

Abstract
Lysophosphatidic acid (LPA) is an endogenous lysophospholipid with signaling properties outside of the cell and it signals through specific G protein-coupled receptors, known as LPA1–6. For one of its receptors, LPA1 (gene name Lpar1), details on the cis-acting elements for transcriptional control have not been defined. Using 5′RACE analysis, we report the identification of an alternative transcription start site of mouse Lpar1 and characterize approximately 3,500 bp of non-coding flanking sequence 5′ of mouse Lpar1 gene for promoter activity. Transient transfection of cells derived from mouse neocortical neuroblasts with constructs from the 5′ regions of mouse Lpar1 gene revealed the region between −248 to +225 serving as the basal promoter for Lpar1. This region also lacks a TATA box. For the region between −761 to −248, a negative regulatory element affected the basal expression of Lpar1. This region has three E-box sequences and mutagenesis of these E-boxes, followed by transient expression, demonstrated that two of the E-boxes act as negative modulators of Lpar1. One of these E-box sequences bound the HeLa E-box binding protein (HEB), and modulation of HEB levels in the transfected cells regulated the transcription of the reporter gene. Based on our data, we propose that HEB may be required for a proper regulation of Lpar1 expression in the embryonic neocortical neuroblast cells and to affect its function in both normal brain development and disease settings.
Keywords: alternative splicing, HeLa E-box binding protein, lysophosphatidic acid receptor 1, transcription repressor
INTRODUCTION
Lysophosphatidic acid [(LPA), 1-acyl-sn-glycerol-3-phosphate] is a bioactive phospholipid affecting various cell types for proliferation, differentiation, and survival (Anliker et al., 2013; Chen et al., 2013; Lapierre et al., 2010; Liu et al., 2010; Sheng et al., 2015; Ye and Chun, 2010; Zhang et al., 2012). There are various forms of LPA, each differing on the fatty acids conjugated at the hydroxy residue at the sn-1 or sn-2 positions (Kano et al., 2008). Numerous biochemical pathways can lead to synthesis of LPA (Aoki, 2004; Yung et al., 2014) with LPA being present in significant amounts in human and mouse serum at concentrations that may exceed tens of micro molars under certain conditions. In addition, LPA is present in the cerebral cortex during development (Aoki, 2004; Yung et al., 2014).
With LPA, being ubiquitously present, varying the expression, selectivity or sensitivity of its family of receptors probably provides a key mode of control for its biology. There are at least six known G protein-coupled receptors (GPCRs) of LPA, namely LPA1, 2, 3, 4, 5, and 6 (D’Souza et al., 2018; Kihara et al., 2014; Yung et al., 2014). These receptors have varying affinities to different forms of LPA and the receptors may mediate differing pathways depending on the nature of the heterodimeric G proteins each receptor activates (Kano et al., 2008; Sheng et al., 2015). LPA’s first cloned receptor with its gene name Lpar1, LPA1 is expressed in multiple organs including brain, lung, heart in mice and humans. However, the expression of LPA1 is strictly controlled depending on the context. From mouse studies, LPA1 is developmentally expressed in the neural progenitor zone of the embryonic cerebral cortex, namely the ‘ventricular zone’ (VZ), and its expression disappears at the end of cortical neurogenesis just before birth (Hecht et al., 1996). From Lpar1 knockout mouse studies, there was altered food consumption in mice lacking Lpar1 due to the effects in the CNS (Contos et al., 2000; D’Souza et al., 2018; Estivill-Torrus et al., 2008). In these mice, there were also effects on the cardiovascular system, lung, intestine, adipose tissue, and bone (Contos et al., 2000; Gennero et al., 2011). Also from studies with Lpar1-null mice, LPA1 has been shown to play a role in the biology of neuropathic pain (Inoue et al., 2008; Uchida et al., 2014).
E-proteins are members of the basic helix-loop-helix (bHLH) protein family and are important in several developmental processes. bHLH proteins are classified into two groups, structurally and functionally. Class-I bHLH proteins which we will refer to as ‘E-proteins’ bind to the DNA E-box motif, CANNTG, as homodimers or as heterodimers with other bHLH proteins. Class-II bHLH proteins are tissue-specific and can only bind DNA as heterodimers with other E-proteins. The mammalian E-proteins are encoded within three separate genes, E2A (as splice variants E12, E47), HEB and E2-2 (Wang and Baker, 2015; Welner et al., 2008). E-proteins are also negatively regulated by the Id (inhibitor of differentiation or DNA binding) class of bHLH proteins. There are four vertebrate Id proteins, Id1, Id2, Id3 and Id4, which comprise a class of HLH proteins that lack a DNA binding domain. Id proteins incorporate E-proteins into E–Id heterodimers, making the E-proteins unable to bind E-box sequences, functioning as negative regulators of E-protein activity (Wang and Baker, 2015; Welner et al., 2008). In cerebral cortical development, bHLH proteins play key roles, affecting the timing of differentiation and the specification of cell fate (Powell and Jarman, 2008; Ross et al., 2003; Wang and Baker, 2015).
In this study, we characterize the mouse Lpar1 promoter region, as there is little information about the mechanism of Lpar1 regulation for potential cis-acting elements. We show that the core promoter lacks a TATA box and the 5′ deletion constructs identify positive and negative cis-elements in regulating Lpar1 expression. We report that the E-protein HEB (gene name Tcf12) represses Lpar1 promoter activity in mouse neocortical neuroblast cells and map its site of interaction as it implies an important role in brain development.
MATERIALS AND METHODS
Materials
2-mercaptoethanol was purchased from Sigma-Aldrich (St. Louis, USA). Fetal bovine serum (FBS) was obtained from HyClone/GE Healthcare (Logan, USA). Lipofectamine 2000, Opti-MEM I, and penicillin/ streptomycin were obtained from Invitrogen/Thermo Fisher (Waltham, USA). The TOPcloner TA vector for sequencing and nPfuforte DNA polymerase were purchased from Enzynomics (Daejeon, Korea). pGL3-Basic, Renilla luciferase reporter plasmid, and Dual luciferase reporter assay system were purchased from Promega (Madison, USA). Antibody against β-actin was obtained from Cell Signaling Technology (Beverly, USA). Antibody specific to HEB was purchased from Santa Cruz Biotechnology (Santa Cruz, USA). The HEB and Id3 expression vector was a gift from Dr. Sung Ho Jeon (Hallym University, Korea). All other chemicals were received from Sigma-Aldrich.
5′ rapid amplification of cDNA ends (5′RACE)
Total RNA was isolated from TR cells, which are a necortical neuroblast clonal cell line of mouse origin transformed by large T antigen and vras (Chun and Jaenisch, 1996). Total RNA extraction was via the guanidine isothiocyanate method (Chomczynski and Sacchi, 1987). TR is a necortical neuroblast clonal cell line of mouse origin transformed by large T antigen and vras (Chun and Jaenisch, 1996). These cells stably express telencephalon-specific gene BF-1 and a gene enriched in the neocortical ventricular zone, vzg-1. An antisense Lpar1 oligodeoxynucleotide (5′-CCGGTTGCTCATTCGT GTATGGAGCTG-3′) corresponding to the center region of exon 3 was synthesized (Contos and Chun, 1998). The synthesis of the first cDNA strand and subsequent amplification of 5′ cDNA end was carried out as detailed in the BD SMART RACE cDNA Amplification kit manual. Total RNA was reverse-transcribed using a modified lock-docking oligonucleotide (dT) primer and BD SMART II oligonucleotide at 42°C for 1.5 h to obtain the first cDNA strand. 5′RACE was performed by incubating the antisense Lpar1 antisense primer with the first cDNA strand, using the following PCR conditions. After an initial denaturation of one cycle at 94°C for 2 min, the mixture was amplified at 94°C for 45 s, at 68°C for 45 s, and at 72°C for 3 min for 30 cycles. The resulting products were cloned into a sequencing vector, TOP cloner TA, and sequenced to determine the transcription start site.
Constructs
The mouse Lpar1 promoter region studied in this paper was isolated by screening a mouse genomic library (Contos and Chun, 1998). The mouse Lpar1 promoter region was further digested and sub-cloned into promoter-less pGL3-BASIC (Promega) vector into restriction enzyme sites listed in Table 1. In generating the −3549/+518 construct, the SacII restriction enzyme fragment of the genomic DNA was blunt ended using the Klenow enzyme and digested using KpnI. This fragment was then subcloned into the KpnI/SmaI site of the pGL3-BASIC vector. The Lpar1 promoter deletion constructs were also generated by restriction enzyme digest and PCR. The −2867/+225 and −1766/+225 constructs were produced by digesting the −3549/+518 construct with the indicated restriction enzymes, ligating, digesting again with SmaI, and re-ligating. The −761/+225, −142/+225, and −3/+225 constructs were also made from the −1766/+225 construct using the same procedure and their specified restriction enzymes (Table 1). The three constructs, −660/+225, −432/+225, −350/+225 were generated by PCR, PstI digestion, and subsequent ligation into the −1766/+225 construct which had been digested with NheI, made blunt, and digested once more with PstI. The −937/+225 construct was generated by PCR, SphI digestion, and subsequent ligation into the 5.5 kb elution product of −1766/+255 construct which had been digested with NheI, made blunt, and partially digested using SphI. The −248/+225 construct was also generated by PCR, XhoI digestion, and subsequent ligation into the −1766/+255 construct, which was digested with NheI, made blunt, and digested with XhoI. The PCR primers are listed in Table 1. All constructs were confirmed by automated DNA sequencing.
Table 1.
| Primers and the restriction enzyme used for generation of promoter reporter constructs. | ||
|---|---|---|
|
| ||
| Constructs | Primers | Restriction enzyme |
| −937/+225 | Forward: ACTATCCAACCCCCAGTGTCTT Reverse: TCTTGCAGAGCTTTGGTGGA |
SphI |
| −248/+225 | Forward: CTTGTTTTCCCCAGGCTCTGT Reverse: GCCGGGCCTTTCTTTATGTT |
XhoI |
| −660/+225 | Forward: GTGTGAAGCAGGTGGGTGAA Reverse: GCCACATCCACAGAGCACAA |
PstI |
| −539/+225 | Forward: CCGCCTACACTTCCAGGTG Reverse: GCCACATCCACAGAGCACAA |
PstI |
| −432/+225 | Forward: AGCTCTAGGGACACAAAGGC Reverse: GCCACATCCACAGAGCACAA |
PstI |
| −350/+225 | Forward: GCTGTGCTGGTTGAAACTTTGT Reverse: GCCACATCCACAGAGCACAA |
PstI |
|
| ||
| Restriction enzyme used for generation of promoter reporter constructs. | ||
|
| ||
| Constructs | Restriction enzyme | |
|
| ||
| −2867/+225* | SacI/KpnI | |
| −1766/+225* | NheI/KpnI | |
| −761/+225 | SauI/NheI | |
| −142/+225 | StuI/NheI | |
| −3/+225 | PstI/NheI | |
Constructs made from restriction enzyme digest of the −3549/+518 construct
All the other constructs were created by PCR and/or restriction enzyme digest of the −1766/+225 construct
Database searches for identification of conserved E-protein sites in 5′ Lpar1 upstream sequences
The 5′ upstream Lpar1 region sequences for human, mouse and rat were obtained from the Gene annotations of the NCBI database for Lpar1: human, https://www.ncbi.nlm.nih.gov/gene/1902; mouse, https://www.ncbi.nlm.nih.gov/gene/14745; and rat, https://www.ncbi.nlm.nih.gov/gene/116744. The region of interest for each gene was identified through Nucleotide BLAST alignment search tool (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Clustal Omega program was then used for identification and annotation of Lpar1 upstream conserved nucleotide residues (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Mutagenesis for E-protein binding sites
The mutations for putative E-protein binding sites on the constructs of mouse Lpar1 promoter were generated by PCR, using the overlap extension method (Heckman and Pease, 2007). Mutant constructs were created by using unique SauI restriction sites (listed in Table 1). All PCR constructs were verified by DNA sequencing. Site-directed mutagenesis was performed with the mega primer PCR and overlap extension PCR method (Ke and Madison, 1997; Urban et al., 1997).
Cell culture
TR mouse cells (Chun and Jaenisch, 1996) were maintained as monolayer cultures in Opti-MEM I reduced–serum medium supplemented with 2.5% heat-inactivated fetal bovine serum, 20 mM glucose, 55 μM 2-mercaptoethanol, and 100 unit penicillin/ 100 μg streptomycin.
Transient transfection and luciferase assay
TR mouse cells (Chun and Jaenisch, 1996) were cultured to 60–80% confluence in 24-well plates for transfection experiments. For each well, Lipofectamine 2000 reagent was used as specified in the manufacturer’s instructions. Plasmids with the mouse Lpar1 promoter were fused to the firefly luciferase expression vector (−3549/+518A1). The plasmid containing the Renilla luciferase gene driven by the TK promoter was included to normalize the transfection efficiency. DNA and the Lipofectamine 2000 reagent were diluted separately in 50 μl of serum-free medium without antibiotics, mixed together, and incubated at room temperature for 30 min. The culture plates were washed with phosphate-buffered saline (PBS) and 400 μl of antibiotic-free medium was added. The 100 μl of the plasmid / Lipofectamine 2000 mixture was then gently added to each well and the plates were incubated at 37°C for 24 h. Thereafter, cell extracts were prepared by rinsing each plate twice with PBS for lysing in 100 μl of Reporter Assay Lysis buffer (Promega). The lysed cells were collected and the supernatants were assayed for firefly luciferase and Renilla luciferase activities. They were measured with a Promega Dual-luciferase reporter assay system using an Autolumat Luminometer (Berthold, USA). Statistical analysis was carried out by Student’s t test.
Quantitative RT-PCR analysis
Total RNA was isolated from cell and tissue, and cDNA was made using 2 μg of total RNA and AMV reverse-transcriptase (Promega) in 20 μl reaction mixtures in the presence of 2.5 μM oligo (dT) primer and 20 μM dNTP mixture for 60 min at 42°C. Real-Time PCR was performed in triplicate in 20 μl using the QuantiTect SYBR Green PCR kit with reads normalized against Gapdh housekeeping control (Qiagen, USA) and following the manufacturer’s instructions. The reactions were carried out in the Rotor-Gene 3000 system (Qiagen).
The Lpar1 exon 3 reverse primer, 5′-CAATCCAGCGAAGA AGTCTGCA-3′, present in exon 3, was common in each reaction as the 3′ primer. Three separate 5′ primers were used corresponding to the three putative primary exons: Lpar1 transcript variant1 (Genbank accession number NM010336) forward primer 5′-TGAACTGCGGAGCTGGACC TA-3′, Lpar1 transcript variant2 (Genbank accession number NM172989) forward primer 5′-CACCAGCCGGTGGAACTCA-3′, Lpar1 transcript variant 3 forward primer 5′-ACAGGAGG CAGAGTCGCTGAG-3′. The thermal cycling conditions included an initial activation step at 95°C for 15 min, followed by 50 cycles of denaturation, annealing, and amplification (94°C for 15 s, 57°C for 30 s, 72°C for 45 s). Fluorescence data collection was performed during the annealing step.
Isolation of total protein and western blot analysis
TR mouse cells (Chun and Jaenisch, 1996) were lysed in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 1 mM Na3VO4, 5 mM NaF, and protease inhibitor cocktail). After incubation on ice for 30 min, the lysates were centrifuged (15,000 g, 15 min). Supernatants were collected and protein concentrations were determined by Bradford assay (Bio-Rad, USA). Equal amounts of protein were loaded and separated by SDS-PAGE transferred to polyvinylidene difluoride membranes (Millipore, USA), and blocked with 5% non-fat milk. Membranes were incubated in primary antibody overnight at 4°C. Membranes were then washed in TBST (10 mM Tris, 140 mM NaCl, 0.1% Tween 20, pH 7.6), incubated with appropriate secondary antibody, and washed again in TBST. Bands were visualized by chemiluminescence and exposed to X-ray film.
RNA interference by siRNA
TR mouse cells (Chun and Jaenisch, 1996) were transiently transfected with the Dicer-substrate siRNA oligonucleotides by using Lipofectamine 2000 (Invitrogen). The most effective siRNA oligonucleotide directed toward HEB gene was chosen among the TriFECTa siRNAs (Integrated DNA Technologies, USA) by testing the efficacy to reduce the levels of specific protein. The specific sequences of the siRNA including the control siRNA were optimized by the manufacturer (Integrated DNA Technologies) as 3 oligonucleotides per target; however, the sequences of the oligoes were not made available by the manufacturer.
Chromatin immuno-precipitation (ChIP) analysis
ChIP analyses were performed according to the manufacturer’s instructions (Upstate Biotechnology, USA). Cells (one 100-mm dish) were cross-linked with 1% formaldehyde for 10 min at 37°C. After washing twice with ice-cold PBS containing protease inhibitors, scraping and centrifugation cell pellets were resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, and protease inhibitor cocktail). After incubation for 10 min at 4°C, cell lysates were sonicated 4 times for 15 sec each on ice. After centrifugation, the sonicated cell supernatant was diluted 10 fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, and protease inhibitor cocktail). The diluted supernatant was precleared by incubation with 75 μl of 50% (v/v) protein A agarose/Salmon sperm DNA beads for 30 min at 4°C. Precleared supernatant was incubated overnight at 4°C with anti-HEB (Santa Cruz Biotechnology) on a rocking platform. Immune complexes were recovered by the addition of 60 μl of 50% (v/v) protein A agarose/Salmon sperm DNA beads and incubation for one h at 4°C. The agarose beads were sequentially washed 5 times with lysis buffer, once with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl), LiCl buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl (pH 8.1)), and finally twice with TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA). The immune complexes were eluted by incubation with 500 μl elution buffer (1% SDS, 0.1 M NaHCO3) for 15 min at room temperature. To reverse the cross-linking of immune complexes, 20 μl of 5 M NaCl was added to the combined eluates, which were incubated for 4 h at 65°C.
After treatment with proteinase K for 1 h at 45°C, DNA was recovered by phenol-chloroform extraction and ethanol precipitation. DNA was detected by 30 cycles of PCR with a pair of primers specific for the Lpar1 promoter region containing E-box. Eb1 forward primer, 5′-TCTTATCAGCTCTGCC CATAGCTG-3′; and Eb1 reverse primer, 5′-AGCGGAGTCCTC CACACATCA-3′; Eb3 forward primer, 5′-CATAGCTCTAGGG ACACAAAGGCA-3′; and Eb3 reverse primer, 5′-AGCCCTGG AAACCATCACAGAG-3′; Lpar1-Exon4 forward primer, 5′-CGGGATTGGTCTTGTTATTGC-3′; and Lpar1-Exon4 reverse primer, 5′-CATCTCTTTGTCGCGGTAGG-3′.
RESULTS
Identification of a novel transcription start site in mouse Lpar1 gene
In our previous study, we showed that the genomic structure of mouse Lpar1 consisted of four exons separated by three introns and postulated that the translational start site and termination signal were located within exon 2 and exon 4, respectively (Contos and Chun, 1998). In this present study, we identified the transcription initiation site for Lpar1 using the 5′-rapid amplification of cDNA ends (5′RACE) method. For 5′RACE, we used specific primers anchored in exon 3 and total RNA from TR mouse neocortical neuroblast cells (Chun and Jaenisch, 1996). TR is a murine neocortical neuroblast cell line transformed by large T antigen and vras (Chun and Jaenisch, 1996). A single band with an approximate size of 700 bp was detected from 5′RACE amplification and it was named transcript variant 3, TR3 (Fig. 1A).
Fig. 1. Identification of a novel transcript of Lpar1 (transcript variant 3) and its transcription start site in neocortical neuroblast cells.

(A) Total RNA isolated from TR mouse cells (Chun and Jaenisch, 1996) was subjected to a 5′RACE reaction using 5′ universal primer mix and 3′ Lpar1-specific primers. Lane 1 represents a 1kb plus ladder and lane 2 represents the transcript variant 3 identified from 5′RACE analysis. (B) A schematic representation of transcript variant 3 with indicated positions of the alternatively spliced sequences of exon 1a, 1b and 1c. The asterisk shows the relative position of translation start site for LPA1. (C) Transcript variant of Lpar1 gene expression was analyzed by quantitative reverse transcriptase polymerase chain reaction. Each cDNA from embryo and adult C57BL/6 mice tissue or TR cells was amplified using specific primers (n=3, versus transcript variant 1 in TR cells). Error bars indicate S.E.
Cloning and subsequent sequencing of TR3 revealed that its 5′-UTR was different from that of transcript variant 1 (TR1) (Contos and Chun, 1998) and transcript variant 2 (TR2) (Genbank accession number NM172989). For TR3, there was a 70 bp 5′UTR corresponding to a novel upstream exon we named exon 1c (Fig. 1B). This transcript is identical to the Genbank accession XM_011249929 predicted sequence of mouse Lpar1. The sequences of the splice acceptor and donor sites complied with the GT-AG rule for splicing. We designated the initiation of transcription site of transcript variant 3 as +1 on exon 1c (Fig. 1B), and sequencing data also detected different transcription start sites at +14, +39 and +48 within exon 1c (data not shown). It is noted that presence of multiple minor transcription start sites in TATA-less promoters is not uncommon (Benson et al., 1999; Burke and Kadonaga, 1997; Zhou and Chiang, 2001).
We next examined expression of TR3 in mouse embryo and adult tissues, and compared it to that of TR1 and TR2. Both TR1 and 2 were expressed in all mouse tissues, with high expression of TR2 in all tissues. TR3 was predominantly expressed in the embryonic brain, adult brain, thymus, testis, stomach, and spleen. The TR mouse cells (Chun and Jaenisch, 1996) expressed all the alternative transcript forms of Lpar1, TR1, TR2 and TR3 (Fig. 1C).
Structural promoter mapping by sequence analysis of mouse Lpar1
In order to identify functionally important cis-acting elements in the 5′-upstream region of the mouse Lpar1, we first determined the DNA sequence of the 5′-flanking region of Lpar1. We then utilized the Transcription Element Search System (TESS) (http://www.cbil.upenn.edu/tess/) to identify specific sites in the Lpar1 promoter region that could serve as important cis-acting elements regulating transcription of Lpar1 (Fig. 2A). Consensus sequences of several transcription factors in −950 to +1 promoter region, which included GATA1, Runx, E-protein, Oct1, AP1, and SP1, were identified through this screen (Fig. 2A).
Fig. 2. Putative cis-acting elements and reporter activity of the mouse Lpar1 promoter.
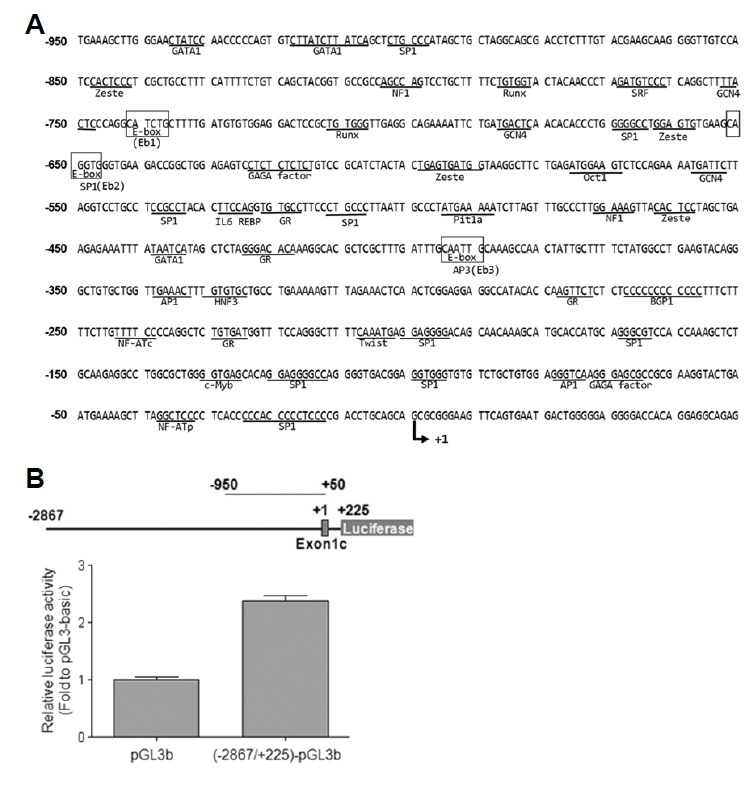
(A) These putative transcriptional regulation elements were identified from the Transcription Element Search System (TESS http://www.cbil.upenn.edu/tess/). (B) TR mouse neocortical neuroblast cells (Chun and Jaenisch, 1996) were transiently transfected with reporter plasmids, pGL3b (pGL3-basic) or (−2867/+225)-pGL3b. The cells were lysed and assayed for luciferase activity twenty-four hours post-transfection. The values represent the mean ± S.E. of increase in luciferase activity in folds with respect to the pGL3-basic construct. The experiment was performed in triplicate and values were normalized to the activity of Renilla luciferase, which was transfected concurrently (40 ng) in all the assays to measure transfection efficiency. **P < 0.01.
Identification of Lpar1 promoter activity in neocortical neuroblast cells
To identify putative cis-acting elements for the mouse Lpar1 promoter, we established a luciferase reporter assay in TR cells. These cells express endogenous Lpar1, thus allowing analysis of the LPA1 machinery. The −2867/+225-Luciferase construct exhibited a 1.5-fold higher promoter activity compared to the pGL3-basic construct control vector, indicating that the −2867/+225 region of Lpar1 does indeed act as a promoter for Lpar1 transcription (Fig. 2B).
Mapping of the Lpar1 promoter by deletion constructs
Based on the location of the transcriptional start site for TR3, we generated a series of 5′ promoter deletion constructs. These were made in pGL3-basic firefly luciferase reporter plasmid and were transfected into TR mouse cells for assay readout. The −248/+225 construct seemed to define the basal promoter activity (Fig. 3A). This region has a high GC content (61.3%) and a putative binding site for transcription factor SP1, but does not contain a TATA-box (Fig. 2A). These features are common for a TATA-less promoter (Chow and Knudson, 2005; Gery and Koeffler, 2003; Zhou and Chiang, 2002; 2001; Zhou et al., 2004).
Fig. 3. Identification of negative cis-acting elements using reporter constructs containing serially deleted 5′ flanking region of the mouse Lpar1.
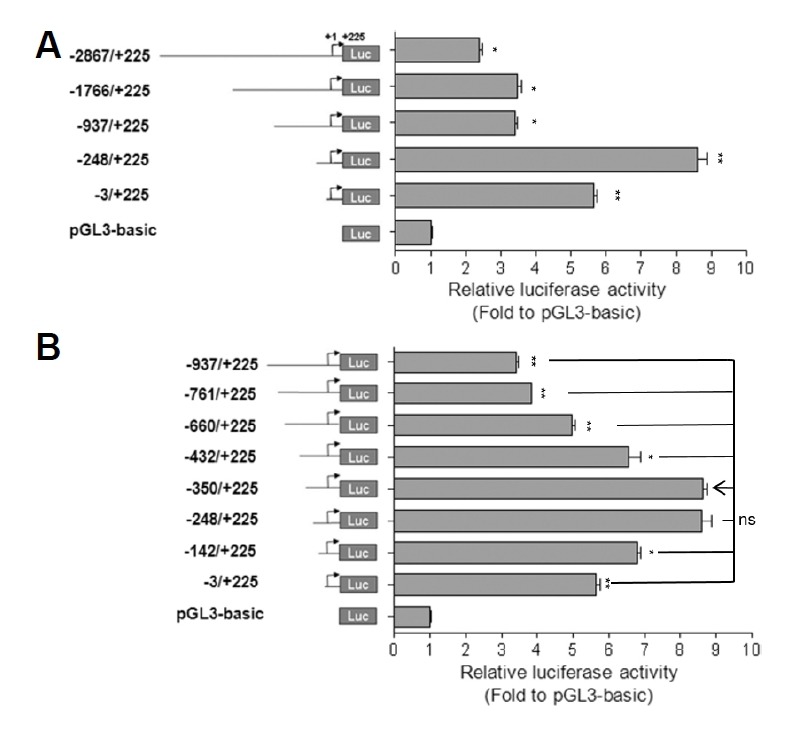
A schematic diagram of several types of Lpar1 promoter deletion constructs is in the left panel. (A–B) TR mouse cells (Chun and Jaenisch, 1996) were transfected with one of the Lpar1 promoter constructs or pGL3-basic vector. The cells were lysed and assayed for luciferase activity twenty-four hours post-transfection. The values represent the mean ± S.E. of luciferase activity increases in folds with respect to the pGL3-basic construct. The experiment was performed in triplicate and the values were normalized to the activity of concurrently transfected Renilla luciferase. *P < 0.05, **P < 0.01. ns is non-significant.
Interestingly, inclusion of the region between −937 to −248 resulted in a substantial (2.5 fold) decrease in promoter activity compared to that of the −937/+225 construct, indicating the presence of a potential negative regulatory element in the region (Fig. 3A). With additional deletion constructs, three negative regulatory regions (−761 to −660, −660 to −432, −432 to −350) could be defined in the region starting from −761 (Fig. 3B). Most predictive were putative three E-protein binding sites within these regions as E-proteins are known to be transcriptional repressors as well as activators (Fig. 2A) (Massari and Murre, 2000).
E-box binding protein HEB in transcriptional regulation of Lpar1
For the three putative E-box elements described above, we tested whether they played a role in regulating transcription of Lpar1 (Figs. 4A, 4B and 4C). The sequences are of the CANNTG motif, although we also checked for known non-classical E-box motifs (Yoshitane et al., 2014). Of these, only E-box 3 (EB3) was present in all three genomes for human, mouse and rat (Fig. 4A). The three mouse E-boxes were individually, doubly or triply mutated and were assessed for their ability to induce expression of the linked luciferase reporter (Fig. 4B). The luciferase activity of Eb1mut (−742~−737) and Eb3mut (−395~−390) were 20% and 28%, respectively, of the Eb1/3mut in TR cells (Fig. 4C). Only mutation of the second E-box (Eb2) significantly decreased promoter activity, indicating the potential activating role of Eb2 in Lpar1 expression. These results suggested that all three E-boxes, whether as activators or repressors, might be involved in regulating the Lpar1 promoter activity. We then investigated the specific effects of E-proteins on the Lpar1 promoter. There are known four E-proteins in mammalian cells, namely E12, E47, HEB, and E2-2. From our analysis, no change in promoter activity was detected in E12, E47 and E2-2 expression vector transfection (data not shown).
Fig. 4. Identification and characterization of negative regulatory cis-acting elements (E-boxes) in the Lpar1 promoter.
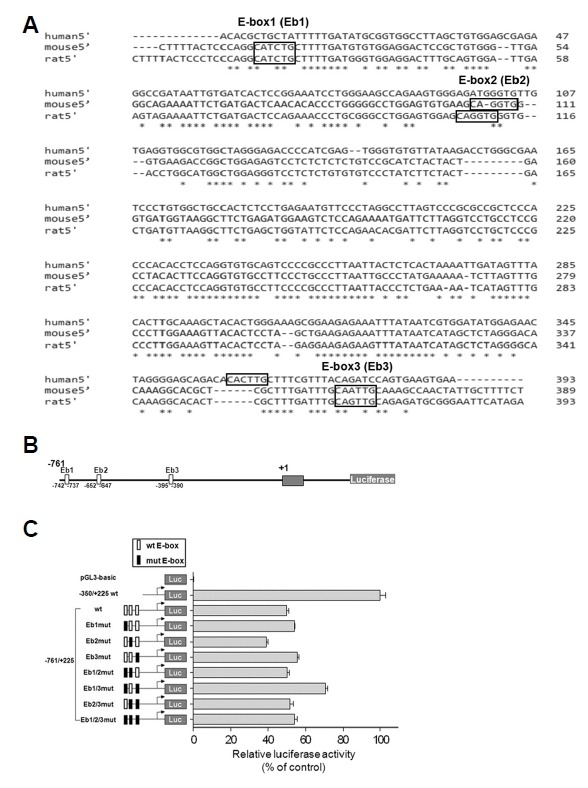
(A) Comparison of the human, mouse and rat Lpar1 promoter sequences with the E-boxes having the CANNTG motif marked and the conserved sequences denoted by asterisks. (B) Putative E-box positions numbered according to their positions and the luciferase construct. (C) Reporter gene assay with promoter constructs containing individually mutated E-boxes. Transient transfection was performed in TR mouse cells (Chun and Jaenisch, 1996) using reporter constructs −761/+225wt, Eb1mut, Eb2mut, Eb3mut, Eb1/2mut, Eb1/3mut, Eb2/3mut and Eb1/2/3mut. Reporter gene activities were normalized to that of the −350/+225 wild-type construct. Each of the mutated E-box sequence is indicated by a dark box (for site Eb1, CATCTG was changed to AGTCTA; for site Eb2, CAGGTG was changed to AGGGTA; and for site Eb3, CAATTG was changed to AGATTA). Error bars indicate S.E. *P < 0.05, ns is non-significant.
We next determined if HEB (gene Tcf12) expression affected Lpar1 promoter activity. HEB is expressed in various regions of the developing and adult mouse brain according to the Allen Brain Atlas (http://www.brain-map.org/). HEB expression was examined by co-transfecting TR cells with the reporter plasmids, −761/+225, and HEB expression vectors. The over-expression of HEB with wild type reporter plasmids resulted in approximately a 50% decrease in the reporter gene activity (Fig. 5A). Since Eb1 and Eb3 acted as inhibitory elements for Lpar1 promoter activity (Fig. 4C), it seemed likely that they are involved in the HEB-induced down regulation of Lpar1 promoter activity. Therefore, we also co-transfected the TR cells with the mutant reporter plasmids and HEB expression vectors. As shown in Fig. 5A, the decrease in the reporter gene activity was abolished in the −761/+225 Eb1/3 mutant. These results suggested that Eb1 and Eb3 might be important in Lpar1 gene transcription.
Fig. 5. Involvement of HEB in negative regulation of the mouse Lpar1 promoter through E-boxes.
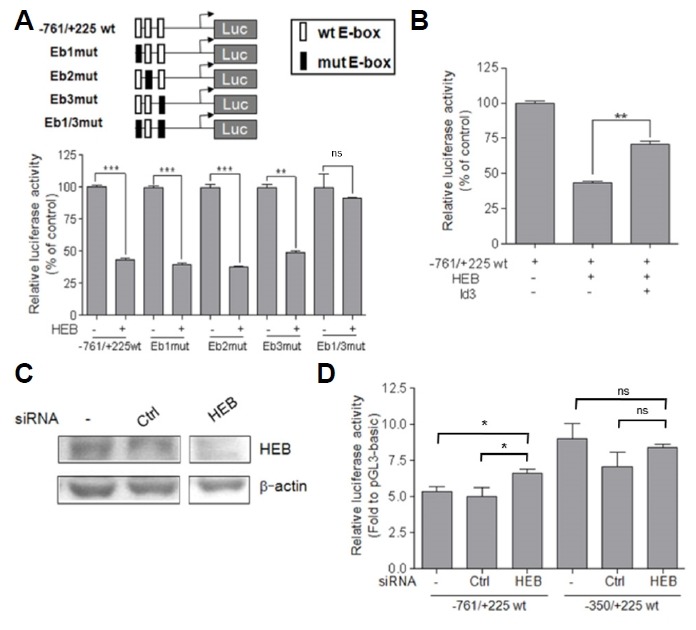
(A) TR cells were co-transfected with luciferase constructs (−761/+225wt, Eb1mut, Eb2mut, Eb3mut, and Eb1/3mut) and the HEB expression vector. (B) TR cells were co-transfected with −761/+225 wt and expression vectors (HEB, Id3). The values represent the mean ± S.E. of the percentage of luciferase activity in the presence of HEB or Id3 expression vector relative to the luciferase activity in the presence of an empty vector. The experiment was performed in triplicate and the values were normalized to the activity of concurrently transfected Renilla luciferase. (C) After forty-eight hours, cells were transfected with siRNAs for no-silencing (Ctrl) and HEB. Cell lysates were prepared by western blot analysis for detection of HEB and β-actin. (D) TR cells were co-transfected with luciferase constructs (−761/+225wt and −350/+225wt) and siRNAs for no-silencing (Ctrl) or HEB. *P < 0.05, **P < 0.01, ***P < 0.001. ns is non-significant.
Id proteins are known endogenous inhibitors of E-proteins (Ross et al., 2003) and we chose to study the effect of Id3 expression on HEB and Lpar1 interaction (Fig. 5B). We chose Id3 as it is known to be expressed in the ventricular zone (VZ) during the brain development (Jen et al., 1997)(BGEM http://www.stjudebgem.org). When co-transfected with Id3, the effect by HEB in −761/+225 wild type promoter construct was blunted to a significant degree (**P < 0.01)(Fig. 5B). Additionally, we examined the effect of HEB knockdown on transcriptional regulation of Lpar1 by transfection of TR cells with siRNA specific for HEB. The HEB siRNA-transfected cells displayed up-regulation of −761/+225 wt Lpar1 promoter activity compared to cells that were untransfected or transfected with a nonsense siRNA (Ctrl) (Figs. 5C and 5D). Taken together, the endogenous E-box protein, HEB, may be involved in negative regulation of the Lpar1 transcription.
Binding of HEB protein to the Lpar1 promoter
Because our luciferase reporter experiment revealed that HEB is a negative regulator of the Lpar1 promoter (Fig. 5), we further tested whether HEB could directly bind to the Lpar1 promoter using the chromatin immunoprecipitation assay (ChIP). We transfected HEB or HEB+Id3 into the TR cells and in addition, observed the behavior of endogenous HEB in the cells. Consistent with the luciferase assay experiment, we found that endogenous HEB was bound to the oligonucleotide containing the Eb3 site on the Lpar1 promoter in the TR cells (in the 224 bp construct for Eb3). Co-expression of Id3 protein decreased this binding: when comparing the ratio of 3rd α-HEB lane to the 3rd total lane and the ratio was decreased compared with the same ratio for the 2nd α-HEB lane to the 2nd total lane (Fig. 6; 224 bp construct for Eb3). As a negative control, we used the oligonucleotide containing a 137 fragment of exon 4 of Lpar1, which lacks an E-box (Fig. 6; exon 4; 137 bp construct). Taken together, we show that HEB directly binds to the Eb3 site on the Lpar1 promoter (224 bp construct) and the binding is partially abolished by presence of transfected Id3.
Fig. 6. Chromatin immunoprecipitation analysis of HEB binding at E-boxes of the mouse Lpar1 promoter in neocortical neuroblast cells.
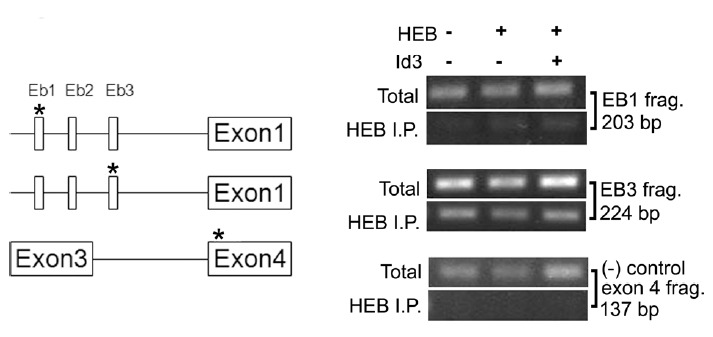
Results of ChIP assay using an antibody for HEB in TR cells are shown. The location of oligonucleotide fragments used for each E-box is displayed on the map to the left. Chromatin immunoprecipitation assays were performed as described in Materials and Methods. Cells were transfected for 24 h with HEB or the Id3 expression vector (+) or without (−) before the experiments. In the ChIP assay, the Lpar1 promoter was analyzed by quantitative PCR with primer pairs. As controls, chromatin samples were analyzed before immunoprecipitation (total) to show the equal amount of starting material. * Denotes the location of the oligonucleotide fragments used for the ChIP assay in the genome map.
DISCUSSION
The main goal of this study was to characterize the transcription initiation site and regulatory elements of Lpar1 in murine neocortical neuroblast cells. Our results provide an understanding of the transcriptional control for Lpar1 relevant to the developing neocortex, and likely other tissues and these results indicate that the E-protein HEB plays a role in Lpar1 transcription. Analysis of Lpar1 upstream region revealed that it does not possess a canonical TATA box, but has a high GC content and contains several putative cis-acting elements, such as multiple SP1 binding sites and three E-protein binding sequences (Fig. 2). All of these features are characteristic of constitutively active housekeeping genes along with developmentally regulated genes.
Previously, we and another laboratory identified two novel Lpar1 transcript variants (transcript variants 1 and 2)(Contos and Chun, 1998)(Genbank accession number NM172989), and this paper reveals a third novel transcript (transcript variant 3)(Fig. 1A). RT-PCR analysis from various tissues revealed that all three specific Lpar1 transcript variants are expressed in most tissues, especially the cerebellum (Fig. 1C). Further investigation is required to determine the significance of high Lpar1 expression in the cerebellum, where much of the signal appears to be present in the oligodendrocytes of the white matter (the Allen Brain Atlas http://www.brainmap.org). Transcript variant 3 has intrinsic promoter activity (Fig. 2B) along with a variety of potential cis-acting elements (Fig. 2A). There were negative regulatory regions present in the promoter region (Fig. 3) and mapping identified three E-box elements (Eb1–3). Deletion of all three potential E-protein binding sequences resulted in full basal activity of the Lpar1 promoter (Fig. 3). With point mutation analysis of these putative E-boxes (Figs. 4A and 4B), the mutation in the second E-box (Eb2) decreased promoter activity whereas mutations in the first and the third E-boxes increased promoter activity. This suggested the possibility of an activator being attached to the second E-box, affecting Lpar1 transcription, and the first and third E-boxes being used for suppression of the promoter activity.
There was enhanced promoter activity upon deletion of the region from −660 to −432 (Fig. 3B) and this region contains a consensus Oct1 sequence, suggesting that Oct1 may be also be involved as a negative regulator of Lpar1 transcription; however, a functional characterization for this possibility is required. In measuring the Lpar1 promoter activity affected by the over-expression of HEB, we found that individual mutation constructs of Eb1 and Eb3 did not show a significant difference in promoter activity from the wild type construct (Fig. 5A). However, in the double mutant construct, Eb1/3mut, we saw that HEB lost its negative regulatory effect on the Lpar1 promoter activity (Fig. 5A). The EB1 and EB3 seem to work in tandem as when individually mutated (in Eb1mut and Eb3mut), they cannot lift the repression of the wild type region, but when both are mutated (in (EB1/3mut), the repression in the wild type region is significantly increased. The possible mechanism may involve HEB binding to EB3 and at the same time to as-yet-an-unknown factor binding to EB1 for maximal repression of the LPAR1 promotor region in a folding of the 330 or so base pairs of DNA between EB1 and EB3. Mutation of both sites seems to be required for the complete de-repression. HEB has recently been shown to maximally affect enhancer elements in a number of developmentally-related genes when interacting with PRC2 in one scenario and with SMAD2/3 in another scenario both in mouse embryonic stem cells (ESCs) shown in ChIP (Yoon et al., 2015). This is an example of HEB being able to interact with other proteins in its interaction with DNA.
E-protein transcription factors as part of the bHLH family with members such as HEB play significant roles in brain development (Fischer et al., 2014; Powell and Jarman, 2008; Ross et al., 2003). In our study, over-expression of HEB decreased the promoter activity supporting the notion of E-proteins modulating Lpar1 transcription. Id family proteins are known as endogenous inhibitors of E-proteins (Ross et al., 2003; Wang and Baker, 2015) and we chose to study Id3 as it is expressed in the ventricular zone (VZ) during brain development (Jen et al., 1997)(BGEM http://www.stjudebgem.org). Co-transfection of Id3 counteracted the effect of HEB and showed an increase in the promoter activity for Lpar1 (Fig. 5). The ChIP assay revealed HEB binding to the third E-box of the Lpar1 promoter (Fig. 6; 224 bp construct), implying that HEB selectively binds to Eb3 and not to Eb1. The faint band in Eb1 site shown in the ChIP assay results can be explained by the fact that the distance between the three E-box sites are too close to be resolved by the assay. The density of the bands decreased in a distance dependent manner (data not shown). Although HEB does not seem to bind directly to Eb1, Eb1 is clearly involved in negatively regulating Lpar1 promoter activity. This effect may be due to another bHLH transcription factor binding to Eb1, and potentially resulting from a HEB-Eb3 interaction.
LPA1 has a critical role in development, and our data indicate that there is a tight regulation of Lpar1 expression at a cis-dependent level exerted by presence of HEB. There also exist other levels of LPA1 regulation, such as DNA methylation observed in rat tumor cell lines as shown by Fukushima and colleagues (Tsujiuchi et al., 2006). Taken together, our data support a previously unrecognized level of transcriptional control that can affect Lpar1 transcription via E-box containing regulatory elements in the Lpar1 promoter, and that these regulatory elements function in part through HEB binding to Eb3.
Lpar1 has been implicated in multiple disease processes, particularly neurological diseases (Choi et al., 2010; Herr et al., 2011; Yung et al., 2011; 2015) and Lpar1-null mice have been reported to have a number of neurochemical alterations in several brain regions (Harrison et al., 2003; Roberts et al., 2005). Notable disease-relevant phenotypes in Lpar1-null mouse indicate altered cerebral cortical organization, changes in perception of neuropathic pain and contextual memory (Birgbauer and Chun, 2006; Estivill-Torrus et al., 2008; Inoue et al., 2004; Ladrón de Guevara-Miranda et al., 2018; Xie et al., 2008), altered response to hypoxic brain damage (Herr et al., 2011) and changes in response to intra-cranial hemorrhage in developing fetal hydrocephalus (Yung et al., 2011). Our findings suggest that regulating Lpar1 through transcriptional activity via HEB may be relevant in developing novel drugs relevant to various neurological indications.
ACKNOWLEDGMENTS
This research was supported by research grants through the National Research Foundation (NRF 2018R1A2A2A05023615 to S.-O.H.; NRF 2017R1D1A3B03030324 to A.S.), Hallym University (HRF-201701-016 to S.-O.H.), South Korea and the National Institutes of Health (NIH NS084398 to J.C.), USA.
REFERENCES
- Anliker B., Choi J.W., Lin M-E, Gardell S.E., Rivera R.R., Kennedy G., Chun J. Lysophosphatidic acid (LPA) and its receptor, LPA1, influence embryonic schwann cell migration, myelination, and cell-to-axon segregation. Glia. 2013;61:2009–2022. doi: 10.1002/glia.22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki J. Mechanisms of lysophosphatidic acid production. Semin Cell Dev Biol. 2004;15:477–489. doi: 10.1016/j.semcdb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Benson L.Q., Coon M.R., Krueger L.M., Han G.C., Sarnaik A.A., Wechsler D.S. Expression of MXI1, a Myc antagonist, is regulated by Sp1 and AP2. J Biol Chem. 1999;274:28794–28802. doi: 10.1074/jbc.274.40.28794. [DOI] [PubMed] [Google Scholar]
- Birgbauer E., Chun J. New developments in the biological functions of lysophospholipids. Cell Mol Life Sci. 2006;63:2695–2701. doi: 10.1007/s00018-006-6155-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke T.W., Kadonaga J.T. The downstream core promoter element, DPE, is conserved from Drosophila to humans and is recognized by TAFII60 of Drosophila. Genes Dev. 1997;11:3020–3031. doi: 10.1101/gad.11.22.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan L.C., Peters W., Xu Y., Chun J., Farese R.V.J., Cases S. LPA3 receptor mediates chemotaxis of immature murine dendritic cells to unsaturated lysophosphatidic acid (LPA) J Leukoc Biol. 2007;82:1193–1200. doi: 10.1189/jlb.0407221. [DOI] [PubMed] [Google Scholar]
- Chen Y., Ramakrishnan D.P., Ren B. Regulation of angiogenesis by phospholipid lysophosphatidic acid. Front Biosci (Landmark Ed) 2013;18:852–861. doi: 10.2741/4148. [DOI] [PubMed] [Google Scholar]
- Choi J.W., Herr D.R., Noguchi K., Yung Y.C., Lee C-W, Mutoh T., Lin M-E, Teo S.T., Park K.E., Mosley A.N., et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol. 2010;50:157–186. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chow G., Knudson W. Characterization of promoter elements of the human HYAL-2 gene. J Biol Chem. 2005;280:26904–26912. doi: 10.1074/jbc.M413845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J., Jaenisch R. Clonal cell lines produced by infection of neocortical neuroblasts using multiple oncogenes transduced by retroviruses. Mol Cell Neurosci. 1996;7:304–321. doi: 10.1006/mcne.1996.0023. [DOI] [PubMed] [Google Scholar]
- Contos J.J., Chun J. Complete cDNA sequence, genomic structure, and chromosomal localization of the LPA receptor gene, lpA1/vzg-1/Gpcr26. Genomics. 1998;51:364–378. doi: 10.1006/geno.1998.5400. [DOI] [PubMed] [Google Scholar]
- Contos J.J., Fukushima N., Weiner J.A., Kaushal D., Chun J. Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proc Natl Acad Sci USA. 2000;97:13384–13389. doi: 10.1073/pnas.97.24.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza K., Paramel G.V., Kienesberger P.C. Lysophosphatidic acid signaling in obesity and insulin resistance. Nutrients. 2018;10:399. doi: 10.3390/nu10040399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel I., Murre C. The function of E- and Id proteins in lymphocyte development. Nat Rev Immunol. 2001;1:193–199. doi: 10.1038/35105060. [DOI] [PubMed] [Google Scholar]
- Estivill-Torrus G., Llebrez-Zayas P., Matas-Rico E., Santin L., Pedraza C., De Diego I., Del Arco I., Fernandez-Llebrez P., Chun J., De Fonseca F.R. Absence of LPA1 signaling results in defective cortical development. Cereb Cortex. 2008;18:938–950. doi: 10.1093/cercor/bhm132. [DOI] [PubMed] [Google Scholar]
- Fischer B., Azim K., Hurtado-Chong A., Ramelli S., Fernández M., Raineteau O. E-proteins orchestrate the progression of neural stem cell differentiation in the postnatal forebrain. Neural Dev. 2014;9:23. doi: 10.1186/1749-8104-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima N., Weiner J.A., Chun J. Lysophosphatidic acid (LPA) is a novel extracellular regulator of cortical neuroblast morphology. Dev Biol. 2000;228:6–18. doi: 10.1006/dbio.2000.9930. [DOI] [PubMed] [Google Scholar]
- Gennero I., Laurencin-Dalicieux S., Conte-Auriol F., Briand-Mesange F., Laurencin D., Rue J., Beton N., Malet N., Mus M., Tokumura A., et al. Absence of the lysophosphatidic acid receptor LPA1 results in abnormal bone development and decreased bone mass. Bone. 2011;49:395–403. doi: 10.1016/j.bone.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gery S., Koeffler H.P. Repression of the TMEFF2 promoter by c-Myc. J Mol Biol. 2003;328:977–983. doi: 10.1016/s0022-2836(03)00404-2. [DOI] [PubMed] [Google Scholar]
- Goetzl E.J., Kong Y., Voice J.K. Cutting edge: differential constitutive expression of functional receptors for lysophosphatidic acid by human blood lymphocytes. J Immunol. 2000;164:4996–4999. doi: 10.4049/jimmunol.164.10.4996. [DOI] [PubMed] [Google Scholar]
- Harrison S.M., Reavill C., Brown G., Brown J.T., Cluderay J.E., Crook B., Davies C.H., Dawson L.A., Grau E., Heidbreder C., et al. LPA1 receptor-deficient mice have phenotypic changes observed in psychiatric disease. Mol Cell Neurosci. 2003;24:1170–1179. doi: 10.1016/j.mcn.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Hecht J.H., Weiner J.A., Post S.R., Chun J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J Cell Biol. 1996;135:1071–1083. doi: 10.1083/jcb.135.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr K.J., Herr D.R., Lee C-W, Noguchi K., Chun J. Stereotyped fetal brain disorganization is induced by hypoxia and requires lysophosphatidic acid receptor 1 (LPA1) signaling. Proc Natl Acad Sci USA. 2011;108:15444–15449. doi: 10.1073/pnas.1106129108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M., Rashid M.H., Fujita R., Contos J.J.A., Chun J., Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Inoue M., Xie W., Matsushita Y., Chun J., Aoki J., Ueda H. Lysophosphatidylcholine induces neuropathic pain through an action of autotaxin to generate lysophosphatidic acid. Neuroscience. 2008;152:296–298. doi: 10.1016/j.neuroscience.2007.12.041. [DOI] [PubMed] [Google Scholar]
- Jen Y., Manova K., Benezra R. Each member of the Id gene family exhibits a unique expression pattern in mouse gastrulation and neurogenesis. Dev Dyn. 1997;208:92–106. doi: 10.1002/(SICI)1097-0177(199701)208:1<92::AID-AJA9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Kano K., Arima N., Ohgami M., Aoki J. LPA and its analogs-attractive tools for elucidation of LPA biology and drug development. Curr Med Chem. 2008;15:2122–2131. doi: 10.2174/092986708785747562. [DOI] [PubMed] [Google Scholar]
- Ke S.H., Madison E.L. Rapid and efficient site-directed mutagenesis by single-tube “megaprimer” PCR method. Nucleic Acids Res. 1997;25:3371–3372. doi: 10.1093/nar/25.16.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara Y., Maceyka M., Spiegel S., Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171:3575–3594. doi: 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladrón de Guevara-Miranda D., Moreno-Fernández R.D., Gil-Rodríguez S., Rosell-Valle C., Estivill-Torrús G., Serrano A., Pavón F.J., Rodríguez de Fonseca F., Santín L.J., Castilla-Ortega E. Lysophosphatidic acid-induced increase in adult hippocampal neurogenesis facilitates the forgetting of cocaine-contextual memory. Addict Biol. 2018 doi: 10.1111/adb.12612. [DOI] [PubMed] [Google Scholar]
- Lapierre D.M., Tanabe N., Pereverzev A., Spencer M., Shugg R.P.P., Dixon S.J., Sims S.M. Lysophosphatidic acid signals through multiple receptors in osteoclasts to elevate cytosolic calcium concentration, evoke retraction, and promote cell survival. J Biol Chem. 2010;285:25792–25801. doi: 10.1074/jbc.M110.109322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazorchak A., Jones M.E., Zhuang Y. New insights into E-protein function in lymphocyte development. Trends Immunol. 2005;26:334–338. doi: 10.1016/j.it.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Liu Y-B, Kharode Y., Bodine P.V.N., Yaworsky P.J., Robinson J.A., Billiard J. LPA induces osteoblast differentiation through interplay of two receptors: LPA1 and LPA4. J Cell Biochem. 2010;109:794–800. doi: 10.1002/jcb.22471. [DOI] [PubMed] [Google Scholar]
- Massari M.E., Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C. Helix-loop-helix proteins and lymphocyte development. Nat Immunol. 2005;6:1079–1086. doi: 10.1038/ni1260. [DOI] [PubMed] [Google Scholar]
- Powell L.M., Jarman A.P. Context dependence of proneural bHLH proteins. Curr Opin Genet Dev. 2008;18:411–417. doi: 10.1016/j.gde.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts C., Winter P., Shilliam C.S., Hughes Z.A., Langmead C., Maycox P.R., Dawson L.A. Neurochemical changes in LPA1 receptor deficient mice--a putative model of schizophrenia. Neurochem Res. 2005;30:371–377. doi: 10.1007/s11064-005-2611-6. [DOI] [PubMed] [Google Scholar]
- Ross S.E., Greenberg M.E., Stiles C.D. Basic helix-loop-helix factors in cortical development. Neuron. 2003;39:13–25. doi: 10.1016/s0896-6273(03)00365-9. [DOI] [PubMed] [Google Scholar]
- Sheng X., Yung Y.C., Chen A., Chun J. Lysophosphatidic acid signalling in development. Development. 2015;142:1390–1395. doi: 10.1242/dev.121723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujiuchi T., Shimizu K., Onishi M., Sugata E., Fujii H., Mori T., Honoki K., Fukushima N. Involvement of aberrant DNA methylation on reduced expression of lysophosphatidic acid receptor-1 gene in rat tumor cell lines. Biochem Biophys Res Commun. 2006;349:1151–1155. doi: 10.1016/j.bbrc.2006.08.159. [DOI] [PubMed] [Google Scholar]
- Uchida H., Nagai J., Ueda H. Lysophosphatidic acid and its receptors LPA1 and LPA3 mediate paclitaxel-induced neuropathic pain in mice. Mol Pain. 2014;10:71. doi: 10.1186/1744-8069-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban A., Neukirchen S., Jaeger K.E. A rapid and efficient method for site-directed mutagenesis using one-step overlap extension PCR. Nucleic Acids Res. 1997;25:2227–2228. doi: 10.1093/nar/25.11.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valet P., Pages C., Jeanneton O., Daviaud D., Barbe P., Record M., Saulnier-Blache J.S., Lafontan M. Alpha2-adrenergic receptor-mediated release of lysophosphatidic acid by adipocytes. A paracrine signal for preadipocyte growth. J Clin Invest. 1998;101:1431–1438. doi: 10.1172/JCI806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L-H, Baker N.E. E Proteins and ID Proteins: Helix-Loop-Helix Partners in Development and Disease. Dev Cell. 2015;35:269–280. doi: 10.1016/j.devcel.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welner R.S., Pelayo R., Kincade P.W. Evolving views on the genealogy of B cells. Nat Rev Immunol. 2008;8:95–106. doi: 10.1038/nri2234. [DOI] [PubMed] [Google Scholar]
- Xie W., Matsumoto M., Chun J., Ueda H. Involvement of LPA1 receptor signaling in the reorganization of spinal input through Abeta-fibers in mice with partial sciatic nerve injury. Mol Pain. 2008;4:46. doi: 10.1186/1744-8069-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X., Chun J. Lysophosphatidic acid (LPA) signaling in vertebrate reproduction. Trends Endocrinol Metab. 2010;21:17–24. doi: 10.1016/j.tem.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S-J, Foley J.W., Baker J.C. HEB associates with PRC2 and SMAD2/3 to regulate developmental fates. Nat Commun. 2015;6:6546. doi: 10.1038/ncomms7546. [DOI] [PubMed] [Google Scholar]
- Yoshitane H., Ozaki H., Terajima H., Du N-H, Suzuki Y., Fujimori T., Kosaka N., Shimba S., Sugano S., Takagi T., et al. CLOCK-controlled polyphonic regulation of circadian rhythms through canonical and noncanonical E-boxes. Mol Cell Biol. 2014;34:1776–1787. doi: 10.1128/MCB.01465-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukiura H., Hama K., Nakanaga K., Tanaka M., Asaoka Y., Okudaira S., Arima N., Inoue A., Hashimoto T., Arai H., et al. Autotaxin regulates vascular development via multiple lysophosphatidic acid (LPA) receptors in zebrafish. J Biol Chem. 2011;286:43972–43983. doi: 10.1074/jbc.M111.301093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung Y.C., Mutoh T., Lin M-E, Noguchi K., Rivera R.R., Choi J.W., Kingsbury M.A., Chun J. Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Sci Transl Med. 2011;3:99ra87. doi: 10.1126/scitranslmed.3002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung Y.C., Stoddard N.C., Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res. 2014;55:1192–1214. doi: 10.1194/jlr.R046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung Y.C., Stoddard N.C., Mirendil H., Chun J. Lysophosphatidic Acid Signaling in the Nervous System. Neuron. 2015;85:669–682. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chen Y-CM, Krummel M.F., Rosen S.D. Autotaxin through lysophosphatidic acid stimulates polarization, motility, and transendothelial migration of naive T cells. J Immunol. 2012;189:3914–3924. doi: 10.4049/jimmunol.1201604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Chiang C-M. Sp1 and AP2 regulate but do not constitute TATA-less human TAF(II)55 core promoter activity. Nucleic Acids Res. 2002;30:4145–4157. doi: 10.1093/nar/gkf537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Chiang C.M. The intronless and TATA-less human TAF(II)55 gene contains a functional initiator and a downstream promoter element. J Biol Chem. 2001;276:25503–25511. doi: 10.1074/jbc.M102875200. [DOI] [PubMed] [Google Scholar]
- Zhou G-P, Wong C., Su R., Crable S.C., Anderson K.P., Gallagher P.G. Human potassium chloride cotransporter 1 (SLC12A4) promoter is regulated by AP-2 and contains a functional downstream promoter element. Blood. 2004;103:4302–4309. doi: 10.1182/blood-2003-01-0107. [DOI] [PubMed] [Google Scholar]