DOI] [PubMed] [Google Scholar]
- 77.Doyle JJ, Doyle AJ, Wilson NK, et al. ; GenTAC Registry Consortium; MIBAVA Leducq Consortium. A deleterious gene-by-environment interaction imposed by calcium channel blockers in Marfan syndrome. Elife 2015;4:e08648 10.7554/eLife.08648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cook JR, Carta L, Bénard L, Chemaly ER, Chiu E, Rao SK, Hampton TG, Yurchenco P, Costa KD, Hajjar RJ, Ramirez F; GenTAC Registry Consortium. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest 2014;124:1329–1339. 10.1172/JCI71059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, Ramirez F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem 2009;284:5630–5636. 10.1074/jbc.M806962200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet 2017;49:97–109. 10.1038/ng.3723 [DOI] [PubMed] [Google Scholar]
- 81.Hirata K, Triposkiadis F, Sparks E, Bowen J, Wooley CF, Boudoulas H. The Marfan syndrome: abnormal aortic elastic properties. J Am Coll Cardiol 1991;18:57–63. [DOI] [PubMed] [Google Scholar]
- 82.Murphy AM, Wong AL, Bezuhly M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015;8:7 10.1186/s13069-015-0023-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Selamet Tierney ES, Levine JC, Sleeper LA, et al. ; Pediatric Heart Network Investigators. Influence of aortic stiffness on aortic-root growth rate and outcome in patients with the Marfan syndrome. Am J Cardiol 2018;121:1094–1101. 10.1016/j.amjcard.2018.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hiratzka LF, Bakris GL, Beckman JA, et al. ; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society forVascular Medicine. Anesth Analg 2010;111:279–315. 10.1213/ANE.0b013e3181dd869b [DOI] [PubMed] [Google Scholar]
- 85.McLoughlin D, McGuinness J, Byrne J, Terzo E, Huuskonen V, McAllister H, Black A, Kearney S, Kay E, Hill AD, Dietz HC, Redmond JM. Pravastatin reduces Marfan aortic dilation. Circulation 2011;124(suppl 11):S168–S173. 10.1161/CIRCULATIONAHA.110.012187 [DOI] [PubMed] [Google Scholar]
- 86.Hibender S, Franken R, van Roomen C, Ter Braake A, van der Made I, Schermer EE, Gunst Q, van den Hoff MJ, Lutgens E, Pinto YM, Groenink M, Zwinderman AH, Mulder BJ, de Vries CJ, de Waard V. Resveratrol inhibits aortic root dilatation in the Fbn1C1039G/+ Marfan mouse model. Arterioscler Thromb Vasc Biol 2016;36:1618–1626. 10.1161/ATVBAHA.116.307841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chung AW, Yang HH, Radomski MW, van Breemen C. Long-term doxycycline is more effective than atenolol to prevent thoracic aortic aneurysm in marfan syndrome through the inhibition of matrix metalloproteinase-2 and −9. Circ Res 2008;102:e73–e85. 10.1161/CIRCRESAHA.108.174367 [DOI] [PubMed] [Google Scholar]
- 88.Mas-Stachurska A, Siegert AM, Batlle M, et al. Cardiovascular benefits of moderate exercise training in Marfan syndrome: insights from an animal model. JAm Heart Assoc 2017;6:e006438 10.1161/JAHA.117.006438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Emrich FC, Okamura H, Dalal AR, et al. Enhanced caspase activity contributes to aortic wall remodeling and early aneurysm development in a murine model of Marfan syndrome. Arterioscler Thromb Vasc Biol 2015;35:146–154. 10.1161/ATVBAHA.114.304364 [DOI] [PubMed] [Google Scholar]
- 90.Xiong W, Knispel RA, Dietz HC, Ramirez F, Baxter BT. Doxycycline delays aneurysm rupture in a mouse model of Marfan syndrome. J Vasc Surg 2008;47:166–172; 10.1016/j.jvs.2007.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guo G, Ott CE, Grünhagen J, Muñoz-García B, Pletschacher A, Kallenbach K, von Kodolitsch Y, Robinson PN. Indomethacin prevents the progression of thoracic aortic aneurysm in Marfan syndrome mice. Aorta (Stamford) 2013;1:5–12. 10.12945/j.aorta.2013.13.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’gara PT, Evangelista A, Fattori R, Meinhardt G, Trimarchi S, Bossone E, Suzuki T, Cooper JV, Froehlich JB, Nienaber CA, Eagle KA; International Registry of Acute Aortic Dissection (IRAD) Investigators. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007;116:1120–1127. 10.1161/CIRCULATIONAHA.107.702720 [DOI] [PubMed] [Google Scholar]
- 93.Parker SJ, Stotland A, MacFarlane E, Wilson N, Orosco A, Venkatraman V, Madrid K, Gottlieb R, Dietz HC, Van Eyk JE. Proteomics reveals Rictor as a noncanonical TGF-β signaling target during aneurysm progression in Marfan mice. Am J Physiol Heart Circ Physiol 2018;315:H1112–H1126. 10.1152/ajpheart.00089.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ. Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med 2011;3:96ra76 10.1126/scitranslmed.3002648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jondeau G, Ropers J, Regalado E, et al. ; Montalcino Aortic Consortium. International registry of patients carrying TGFBR1 or TGFBR2 mutations: results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet 2016;9:548–558. 10.1161/CIRCGENETICS.116.001485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Regalado ES, Guo DC, Prakash S, et al. ; Montalcino Aortic Consortium. Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet 2015;8:457–464. 10.1161/CIRCGENETICS.114.000943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wallace SE, Regalado ES, Gong L, et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants [published online June 20, 2018]. Genet Med 10.1038/s41436-018-0038-0 [DOI] [PMC free article] [PubMed]
- 98.Prakash SK, Bossé Y, Muehlschlegel JD, Michelena HI, Limongelli G, Della Corte A, Pluchinotta FR, Russo MG, Evangelista A, Benson DW, Body SC, Milewicz DM; BAVCon Investigators. A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications: insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J Am Coll Cardiol 2014;64:832–839. 10.1016/j.jacc.2014.04.073 [DOI] [PMC free article] [PubMed] [Google Scholar]