

Abstract
Measles remains one of the leading causes of child mortality worldwide and is re-emerging in some countries due to poor vaccine coverage, concomitant with importation of measles virus (MV) from endemic areas. The lack of specific chemotherapy contributes to negative outcomes, especially in infants or immunodeficient individuals. Fusion inhibitor peptides derived from the MV Fusion protein C-terminal Heptad Repeat (HRC) targeting MV envelope fusion glycoproteins block infection at the stage of entry into host cells, thus preventing viral multiplication. To improve efficacy of such entry inhibitors, we have modified a HRC peptide inhibitor by introducing properties of self-assembly into nanoparticles (NP) and higher affinity for both viral and cell membranes. Modification of the peptide consisted of covalent grafting with tocopherol to increase amphipathicity and lipophilicity (HRC5). One additional peptide inhibitor consisting of a peptide dimer grafted to tocopherol was also used (HRC6). Spectroscopic, imaging, and simulation techniques were used to characterize the NP and explore the molecular basis for their antiviral efficacy. HRC5 forms micellar stable NP while HRC6 aggregates into amorphous, loose, unstable NP. Interpeptide cluster bridging governs NP assembly into dynamic metastable states. The results are consistent with the conclusion that the improved efficacy of HRC6 relative to HRC5 can be attributed to NP instability, which leads to more extensive partition to target membranes and binding to viral target proteins.
Keywords: self-assembling, peptide, nanoparticle, metastable, antiviral, fusion inhibitor, measles virus
Graphical Abstract
Measles is a highly contagious acute viral pathology that has undergone considerable re-emergence in recent years. It remains one of the leading causes of child mortality worldwide and a serious global health concern among vaccine-preventable diseases.1,2 Despite the efforts to eradicate the pathogenic measles virus (MV), the declining vaccine coverage and sporadic outbreaks highlight the need to reappraise antiviral drug development.3 The current absence of measles-specific treatment options threatens the prognosis in infants and immunocompromised patients with severe cases of infection.4
MV, a member of the Paramyxoviridae family, infects host immune and epithelial cells through the coordinated action of the envelope glycoproteins, Hemagglutinin (H) and Fusion (F) proteins. The F protein promotes viral and target cell membrane fusion.5,6 H mediates cell receptor recognition and attachment7 and, once receptor engaged, is responsible for activating F to its fusion-capable state. MV F-derived fusion inhibitor peptides possess promising antiviral potency against MV infection and spread.8–11 These act at the extracellular level by blocking the natural association between the N- and C-terminal helical heptad repeats (HRN and HRC, respectively) and thereby interfering with the F protein structural transitions that drive membrane fusion. It was recently shown that the inhibitory potency of HRC-derived fusion inhibitor peptides can be enhanced through lipophilic moiety conjugation and dimerization, with the addition of flexible polyethylene glycol (PEG) linkers.12 Biophysical characterization of the behavior of the conjugated inhibitor peptides in aqueous solution and in the presence of lipid membranes correlated to the efficacy and biodistribution data for the same peptides, and identified both self-assembly into nanoparticles (NP) and lipid membrane partition/retention as key modulators of activity in vivo.11,13 Tocopherol (Toc)-conjugated HRC5 (monomer) and HRC6 (dimer) peptides (Figure S1) show structural and functional differences in parallel with their antiviral efficacy. HRC5 is overall less active and forms smaller NP that insert less efficiently into lipid membranes; HRC6 is highly active, aggregates in a concentration-dependent fashion into larger NP and inserts extensively into lipid membranes.11 These data suggest that cell membrane integration, enhanced biodistribution and longer half-life are important for the inhibitory action of these conjugated peptides.10,11
In the present work, we explore the role of peptide dimerization in the morphological and biophysical properties of MV F-derived fusion inhibitor peptide NP to determine how these correlate with antiviral efficacy. We evaluated HRC5 and HRC6 peptide NP structure and organization through the use of microscopy and spectroscopy techniques. Additionally, we performed coarse-grained molecular dynamics (CG MD) simulations of peptide systems to characterize the self-assembly process, at the molecular level. We show that HRC5 forms compact particles with typical packed micellar assembly, with high stability and monodispersed in size. Conversely, HRC6 assembles into polydispersed loose spheroid particles with an amorphous internal space, more permeable to small polar molecules. Peptide intra- and intercluster bridge formation drive the self-assembly process. Our results suggest that dimerization introduces a degree of structural instability (metastability) and steric restrictions within the NP architecture that translate into enhanced antiviral efficacy, whereas monomeric peptide assemblies are more compact and stable, which reduces the peptides’ antiviral activity. These findings challenge current paradigms linking the stability and efficacy of NP drug delivery systems14–16 and may help guide design strategies for self-deliverable peptide/protein therapeutics.
RESULTS AND DISCUSSION
NP Morphology Suggests Distinct Packing Architectures between HRC5 and HRC6.
Both HRC5 and HRC6 are strongly amphipathic due to the hydrophilic peptide chain(s) and hydrophobic Toc domain (Figure S1). Peptide amphiphiles (PA) such as these share an entropic tendency to self-associate into supramolecular higher-order structures.17 Depending on the peptide sequence length, amino acid composition, hydrophobic moiety, concentration, and properties of the surrounding medium, these may assume variable sizes and shapes,18–22 which will impact their biological function.
Using transmission electron microscopy (TEM), we measured the length of the symmetry axis of the imaged NP (Figure 1A). Single HRC5 NP, imaged at ×12 000 magnification, have rough geometrical shapes. The longest particle axis ranged from 100 to 150 nm. In comparison, HRC6 NP showed a larger spherical-shaped morphology. Small budding motifs protruding from the nanoparticle surface were observed. Additionally, these particles seemed to incorporate more contrasting agent as they had a darker tonality. Particle longest axis measured ranged between 200 and 350 nm. In both cases, nanoparticles were occasionally part of larger clusters. These observations were complemented with atomic force microscopy (AFM) topographic images (Figure 1B). AFM images of HRC6 deposited in mica show spherical-shaped particles with average heights of 50–100 nm. HRC6 NP average Feret’s radius is 111 ± 41 nm. Larger and more irregular structures were also found (Figure 1C). We were not able to detect HRC5 NP deposited in mica surfaces (Figure S3). This could have been a result of particle diffusion without efficient attachment to the mica, probably reflecting structural differences between the two peptide assemblies.
Figure 1.

HRC5 and HRC6 NP morphology imaged using TEM and AFM. (A) TEM images of deposited HRC5 and HRC6 peptides (30 μM) self-assembled NP. NP selected for measurement are highlighted in 1 × 1 (HRC5) and 2 × 2 μm2 (HRC6) zoomed images by a white arrow. The measured NP longest axis are highlighted by a white line in individual zoomed insets (a−f). (B) AFM topographic imaging of deposited HRC6 (30 μM) self-assembled NP. The analysis of HRC5 (30 μM) and control images are included in Figure S3. (C) Height profiles of selected linear sections a−c, highlighted in B (left); NP Feret’s radius frequency distribution, grouped in 12.5 nm intervals (right). Error bars correspond the standard deviation of the mean from three independent experiments. (D) Abnormal HRC6 NP morphologies detected by AFM. Representative height images emphasizing uncharacteristic HRC6 NP clusters, respective 3D projections and height profiles of selected linear sections, highlighted in black, are presented for three independent cases. h, height.
Spherical-shaped nanoassemblies are usually associated with long peptide sequences (>20 amino acid residues) and high ionic strength.23,24 The HRC5 and HRC6 common peptide sequence (42 amino acid residues) and the salt content in solution (mimicking physiological conditions) is in agreement with the spheroid morphology observed. Nonetheless, the described nanoparticle sizes are considerably larger than typical PA micellar aggregates (10–50 nm diameters),24–27 though other cases with larger sizes have been reported.18,28 Moreover, the occurrence of sporadic and aberrant large clusters of HRC6 detected through TEM and AFM, characteristic of metastable polymeric assemblies,29 cannot be ruled out. In Figure 1A, a phenomenon similar to budding/fusion between HRC6 nanoparticles is observed. Although spurious aggregation during sample preparation cannot be ruled out, it is expected that less densely packed structures would be more avid to collapse into each other forming aggregate clusters.
HRC6, but Not HRC5, NP Form Metastable Poly-dispersed Aggregates in Solution.
To investigate NP mean size and structure in solution, we carried out time-resolved dynamic light scattering (DLS) experiments. Micellar-like stable aggregates are expected to have monodispersed concentration independent size distributions. In contrast, disordered unstable NP have typically polydispersed concentration-dependent size distributions. During a period of 9 h after NP preparation, we followed HRC5 and HRC6 number-averaged particle size distributions, calculating the average hydrodynamic radius (RH) and polydispersity index (PDI; Figure 2). HRC5 NP have a monodispersed size distribution with average RH of 50–65 nm (Figure 2A and B) and PDI values ranging between 10 and 15% (Figure 2C). HRC5 NP are highly stable over time. In contrast, HRC6 NP are characterized by a complex time- and concentration-dependent size distribution. Although a monomodal particle size population is observed for the initial 25 min of the experiment (average RH ~ 55 nm), another subpopulation is also detected for the remaining experimental time interval (RH ~ 150–250 nm; Figure 2D). This is followed by an increasing average RH up to 2 h (Figure 2E). NP PDI also increased, from an initial ~23% to a very significant ~70% (Figure 2F), indicative of severe polydispersity. This observation supports the existence of cluster-type aggregates in solution.
Figure 2.

HRC5 and HRC6 NP time-dependent size distributions. Time-resolved DLS measurements of HRC5 (A−C) and HRC6 (D−F) NP. Number-averaged size distribution profiles (A and D) of each sample (30 μM), collected for a 9 h period (0, 3, 6, and 9 h profiles are presented), were used to determine the respective average RH (B and E) and sample PDI (C and F) values, plotted for each time point. The dashed line is a guide to the eye. RH and PDI data sets are the average of three independent replicates. Error bars correspond to the standard deviation of the mean.
Although NP may eventually behave differently in vivo due to different factors such as the interaction with host circulating proteins,30 metastable HRC6 NP stability properties correlate directly with the antiviral pharmacokinetics and pharmacodynamics. HRC6 was still available in the serum (~3 nM) and lungs (~2 μM) 8 h post intranasal administration and was highly potent when administrated 4 h prior to MV infection, in a cotton rat model.11 It is worth highlighting that, at the reported in vivo lung concentration (≥1 μM),11 HRC6 form detectable NP. Under the same conditions, HRC5 was not effective.
Peptide Dimerization Influences HRC5 and HRC6 NP Internal Structure but Not Surface Charge.
After studying the NP as a whole, we focused on the internal structure and organization of the peptides assemblies. The internal peptide packing organization was characterized using a methodology based on pyrene fluorescence. Pyrene is a small fluorescent molecule used to probe the assembly of micellar structures in solution, through oriented excimer formation when inserted in hydrophobic pockets of those structures. The characteristic excimer emission peak (~383 nm) was detected for pyrene inserted within HRC5 nanoparticles, but not when inserted in HRC6 ones (Figure 3A). The critical micelle concentration (CMC) value for HRC5, determined through the 3:1 peak ratio method,31 was 1.83 ± 0.22 μM. CMCs in the low micromolar range are common for micellar PAs.23,32,33 The absence of pyrene excimer formation within NP has been associated with amorphous internal arrangements,23 as ordered hydrophobic pockets are not formed. Amorphous clusters are characterized by higher contribution of hydrogen bonding and lower entropic attraction.34
Figure 3.

Biophysical characterization of HRC5 and HRC6 NP structure. (A) HRC5 and HRC6 CMC determination through the pyrene 3:1 peak ratio method: pyrene (5 μM) fluorescence emission intensity ratio I3/I1 (I3, 383 nm; I1, 372 nm) was plotted as a function of the peptide concentration (0.03–30 μM). Lines correspond to linear regressions fitted to the experimental data regimes in the presence (high slope) and absence (low slope) of pyrene excimers. CMC is the x-axis value at the intersecion. Presented data sets are one of three independent replicates. (B) HRC5 and HRC6 NP internal accessibility probed by ANS fluorescence quenching. Stern−Volmer plots of ANS (12.8 μM) fluorescence emission intensity while inserted in HRC5 or HRC6 NP (30 μM), quenched by increasing acrylamide concentrations (0–600 mM). Lines correspond to the best fit of the Stern−Volmer equation (eq 1) to one of three independent replicates for each peptide. (C) HRC1, HRC5, and HRC6 peptide (30 μM) secondary structure in aqueous solution evaluated through CD spectroscopy. HRC1 was studied in aqueous solution and in the presence of 20% (v/v) TFE. Peptide mean residue ellipticity values ([θ]) were calculated according to eq 4. Presented spectra are one of three independent replicates. (D) HRC5 and HRC6 NP ζ-potential. Independent replicates correspond to the average of 15 measurements performed for each peptide sample (30 μM). Error bars correspond to the standard deviation of the mean.
To further explore the compactness/order vs loose/disorder organization of the NP, we used an assay based on permeation of the NP. 1-Anilino-8-naphthalenesulfonate (ANS) is a probe that inserts in the hydrophobic pockets of the NP with concomitant increase in the emission quantum yield. Acrylamide was then used to quench ANS fluorescence emission within the NP structure, enabling to conclude on the permeation of the NP to this small hydrophilic neutral molecule. ANS fluorescence emission was more efficiently quenched by acrylamide when inserted within HRC6 NP, comparatively to when inserted within HRC5 ones (Figure 3B). In both cases, linear Stern−Volmer relationships excluded the existence of nonaccessible ANS populations within the NP. The ⟨kq⟩ values, used as a measure of quenching efficiency, were 1.65 ± 0.17 and 2.77 ± 0.05 × 108 M·s−1 for ANS within HRC5 and HRC6, respectively (Table 1). ANS has the same ⟨τ⟩0 within HRC5 or HRC6 NP (Figure S4). These observations suggest that both HRC5 and HRC6 NP internal space is accessible to polar acrylamide molecules, i.e., the aggregates are internally solvated by polar molecules in solution, although to different extents.35 Differences in ANS ⟨kq⟩ values are associated with the respective acrylamide diffusion efficiency within the NP, and imply that HRC6 exhibits a more permeable lattice when compared to HRC5.35,36 This would explain why HRC6 NP seem to better incorporate the contrasting agent used in TEM studies (Figure 1A).
Table 1.
ANS Fluorescence Emission Quenching Parameters in HRC5 or HRC6 Peptides NP
| peptide | ⟨τ⟩ANS (ns) | KSV × 10−3 (M−1) | ⟨kq⟩ × 108 (M·s−1) |
|---|---|---|---|
| HRC5 | 8.69 ± 0.03 | 1.43 ± 0.14 | 1.65 ± 0.17 |
| HRC6 | 8.42 ± 0.23 | 2.33 ± 0.10 | 2.77 ± 0.05 |
Overall, the results suggest that the HRC6 double peptide chain leads to steric hindrance within the NP, breaking the typical micellar architecture, conserved in HRC5. Steric hindrance results from the spatial arrangement of peptide structure, in addition to the obvious differences in volume inherent to having a monomer (HRC5) or a dimer (HRC6). Using circular dichroism (CD) spectroscopy, we analyzed peptides secondary structure within the respective NP. The MV F protein HRC domain is natively α-helical (Figure S1), so as a control for the HRC domain structure, we included in our studies a monomeric unconjugated peptide, HRC1, which has a CD spectrum in aqueous solution that is typical of random coils (Figure 3C). A characteristic α-helical spectrum is obtained in the presence of 20% (v/v) trifluoroethanol (TFE), with the associated spectral minimum shifting from 200 to 208 nm (Figure 2C). TFE has been commonly used as a α-helix-stabilizing cosolvent.37 Both HRC5 and HRC6 peptides have α-helical type spectra with the characteristic minima at 222 nm, and not so pronounced minima at 208 nm. The corresponding peptide α-helical content (XH), summarized in Table 2, showed a significant variance in peptide secondary structure between HRC5 (42.1 ± 0.9%) and HRC6 (17.7 ± 2.2%). Additionally, the [θ]222/[θ]208 ratio, related to the balance between n−π* and π−π* amide backbone transitions, was used as a measure of α-helical backbone flexibility (Table 2).38,39 TFE-stabilized helices were considerably less flexible (0.68 ± 0.02) than HRC5 (1.56 ± 0.10) and HRC6 (1.61 ± 0.08) self-assembling peptides. Even though both peptide helices display high flexibility (high [θ]222/[θ]208), HRC6 dimeric peptides attain considerably lower total helical content compared to HRC5. Both dimerization and less extended (helical) secondary structure may contribute to bulkier HRC6 peptides when compared to HRC5, therefore explaining why NP packaging is not so ordered and dense in HRC6 NP as in HRC5 NP. Interestingly, previous studies associate lipidation-driven peptide secondary structure to improved biological activity and function.40,41 Our findings challenge this premise: potent antiviral efficacy, documented for HRC6, is unrelated to high peptide secondary structure content within NP. Instead, aggregate structure and dynamics rules pharmacodynamics. However, it should be stressed that due to differences in NP internal organization, permeability and secondary structure, HRC5 and HRC6 peptides have distinct sensitivity to proteolytic digestion within NP (Figure S5). Though more potent in vivo, HRC6 displays a shorter half-life (t1/2) of ~6 min compared to the t1/2 of HRC5 of ~50 min in the presence of trypsin, a well-studied endogenous protease. This further emphasizes that antiviral efficacy is unrelated to properties such as resistance to digestion and longer expected t1/2 in circulation, suggesting aggregate structure and dynamics dictate pharmacokinetics in vivo.
Table 2.
HRC5 and HRC6 Peptide α-Helix Parameters Calculated through CD Data Analysis
| peptide | solvent | XH (%) | [θ]222/[θ]208 |
|---|---|---|---|
| HRC1 | sample buffer | 8.5 ± 1.6 | 0.37 ± 0.01 |
| sample buffer, 20% (v/v) TFE | 28.4 ± 1.5 | 0.68 ± 0.02 | |
| HRC5 | sample buffer | 42.1 ± 0.9 | 1.56 ± 0.10 |
| HRC6 | sample buffer | 17.7 ± 2.2 | 1.61 ± 0.08 |
Increased tendency to aggregate of HRC6 NP may also hypothetically result from a decreased surface charge density, which in turn implies decreased electrostatic repulsion. Although both HRC5 and HRC6 have similar net charges, the conformation and supra-molecular organization inside NP may determine important differences in surface charge density. ζ-potential were determined to ascertain eventual differences in the surface charge density of HRC5 and HRC6 NP. ζ-potential values were, respectively, −17.6 ± 0.2 and −15.4 ± 0.3 mV for HRC5 and HRC6 (Figure 3D). Anionic NP have been associated with lower cell membrane disruption compared to cationic ones, albeit other potential undesired effects may occur.42 Although a small contribution of ζ-potential to HRC5 stability cannot be completely excluded, it is not expected that inter-NP repulsion accounts for the differences observed with HRC5 and HRC6.
Interpeptide Cluster Bridging Explains the Structural Dynamics of Metastable HRC6 NP.
PAs self-assembling mechanisms at the molecular level remain highly elusive. Though there is clear evidence of HRC5 and HRC6 contrasting behavior, which we attribute to peptide dimerization and secondary structure, the insight into the individual peptide−peptide interactions that drive assembly and modulate the NP structure needs further investigation. MD simulations are an alternative approach for this purpose. These provide a controlled environment, under fine-tuned conditions, to simulate the molecular behavior, probe interactions and evaluate system evolution trends over time. MD simulations have been previously used to probe PA self-assembly, with promising mechanistic and structural results.43–45
Since time-resolved DLS suggested that HRC6 peptide NP may reach metastable equilibria, in contrast to the highly stable HRC5 NP, we examined the molecular HRC peptide−peptide association modes driving self-assembly. To analyze the assembly of HRC5 and HRC6 we chose a CG methodology instead of a more common atomistic model simulation. Since we intended to analyze a time-dependent process, CG MD allows simulation of longer time scales (μs range) without the significant drawback of longer computational processing times. Detailed atomistic information is also less relevant in this case, as we seek to understand intermolecular association of large peptides into larger clusters. CG MD simulations of 8 HRC5 or HRC6 Martini CG peptide models (Figure 4A), lasting 10 μs, allowed the assessment of aggregation evolution from nonassembled initial states. The respective quantitative analysis of preferential contact regions between peptides (Figure 4) and preferential contact modes (Table 3) was performed. The eight simulated HRC5 peptides become associated in the first few nanoseconds of the simulation (Figure 4B top panel). These aggregates were stable and long-lasting throughout the simulation, with sporadic release of 1–2 single peptides from the ensemble. Simulations and contact maps revealed that the majority of the contacts between peptides were Toc−Toc interactions (Figure 4C top panel). Since the simulation medium is representative of physiological polarity and ionic strength, this observation was expected. Similarly, HRC6 peptides associate very quickly during the simulation but form smaller aggregates clusters composed by two to three peptides (Figure 4B bottom panel). During the simulation, these seem to contact and interchange peptides with each other, eventually coalescing into larger, bridged particles. We did not observe considerable contact between the amino acid residue chains of the same or different peptides. Contact maps revealed that both Toc−Toc and Toc−peptide backbone interactions are possible, although the latter was less frequent (Figure 4C bottom panel). Toc−peptide interaction within HRC6 NP could help explain a less ordered internal organization, the permeable internal space, and the lower content in α-helix of the peptide secondary structure. Instead of a typical micelle, the NP would represent a mesh of nonoriented peptides, connected through Toc−Toc and Toc−peptide interactions.
Figure 4.

CG MD simulations of HRC5 and HRC6 assembly into NP. Coarse-grained molecular dynamics simulations of 8 HRC5 or HRC6 peptides, followed for 10 μs. (A) Representative image of the peptide models and most characteristic peptide assembling behavior. The peptide chain backbone is represented in red, the PEG4 linker in gray, and the Toc moiety in yellow (all other system components were omitted for clarity). Black arrows indicate different association clusters. (B) Stills of each peptide simulation, captured at 0, 2, 4, 6, 8, and 10 μs, collected to emphasize the progression of the peptides assembly during the experiment. (C) Preferential contact maps reporting favored interaction regions between peptides, built from full simulation data. For HRC5, the CG bead numbers correspond to peptide chain −0 to 80; PEG4 linker −81 to 89; and Toc −90 to 95. For HRC6, CG bead numbers are the same as those for HRC5, with the second peptide chain and PEG4 linker corresponding to beads 96 through 180. Darker tones in the contact map gradient scale indicate a higher number of detected contacts. For both HRC5 and HRC6, Toc−Toc interactions can be seen to predominate; additional Toc−peptide interactions are visible for HRC6 but not for HRC5.
Table 3.
HRC5 and HRC6 Peptide Preferential Contacts Modes Assessed through CG Simulation Times
| tCGS (%) | |||||
|---|---|---|---|---|---|
| peptide | no contacts | HD−HDa | HD−PAb | HD−PA-Ac | RA-A |
| HRC5 | 9.0 ± 0.5 | 90.1 ± 0.5 | 0.9 ± 0.1 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| HRC6 | 67.1 ± 17.3 | 31.5 ± 8.4 | 0.5 ± 0.2 | 0.9 ± 0.3 | 5.0 ± 1.6 |
HD−HD: interactions through hydrophobic domains.
HD−PA: interactions through hydrophobic domains and peptide chains in the same aggregation cluster.
HD−PA−A: interactions through hydrophobic domains and peptide chains in different aggregation clusters.
Through contact analysis, we quantified the aggregates bridge ratio (RA−A). This parameter corresponds to the simulation time involved in interactions between different clusters as a fraction of the total interactions time. RA−A values (Table 3) directly correlate with the potential NP metastability and tendency to collapse into larger clusters. HRC5 and HRC6 RA−A were, respectively, ~0 and 5%. Stable HRC5 clusters have practically no contacts between peptides from different clusters. We cannot exclude that the relatively low number of simulated peptides may prevent formation of additional clusters in this case; nonetheless the formation of a single eight peptide cluster suggests high stability. Multiple HRC6 clusters contact between themselves through interpeptide bridging, in which the central portion of a peptide sequence from one cluster interacts with the Toc moiety of a different cluster. If propagated in solution, this behavior may lead smaller particles to mix into larger ones and eventually revert back into the smaller, more stable clusters, thus emulating the metastable behavior suggested by time-resolved DLS. A similar clustering behavior has been observed in simulated PA nanofibers.45 The PAs initially assemble into spherical shaped aggregates that gradually collapse into cylindrical nanofibers. There was no evidence of relapse into smaller structures, probably due to the small peptide sequence and high stability of the resulting fiber. In an independent study, the authors observed the same stable fiber structures, forming within 16 μs.44 In both studies, the number of simulated particles was significantly larger, which may provide more refined macromolecular details.
CONCLUSION
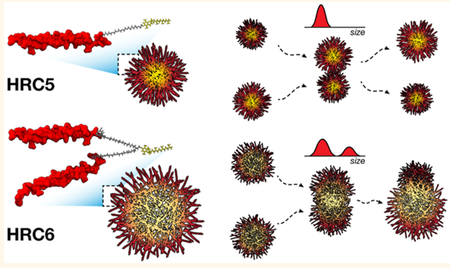
Antiviral drug development in the era of reemerging viruses, such as measles, is critical. MV fusion inhibitor peptides, designed as amphiphilic lipid-tagged conjugates, are a promising class of antivirals for acute (and perhaps for persistent) measles.9,10 The strong evidence for self-assembling and self-delivery to relevant infection sites, resulting in high potency, prompted an exploration of the physical and chemical properties that guide these phenomena. In the present study, we have shown that properties of self-assembled NP are influenced by dimerization of the peptide subunits. Dimerization modulates NP morphology, internal organization, solvent permeation, peptide secondary structure, and, ultimately, particle stability. In contrast, dimerization by itself, without aggregation, does not significantly improve the effectiveness of unconjugated HRC-derived peptides against MV fusion and spread.9,10 Dimeric HRC6 peptide NP, highly potent in vitro and in vivo, achieve metastable equilibria in solution through nonmicellar peptide−peptide association. At variance, HRC5 monomers assemble into very stable NP that have little antiviral efficacy. In sum, we have identified several molecular mechanisms that link dimerization to improved MV antiviral efficacy (Figure 5).
Figure 5.

Schematic simplified representation of the effect of peptide dimerization in HRC5 and HRC6 NP structural dynamics. (A) Peptide dimerization impacts HRC5 and HRC6 NP internal and surface-related properties. NP formed by HRC5 monomer peptides are small, tightly packed micellar structures. Because internal accessibility to polar solvents is low, HRC5 NP core is expected to be considerably hydrophobic. HRC6 forms larger and looser NP deprived of a well-organized internal architecture. The internal space is permeable to polar molecules, which translates into a more hydrophilic interior. Dimerization introduces repulsion between the two peptide chains and partial loss of extended (helical) conformation of the peptide causing high internal steric hindrance to ordered packing. (B) Stable HRC5 NP collide elastically with very low impact in the overall particle structure (size and dispersity are constant); in distinction, HRC6 NP collisions are more inelastic, leading to the formation of larger aggregated clusters, over time (increase in size and polydispersion). The metastable behavior correlates with the peptides mode of action, as a determining factor for efficacy.
We speculate that intrinsic NP stability is an important determinant of the HRC peptides’ membrane interaction.11,13 In the presence of lipid membranes, stochastic NP collisions with the bilayer surface may result in different outcomes. Highly stable structures, such as HRC5 NP, are expected to contact more elastically with the lipid polar heads, resulting in inefficient disassembling and Toc insertion. However, metastable HRC6 NP are expected to disassemble more easily upon contact with membranes. Since NP interaction with cell membranes precedes binding of individual peptides to MV F glycoproteins, the consequences of dimerization on NP structural stability are essentially more stringent to antiviral efficacy than the inherent avidity to its target. Additional properties of airway epithelium translocation upon intranasal delivery (by means of mucoadhesion, for instance), high concentrations in relevant infection sites, and prolonged in vivo circulation lifetimes contribute toward explaining HRC6 NP antiviral efficacy. Anti-MV fusion inhibitor peptide dimers were shown to translocate human airway epithelium barriers 24 h sooner than monomeric peptides,10 which contributes positively to bioavailability and efficacy. When NP are administrated intranasally, they must interact with the airway mucosa and mucus secretions, prior to translocation, as proposed by other authors.46–48 Due to its morphological dynamics here reported, it is possible that HRC6 NP clusters establish strong adhesion to the mucus layer with concomitant peptide diffusion and slowed mucociliary clearance, thereby enhancing penetration and translocation.
MATERIALS AND METHODS
Chemicals and Reagents.
HRC1, HRC5, and HRC6 peptides (Figure S1) having ≥95% purity were purchased from American Peptide Company (Sunnyvale, CA). Peptides were chemically synthesized by standard 9-fluorenylmethoxy carbonyl (Fmoc) solid-phase methods; HRC5 and HRC6 were conjugated with tocopherolcore functional lipophilic moieties, as described elsewhere.11 Ammonium heptamolybdate, pyrene, ANS, and acrylamide were purchased from Sigma-Aldrich (St. Louis, MO). N-2-Hydroxyethylpi perazine-N′−2-ethanesulfonic acid (HEPES), NaCl, MgCl2, NaF, ethanol, dimethyl sulfoxide (DMSO), and TFE (the last three with spectroscopic grade) were purchased from Merck (Darmstadt, Germany).
Sample Preparation.
Lyophilized HRC1, HRC5, and HRC6 peptides were dissolved in 100% DMSO to final concentrations of 10, 10, and 50 mg/mL, respectively, sonicated in an ultrasonic bath for 10 min, and stored at −20 °C. For experiments, stock solutions were diluted in 10 mM HEPES, 150 mM NaCl, pH 7.4 (sample buffer) to final working concentrations and sonicated for 2 min. For CD studies, DMSO and NaCl were replaced by ethanol and NaF, respectively, to avoid absorbance in the far UV. For all samples, DMSO or ethanol content was maintained below 2% (v/v). Working samples were used immediately after preparation.
Instrumentation.
TEM was performed on a H-7650 electron microscope from Hitachi (Tokyo, Japan) at an acceleration voltage of 100 kV; acquisitions and measurements were performed using a XR41 M mid mount digital camera from Advanced Microscopy Techniques (Woburn, MA). AFM imaging was performed with a Nano Wizard IV from JPK (Berlin, Germany) mounted on a Axiovert 200 inverted microscope from Zeiss (Oberkochen, Germany). The AFM head is equipped with a 15 μm z-range linearized piezoelectric scanner and an infrared laser. Steady-state and time-resolved fluorescence measurements were performed in FLS920 and Lifespec II fluorometers, respectively, both from Edinburgh Instruments (Livingston, UK). Dynamic light scattering particle size and ζ-potential measurements were performed in Zetasizer Nano ZS equipment from Malvern (Worcestershire, UK). CD spectroscopy was performed in a J-815 spectrophotometer from Jasco (Oklahoma City, OK). All measurements were performed at 25 °C.
Transmission Electron Microscopy.
For TEM analysis, 10 μL of either HRC5 or HRC6 solution (30 μM) was deposited onto a Formvar/carbon-coated 400 mesh copper grid from Agar Scientific (Standsted Essex, UK). After 30 s, the excess fluid was removed and the grid was negatively stained with 2% (w/v) aqueous ammonium heptamolybdate for approximately 5 min. The copper grid was air-dried prior to insertion into the microscope.
Atomic Force Microscopy.
For AFM analysis, 70 μL of either HCR5 or HCR6 solution (30 μM) was deposited for 2 h on freshly cleaved mica substrate, previously treated with 2 mM MgCl2 prepared in sample buffer and glued to a glass coverslip. For HRC5, an additional 24 h incubation time was tested. The samples were then rinsed at least four times with ultrapure water and left to air-dry. Images were obtained in air using intermittent contact mode. Uncoated silicon ACL cantilevers from AppNano (Mountain View, CA) with typical resonance frequencies ranging between 145 and 230 kHz and an average spring constant of 45 N/m were used. Scan speeds were lower than 1 Hz and total areas with 2.5 × 2.5 to 10 × 10 μm2 were scanned with 512 × 512 pixel resolution. Height and error images were recorded and line fitted as required.
Dynamic Light Scattering.
RH and PDI over time were characterized using DLS in particle size analysis mode. HRC5 and HRC6 samples (30 μM) were preincubated for 5 min at 25 °C before starting each measurement. Time-resolved experiments consisted in consecutive single measurements, each corresponding to an autocorrelation curve averaged from a minimum of 12 runs, performed every 5 min, over a 9 h period. Measurement time (~2.5 min) was accounted for when analyzing data. Peptide NP diffusion coefficients (D) were calculated from autocorrelation curves at each time-point through a CONTIN-like method,49 and used to plot number-averaged RH profiles (10–1000 nm) by application of the Stokes−Einstein−Sutherland equation.50 Average RH and PDI values were obtained for each size distribution profile.
DLS in ζ-potential mode was used for peptide NP global surface charge density characterization. HRC5 and HRC6 samples (30 μM) were preincubated for 15 min at 25 °C before starting each measurement. Single independent experiments consisted of 15 measurements, each averaged from ~20–50 runs performed at constant voltage (40 V). ζ-potential values were calculated from the mean value of each measurement set.
Fluorescence Studies.
To determine the HRC5 and HRC6 apparent CMC, pyrene fluorescent probe (5 μM) was added to peptide samples at concentrations ranging from 0.03 to 30 μM. After a 10 min incubation period, pyrene fluorescence emission intensity was collected from 350 to 450 nm, using a fixed excitation wavelength, λexc, of 330 nm. Excitation and emission slits were 4 and 10 nm, respectively. Characteristic I1 and I3 (excimer) pyrene fluorescence emission maxima, detected at 372 and 383 nm, respectively, were used to calculate the peptide CMC through the pyrene 3:1 peak ratio method.31 Briefly, the CMC was determined at the inflection point of I3/I1 vs peptide concentration plots, through the intersection of linear regression curves fitted to the two coherently linear data set regimes: in the presence (high slope) or absence (low slope) of pyrene excimer formation.
HRC5 and HRC6 NP density was evaluated through internal accessibility by a small fluorescent probe, ANS (12.8 μM), which was preincubated with peptides (30 μM) for 10 min, to enable complete internalization into the NP internal space.11 ANS fluorescence emission was then quenched through the addition of small volumes of acrylamide solution, to final concentrations between 0 and 600 mM. After each addition, a 10 min incubation time was allowed before performing measurements. ANS fluorescence emission intensity was collected from 400 to 600 nm, with a fixed λexc of 365 nm. Excitation and emission slits were 5 and 10 nm, respectively. Fluorescence intensity was corrected for sample dilution and background. Average bimolecular quenching rate constants, ⟨kq⟩, were determined through the Stern−Volmer relationship:51,52
| (1) |
| (2) |
in which,= I0 and I correspond to the ANS fluorescence emission spectrum integral in the absence and presence of quencher, respectively, KSV is the Stern−Volmer constant, [Q] is the molar concentration of quencher, and ⟨τ⟩0 is the average ANS fluorescence lifetime in the absence of quencher. To experimentally determine the ⟨τ⟩0 parameter, fluorescence lifetime decays of ANS inserted in HRC5 or HRC6 were collected between 0 and 20 ns, using a 103 photon count acquisition stop condition. ANS was excited with a 368.8 nm picosecond pulsed LED, and emission was collected at 480 nm with emission slits of 35 nm. Lifetime decays were corrected with the instrument response function and fitted with a double exponential curve using the FAST software from Edinburgh. Average fluorescence lifetimes were obtained through the following relationship:51
| (3) |
in which ai and τi correspond to the amplitude and lifetime associated with the ith mode of the multiexponential distribution, respectively.
Circular Dichroism.
To evaluate the peptides secondary structure, CD spectra of HRC1, HRC5 and HRC6 (30 μM) were collected between 190 and 260 nm, averaged from 10 consecutive scans, and corrected for the background noise. A HRC1 sample containing 20% (v/v) TFE was also analyzed. Peptide mean residue ellipticity, [θ], was calculated using the following equation:53
| (4) |
in which θ corresponds to the observed ellipticity, N is the number of amino acid residues, l is the optical path length, and c is the molar concentration of peptide.
Peptide helical content, XH, was determined using the following relationship:54
| (5) |
where [θ]222 is the mean residue ellipticity at 222 nm, is the mean residue ellipticity of a putative helical peptide with infinite amino acid residues, k is a peptide length-dependent factor, and n is the number of amino acid residues. and k, respectively, −39 500 deg·cm2·dmol−1 and 2.57, were obtained from theoretically derived CD data of a putative helical peptide composed of an infinite amino acid residue sequence.54
Coarse-Grained Simulations.
HRC5 and HRC6 peptide CG molecular structure was built in three steps: (i) Homology modeling was used to obtain the atomistic structures of HRC5 and HRC6, based on the atomistic structure of the human type 3 Parainfluenza Virus (hPIV3) fusion protein (PDB: 1ZTM). The Martini model, for which proteins and PEG representations are available and readily adaptable to our system, was used as CG force field. The atomistic structures were converted to the corresponding CG models using the martinize.py script,55 for subsequent simulation under GROMACS 5.1.56 (ii) The PEG CG model57 was added to the peptide CG models as PEG4. Structural parameters of peptide−PEG links follow existing Martini topologies. For simplicity, the maleimide group in HRC6 was replaced by three PEG beads in the final models. (iii) Toc CG topology was parametrized based on atomistic data, validated as described in the Supporting Information, and added to the final peptide CG models.
For peptide aggregation simulations, boxes containing 8 peptide molecules and either 23 075 Martini waters, 271 Na+ and 255 Cl− beads for HRC5 or 23 051 Martini waters, 295 Na+ and 255 Cl− beads for HRC6 were used. Each box had a net neutral charge and recreated the in vitro solvent saline concentration. The Martini model has been reported to overly represent protein−protein interactions in the aqueous phase; to prevent this from causing the system to become caught in initial-contact configurations, peptide−water interaction forces were increased by 25% relative to the standard Martini interactions. The increased solvation allows peptides to associate and dissociate in the studied microsecond scale. CG data were generated with the tuned peptide−water Martini model and simulated at 20 fs integration steps, for trajectories at least 10 μs long. Temperature was coupled to 300 K using the v-rescale thermostat with 1 ps coupling time, and the pressure was isotropically coupled to 1 bar using the Parrinello−Rahman barostat with a 2 ps coupling time.
For each simulation, contact mapping of preferential interaction regions between pairs of peptides and contact counting of three different types of interactions (HD−HD, interactions through hydrophobic domains; HD−PA, interactions through hydrophobic domains and peptide chains in the same aggregation cluster; HD− PA−A, interactions through hydrophobic domains and peptide chains in different aggregation clusters) were performed. Aggregate cluster bridge ratio, RA−A, was calculated through the following relationship:
| (6) |
in which tCGS(HD−HD), tCGS(HD−PA), and tCGS(HD−PA−A) correspond to the respective coarse-grained simulation contact times for each interaction type, quantified as a percentage of the total simulation time.
These procedures were implemented in Python with extensive use of MDAnalysis58 and NumPy packages. Both analyses discarded the first microsecond of initial trajectory time as peptide equilibration and approximation time. Visualization and image generation were carried out using the VMD 1.9.359 package and the Tachyon ray-tracer.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge Andreia Pinto for TEM micrograph acquisition. This work was supported by Fundação para a Ciência e a Tecnologia (FCT-MCTES) Project PTDC/QEQMED/4412/2014. T.N.F., D.A.M,. and D.G. acknowledge individual fellowships SFRH/BD/5283/2013, PD/BD/136752/2018 and SFRH/BPD/109010/2015 funded by FCT-MCTES. A.S.V. acknowledges funding under the Investigator Programme (IF/00803/2012) from FCTMCTES. M.N.M. acknowledges Grant LISBOA-01–0145-FEDER-007660 (Microbiologia Molecular, Estrutural e Celular) funded by Fundo Europeu de Desenvolvimento Regional (FEDER) and FCT-MCTES. A.M. acknowledges Grants RO1AI114736, R33AI101333, and RO1AI031971, and M.P. acknowledges grants R01AI119762, R01AI121349, and R01NS105699 funded by the National Institutes of Health (NIH).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b01422.
Description of the studied molecules and related structural features; tocopherol coarse-grained molecular model development and validation; complementary AFM micrographs; ANS fluorescence lifetime data; and peptide sensitivity to proteolysis (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Moss WJ; Griffin DE Measles. Lancet 2012, 379, 153–164. [DOI] [PubMed] [Google Scholar]
- (2).WHO. 2016 Midterm Review of the Global Vaccine Action Plan; 2016; pp 1–26. [Google Scholar]
- (3).Holzmann H; Hengel H; Tenbusch M; Doerr HW Eradication of Measles: Remaining Challenges. Med. Microbiol. Immunol 2016, 205, 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Saso A; Kampmann B Vaccine Responses in Newborns. Semin. Immunopathol 2017, 39, 627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chang A; Dutch RE Paramyxovirus Fusion and Entry: Multiple Paths to a Common End. Viruses 2012, 4, 613–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Plattet P; Alves L; Herren M; Aguilar HC Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hashiguchi T; Maenaka K; Yanagi Y Measles Virus Hemagglutinin: Structural Insights Into Cell Entry and Measles Vaccine. Front. Microbiol 2011, 2, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wild TF; Buckland R Inhibition of Measles Virus Infection and Fusion with Peptides Corresponding to the Leucine Zipper Region of the Fusion Protein. J. Gen. Virol 1997, 78, 107–111. [DOI] [PubMed] [Google Scholar]
- (9).Welsch JC; Talekar A; Mathieu C; Pessi A; Moscona A; Horvat B; Porotto M Fatal Measles Virus Infection Prevented by Brain-Penetrant Fusion Inhibitors. J. Virol 2013, 87, 13785–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mathieu C; Huey D; Jurgens E; Welsch JC; DeVito I; Talekar A; Horvat B; Niewiesk S; Moscona A; Porotto M Prevention of Measles Virus Infection by Intranasal Delivery of Fusion Inhibitor Peptides. J. Virol 2015, 89, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Figueira TN; Palermo LM; Veiga AS; Huey D; Alabi CA; Santos NC; Welsch JC; Mathieu C; Horvat B; Niewiesk S; Moscona A; Castanho MARB; Porotto M In Vivo Efficacy of Measles Virus Fusion Protein-Derived Peptides Is Modulated by the Properties of Self-Assembly and Membrane Residence. J. Virol 2017, 91, e01554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pessi A; Langella A; Capitò E; Ghezzi S; Vicenzi E; Poli G; Ketas T; Mathieu C; Cortese R; Horvat B; Moscona A; Porotto M A General Strategy to Endow Natural Fusion-Protein-Derived Peptides with Potent Antiviral Activity. PLoS One 2012, 7, e36833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Figueira TN; Freire JM; Cunha-Santos C; Heras M; Goncalves J; Moscona A; Porotto M; Salomé Veiga A; Castanho MARB Quantitative Analysis of Molecular Partition Towards Lipid Membranes Using Surface Plasmon Resonance. Sci. Rep 2017, 7, 45647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dawidczyk CM; Kim C; Park JH; Russell LM; Lee KH; Pomper MG; Searson PC State-of-the-Art in Design Rules for Drug Delivery Platforms: Lessons Learned From FDA-Approved Nanomedicines. J. Controlled Release 2014, 187, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ernsting MJ; Murakami M; Roy A; Li S-D Factors Controlling the Pharmacokinetics, Biodistribution and Intratumoral Penetration of Nanoparticles. J. Controlled Release 2013, 172, 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sakai-Kato K; Nishiyama N; Kozaki M; Nakanishi T; Matsuda Y; Hirano M; Hanada H; Hisada S; Onodera H; Harashima H; Matsumura Y; Kataoka K; Goda Y; Okuda H; Kawanishi T General Considerations Regarding the in Vitro and in Vivo Properties of Block Copolymer Micelle Products and Their Evaluation. J. Controlled Release 2015, 210, 76–83. [DOI] [PubMed] [Google Scholar]
- (17).Dehsorkhi A; Castelletto V; Hamley IW Self-Assembling Amphiphilic Peptides. J. Pept. Sci 2014, 20, 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Liu L; Xu K; Wang H; Jeremy Tan PK; Fan W; Venkatraman SS; Li L; Yang Y-Y Self-Assembled Cationic Peptide Nanoparticles as an Efficient Antimicrobial Agent. Nat. Nanotechnol 2009, 4, 457–463. [DOI] [PubMed] [Google Scholar]
- (19).Fields GB Induction of Protein-Like Molecular Architecture by Self-Assembly Processes. Bioorg. Med. Chem 1999, 7, 75–81. [DOI] [PubMed] [Google Scholar]
- (20).van Hell AJ; Costa CICA; Flesch FM; Sutter M; Jiskoot W; Crommelin DJA; Hennink WE; Mastrobattista E Self-Assembly of Recombinant Amphiphilic Oligopeptides Into Vesicles. Biomacromolecules 2007, 8, 2753–2761. [DOI] [PubMed] [Google Scholar]
- (21).Hartgerink JD; Beniash E; Stupp SI Peptide-Amphiphile Nanofibers: a Versatile Scaffold for the Preparation of Self-Assembling Materials. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 5133–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ariga K; Kikuchi J; Naito M; Koyama E; Yamada N Modulated Supramolecular Assemblies Composed of Tripeptide Derivatives: Formation of Micrometer-Scale Rods, Nanometer-Size Needles, and Regular Patterns with Molecular-Level Flatness From the Same Compound. Langmuir 2000, 16, 4929–4939. [Google Scholar]
- (23).Meng Q; Kou Y; Ma X; Liang Y; Guo L; Ni C; Liu K Tunable Self-Assembled Peptide Amphiphile Nanostructures. Langmuir 2012, 28, 5017–5022. [DOI] [PubMed] [Google Scholar]
- (24).Meng Q; Kou Y; Ma X; Guo L; Liu K Nanostructures From the Self-Assembly of A-Helical Peptide Amphiphiles. J. Pept. Sci 2014, 20, 223–228. [DOI] [PubMed] [Google Scholar]
- (25).Gore T; Dori Y; Talmon Y; Tirrell M; Bianco-Peled H Self-Assembly of Model Collagen Peptide Amphiphiles. Langmuir 2001, 17, 5352–5360. [Google Scholar]
- (26).Makovitzki A; Baram J; Shai Y Antimicrobial Lipopolypeptides Composed of Palmitoyl Di- and Tricationic Peptides: in Vitro and in Vivo Activities, Self-Assembly to Nanostructures, and a Plausible Mode of Action. Biochemistry 2008, 47, 10630–10636. [DOI] [PubMed] [Google Scholar]
- (27).Han S; Cao S; Wang Y; Wang J; Xia D; Xu H; Zhao X; Lu JR Self-Assembly of Short Peptide Amphiphiles: the Cooperative Effect of Hydrophobic Interaction and Hydrogen Bonding. Chem. - Eur. J 2011, 17, 13095–13102. [DOI] [PubMed] [Google Scholar]
- (28).Zhang CY; Chen Q; Wu WS; Guo XD; Cai CZ; Zhang LJ Colloids and Surfaces B: Biointerfaces. Colloids Surf., B 2016, 142, 55–64. [DOI] [PubMed] [Google Scholar]
- (29).Foster EJ; Berda EB; Meijer EW Metastable Supramolecular Polymer Nanoparticles via Intramolecular Collapse of Single Polymer Chains. J. Am. Chem. Soc 2009, 131, 6964–6966. [DOI] [PubMed] [Google Scholar]
- (30).Aggarwal P; Hall JB; McLeland CB; Dobrovolskaia MA; McNeil SE Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Adv. Drug Delivery Rev 2009, 61, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Aguiar J; Carpena P; Molina-Bolívar JA; Carnero Ruiz CC On the Determination of the Critical Micelle Concentration by the Pyrene 1:3 Ratio Method. J. Colloid Interface Sci 2003, 258, 116–122. [Google Scholar]
- (32).Ghosh A; Buettner CJ; Manos AA; Wallace AJ; Tweedle MF; Goldberger JE Probing Peptide Amphiphile Self-Assembly in Blood Serum. Biomacromolecules 2014, 15, 4488–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wakabayashi R; Abe Y; Kamiya N; Goto M The Self-Assembly and Secondary Structure of Peptide Amphiphiles Determine the Membrane Permeation Activity. RSC Adv. 2014, 4, 30654–30657. [Google Scholar]
- (34).Velichko YS; Stupp SI; de la Cruz MO Molecular Simulation Study of Peptide Amphiphile Self-Assembly. J. Phys. Chem. B 2008, 112, 2326–2334. [DOI] [PubMed] [Google Scholar]
- (35).Tovar JD; Claussen RC; Stupp SI Probing the Interior of Peptide Amphiphile Supramolecular Aggregates. J. Am. Chem. Soc 2005, 127, 7337–7345. [DOI] [PubMed] [Google Scholar]
- (36).Jiang H; Zhao X; Schanze KS Effects of Polymer Aggregation and Quencher Size on Amplified Fluorescence Quenching of Conjugated Polyelectrolytes. Langmuir 2007, 23, 9481–9486. [DOI] [PubMed] [Google Scholar]
- (37).Su JY; Hodges RS; Kay CM Effect of Chain Length on the Formation and Stability of Synthetic Alpha-Helical Coiled Coils. Biochemistry 1994, 33, 15501–15510. [DOI] [PubMed] [Google Scholar]
- (38).Cooper TM; Woody RW The Effect of Conformation on the CD of Interacting Helices: a Theoretical Study of Tropomyosin. Biopolymers 1990, 30, 657–676. [DOI] [PubMed] [Google Scholar]
- (39).Tatulian SA Structural Effects of Covalent Inhibition of Phospholipase A2 Suggest Allosteric Coupling Between Membrane Binding and Catalytic Sites. Biophys. J 2003, 84, 1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Malina A; Shai Y Conjugation of Fatty Acids with Different Lengths Modulates the Antibacterial and Antifungal Activity of a Cationic Biologically Inactive Peptide. Biochem. J 2005, 390, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ward BP; Ottaway NL; Perez-Tilve D; Ma D; Gelfanov VM; Tschöp MH; DiMarchi RD Peptide Lipidation Stabilizes Structure to Enhance Biological Function. Mol. Metab 2013, 2, 468–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Fröhlich E The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomed 2012, 7, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Lee O-S; Stupp SI; Schatz GC Atomistic Molecular Dynamics Simulations of Peptide Amphiphile Self-Assembly Into Cylindrical Nanofibers. J. Am. Chem. Soc 2011, 133, 3677–3683. [DOI] [PubMed] [Google Scholar]
- (44).Lee O-S; Cho V; Schatz GC Modeling the Self-Assembly of Peptide Amphiphiles Into Fibers Using Coarse-Grained Molecular Dynamics. Nano Lett. 2012, 12, 4907–4913. [DOI] [PubMed] [Google Scholar]
- (45).Fu IW; Nguyen HD Sequence-Dependent Structural Stability of Self-Assembled Cylindrical Nanofibers by Peptide Amphiphiles. Biomacromolecules 2015, 16, 2209–2219. [DOI] [PubMed] [Google Scholar]
- (46).Gao X; Tao W; Lu W; Zhang Q; Zhang Y; Jiang X; Fu S Lectin-Conjugated PEG-PLA Nanoparticles: Preparation and Brain Delivery After Intranasal Administration. Biomaterials 2006, 27, 3482–3490. [DOI] [PubMed] [Google Scholar]
- (47).Xu Q; Ensign LM; Boylan NJ; Schön A; Gong X; Yang J-C; Lamb NW; Cai S; Yu T; Freire E; Hanes J Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus Ex Vivo and Distribution in Vivo. ACS Nano 2015, 9, 9217–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Murgia X; Loretz B; Hartwig O; Hittinger M; Lehr C-M The Role of Mucus on Drug Transport and Its Potential to Affect Therapeutic Outcomes. Adv. Drug Delivery Rev 2018, 124, 82–97. [DOI] [PubMed] [Google Scholar]
- (49).Provencher SW CONTIN: a General Purpose Constrained Regularization Program for Inverting Noisy Linear Algebraic and Integral Equations. Comput. Phys. Commun 1982, 27, 229–242. [Google Scholar]
- (50).Berne BJ; Pecora R Dynamic Light Scattering: with Applications to Chemistry, Biology and Physics; 1st ed; Dover Publications Inc., 2000. [Google Scholar]
- (51).Lakowicz JR Principles of Fluorescence Spectroscopy; 3rd ed; Springer, 2007. [Google Scholar]
- (52).Sillen A; Engelborghs Y The Correct Use of “Average” Fluorescence Parameters. Photochem. Photobiol. 1998, 67, 475–486. [Google Scholar]
- (53).Kelly SM; Jess TJ; Price NC How to Study Proteins by Circular Dichroism. Biochim. Biophys. Acta, Proteins Proteomics 2005, 1751, 119–139. [DOI] [PubMed] [Google Scholar]
- (54).Chen YH; Yang JT; Chau KH Determination of the Helix and Beta Form of Proteins in Aqueous Solution by Circular Dichroism. Biochemistry 1974, 13, 3350–3359. [DOI] [PubMed] [Google Scholar]
- (55).de Jong DH; Singh G; Bennett WFD; Arnarez C; Wassenaar TA; Schäfer LV; Periole X; Tieleman DP; Marrink SJ Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput 2013, 9, 687–697. [DOI] [PubMed] [Google Scholar]
- (56).Pronk S; Páll S; Schulz R; Larsson P; Bjelkmar P; Apostolov R; Shirts MR; Smith JC; Kasson PM; van der Spoel D; Hess B; Lindahl E GROMACS 4.5: a High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Lee H; de Vries AH; Marrink S-J; Pastor RW A Coarse-Grained Model for Polyethylene Oxide and Polyethylene Glycol: Conformation and Hydrodynamics. J. Phys. Chem. B 2009, 113, 13186–13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O MDAnalysis: a Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem 2011, 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.