

Abstract
Tracking the fate of individual cells and their progeny through lineage tracing has been widely used to investigate various biological processes including embryonic development, homeostatic tissue turnover, and stem cell function in regeneration and disease. Conventional lineage tracing involves the marking of cells either with dyes or nucleoside analogues or genetic marking with fluorescent and/or colorimetric protein reporters. Both are imaging-based approaches that have played a crucial role in the field of developmental biology as well as adult stem cell biology. However, imaging-based lineage tracing approaches are limited by their scalability and the lack of molecular information underlying fate transitions. Recently, computational biology approaches have been combined with diverse tracing methods to overcome these limitations and so provide high-order scalability and a wealth of molecular information. In this review, we will introduce such novel computational methods, starting from single-cell RNA sequencing-based lineage analysis to DNA barcoding or genetic scar analysis. These novel approaches are complementary to conventional imaging-based approaches and enable us to study the lineage relationships of numerous cell types during vertebrate, and in particular human, development and disease.
Keywords: genetic barcoding and genetic scar, lineage tracing, natural DNA-scar based lineage tracing, scRNA-sequencing
INTRODUCTION
Cells can occupy one of two states, steady or transitioning, during postnatal development, homeostatic turnover and regeneration upon injury. During homeostatic turnover in mature organs, multipotent adult stem cells give rise to additional stem cells (self-renewal) or to committed progenitors which will become terminally differentiated cells (differentiation), with the cells tending towards occupancy of the steady state (stem cells and terminally differentiated cells) rather than the transitioning state (differentiating cells) (Clevers, 2013; Gehart and Clevers, 2019). In contrast, during development or regeneration, occupancy of the transitioning state may be more common (Olsson et al., 2016). For example, the fertilized egg begins development as a totipotent zygote, competent to form both embryonic and extraembryonic tissue, which undergoes multiple rounds of cleavage and gives rise to pluripotent cells, which can give rise to all three germ layers of the embryo. Subsequently, pluripotent cells differentiate and give rise to patterned tissues and organs with distinct functions (Arnold and Robertson, 2009). Changes in morphology, gene expression, epigenetic marks and metabolic state can be observed in nearly all cases of cell fate transition and differentiation. Understanding how a cell changes fate and what factors determine lineage hierarchy during development, homeostasis and regeneration would allow researchers to understand the overall kinetics of these fundamental dynamic processes.
Lineage tracing is the term for a set of methods that allow us to follow the fate of individual cells and their progeny with minimal disturbance of their physiological function. It has been widely used to delineate complex biological processes involving multiple cell types with different lineage hierarchies. Historically, lineage tracing has been carried out by careful microscopic observation of the developing embryo in order to determine the lineage tree (Sulston et al., 1983) and microinjection of dyes into single cells or groups of cells to observe cell migration (Thomas et al., 1998) and proliferation (Kit et al., 1958). Although many other methods (reviewed in our previous article (Fink et al., 2015)) have been developed, in the last decade genetic reporters based on the Cre-LoxP recombinase system have emerged as a gold standard in lineage tracing.
Such systems allow exquisite specificity of labeling: as an example, expression of the tamoxifen-inducible CreER recombinase can be under the control of a tissue-specific promoter (Murray et al., 2012) to provide temporal control of activation. Following administration of tamoxifen, the CreER recombinase can remove a LoxP-STOP-LoxP cassette from a reporter to allow expression of a fluorescent or colorimetric protein to genetically label the cell and all its subsequent progeny, as the genetic change will be passed down the lineage tree (Fink et al., 2015). Fluorescent reporters can be used individually or in combinations (multicolor labeling) to achieve cell labeling in living organisms, such methods becoming more readily available with the advent of tissue clearing methods and confocal/lightsheet microscopy (Fink et al., 2015). Alternatively, the live-tracing of individual cells in living animals has been reported through the use of intravital imaging, where an optical window is surgically implanted into living animals (Alieva et al., 2014). It is also possible to live-image developing zebrafish and mouse embryos at single-cell resolution as they undergo gastrulation and morphogenesis (Briggs et al., 2018; Farrell et al., 2018; Keller et al., 2008; McDole et al., 2018). Although imaging approaches provide valuable spatio-temporal and histological information in combination with the hierarchy of individual cells or clones, in order to uncover the full details of lineage relationships and cell fate regulation, we require additional strategies to reveal the molecular information underlying fate transitions.
Recently, next-generation sequencing, deep sequencing, whole genome/exome sequencing (WGS/WES) and single-cell messenger RNA sequencing (scRNA-seq) have become available as new methods to trace or reconstruct cellular lineages at an unprecedented scale, and also simultaneously profile gene expression patterns in the case of scRNA-seq. Therefore, currently available lineage tracing strategies can be broadly classified into imaging- and computational-based methods, which can be further divided into prospective and retrospective approaches (Kester and van Oudenaarden, 2018; Winters et al., 2018). Our previous review dealt with imaging-based lineage tracing (Fink et al., 2015); in the current review, we will focus on computational-based approaches, starting with scRNA-seq and describing prospective scarring methods via genetic engineering and genetic barcoding, and finishing with retrospective lineage tracing through analysis of somatic mutations.
INFERRING CELL FATE TRANSITIONS BY QUANTITATIVE RNA PROFILING
In both developing and mature tissues, there exist distinct populations of cells with different functions, potency, and lineage hierarchy. Differences between cell types can be assessed by comparing their gross morphologies, epigenomes, transcriptomes and proteomes. While morphology is mostly descriptive, and epigenetic descriptions can only indirectly imply function, transcriptomic and proteomic analyses serve as more reliable readouts of cellular function (Ye and Sarkar, 2018). While quantitative proteomic methods remain greatly challenging, especially with limited starting materials, despite recent advances (Swaminathan et al., 2018), RNA quantification can be used reliably in most cases to infer cell identities and functions (Edfors et al., 2016). With advances in scRNA-seq, it is possible to distinguish populations and subpopulations of cells at single cell resolution, thereby giving more comprehensive information about cellular heterogeneity and dynamic gene expression patterns (Kolodziejczyk et al., 2015; Svensson et al., 2018). In particular, scRNA-seq also allows the detection of infrequently-represented transcripts of rare cell types, which would otherwise be missed in bulk-level transcriptome analyses (Grün et al., 2015; Haber et al., 2017). Being able to profile gene expression in a given population and for cells in transition has greatly increased our understanding of the molecular mechanisms underlying cell fate transition and differentiation.
Whilst traditional lineage tracing with genetic reporters has been informative for revealing the potential of particular cell type(s), with clear directional information between lineages, scRNA-seq is useful in studying how particular transitions from given cell type(s) occur, but with only a relatively rough idea of the directionality of those cellular transitions. The basic workflow involves first isolating single cells and lysing them separately, followed by reverse transcription to generate cDNA and amplification of that cDNA. The resulting pool of cDNA is subsequently prepared for sequencing (Baran-Gale et al., 2018). Since the first report of scRNA-seq (Tang et al., 2009), several labs have improved the technology by various means (see comparison: Ziegenhain et al., 2017), such as by incorporating fluidic devices to capture single cells and the incorporation of unique molecular identifiers (UMI) to resolve technical noise signals. With commercialized library preparation and sequencing pipelines and the ready availability of analysis algorithms, scRNA-seq has become popular in many research labs across a variety of research fields.
scRNA-seq can reveal the gene expression profiles of both the steady and transitioning states of captured cells (Figs. 1A and 1B). Assuming that the captured cells include cells not only at the start or end of the transition but also those in intermediate phases, one could create a lineage trajectory map along a pseudotime scale and subsequently elucidate candidate factors associated with the transition (Kester and van Oudenaarden, 2018) (Fig. 1C). Numerous trajectory inference algorithms have been developed in recent years (Kester and van Oudenaarden, 2018; Ye and Sarkar, 2018) and applied to analyze various biological transitions in different contexts. A comprehensive study recently aimed to benchmark 29 reported lineage inference methods (Saelens et al., 2018): depending on the type of data generated, the practical guideline can be used to choose the most suitable algorithm for trajectory inference analysis. In general, pseudotime trajectory inference methods are useful in identifying genes underlying state transitions, however, it should be noted that the true directionality of gene expression changes over time is not completely present in the ‘snapshot’ of scRNA-seq data, necessitating the use of additional, complementary strategies to overcome this limitation (Weinreb et al., 2018).
Fig. 1. Analysis of cell populations using single-cell RNA-sequencing.
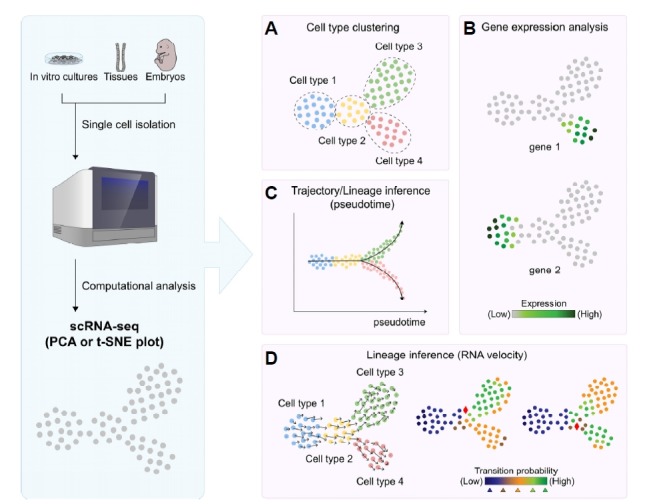
Individual cells isolated from cell culture, embryos or tissues are subjected to scRNA-seq to profile gene expression. Analysis of scRNA-seq results using principal component analysis (PCA) or t-distributed stochastic neighbor embedding (t-SNE) allows the clustering of (sub)populations of cells and identification of cell types (A). The same plot can be used to visualize gene expression levels on a color scale to assess cell type-specific transcripts (B). To investigate the transition between different cell types in a biological context, pseudotime trajectory inference algorithms allow the mapping of transitions on an arbitrary time scale (C). Another lineage inference method, called RNA velocity, calculates the proportion of unspliced and spliced transcripts, thereby allowing prediction of the prospective fate of individual cells (D).
Recently, a different lineage inference approach called RNA velocity was reported which infers the state (transitioning vs steady) and directionality (trajectory) of cell fate by comparing the ratio between immature, unspliced transcripts and mature, spliced transcripts (La Manno et al., 2018). In available scRNA-seq datasets (The Tabula Muris Consortium, 2018), a notable portion of total reads (~20%) contain intronic sequences which correspond to unspliced transcripts. In the RNA velocity approach, the balance between unspliced and spliced mRNA is taken to be informative of the future state of cells. Therefore, one can determine probabilistic directional information from the ‘snapshot’ of gene expression profiles of single cells, which can help in identifying the correct lineage specification and hierarchy (Fig. 1D). For example, for differentiating progenitor cells that are located at the branch point of two lineages, RNA velocity gives a probabilistic value as to which lineage the cell will commit to, thereby also identifying candidate genetic factors for cell fate determination (La Manno et al., 2018). It is likely that RNA velocity will be particularly useful in the analysis of human samples, where the ability to implement complementary experimental strategies is limited.
LINEAGE TRACING WITH GENETIC BARCODING AND GENETIC SCARS
Fluorescent reporter-based lineage tracing methods, which mark each cell with various color combinations, have been fundamental to our understanding of developmental biology and stem cell research. However, practically speaking the number of available combinations is limited to a size of dozens of color codes (Livet et al., 2007; Weissman and Pan, 2015). This limits the possibility of tracing a large number of cells in parallel and potentially complicates lineage analysis due to the high probability of having two independent clones bearing the same color code in close proximity. To overcome this limitation, several methods have been introduced which rely on generating DNA fingerprints in each cell at the cost of the loss of imaging information. Several types of DNA fingerprints have been used, including DNA barcoding, Polylox and CRISPR/Cas9-based scar generation strategies (Fig. 2).
Fig. 2. Overview of modern lineage tracing methods.
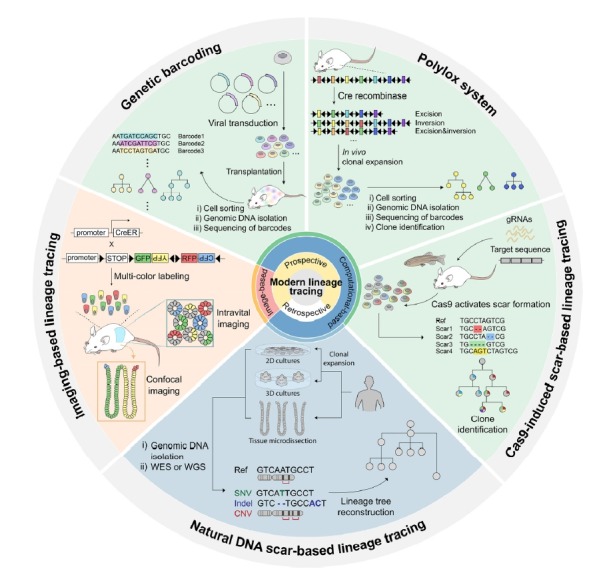
Various strategies to reconstruct cell lineage trees have been developed in line with advances in next-generation sequencing. Modern lineage tracing can be divided into prospective and retrospective methods. Prospective tracing methods mark cells using fluorescence for imaging-based, and genetic barcoding or genetic scars for computational-based methods, whereas retrospective methods use somatic mutations which occur naturally throughout the lifetime of the organism. Every method using genetic information to reconstruct cell hierarchies is computational-based lineage tracing which needs advanced NGS technologies. The figure is adapted from Fig. 3 of Kester and van Oudenaarden (2018).
DNA barcoding with unique nucleotide sequences can label a large number of cells which can then be deconvoluted by DNA sequencing. In the case of 10-bp barcoding, 410 (~106) combinations can be generated, meaning that, theoretically, one million cells can be labelled with different DNA barcodes. Once introduced into the genome of an individual cell, the DNA barcode is passed down to its progeny, allowing the identification of lineage relationships in a large number of cells. With the advent of next-generation sequencing technology, it is now possible to elucidate which cells have which barcodes though standard library preparation and deep sequencing protocols (Kebschull and Zador, 2018). Retro/lentiviruses have been used to integrate a pool of unique DNA barcode sequences into the genome. Virus-encoded genetic barcodes were introduced into hematopoietic cells in vitro and the barcode-labelled cells were subsequently transplanted into a host mouse in order to investigate the diverse clonal differentiation pattern of hematopoietic stem cells or multipotent progenitors in vivo (Gerrits et al., 2010; Lu et al., 2011; Naik et al., 2013; Schepers et al., 2008; Verovskaya et al., 2013). A similar barcoding strategy has also been applied to cancer cells to investigate the heterogeneity and clonal evolution of cancer stem cells and progenitors. Sequencing analysis of barcoded cancer cells after xenotransplantation or after serial xenografts showed growth diversity dependent upon the different cancer subtype used (Nguyen et al., 2014; Nolan-Stevaux et al., 2013).
More recently, a ‘Polylox’ labeling strategy has been published (Pei et al., 2017) which, utilizing a unique design of the Cre-LoxP system, allows the generation of numerous combinations of LoxP barcodes upon Cre activation. The cassette has 10 loxP sites which alternate with 9 stretches of DNA with unique sequences, which in theory allows the generation of 1.8 million different barcodes through the 10 repetitive rounds of Cre excision and inversion. The authors identified 849 barcoded cells generated from up to 6 recombination events in mouse, that number being around one-third of the figure as predicted by computational methods (Pei et al., 2017). The Polylox system has an advantage over the viral barcoding method as the DNA labeling can be controlled spatiotemporally in vivo by using tissue-specific Cre or inducible CreER lines. Nevertheless, the recombination efficiency may still need further improvement in order to draw a lineage tree with the required confidence at larger scale.
The CRISPR/Cas9-based genome editing system has been used in another interesting strategy, where cells are marked by unique scar sequences generated through DNA repair of Cas9-induced double strand breaks (DSBs). This novel strategy has become a powerful tool for high-throughput lineage tracing in many different organisms (Junker et al., 2017; Kalhor et al., 2017; 2018; McKenna et al., 2016; Perli et al., 2016; Spanjaard et al., 2018). CRISPR/Cas9 is a bacterial endonuclease which can generate a DNA DSB at a specific target sequence (Jinek et al., 2012). Unless the cell uses a template for homology-directed repair or microhomology-mediated repair, DSBs will be repaired by an error-prone process which often results in various errors at the target site (Lee et al., 2018). These errors can be short insertions or deletions (indel mutations) of varying length and sequence; genetic scars that can serve as a genetic barcode in lineage tracing.
The CRISPR/Cas9-induced genetic scar method has been used to delineate a lineage tree of cells during zebrafish development (Alemany et al., 2018; McKenna et al., 2016; Spanjaard et al., 2018). Several methods have been used which generate genetic scars in multiple arrays of synthetic target sequences (GESTALT) or transgenes such as GFP (ScarTrace) or RFP (LINNAEUS). Upon co-injection of Cas9 and target-specific gRNA to 1-cell stage zebrafish embryos, multiple indel mutations form in the cells of the embryo during several rounds of division. As a result, newly generated cells can have an accumulation of various indels at the target site in addition to previous indels passed down from ancestor cells. With this information, it was possible to reconstruct the lineage tree for cells from each organ in the adult fish and so visualize how each organ of the adult body is formed from a few progenitor cells. This method has also been applied to murine development with a few modifications. Kalhor and colleagues generated a mouse line harboring specific gRNAs (homing gRNA or hgRNA library) where the target sequence was present in 60 genomic regions (Kalhor et al., 2017; 2018). Mating this line with a Cas9 knock-in line enabled the hgRNAs to start causing mutations in their target loci soon after the introduction of Cas9 and 41 out of the 60 regions were mutated to generate unique genetic scar barcodes. Theoretically, more than 1074 different combinations are possible, which is more than enough to cover the entire lineage tree of mouse development.
TRACING BY NATURALLY OCCURRING SOMATIC MUTATIONS
Mutations occur in the genome during every cell division due to the limited precision of DNA polymerase activity and repair machineries. These naturally-formed mutations in somatic cells are termed somatic mutations. Somatic mutations serve as a natural mark during our development and postnatal growth, and can be utilized as a marker for retrospective lineage tracing (Dou et al., 2018), whereas all previously mentioned barcoding or scar-forming methods are prospective tracers that are introduced intentionally (Fig. 2). Somatic mutations occur stochastically, accumulate throughout the lifetime of the organism and are inherited by all daughter cells. Albeit possible theoretically for a long time, this hidden information about the lineage of each cell has only been decoded relatively recently as a result of the advent of high quality next-generation sequencing technology (Shapiro et al., 2013).
One technical limitation has been the high error rate of sequencing technology, while the presence of somatic mutations in the genome is rare. The first reported strategy to overcome this limitation focused on copy-number variants (CNVs), since CNVs, microsatellites (MSs) and retrotransposition are relatively easy to detect with low genome coverage in comparison to single nucleotide variants (SNVs). CNVs have been used for the reconstruction of cancer cell lineage trees because CNVs frequently occur in cancer cells. A bulk WGS dataset from 21 breast cancer samples revealed the evolutionary tree of each cancer sample based on CNV analysis in combination with analysis of oncogene mutations occurring among subclones (Nik-Zainal et al., 2012). Recently, single cell WGS performed on laser-dissected single cells has enabled the reconstruction of the lineage tree of cancer evolution by CNV profiles to be combined with spatial information (Casasent et al., 2018). MSs, for which mutation sites are relatively well-defined, have been used to delineate lineage trees for many years (Frumkin et al., 2005; Reizel et al., 2011, 2012; Salipante and Horwitz, 2006). Retrotransposition of the LINE1 element was also used as a lineage tracer in order to delineate the lineage tree of the brain (Evrony et al., 2012). However, the use of these markers is specifically suited to the study of cancer (CNVs and MSs) and brain development (retrotransposition) as they occur more frequently in tumorigenesis and the development of specific organs.
To analyze SNVs with a meaningful sequencing depth, some studies utilized targeted deep sequencing on specific gene sets. For example, ultradeep targeted sequencing of 74 oncogenes (870X of median on-target coverage) in normal human esophagus epithelium from patients of various ages showed that mutation number correlates with sample age. By combining the spatial information from the samples with their SNV profiles, it was shown that cells accumulating mutations in specific genes (NOTCH and TP53 ) form rapidly expanding clones within the esophageal epithelium (Martincorena et al., 2018). A similar strategy was used in cancer-prone human skin, where targeted deep sequencing of frequently mutated genes in cutaneous squamous cells revealed that NOTCH1 was the most frequently mutated gene, and that neutral drift and stochastic nucleation of mutations together affected the clonal expansion of mutant clones (Lynch et al., 2017). In addition, two studies have also used targeted deep sequencing to reveal small subpopulations within tumors (Leung et al., 2017; Roerink et al., 2018).
Finally, several studies have used bulk WGS following clonal derivation from a single, sorted cell. In order to generate clones of cells derived from liver, small intestine and colon, single cells sorted from each tissue were seeded into 3D culture conditions to generate organoids. WGS of these clonal organoids provides high quality genome coverage with precise sequence information. In this study, the accumulation and type of mutations present in adult stem cells were found to differ according to tissue type (Blokzijl et al., 2016). Subclones from a single tumor mass have also been cultured as clonal organoids and sequenced to investigate intra-tumor heterogeneity in colorectal cancer (Roerink et al., 2018). Similarly, blood cells have been cultured as single cell-derived colonies and analyzed to delineate the lineage tree of human blood cells (Lee-Six et al., 2018). Whole genome sequences of human fetal forebrains were analyzed after the derivation of clones from single cells and compared with the genome of spleen cells to reveal the origin of each somatic mutation (Bae et al., 2018). Besides clonal derivation, one study used variant allele fractions of somatic mutations, which reveals the proportional frequency of mutation reads, from deep, bulk WGS of adult tissues to deduce early embryonic cell lineage diversification (Ju et al., 2017).
DISCUSSION AND FUTURE DIRECTIONS
In this review, we have introduced conventional imaging-based strategies (reviewed in Fink et al., 2015) as well as recently developed computational approaches (Fig. 2). Each method has its own pros and cons (Table 1). Thus, utilizing an appropriate method or combined strategy for a given biological question is key.
Table 1.
Comparison of each lineage tracing method
| Pros | Cons | Requirement | |
|---|---|---|---|
| Imaging-based lineage tracing | Completely retains spatial information; does not need complicated algorithm for analysis; potential for multiple timepoint tracing/retracing; applicable to various tissues | Limits scalability of traced progeny; variation in marking is limited; generation of new (mouse) lines may be time-consuming; not easily coupled with scRNA-seq | Inducible CreER lines in desired tissue; tissue processing (sectioning or clearing); 3D microscopy (confocal, lightsheet, intravital) |
| Genetic barcoding | Relatively easy to assign barcodes to each cell; high scalability; can be easily coupled with scRNA-seq | Limited targetable tissues; lack of spatial information; single timepoint tracing | Barcode library, delivery methods, implantation techniques; library preparation for NGS; computational reconstruction analysis |
| Polylox system | Relatively easy to assign barcodes to each cell; high scalability; applicable to various tissues; can be easily coupled with scRNA-seq | Only available in mouse, currently; single timepoint tracing | Various Cre lines; library preparation for NGS; computational reconstruction analysis |
| CRISPR/Cas9-induced scar-based lineage tracing | Relatively easy to assign genetic scars to each cell; available in various model organisms; high scalability; potential for multiple timepoint tracing; can be easily coupled with scRNA-seq | Off-target effects and multiple DSBs could result in genotoxicity | Integrating target sequences and gRNAs for target sites; induction of Cas9 endonuclease; library preparation for NGS; computational reconstruction analysis |
| Natural DNA scar-based lineage tracing | Can be applied to human patient samples; least artificial set-up because it does not need any molecular or genetic intervention | High costs; needs high computational power to distinguish between clones; unknown origin of progeny; may require clonal derivation to improve coverage | In vitro cultures to amplify single clones or laser dissection of tissues; library preparation for NGS; computational reconstruction analysis |
The imaging-based approach powered by multicolor fluorescent reporter systems often provides multifaceted visual information, including clone size, structure, distribution and cell types within the clone. Measuring these at different timepoints enables us to reconstruct in detail how a clone grows and is maintained in a tissue or developing organ. Multicolor-based mosaic genetic analysis has also become possible, combining imaging-based lineage tracing analysis with analysis of the genetic perturbations present in each colored clone (Pontes-Quero et al., 2017). However, a clear downside of imaging-based approaches is the limited number of clones that can be labelled by these systems.
Genetic barcoding strategies can overcome this limitation, but at the cost of the spatial information provided by imaging. Retro/lentiviral barcoding has been widely used in order to simultaneously analyze the clonal behavior of hundreds to millions of cells. This method is very simple to apply from a design perspective but it is limited by the accessibility of target cells for viral infection. Although complicated, Cre-LoxP-based Polylox barcoding is a powerful alternative as it combines genetic labeling in vivo with spatiotemporal control of cell labeling. As there are many Cre and tamoxifen-inducible CreER mouse lines readily available, the Polylox method is a flexible system for addressing different biological problems in mice. While the Cre-LoxP system is popular in mouse genetics, it is not widely used in other model organisms. A novel lineage tracing method utilizing CRISPR/Cas-induced genetic scars is widely considered to be more versatile and easy to apply in different model organisms. Nevertheless, the CRISPR system may cause off-target effects and inducing multiple DSBs simultaneously in a cell can cause adverse effects driven by genotoxic response.
Retrospective lineage analysis based on naturally occurring somatic variants is another promising method, which can even be applied to the analysis of human development and disease progression. This method does not employ any kind of molecular or genetic intervention, meaning that it has the least artificial experimental set up. Although there are multiple ways to circumvent associated problems (Dou et al., 2018), it is still challenging to utilize this method in delineating the entire lineage tree of an organism due to limitations such as sequencing costs, sequencing errors, required computational power, etc. As outlined above, there is no single catchall method applicable to all study types and therefore it is key to consider the requirements of individual experiments or combination strategies.
In addition to the methods described above, state-of-the-art scRNA-seq technology allows gene expression profiling at high resolution to generate a close approximation of lineage information. With scRNA-seq, it is now possible to dissect differences between (sub)populations of cells and to predict a theoretical lineage trajectory along a pseudotime scale. The recently developed RNA velocity protocol predicts each cell’s future state by quantifying unspliced and spliced transcripts, so improving the level of confidence in lineage analysis. In addition, several protocols for measuring the transcriptome, methylome and/or chromatin accessibility in single cells have also been introduced, whereby methylome and chromatin accessibility provide additional clues as to directionality (Cao et al., 2018; Clark et al., 2018; Lake et al., 2018). A multiomics single cell profiling method with genetic barcoding for lineage tracing will soon become available. Finally, in order to avoid the loss of spatial information from computational-based approaches, alternative imaging-based approaches such as single-molecule fluorescent in situ hybridization (smFISH) (Frieda et al., 2017) or optical sequencing (Feldman et al., 2018) can be applied (Moor and Itzkovitz, 2017).
Lineage tracing now comprises both imaging- and computational-based approaches. High throughput approaches are now in place for the tracing and profiling of large quantities of clones. It is expected that combinatorial approaches will allow more robust and accurate investigation of lineage transitions under various biological contexts.
ACKNOWLEDGMENTS
We are grateful to Dr. Christopher J. Hindley for critical reading of the manuscript and to Sung-hwan Bae for graphic illustration. This work was supported by the ERC starting grant (Troy Stem Cells, 639050).
REFERENCES
- Alemany A., Florescu M., Baron C.S., Peterson-Maduro J., Van Oudenaarden A. Whole-organism clone tracing using single-cell sequencing. Nature. 2018;556:108–112. doi: 10.1038/nature25969. [DOI] [PubMed] [Google Scholar]
- Alieva M., Ritsma L., Giedt R.J., Weissleder R., van Rheenen J. Imaging windows for long-term intravital imaging: general overview and technical insights. IntraVital. 2014;3:e29917. doi: 10.4161/intv.29917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S.J., Robertson E.J. Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat Rev Mol Cell Biol. 2009;10:91–103. doi: 10.1038/nrm2618. [DOI] [PubMed] [Google Scholar]
- Bae T., Tomasini L., Mariani J., Zhou B., Roychowdhury T., Franjic D., Pletikos M., Pattni R., Chen B-J, Venturini E., et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science. 2018;359:550–555. doi: 10.1126/science.aan8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran-Gale J., Chandra T., Kirschner K. Experimental design for single-cell RNA sequencing. Brief Funct Genomics. 2018;17:233–239. doi: 10.1093/bfgp/elx035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F., De Ligt J., Jager M., Sasselli V., Roerink S., Sasaki N., Huch M., Boymans S., Kuijk E., Prins P., et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs J.A., Weinreb C., Wagner D.E., Megason S., Peshkin L., Kirschner M.W., Klein A.M. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science. 2018;360:eaar5780. doi: 10.1126/science.aar5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J., Cusanovich D.A., Ramani V., Aghamirzaie D., Pliner H.A., Hill A.J., Daza R.M., McFaline-Figueroa J.L., Packer J.S., Christiansen L., et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:1380–1385. doi: 10.1126/science.aau0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasent A.K., Schalck A., Gao R., Sei E., Long A., Pangburn W., Casasent T., Meric-Bernstam F., Edgerton M.E., Navin N.E. Multiclonal invasion in breast tumors identified by topographic single cell sequencing. Cell. 2018;172:205–217.e12. doi: 10.1016/j.cell.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S.J., Argelaguet R., Kapourani C-A, Stubbs T.M., Lee H.J., Alda-Catalinas C., Krueger F., Sanguinetti G., Kelsey G., Marioni J.C., et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9:781. doi: 10.1038/s41467-018-03149-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–284. doi: 10.1016/j.cell.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Dou Y., Gold H.D., Luquette L.J., Park P.J. Detecting somatic mutations in normal cells. Trends Genet. 2018;34:545–557. doi: 10.1016/j.tig.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfors F., Danielsson F., Hallström B.M., Käll L., Lundberg E., Pontén F., Forsström B., Uhlén M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016;12:883. doi: 10.15252/msb.20167144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrony G.D., Cai X., Lee E., Hills L.B., Elhosary P.C., Lehmann H.S., Parker J.J., Atabay K.D., Gilmore E.C., Poduri A., et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151:483–496. doi: 10.1016/j.cell.2012.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell J.A., Wang Y., Riesenfeld S.J., Shekhar K., Regev A., Schier A.F. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science. 2018;360:eaar3131. doi: 10.1126/science.aar3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman D., Singh A., Schmid-Burgk J.L., Mezger A., Garrity A.J., Carlson R.J., Zhang F., Blainey P.C. Pooled optical screens in human cells. BioRxiv. 2018 doi: 10.1016/j.cell.2019.09.016. 383943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink J., Andersson-Rolf A., Koo B.K. Adult stem cell lineage tracing and deep tissue imaging. BMB Rep. 2015;48:655–667. doi: 10.5483/BMBRep.2015.48.12.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieda K.L., Linton J.M., Hormoz S., Choi J., Chow K-HK, Singer Z.S., Budde M.W., Elowitz M.B., Cai L. Synthetic recording and in situ readout of lineage information in single cells. Nature. 2017;541:107–111. doi: 10.1038/nature20777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumkin D., Wasserstrom A., Kaplan S., Feige U., Shapiro E. Genomic variability within an organism exposes its cell lineage tree. PLoS Comput Biol. 2005;1:e50. doi: 10.1371/journal.pcbi.0010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehart H., Clevers H. Tales from the crypt: new insights into intestinal stem cells. Nat Rev Gastroenterol Hepatol. 2019;16:19–34. doi: 10.1038/s41575-018-0081-y. [DOI] [PubMed] [Google Scholar]
- Gerrits A., Dykstra B., Kalmykowa O.J., Klauke K., Verovskaya E., Broekhuis M.J.C., de Haan G., Bystrykh L.V. Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood. 2010;115:2610–2618. doi: 10.1182/blood-2009-06-229757. [DOI] [PubMed] [Google Scholar]
- Grün D., Lyubimova A., Kester L., Wiebrands K., Basak O., Sasaki N., Clevers H., van Oudenaarden A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- Haber A.L., Biton M., Rogel N., Herbst R.H., Shekhar K., Smillie C., Burgin G., Delorey T.M., Howitt M.R., Katz Y., et al. A single-cell survey of the small intestinal epithelium. Nature. 2017;551:333–339. doi: 10.1038/nature24489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y.S., Martincorena I., Gerstung M., Petljak M., Alexandrov L.B., Rahbari R., Wedge D.C., Davies H.R., Ramakrishna M., Fullam A., et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–718. doi: 10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junker J.P., Spanjaard B., Peterson-Maduro J., Alemany A., Hu B., Florescu M., van Oudenaarden A. Massively parallel clonal analysis using CRISPR/Cas9 induced genetic scars. BioRxiv. 2017 056499. [Google Scholar]
- Kalhor R., Mali P., Church G.M. Rapidly evolving homing CRISPR barcodes. Nat Methods. 2017;14:195–200. doi: 10.1038/nmeth.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalhor R., Kalhor K., Mejia L., Leeper K., Graveline A., Mali P., Church G.M. Developmental barcoding of whole mouse via homing CRISPR. Science. 2018;361:eaat9804. doi: 10.1126/science.aat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull J.M., Zador A.M. Cellular barcoding: lineage tracing, screening and beyond. Nat Methods. 2018;15:871–879. doi: 10.1038/s41592-018-0185-x. [DOI] [PubMed] [Google Scholar]
- Keller P.J., Schmidt A.D., Wittbrodt J., Stelzer E.H.K. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 2008;322:1065–1069. doi: 10.1126/science.1162493. [DOI] [PubMed] [Google Scholar]
- Kester L., van Oudenaarden A. Single-cell transcriptomics meets lineage tracing. Cell Stem Cell. 2018;23:166–179. doi: 10.1016/j.stem.2018.04.014. [DOI] [PubMed] [Google Scholar]
- Kit S., Beck C., Graham O.L., Gross A. Effect of 5-bromodeoxyuridine on deoxyribonucleic acid-thymine synthesis and cell metabolism of lymphatic tissues and tumors. Cancer Res. 1958;18:598–602. [PubMed] [Google Scholar]
- Kolodziejczyk A.A., Kim J.K., Svensson V., Marioni J.C., Teichmann S.A. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Lake B.B., Chen S., Sos B.C., Fan J., Kaeser G.E., Yung Y.C., Duong T.E., Gao D., Chun J., Kharchenko P.V., et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H., Øbro NF, Shepherd M.S., Grossmann S., Dawson K., Belmonte M., Osborne R.J., Huntly B.J.P., Martincorena I., Anderson E., et al. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561:473–478. doi: 10.1038/s41586-018-0497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Kim S., Hur J.K. CRISPR and target-specific DNA endonucleases for efficient DNA knock-in in eukaryotic genomes. Mol Cells. 2018;41:943–952. doi: 10.14348/molcells.2018.0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung M.L., Davis A., Gao R., Casasent A., Wang Y., Sei E., Vilar E., Maru D., Kopetz S., Navin N.E. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017;27:1287–1299. doi: 10.1101/gr.209973.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livet J., Weissman T.A., Kang H., Draft R.W., Lu J., Bennis R.A., Sanes J.R., Lichtman J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature. 2007;450:56–62. doi: 10.1038/nature06293. [DOI] [PubMed] [Google Scholar]
- Lu R., Neff N.F., Quake S.R., Weissman I.L. Tracking single hematopoietic stem cells in vivo using high-throughput sequencing in conjunction with viral genetic barcoding. Nat Biotechnol. 2011;29:928–933. doi: 10.1038/nbt.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M.D., Lynch C.N.S., Craythorne E., Liakath-Ali K., Mallipeddi R., Barker J.N., Watt F.M. Spatial constraints govern competition of mutant clones in human epidermis. Nat Commun. 2017;8:1–11. doi: 10.1038/s41467-017-00993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., Lidschreiber K., Kastriti M.E., Lönnerberg P., Furlan A., et al. RNA velocity of single cells. Nature. 2018;560:494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I., Fowler J.C., Wabik A., Lawson A.R.J., Abascal F., Hall M.W.J., Cagan A., Murai K., Mahbubani K., Stratton M.R., et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–917. doi: 10.1126/science.aau3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDole K., Guignard L., Amat F., Berger A., Malandain G., Royer L.A., Turaga S.C., Branson K., Keller P.J. In toto imaging and reconstruction of post-implantation mouse development at the single-cell level. Cell. 2018;175:859–876.e33. doi: 10.1016/j.cell.2018.09.031. [DOI] [PubMed] [Google Scholar]
- McKenna A., Findlay G.M., Gagnon J.A., Horwitz M.S., Schier A.F., Shendure J. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016;353:aaf7907. doi: 10.1126/science.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor A.E., Itzkovitz S. Spatial transcriptomics: paving the way for tissue-level systems biology. Curr Opin Biotechnol. 2017;46:126–133. doi: 10.1016/j.copbio.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Murray S.A., Eppig J.T., Smedley D., Simpson E.M., Rosenthal N. Beyond knockouts: cre resources for conditional mutagenesis. Mamm Genome. 2012;23:587–599. doi: 10.1007/s00335-012-9430-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S.H., Perié L., Swart E., Gerlach C., van Rooij N., de Boer R.J., Schumacher T.N. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature. 2013;496:229–232. doi: 10.1038/nature12013. [DOI] [PubMed] [Google Scholar]
- Nguyen L.V., Cox C.L., Eirew P., Knapp D.J.H.F., Pellacani D., Kannan N., Carles A., Moksa M., Balani S., Shah S., et al. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat Commun. 2014;5:5871. doi: 10.1038/ncomms6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S., Van Loo P., Wedge D.C., Alexandrov L.B., Greenman C.D., Lau K.W., Raine K., Jones D., Marshall J., Ramakrishna M., et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan-Stevaux O., Tedesco D., Ragan S., Makhanov M., Chenchik A., Ruefli-Brasse A., Quon K., Kassner P.D. Measurement of cancer cell growth heterogeneity through lentiviral barcoding identifies clonal dominance as a characteristic of in vivo tumor engraftment. PLoS One. 2013;8:e67316. doi: 10.1371/journal.pone.0067316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A., Venkatasubramanian M., Chaudhri V.K., Aronow B.J., Salomonis N., Singh H., Grimes H.L. Single-cell analysis of mixed-lineage states leading to a binary cell fate choice. Nature. 2016;537:698–702. doi: 10.1038/nature19348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W., Feyerabend T.B., Rössler J., Wang X., Postrach D., Busch K., Rode I., Klapproth K., Dietlein N., Quedenau C., et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature. 2017;548:456–460. doi: 10.1038/nature23653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perli S.D., Cui C.H., Lu T.K. Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science. 2016;353:aag0511. doi: 10.1126/science.aag0511. [DOI] [PubMed] [Google Scholar]
- Pontes-Quero S., Heredia L., Casquero-García V., Fernández-Chacón M., Luo W., Hermoso A., Bansal M., Garcia-Gonzalez I., Sanchez-Muñoz M.S., Perea J.R., et al. Dual ifgMosaic: a versatile method for multispectral and combinatorial mosaic gene-function analysis. Cell. 2017;170:800–814.e18. doi: 10.1016/j.cell.2017.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizel Y., Chapal-Ilani N., Adar R., Itzkovitz S., Elbaz J., Maruvka Y.E., Segev E., Shlush L.I., Dekel N., Shapiro E. Colon stem cell and crypt dynamics exposed by cell lineage reconstruction. PLoS Genet. 2011;7:e1002192. doi: 10.1371/journal.pgen.1002192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reizel Y., Itzkovitz S., Adar R., Elbaz J., Jinich A., Chapal-Ilani N., Maruvka Y.E., Nevo N., Marx Z., Horovitz I., et al. Cell lineage analysis of the mammalian female germline. PLoS Genet. 2012;8:e1002477. doi: 10.1371/journal.pgen.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roerink S.F., Sasaki N., Lee-Six H., Young M.D., Alexandrov L.B., Behjati S., Mitchell T.J., Grossmann S., Lightfoot H., Egan D.A., et al. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature. 2018;556:457–462. doi: 10.1038/s41586-018-0024-3. [DOI] [PubMed] [Google Scholar]
- Saelens W., Cannoodt R., Helena T., Saeys Y. A comparison of single-cell trajectory inference methods: towards more accurate and robust tools. BioRxiv. 2018 doi: 10.1038/s41587-019-0071-9. 276907. [DOI] [PubMed] [Google Scholar]
- Salipante S.J., Horwitz M.S. Phylogenetic fate mapping. Proc Natl Acad Sci. 2006;103:5448–5453. doi: 10.1073/pnas.0601265103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers K., Swart E., van Heijst J.W.J., Gerlach C., Castrucci M., Sie D., Heimerikx M., Velds A., Kerkhoven R.M., Arens R., et al. Dissecting T cell lineage relationships by cellular barcoding. J Exp Med. 2008;205:2309–2318. doi: 10.1084/jem.20072462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro E., Biezuner T., Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet. 2013;14:618–630. doi: 10.1038/nrg3542. [DOI] [PubMed] [Google Scholar]
- Spanjaard B., Hu B., Mitic N., Olivares-Chauvet P., Janjuha S., Ninov N., Junker J.P. Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat Biotechnol. 2018;36:469–473. doi: 10.1038/nbt.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston J.E., Schierenberg E., White J.G., Thomson J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Svensson V., Vento-Tormo R., Teichmann S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc. 2018;13:599–604. doi: 10.1038/nprot.2017.149. [DOI] [PubMed] [Google Scholar]
- Swaminathan J., Boulgakov A.A., Hernandez E.T., Bardo A.M., Bachman J.L., Marotta J., Johnson A.M., Anslyn E.V., Marcotte E.M. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat Biotechnol. 2018;36:1076–1082. doi: 10.1038/nbt.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F., Barbacioru C., Wang Y., Nordman E., Lee C., Xu N., Wang X., Bodeau J., Tuch B.B., Siddiqui A., et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- The Tabula Muris Consortium. Single-cell transcriptomics of 20 mouse organs creates a tabula muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P.Q., Brown A., Beddington R.S. Hex: a homeobox gene revealing peri-implantation asymmetry in the mouse embryo and an early transient marker of endothelial cell precursors. Development. 1998;125:85–94. doi: 10.1242/dev.125.1.85. [DOI] [PubMed] [Google Scholar]
- Verovskaya E., Broekhuis M.J.C., Zwart E., Ritsema M., van Os R., de Haan G., Bystrykh L.V. Heterogeneity of young and aged murine hematopoietic stem cells revealed by quantitative clonal analysis using cellular barcoding. Blood. 2013;122:523–532. doi: 10.1182/blood-2013-01-481135. [DOI] [PubMed] [Google Scholar]
- Weinreb C., Wolock S., Tusi B.K., Socolovsky M., Klein A.M. Fundamental limits on dynamic inference from single-cell snapshots. Proc Natl Acad Sci. 2018;115:E2467–E2476. doi: 10.1073/pnas.1714723115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman T.A., Pan Y.A. Brainbow: new resources and emerging biological applications for multicolor genetic labeling and analysis. Genetics. 2015;199:293–306. doi: 10.1534/genetics.114.172510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters I.P., Murray C.W., Winslow M.M. Towards quantitative and multiplexed in vivo functional cancer genomics. Nat Rev Genet. 2018;19:741–755. doi: 10.1038/s41576-018-0053-7. [DOI] [PubMed] [Google Scholar]
- Ye Z., Sarkar C.A. Towards a quantitative understanding of cell identity. Trends Cell Biol. 2018;28:1030–1048. doi: 10.1016/j.tcb.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegenhain C., Vieth B., Parekh S., Reinius B., Guillaumet-Adkins A., Smets M., Leonhardt H., Heyn H., Hellmann I., Enard W. Comparative analysis of single-cell RNA sequencing methods. Mol Cell. 2017;65:631–643.e4. doi: 10.1016/j.molcel.2017.01.023. [DOI] [PubMed] [Google Scholar]