

Abstract
Bacterial species in the genus Xanthomonas infect virtually all crop plants. Although many genes involved in Xanthomonas virulence have been identified through molecular and cellular studies, the elucidation of virulence-associated regulatory circuits is still far from complete. Functional gene networks have proven useful in generating hypotheses for genetic factors of biological processes in various species. Here, we present a genome-scale co-functional network of Xanthomonas oryze pv. oryzae (Xoo) genes, XooNet (www.inetbio.org/xoonet/), constructed by integrating heterogeneous types of genomics data derived from Xoo and other bacterial species. XooNet contains 106,000 functional links, which cover approximately 83% of the coding genome. XooNet is highly predictive for diverse biological processes in Xoo and can accurately reconstruct cellular pathways regulated by two-component signaling transduction systems (TCS). XooNet will be a useful in silico research platform for genetic dissection of virulence pathways in Xoo.
Keywords: co-functional network, plant bacterial pathogen, Xanthomonas oryzae pv. oryzae
INTRODUCTION
Xanthomonas is a large genus of Gram-negative plant pathogenic bacteria comprised of more than 30 species that collectively infect approximately 400 plants (Hayward, 1993; Parkinson et al., 2007). Xanthomonas species cause serious diseases in diverse hosts including many economically important crops such as rice, tomato, citrus and banana (Boch and Bonas, 2010). An important goal of bacterial research is to identify genes and networks that facilitate infection. For example, it is now well known that bacteria sense and respond to dynamic environmental changes using two-component signaling transduction systems (TCSs) (Bae et al., 2017; Kofoid and Parkinson, 1988). TCSs consist of two proteins: a membrane-bound histidine kinase (HK) that recognizes a specific stimulus and a corresponding response regulator (RR) that is phosphorylated by a HK and transcriptionally regulates the downstream gene expression (Hoch, 2000). Xanthomonas genomes encode a large number of TCSs (>100) (Barakat et al., 2009).
TCSs control diverse aspects of biological function in bacteria. For example, RpfC and RpfG (regulation of pathogenicity factors C and G) are well characterized TCSs in Xanthomonas (Slater et al., 2002). At high cell density, RpfC perceives the quorum sensing diffusible signal factor and RpfG transduces the signal to activate gene expression (Slater et al., 2002). The RpfC/RpfG system has been associated with biofilm formation, motility, extracellular enzyme and extracellular polysaccharide production, and virulence (He and Zhang, 2008).
Although many genes involved in bacterial virulence have been identified through molecular and cellular studies, elucidation of virulence-associated processes of Xanthomonas is still far from complete. For example, we recently demonstrated that Xanthomonas oryzae pv. oryzae (Xoo) carries a set of “rax” genes that are required for activation of XA21-mediated immunity and also for virulence (Pruitt et al., 2017; Pruitt et al., 2015). These genes include raxX, which encodes a small sulfated protein that activates XA21 immune receptor (Pruitt et al., 2015; Song et al., 1995). RaxX is sulfated by the tyrosine sulfotransferase RaxST (da Silva et al., 2004; Han et al., 2012; Pruitt et al., 2015) and secreted by the RaxABC type I secretion system that consists of a membrane fusion protein RaxA, an ATP-binding cassette (ABC) transporter RaxB, and an outer membrane protein RaxC (da Silva et al., 2004; Luu et al., 2018). Xoo strains carrying a single knockout in any of these genes compromise the ability of Xoo to activate rice XA21-mediated immunity (da Silva et al., 2004; Dee et al., 2018; Pruitt et al., 2015; Song et al., 1995). Despite detailed knowledge on the genes controlling the biosynthesis, processing and secretion of the active form of RaxX, the regulatory circuits controlling gene expression have not yet been identified.
A comprehensive understanding of regulatory circuits controlling bacterial virulence requires insight into the collaborative and regulatory interactions among multiple genes. Molecular interaction networks have proven useful in such endeavors (Cowen et al., 2017). Large-scale molecular networks have been constructed for many organisms, spanning from unicellular microbes to human and crops, by both experimental and computational approaches. However, experimental mapping of molecular interactions has been conducted in only limited number of species.
The availability of data from functional genomics, comparative genomics, and proteomics facilitates a genome-wide scale analysis of gene function. Because datasets from each technique are incomplete, error-prone, and limited in sensitivity, a single dataset alone is insufficient to fully describe a particular biological process. However, such datasets can be integrated to generate a more accurate and comprehensive view of gene function than is contained in any single dataset. For example, genome-scale co-functional networks have enabled effective integration of heterogeneous genomics data, significantly enhancing both accuracy and comprehensiveness of the molecular network models (Shim et al., 2017). These networks can then be utilized for prioritizing candidate genes for biological processes or complex traits of interest. For example, genome-scale co-functional networks for Pseudomonas aeruginosa, a human pathogenic bacterium, were used to identify novel genes for virulence and antibiotic-resistance (Hwang et al., 2016). However, to the best of our knowledge, an experimentally validated genome-scale co-functional network for Xoo has not yet been reported.
In this study, we present a genome-scale co-functional network of Xoo, XooNet, which was constructed by integrating 10 distinct types of genomics data derived from Xoo as well as several other bacterial species. XooNet contains 106,000 functional links among 3,615 Xoo genes, which covers approximately 83% of the coding genome. We found that XooNet is predictive for many biological processes and could reconstruct regulatory circuits that are controlled by TCSs. Network information and network-based hypothesis generation tools are freely available from a web server (www.inetbio.org/xoonet/), which will be a useful resource for the research community.
MATERIALS AND METHODS
Xanthomonas oryzae pv. oryzae genome and functional annotation data
The genome set of Xoo for the network construction was obtained from National Center for Biotechnology Information genome database (ftp://ftp.ncbi.nlm.nih.gov/genomes/) as of April 2015. Xoo genome had a total of 4,798 genes, of which 4,375 was protein coding genes. We used Gene Ontology (The Gene Ontology Consortium, 2015) terms for functional annotations of Xoo protein coding genes.
Gold-standard co-functional gene pairs for network construction
Co-functional gene networks were constructed by supervised machine learning processes, which require gold-standard data for benchmarking inferred models. Gold-standard data play critical roles in error-tolerant and unbiased learning. We compiled 11,669 positive gold-standard co-functional gene pairs from Gene Ontology Biological Process (GOBP) annotations (http://www.jcvi.org/cms/research/past-projects/cmr/overview/), by pairing genes annotated for the same GOBP terms. Gold standard gene pairs were derived from all GOBP terms except five terms for broadly inclusive pathway concepts—biological process (GO:0008150), DNA-mediated transposition (GO:0006313), metabolic process (GO:0008152), transport (GO:0006810), DNA-dependent regulation of transcription (GO:0006355)—to avoid between-pathway gene pairs. In addition, we compiled 18,174 gene pairs from Kyoto Encyclopedia of Genes and Genomes (KEEG) pathway annotations (Kanehisa et al., 2016) with exclusion of three KEGG pathways that are inclusive pathway concepts—metabolic pathway (xoo: 01100), biosynthesis of secondary metabolites (xoo: 01110), microbial metabolism in diverse environments (xoo: 01120). We then combined these two sets of gold-standard gene pairs into a total of 27,250 positive gold-standard gene pairs. We also generated 1,167,035 negative gold-standard gene pairs by pairing the 1,546 annotated genes that have no shared GOBP or KEGG terms. These gold-standard gene pairs were used for machine learning processes to construct XooNet.
To evaluate the constructed network, we generated another gold standard functional gene pairs based on MetaCyc pathway database (Caspi et al., 2016) independently. We generated 7457 and 250,664 gene pairs for positive and negative gold standard data sets, respectively. For the evaluation of network capacity to retrieve known genes for each pathway, we used only 145 MetaCyc pathway terms that have no less than five member genes.
Benchmarking and integrating co-functional networks
We benchmarked inferred co-functional gene pairs for given genomics data (D ) for likelihood of being involved in the same process using log-likelihood score (LLS ) scheme, which is based on Bayesian statistics (Lee et al., 2004).
Where P (L|D) and P (¬L|D) represent the frequencies of positive and negative gold-standard gene pairs for the given evidence of genomics data such as protein-protein interaction and co-expression, respectively. P (L) and P (¬L) represent the total frequencies of positive and negative gold-standard gene pairs, respectively.
If there was no data intrinsic score associated with the inferred gene pairs, the calculated LLS for the entire set of gene pairs was assigned to each of them. For data sets in which each gene pair is associated with a continuous score (e.g., correlation coefficient, mutual information, etc.), we calculated LLS scores for bins containing equal numbers of gene pairs. Those LLS scores and their corresponding data intrinsic scores (as the mean data scores for a bin) were used to find regression models, which were then used to map individual data intrinsic scores to LLS scores in a continuous manner.
Inferred co-functional links with assigned LLS were then integrated by a weighted sum (WS ) scheme (Lee et al., 2004), a variant of naïve Bayesian integration approach that can handle data correlation to some extend by integrating data with differential weights.
where L0 represents the highest LLS for the given link, i is rank order index for the remained LLSs, and W is a free parameter accounting for the relative degree of independence among the data sets. T indicates the threshold of minimum LLS to be considered for integration. The free parameter W ranges from 1 to +ɛ, and is optimized to maximize overall performance (measured by the area under a precision-recall curve) of the integrated network.
Co-functional links inferred from co-expression in Xoo (XO-CX)
Co-expressed genes across various biological conditions are likely to be co-regulated genes for a process. We inferred co-functional links from co-expression across Xoo microarray samples deposited in Gene Expression Omnibus (GEO) (Barrett et al., 2013). We analyzed eight GEO series and could infer gene pairs that are highly likely to be co-functional by co-expression pattern from three GEO series comprising 60 expression samples in total: GSE9640, GSE12099, and GSE24989. These three co-expression networks for each data sets were then integrated in a single network for co-expression analysis.
Co-functional links inferred from domain profile associations (XO-DP)
Protein domain is a structural and functional unit of protein. Therefore, proteins for similar function tend to have similar domain composition. We constructed domain profiles for coding genes of Xoo using InterPro database (Mitchell et al., 2015). We previously developed weighted mutual information (WMI ) measure, which robustly infers co-functional links based on domain profile associations by giving more weights to the rarer domains during mutual information (MI ) computation (Shim and Lee, 2016). The WMI measure takes more weight for rarer domains on the assumption that rarer domains are likely to have higher pathway specificity.
Co-functional links inferred from gene neighborhood (XO-GN)
In prokaryotic genomes, genes operating for the same process are often encoded as a co-transcriptional gene cluster, called operon. Therefore, we may infer co-functional gene pairs by their genomic proximity across prokaryotic genomes (Dandekar et al., 1998). We previously found that two measures of gene neighborhood, distance-based gene neighborhood (DGN) and probability-based gene neighborhood (PGN), are complementary and their integration can increase coverage and accuracy of co-functional network based on gene neighborhood (Shin et al., 2014). We inferred two co-functional networks by DGN and PGN across 1,626 prokaryotic genomes as described in our previous work (Shin et al., 2014), then integrated them into a single network for gene neighborhood.
Co-functional links inferred from phylogenetic profile associations (XO-PG)
Functionally coupled genes are often gained and lost during speciation by their functional constraints. Therefore, we may infer co-functional links by similarity of phylogenetic profiles which are patterns of presence and absence of homologous genes in many other species genomes (Kensche et al., 2008). We refer to these species used to construct the phylogenetic profiles as reference species. We previously found that this network inference could be more effective with phylogenetic profiles for each domain of tree of life: Archaea, Bacteria, and Eukarya (Shin and Lee, 2015). Therefore, we constructed phylogenetic profiles for each of the three domains based on the best BLASTP hit score of all Xoo coding genes in a given reference species. We measured association between phylogenetic profiles by mutual information (MI ) analysis (Shin and Lee, 2017) and could infer co-functional networks that pass benchmarking analysis from two domains: Archaea comprising 122 species and Bacteria comprising 1,626 species. The two co-functional networks were then integrated to a single functional gene network for phylogenetic profiles.
Co-functional links transferred from other species (Associalogs)
As functional genes are evolutionarily conserved between species (orthologs), functional associations between genes can also be evolutionarily conserved between species (associalogs)(Kim et al., 2013). To identify orthology relationships, we used inparanoid (Sonnhammer and Ostlund, 2015) algorithm which includes inparalogous relationships for gene pairs with similar functions. We then identified evolutionarily conserved co-functional links between two species with the following inparanoid weighted LLS (IWLLS ): IWLLS (A–B) = LLS (A′-B′) + ln(inparanoid score of A-A′) + ln(inparanoid score of B-B′), where A and B are Xoo genes and A′ and B′ are orthologous genes in other species. Using associalog concept, we could transfer five co-functional networks from high-throughput protein-protein interactions for five bacterial species: Escherichia coli (Kim et al., 2015), Campylobacter jejuni (Parrish et al., 2007), Helicobacter pylori (Rain et al., 2001), Mycoplasma pneumoniae (Kuhner et al., 2009), Synechocystis sp. strain PCC6803 (Sato et al., 2007). These five networks were then integrated into a single network for bacterial high-throughput protein-protein interactions (BA-HT). We transferred three co-functional networks between E. coli genes from our previously published EcoliNet (Kim et al., 2015): co-citation (EC-CC), co-expression (EC-CX), literature curated protein-protein interaction (EC-LC). We also transferred two co-functional networks between Pseudomonas genes from our published PseudomonasNet (Hwang et al., 2016): co-citation (PA-CC), co-expression (PA-CX).
Bacterial culture and growth conditions
Xoo Philippine strain PXO99, hereafter called Xoo, were grown on peptone sucrose agar (PSA) plates [1% peptone, 1% sucrose, 0.1% sodium L-glutamate, 1.5% agar] at 28°C with appropriate antibiotic(s) during 3 days before further assays. For the Xoo gene expression analysis, full grown Xoo strains were harvested from PSA plates and resuspended into Peptone sucrose broth (PSB) medium [1% peptone, 1% sucrose, 0.1% sodium L(+)-glutamate] at a density of 107 CFU/ml. Xoo strains in 5 ml of liquid culture were incubated for 24 hours at 28°C with shaking at 230 rpm (≥108 CFU/ml). The antibiotics were used at the following concentrations (μg/ml): cephalexin, 20; spectinomycin, 50.
Xoo mutation
To generate a Xoo mutant, a partial gene fragment was amplified by Polymerase chain reaction (PCR) and cloned in to the suicide vector pJP5603 carrying a modified antibiotic resistance gene from KanR to SpecR (Pruitt et al., 2015). Each construct was introduced in Philippine race 6 strain PXO99 by electroporation for single crossover and the transformed cells were plated to PSA with spectinomycin. Colonies with spectinomycin-resistant were selected and validated by PCR. The primers used for these cloning are listed in Supplementary Table S1.
Gene expression analysis by RT-qPCR
To analyze the differential expression levels of PXO_RS05990 (raxX ), PXO_RS06005 (raxST ), PXO_RS06010 (raxA), PXO_RS06015 (raxB ), PXO_RS20460 (raxC ), and PXO_RS14825 (pctB ) genes in Xoo PXO99 strains, quantitative reverse transcription PCR (RT-qPCR) was carried out. Xoo strains were cultured in the indicated liquid media and harvested by centrifugation. RNA was extracted from the cell pellets using TRIzol™ Max™ bacterial RNA isolation kit following the manufacturer’s instructions (Invitrogen Corp., USA). The RNA samples were reverse-transcribed to cDNA using a cDNA synthesis kit (Invitrogen Corp.). For quantitative PCRs, cDNA was analyzed using the Bio-Rad SsoFast EvaGreen Supermix. All primer pairs (Supplementary Table S1) were run using the same cycling parameters: initial denaturation at 95°C for 3 min, followed by 40 cycles with annealing and amplification at 62°C for 20 s and denaturation at 95°C for 5 s. The expression levels of Xoo genes were normalized to PXO_RS14730 gene expression levels.
Rice growth and Xoo inoculation
Oryza sativa ssp. Japonica rice variety Taipei309 (TP309) and transgenic line of TP309 carrying XA21 driven by its own promoter (XA21-TP309) were used for rice inoculations. Rice seeds were germinated in the germination paper (Nasco, USA) with distilled water at 28°C for 1 week. The seedlings were transplanted into 5.5-inch square pots with sandy soil (80% sand and 20% peat (Redi-Gro)) in a greenhouse. Plants were grown in tubs filled with fertilizer water [N, 58 ppm (parts per million); P, 15 ppm; K, 55 ppm; Ca, 20 ppm; Mg, 13 ppm; S, 49 ppm; Fe, 1 ppm; Cu, 0.06 ppm; Mn, 0.4 ppm; Mo, 0.02 ppm; Zn, 0.1 ppm; B, 0.4 ppm] for 4 weeks, followed by water for 2 weeks. Six weeks old plants were transferred to a growth chamber (28°C/24°C, 80%/85% humidity, and 14/10-hour lighting for the day/night cycle) for Xoo inoculation.
Plants were inoculated by Xoo strains using the scissors clipping method. Xoo strains from PSA plates were resuspended into water at a density of 108 CFU/ml. Water-soaked lesions were measured 14 days after inoculation.
RESULTS AND DISCUSSION
Construction of co-functional networks for Xoo
The workflow of the construction of XooNet is summarized in Fig. 1A and described in detail in the Material and Methods section. We constructed four component networks from Xoo-specific genomics sources that reflect functional association between genes: co-expression in Xoo (XO-CX), domain profile associations between Xoo coding genes (XO-DP), gene neighborhood (XO-GN), and phylogenetic profile associations between Xoo genes (XO-PG). To enhance accuracy and coverage of the network, we also utilized orthology-based transferred associalogs from other bacteria: high throughput protein-protein interactions in five bacterial species (BA-HT), co-citation of Escherichia coli (E. coli) gene names in Pubmed articles (EC-CC), co-expression of E. coli genes (EC-CX), literature-curated protein-protein interactions in E. coli (EC-LC), co-citation of P. aeruginosa gene names in Pubmed articles (PA-CC), co-expression of P. aeruginosa genes (PA-CX). We then benchmarked all ten component networks using Bayes statistics framework (Lee et al., 2004) with gold-standard functional gene pairs as described in Material and Methods section. The unified scoring scheme enabled integration of the functional links from heterogeneous data, resulting in the final integrated co-functional network, XooNet, which contains 106,000 links and 3,615 Xoo genes (~83% of all coding genes). The integrated XooNet and the ten component networks are summarized in Table 1.
Fig. 1. The construction and quality assessment of XooNet.
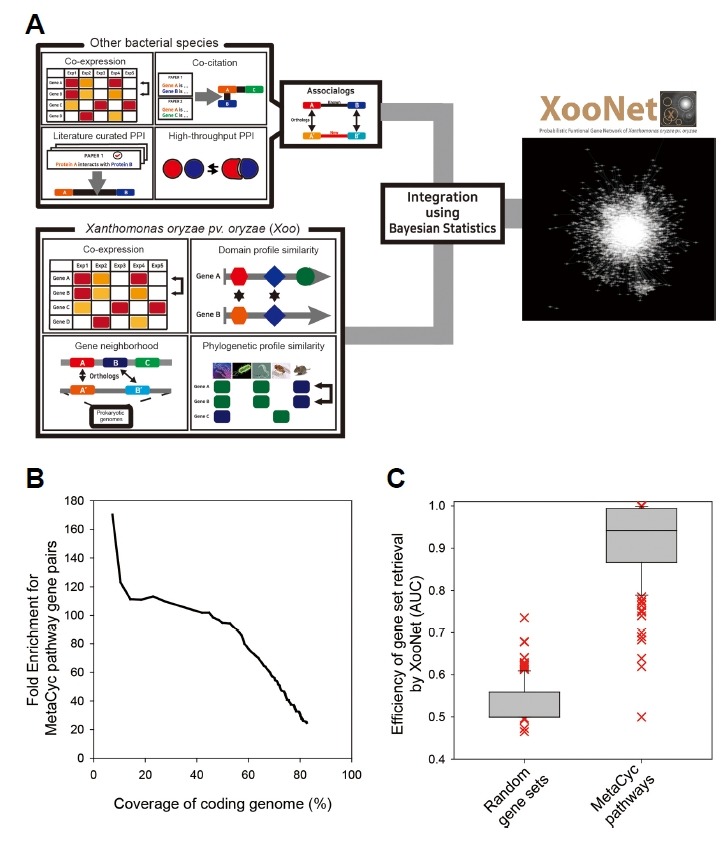
(A) Schematic overview of the construction process of XooNet. The functional associations are inferred from the ten distinct data types and integrated into the XooNet. (B) XooNet was assessed against test gene pairs based on MetaCyc pathway annotation. The graph represents the fold enrichment for the test gene pairs compared with random gene pairs and its corresponding coding genome coverage for every 1,000 links. (C) Area under ROC curve (AUC) was measured to evaluate retrieval efficacy of known genes for each of MetaCyc pathway terms or random gene sets using XooNet. We used only 145 MetaCyc pathway terms that have no less than five member genes.
Table 1.
Component networks from ten distinct data type in XooNet
| Code | Description | #links |
|---|---|---|
| XO-CX | Inferred co-functional network from co-expression in Xoo | 12,269 |
| XO-DP | Inferred co-functional network from similarity of domain profiles between two Xoo coding genes | 3,078 |
| XO-GN | Inferred co-functional network from gene neighborhood of Xoo orthologs across prokaryotic genomes | 18,487 |
| XO-PG | Inferred co-functional network from similarity of phylogenetic profiles between two Xoo genes | 36,244 |
| BA-HT | Associalogs transferred from high-throughput protein-protein interactions in five bacterial species | 5,522 |
| EC-CC | Associalogs transferred from co-citation of E. coli genes in Pubmed articles | 19,210 |
| EC-CX | Associalogs transferred from co-expression of E. coli genes | 20,283 |
| EC-LC | Associalogs transferred from literature-curated protein-protein interactions in E. coli | 828 |
| PA-CC | Associalogs transferred from co-citation of P. aeruginosa genes in Pubmed articles | 10,929 |
| PA-CX | Associalogs transferred from co-expression of P. aeruginosa genes | 6,808 |
| XooNet | An integrated co-functional network for Xoo | 106,000 |
XooNet is highly predictive for cellular pathways
To evaluate the quality of XooNet, we need to use another co-functional gene pairs as a test data set to avoid over-fitting models. For the network assessment, we compiled co-functional gene pairs from MetaCyc pathway annotations, an independent annotation database from GOBP and KEGG that were used for generating co-functional gene pairs to train XooNet. Consistent with their independent origin, the test gene pairs based on MetaCyc pathway annotations overlap with only 9% of the gold-standard gene pairs used for network training. We observed a 20-fold enrichment of MetaCyc pathway gene pairs for the integrated XooNet compared with random gene pairs. This result indicates that integration of diverse genomics data effectively improved the quality of XooNet (Fig. 1B).
Next, we assessed the capability of XooNet to predict pathways in Xoo by measuring the retrieval efficacy of known genes for each pathway using the “guilt-by-association” approach. The underlying logic of this approach is that if genes known for a specific biological process are effectively retrieved by network connections to other member genes of the same pathway, novel genes for the pathway are also likely to be retrieved by connections to the known genes for the pathway. We therefore measured the retrieval rate of known genes for each of 145 MetaCyc pathway terms that have no less than five member genes, using receiver operating characteristic (ROC) analysis. The ROC curve behavior can be summarized as a simple score, the area under the ROC curve (AUC), which would be close to 0.5 for a random predictor, and approaching 1 for a perfect predictor. We found that retrieval efficacy of XooNet for the MetaCyc pathway terms was significantly higher than that for random gene sets (P < 2.2e-16, Wilcoxon signed rank sum test) (Fig. 1C). These results indicate that XooNet accurately mapped co-functional links between genes and would also be able to predict novel genes for a pathway based on connections to the genes already known to function in the same pathway.
XooNet can reconstruct pathways regulated by two-component systems in Xoo
To survive in ever changing environment, bacteria have evolved regulatory circuits that coordinate expression of one set of genes in one environment and a different set of genes in another environment. These regulatory circuits are generally operated by TCSs. Perturbation of key regulators for TCSs can cause differential expression of other genes, providing clues for the cellular pathways regulated by those TCSs. For example, mutations of two TCS regulators, StoS and SreK, which positively regulate extracellular polysaccharide production and swarming in Xoo, led to the identification of differentially expressed proteins (DEP)(Zheng et al., 2016). Such DEPs were divided into two classes: up-regulated DEPs and down-regulated DEPs. If positive regulators have loss-of-function mutation, down-regulated DEPs are likely to be their directly regulated targets, which are often associated with the same pathway. In contrast, up-regulated DEPs might be genes of which expressions were affected by the regulators in an indirect manner. We hypothesized that if XooNet is highly predictive for pathways regulated by TCSs, it would effectively retrieve pathways regulated by StoS and SreK as well-connected networks of the down-regulated DEP (i.e., directly regulated targets). To test the hypothesis, we measure significance of within-group-connectivity of both up-regulated DEPs and down-regulated DEPs using 1,000 random permutations. Consistent with our hypothesis, we found that down-regulated DEPs were significantly connected to one another by XooNet (blue edges; P < 0.001 and P < 0.001 for △sreK and △stoS, respectively) whereas up-regulated DEPs were not (red edges; P = 0.261 and P = 0.139 for △stoS and △sreK, respectively) (Figs. 2A and 2B). These results demonstrate the utility of XooNet for the study of cellular pathways under control of TCS regulators with expression profiles in their mutant lines.
Fig. 2. XooNet can reconstruct regulatory circuits under control of two-component systems in Xoo.
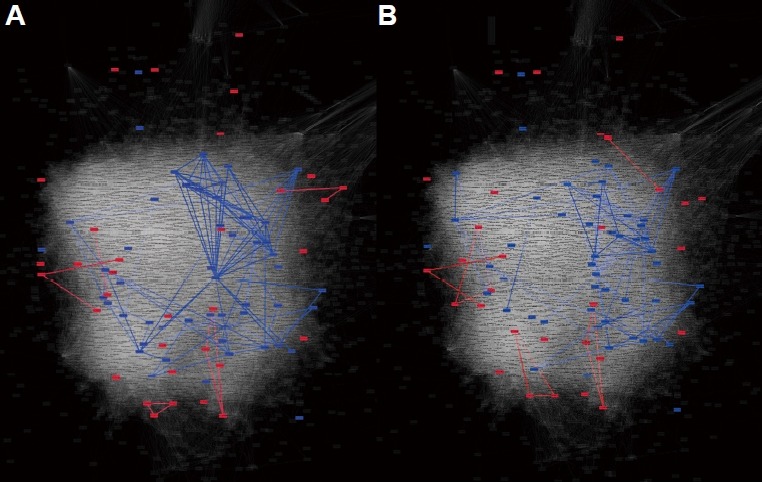
XooNet with highlights for subnetworks of differentially expressed proteins (DEPs). XooNet links between down-regulated DEPs (blue nodes and edges) and those between up-regulated DEPs (red nodes and edges) in (A) △sreK and (B) △stoS are overlaid on the background of the whole XooNet (white nodes and edges). There were 55 and 53 down-regulated DEPs for △sreK and △stoS, respectively, and 46 and 39 upregulated DEPs for △sreK and △stoS, respectively.
XooNet-based web tools for the prediction of novel pathway genes
We implemented two network-based algorithms for generating functional hypotheses, which can be accessed on the XooNet “network-search” page (https://www.inetbio.org/xoonet/search.php). There are two network search options available in XooNet web server: (I) Find new members of a pathway and (II) Infer functions from network neighbors. For search option (I), users can initiate searching for new candidate genes for a pathway by submitting a set of known genes for the desired pathway. These submitted genes are used as “guide genes” to prioritize new candidate genes by total edge weight score to them. The resulting highly-ranked genes are good candidates to be new members of the same pathway. For search option (II), users are able to predict functions for a query gene by collecting all known functional annotations (e.g. GOBP terms) for each of the network neighbors for the query gene. The collected functional annotation terms then are listed by the order of enrichment score.
XooNet-based search for novel regulators of rax
As mentioned above, rax genes are required for activation of XA21-mediated immunity and virulence of Xoo. We next used the network guided prediction to identify regulators that govern expression of the rax genes raxX, raxST, raxA, raxB, raxC, as well as pctB. PctB is an ABC transporter, which serves as a functionally redundant homolog of RaxB (Dee et al., 2018; Luu et al., 2018). Given our observation that XooNet is useful for studying bacterial regulatory circuits, we hypothesized that XooNet could identify regulators that govern expression of rax genes as well.
To identify such regulators, we submitted the five rax genes and pctB gene then launched the “Infer functions from network neighbors” tool of XooNet. Consistent with previous reports, all rax genes and pctB gene have either “protein secretion system” or “transport” as the most enriched GOBP terms among their neighbors (Supplementary Table S2). Next, we launched the “Find new members of a pathway” tool using the 5 rax genes and pctB gene as query genes. From this analysis we identified 273 Xoo genes as candidate network partners (Supplementary Table S3). Since we look for regulators of rax gene transcription, we focused on the genes annotated as “regulation of transcription, DNA-dependent” among the 273 candidate genes, as these were likely to be transcriptional regulators. In this subclass, we identified five candidate genes that have a sum of log likelihood score higher than 1.9: PXO_RS17440 (rank 19), PXO_RS18635 (soxR, rank 87), PXO_RS22160 (rank 103), PXO_RS18575 (oryR, rank 120), and PXO_RS20450 (rank 127) (Supplementary Table S3).
To access the function of the five candidate genes in regulation of rax and pctB gene expression, we generated knockout mutants in Xoo, in which each candidate gene was disrupted by single homologous recombination using the pJP5603 suicide vector (Penfold and Pemberton, 1992). We successfully generated knock-out mutants for PXO_RS18575, PXO_RS18635, PXO_RS20450, and PXO_RS22160. Despite many attempts, we were not able to isolate a PXO_RS17440 mutant, suggesting that the designated crossing over between constructs and genomic DNA were hardly occured or that the lack of PXO_RS17440 is lethal for Xoo. We then analyzed transcript levels of raxX, raxST, raxC and pctB in the four mutant strains. For these experiments, Xoo cells grown on PSA plates were inoculated into liquid culture media, a rich medium PSB (peptone-sucrose broth), at a density of 107 colony-forming units (CFU)/ml. The Xoo cells in liquid media were grown for 24 h until bacterial cell density reached at 108 CFU/ml and then harvested by a centrifugation. Xoo RNA was isolated from the cell pellets then, transcript levels of raxX, raxST, raxC and pctB genes were analyzed by RT-qPCR. None of the knock-out strains for candidate genes showed significant changes in expression of any of the rax genes or pctB as compared with their expression in the wild-type Xoo (Fig. 3).
Fig. 3. Gene expression analysis of rax genes in mutant strains for candidate rax regulators.
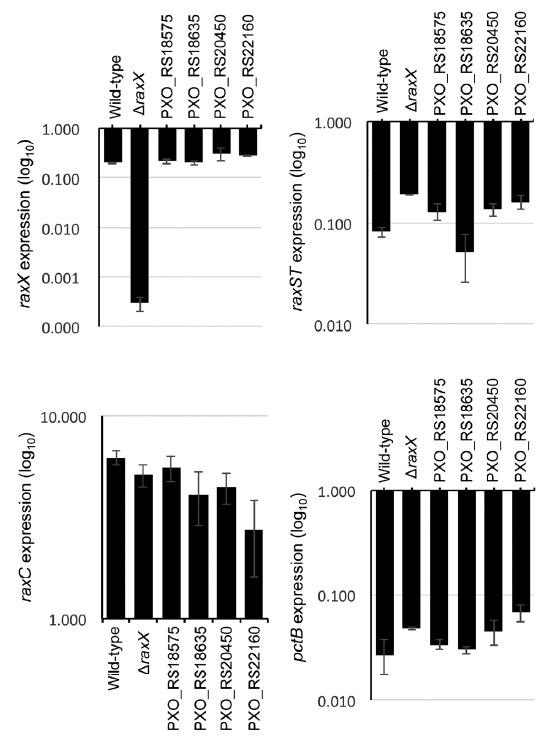
The transcript levels of raxX, raxST, raxC, and pctB were analyzed in the indicated strains that were grown in PSB by qRT-PCR. Data shown here is normalized to the PXO99. Bars depict average expression level ± S.D of three technical replicates. This experiment was repeated at least twice with similar results.
We also tested if the four mutant strains produce functionally active Rax/PctB proteins and activate the XA21 immune receptor in rice. We inoculated the mutant strains on both TP309 and XA21-TP309 rice plants. All mutant strains are virulent on TP309 plant as much as the wild-type strain thus, formed long lesion (>13 cm) (Fig. 4A). On XA21-TP309 plant, those mutants still induced the XA21-mediated immune response and formed a short lesion (6 cm<) like the wild-type strain (Fig. 4B) indicating that all Rax/PctB proteins are functional in the mutants.
Fig. 4. Analysis of XA21-mediated immune response by Xoo inoculation assay.
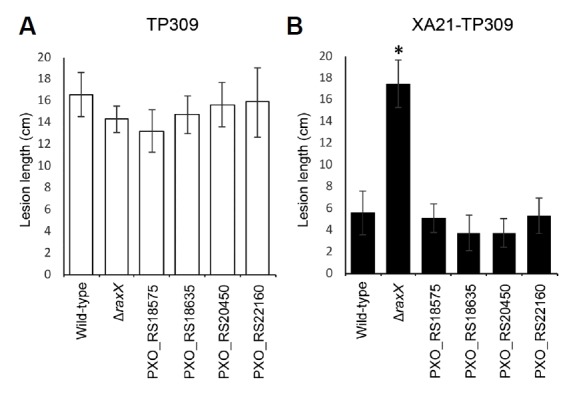
TP309 (A) and XA21-TP309 (B) were inoculated by clipping with scissors dipped in the indicated Xoo suspensions at a density of 108 colony forming units (CFU) per mL. Bars indicate the mean lesion length ± standard error (SE) measured 14 days after inoculation (n ≥ 20). The ‘*’ indicates statistically significant difference from PXO99 within each plant genotype using Dunnett’s test (α = 0.01). Experiments were performed at least two times with similar results.
We have demonstrated XooNet’s ability to predict a wide variety of cellular pathways by high retrieval efficacy of known genes for the same MetaCyc pathways. In addition, we successfully showed that XooNet could reconstruct pathways regulated by two TCS regulators, StoS and SreK, which are for extracellular polysaccharide production and swarming in Xoo. Therefore, results of insignificant changes of rax genes in mutants for the rax regulator candidates may be attributable to the following reasons: (i) regulatory defect of the mutations were compensated by functional redundancy by other regulators, (ii) we did not carry out the experimental tests in appropriate biological environment in which significant expression changes of rax genes could be detected, (iii) candidate genes were pseudogenes, (iv) XooNet is highly predictive for peer-to-peer relationships between regulated targets but not for regulator-target relationship.
CONCLUSIONS
In this report, we present XooNet, a genome-scale co-functional network of Xoo genes, constructed by integration of heterogeneous genomics data derived from Xoo and other bacterial species. We find that XooNet is highly predictive for a wide variety of cellular pathways in Xoo and can reconstruct pathways directly regulated by TCSs involved in bacterial fitness and infection. Based on the network, we also developed web tools to identify novel genes for pathways and to predict cellular functions for query genes. Users can freely access to XooNet edge information and prediction tools from a public web server located at www.inetbio.org/xoonet. Therefore, XooNet will be a useful in silico platform for the study of cellular pathways in Xoo.
Supplementary data
ACKNOWLEDGMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (NRF-2018M3C9A5064709, NRF-2018R1A5A 2025079) to I.L. and NIH GMS 3R01GM122968 to P.C.R.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Bae H.J., Lee H.N., Baek M.N., Park E.J., Eom C.Y., Ko I.J., Kang H.Y., Oh J.I. Inhibition of the DevSR two-component system by overexpression of mycobacterium tuberculosis PknB in mycobacterium smegmatis. Mol Cells. 2017;40:632–642. doi: 10.14348/molcells.2017.0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat M., Ortet P., Jourlin-Castelli C., Ansaldi M., Méjean V., Whitworth D.E. P2CS: a two-component system resource for prokaryotic signal transduction research. BMC Genomics. 2009;10:315. doi: 10.1186/1471-2164-10-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M., et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J., Bonas U. Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol. 2010;48:419–436. doi: 10.1146/annurev-phyto-080508-081936. [DOI] [PubMed] [Google Scholar]
- Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Mueller L.A., et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016;44:D471–480. doi: 10.1093/nar/gkv1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen L., Ideker T., Raphael B.J., Sharan R. Network propagation: a universal amplifier of genetic associations. Nat Rev Genet. 2017;18:551–562. doi: 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- da Silva F.G., Shen Y., Dardick C., Burdman S., Yadav R.C., de Leon A.L., Ronald P.C. Bacterial genes involved in type I secretion and sulfation are required to elicit the rice Xa21-mediated innate immune response. Mol Plant Microbe Interact. 2004;17:593–601. doi: 10.1094/MPMI.2004.17.6.593. [DOI] [PubMed] [Google Scholar]
- Dandekar T., Snel B., Huynen M., Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci. 1998;23:324–328. doi: 10.1016/s0968-0004(98)01274-2. [DOI] [PubMed] [Google Scholar]
- Han S.W., Lee S.W., Bahar O., Schwessinger B., Robinson M.R., Shaw J.B., Madsen J.A., Brodbelt J.S., Ronald P.C. Tyrosine sulfation in a Gram-negative bacterium. Nat Commun. 2012;3:1153–1153. doi: 10.1038/ncomms2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward A.C. The hosts of Xanthomonas. In: Swings J.G., Civerolo E.L., editors. Xanthomonas. Chapman & Hall; London: 1993. pp. 1–119. [Google Scholar]
- He Y.W., Zhang L.H. Quorum sensing and virulence regulation in Xanthomonas campestris. FEMS Microbiol Rev. 2008;32:842–857. doi: 10.1111/j.1574-6976.2008.00120.x. [DOI] [PubMed] [Google Scholar]
- Hoch J.A. Two-component and phosphorelay signal transduction. Curr Opin Microbiol. 2000;3:165–170. doi: 10.1016/s1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- Hwang S., Kim C.Y., Ji S.G., Go J., Kim H., Yang S., Kim H.J., Cho A., Yoon S.S., Lee I. Network-assisted investigation of virulence and antibiotic-resistance systems in Pseudomonas aeruginosa. Sci Rep. 2016;6:26223. doi: 10.1038/srep26223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensche P.R., van Noort V., Dutilh B.E., Huynen M.A. Practical and theoretical advances in predicting the function of a protein by its phylogenetic distribution. J R Soc Interface. 2008;5:151–170. doi: 10.1098/rsif.2007.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E., Kim H., Lee I. JiffyNet: a web-based instant protein network modeler for newly sequenced species. Nucleic Acids Res. 2013;41:W192–197. doi: 10.1093/nar/gkt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Shim J.E., Shin J., Lee I. EcoliNet: a database of cofunctional gene network for Escherichia coli. Database (Oxford) 2015. 2015:bav001. doi: 10.1093/database/bav001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofoid E.C., Parkinson J.S. Transmitter and receiver modules in bacterial signaling proteins. PNAS. 1988;85:4981–4985. doi: 10.1073/pnas.85.14.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhner S., van Noort V., Betts M.J., Leo-Macias A., Batisse C., Rode M., Yamada T., Maier T., Bader S., Beltran-Alvarez P., et al. Proteome organization in a genome-reduced bacterium. Science. 2009;326:1235–1240. doi: 10.1126/science.1176343. [DOI] [PubMed] [Google Scholar]
- Lee I., Date S.V., Adai A.T., Marcotte E.M. A probabilistic functional network of yeast genes. Science. 2004;306:1555–1558. doi: 10.1126/science.1099511. [DOI] [PubMed] [Google Scholar]
- Luu D.D., Joe A., Chen Y., Parys K., Bahar O., Pruitt R., Jade G., Chen L., Petzold C., Long K., et al. Sulfated RaxX, which represents an unclassified group of ribosomally synthesized post-translationally modified peptides, binds a host immune receptor. bioRxiv. 2018 doi: 10.1101/442517. [DOI] [Google Scholar]
- Mitchell A., Chang H.Y., Daugherty L., Fraser M., Hunter S., Lopez R., McAnulla C., McMenamin C., Nuka G., Pesseat S., et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–221. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson N., Aritua V., Heeney J., Cowie C., Bew J., Stead D. Phylogenetic analysis of Xanthomonas species by comparison of partial gyrase B gene sequences. Int J Syst Evol Microbiol. 2007;57:2881–2887. doi: 10.1099/ijs.0.65220-0. [DOI] [PubMed] [Google Scholar]
- Parrish J.R., Yu J., Liu G., Hines J.A., Chan J.E., Mangiola B.A., Zhang H., Pacifico S., Fotouhi F., DiRita V.J., et al. A proteome-wide protein interaction map for Campylobacter jejuni. Genome Biol. 2007;8:R130. doi: 10.1186/gb-2007-8-7-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold R.J., Pemberton J.M. An improved suicide vector for construction of chromosomal insertion mutations in bacteria. Gene. 1992;118:145–146. doi: 10.1016/0378-1119(92)90263-o. [DOI] [PubMed] [Google Scholar]
- Pruitt R.N., Joe A., Zhang W., Feng W., Stewart V., Schwessinger B., Dinneny J.R., Ronald P.C. A microbially derived tyrosine sulfated peptide mimics a plant peptide hormone. New Phytol. 2017;215:725–736. doi: 10.1111/nph.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt R.N., Schwessinger B., Joe A., Thomas N., Liu F., Albert M., Robinson M.R., Chan L.J.G., D.L.D, Chen H., et al. The rice immune receptor XA21 recognizes a tyrosine-sulfated peptide from a Gram-negative bacterium. Sci Adv. 2015;1:e1500245. doi: 10.1126/sciadv.1500245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rain J.C., Selig L., De Reuse H., Battaglia V., Reverdy C., Simon S., Lenzen G., Petel F., Wojcik J., Schachter V., et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- Sato S., Shimoda Y., Muraki A., Kohara M., Nakamura Y., Tabata S. A large-scale protein protein interaction analysis in Synechocystis sp. PCC6803 DNA Res. 2007;14:207–216. doi: 10.1093/dnares/dsm021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J.E., Lee I. Weighted mutual information analysis substantially improves domain-based functional network models. Bioinformatics. 2016;32:2824–2830. doi: 10.1093/bioinformatics/btw320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim J.E., Lee T., Lee I. From sequencing data to gene functions: co-functional network approaches. Animal Cells Syst. 2017;21:77–83. doi: 10.1080/19768354.2017.1284156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Lee I. Co-inheritance analysis within the domains of life substantially improves network inference by phylogenetic profiling. PLoS One. 2015;10:e0139006. doi: 10.1371/journal.pone.0139006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J., Lee I. Construction of functional gene networks using phylogenetic profiles. Methods Mol Biol. 2017;1526:87–98. doi: 10.1007/978-1-4939-6613-4_5. [DOI] [PubMed] [Google Scholar]
- Shin J., Lee T., Kim H., Lee I. Complementarity between distance- and probability-based methods of gene neighbourhood identification for pathway reconstruction. Mol Biosyst. 2014;10:24–29. doi: 10.1039/c3mb70366e. [DOI] [PubMed] [Google Scholar]
- Slater H., Alvarez-Morales A., Barber Christine E., Daniels Michael J., Dow J.M. A two-component system involving an HD-GYP domain protein links cell–cell signalling to pathogenicity gene expression in Xanthomonas campestris. Mol Microbiol. 2002;38:986–1003. doi: 10.1046/j.1365-2958.2000.02196.x. [DOI] [PubMed] [Google Scholar]
- Song W.Y., Wang G.L., Chen L.L., Kim H.S., Pi L.Y., Holsten T., Gardner J., Wang B., Zhai W.X., Zhu L.H., et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995;270:1804–1806. doi: 10.1126/science.270.5243.1804. [DOI] [PubMed] [Google Scholar]
- Sonnhammer E.L., Ostlund G. InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015;43:D234–239. doi: 10.1093/nar/gku1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D., Yao X., Duan M., Luo Y., Liu B., Qi P., Sun M., Ruan L. Two overlapping two-component systems in Xanthomonas oryzae pv. oryzae contribute to full fitness in rice by regulating virulence factors expression. Sci Rep. 2016;6:22768. doi: 10.1038/srep22768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.