

Abstract
Objective:
APOA5 variants are strongly associated with hypertriglyceridemia (HTG) as well as increased risks of cardiovascular disease and acute pancreatitis. HTG in apoA-V dysfunction often aggravates by environmental factors such as high-carbohydrate diets or aging. To date, the molecular mechanisms by which these environmental factors induce HTG are poorly defined, leaving the high-risk HTG condition undertreated. Previously, we reported that LXR-SREBP-1c pathway regulates large VLDL production induced by LXR agonist. However, the pathophysiological relevance of the finding remains unknown.
Approach and Results:
Here, we reconstitute the environment-induced HTG phenotype of human APOA5 deficiency in Apoa5−/− mice and delineate the role of SREBP-1c in vivo by generating Apoa5−/−;Srebp-1c−/− mice. The Apoa5−/− mice, which showed moderate HTG on a chow diet, developed severe HTG upon high-carbohydrate feeding or aging as seen in human apoA-V deficient patients. These responses were nearly completely abolished in the Apoa5−/−;Srebp-1c−/− mice. Further mechanistic studies revealed that in response to these environmental factors, SREBP-1c was activated to increase TG synthesis and to permit the incorporation of TG into abnormally large VLDL particles, which require apoA-V for efficient clearance.
Conclusions:
Severe HTG develops only when genetic factors (apoA-V deficiency) and environmental effects (SREBP-1c activation) coexist. We demonstrate that the regulated production of large-sized VLDL particles via SREBP-1c determines plasma TG levels in apoA-V deficiency. Our findings explain the long-standing enigma of the late-onset HTG phenotype of apoA-V deficiency and suggest a new approach to treat HTG by targeting genes that mediate environmental effects.
Keywords: hypertriglyceridemia, apolipoprotein A-V, atherogenic dyslipidemia, VLDL, SREBP-1c
Graphical Abstract
Introduction
Hypertriglyceridemia (HTG) arises from the accumulation of TG-rich lipoproteins (TRLs) in the circulation 1. Triglycerides (TGs) of exogenous and endogenous origins are secreted from intestine and liver in the form of chylomicrons (CMs) and very low density lipoproteins (VLDLs), respectively. These lipoprotein-TGs are hydrolyzed by lipoprotein lipase (LPL) in the circulation to liberate free fatty acids as energy sources for peripheral tissues. Severe HTG increases the risk of acute pancreatitis, while moderate HTG increases the risk of cardiovascular diseases (CVD). Recent genome-wide association studies uncovered an association between plasma TG levels and CVD risk with the HTG pathway genes, such as apolipoprotein C-III (APOC3), angiopoietin-like 3 (ANGPTL3), angiopoietin-like 4 (ANGPTL4), asialoglycoprotein receptor 1 (ASGR1), LPL, and apolipoprotein A-V (APOA5) 2–4. In spite of these advances, effective therapeutic modalities to rescue HTG are limited, and a better understanding of plasma TG metabolism is warranted 5.
Severe HTG, defined as plasma TG levels greater than 1,000 mg/dl, can be classified into monogenic and polygenic types 5. Monogenic HTG (MIM 238600) is a rare (1 in 1,000,000) disorder with early-onset in infancy or childhood, and caused by monogenic defects in LPL pathway genes such as LPL, apolipoprotein C-II (APOC2), glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1), LPL chaperone lipase maturation factor 1 (LMF1), and APOA5. Polygenic HTG (MIM 144650) is a more common (1 in 600), late-onset disorder, which typically results from the accumulation of multiple genetic variants in LPL pathway genes. In either type, the severe HTG develops by the combination of genetic plus environmental/secondary factors (e.g., high-carbohydrate, lipid-rich diets, aging, obesity, diabetes mellitus, and excess alcohol intake) 6. Previous work has demonstrated that the accumulation of VLDL in response to these factors contributes to the development of severe HTG 7. In vivo kinetic studies have further implicated the contribution of the increased production of large-sized VLDL particles8,9. However, a molecular understanding of how environmental factors induce VLDL accumulation remains elusive in the sense that few studies have successfully identified a single gene that dominantly mediates the environmental effects of severe HTG.
APOA5 encodes apolipoprotein A-V (apoA-V), which is secreted from the liver and circulates on CMs, VLDLs and high-density lipoproteins (HDLs) 10,11. ApoA-V reduces plasma TG levels primarily by stimulating the LPL-mediated plasma TG clearance 12–15. ApoA-V binds both TRL and HSPG/GPIHBP1 that tethers LPL on endothelial surfaces, thereby facilitating the interaction between TRLs and LPL 12–14. ApoA-V has alternative functions to enhance the whole cell uptake of TRLs via its ligand activities for lipoprotein receptors 13,16,17, or to inhibit VLDL synthesis via its intracellular role to promote lipid droplet formation 18,19. The dominant role of apoA-V in regulating plasma TG levels in humans has been documented from the severe HTG phenotype in APOA5 deficient patients (MIM 606368) 20,21. As in severe HTG of other genetic causes 6, severe HTG in APOA5 deficiency often develops in response to environmental/secondary factors such as high-carbohydrate diets, lipid-rich diets, diabetes, and aging 20–23. Molecular understanding of the environment-induced HTG in apoA-V deficiency could lead to the development of new therapeutic options to treat severe HTG by intervening the gene-environment interactions.
Mice deficient in apoA-V (Apoa5−/−) has been described previously11. Apoa5−/− mice manifest only moderate HTG (~400 mg/dl) on a chow diet. In humans, APOA5 deficient patients manifest variable levels of plasma TG, and severe HTG (~6,000 mg/dl) often develops in response to environmental/secondary factors as discussed above20–23. Although this discrepancy has been commonly interpreted as a species difference, we rather reasoned that Apoa5−/− mice might serve as a useful model to identify the environmental/secondary factors that induce severe HTG and to explore its underlying mechanisms in vivo.
Here we thoroughly examined the environmental factors that induce severe HTG in human APOA5 deficient subjects in Apoa5−/− mice. We found that severe HTG was induced in Apoa5−/− mice either by (i) administration of T0901317, a synthetic pharmacological activator of liver X receptor (LXR); (ii) feeding with high-carbohydrate diets; or (iii) aging. These severe HTG responses were all prevented in Apoa5−/− mice that lacked SREBP-1c (Apoa5−/−;Srebp-1c−/−). Further study demonstrated that the activation of SREBP-1c is required to increase TG synthesis and to permit the incorporation of TG into abnormally large VLDL particles, which require apoA-V for efficient clearance. The apoA-V-SREBP-1c interplay expands our understanding of the gene-environment interaction in the pathogenesis of type V HLP, suggesting the potential therapeutic approach to treat severe HTG by targeting the SREBP-1c large-VLDL pathway that mediate the environmental effects.
Methods
The authors declare that all supporting data are available within the article (and its Online Data Supplement).
Animals
Apoa5−/− mice 11 were obtained from Mutant Mice Regional Resource Centers (Stock #011467-UCD, FVB.129X1(B6)-Apoa5tm1Hgc/Mmucd). Srebp-1c−/− mice 24 were obtained from the Jackson Laboratory. The Apoa5−/−;Srebp-1c−/− double knockout mice were generated by breeding Apoa5−/− mice with Srebp-1c−/− mice. C57BL/6J and Ldlr−/− mice (B6.129S7-Ldlrtm1Her/J) were obtained from the Jackson Laboratory. Vldlr−/− mice 25 and MX1 cre+;Lrp1fl/fl mice 26 have been described previously. Mice were used for the experiments with age and sex-matched controls on the same genetic background. Animals were housed in colony cages with a 12-h light/12-h dark cycle and fed a standard chow diet (Teklad Mouse/Rat Diet 7002 from Harlan Teklad Premier Laboratory Diets, Madison, WI; CLEA Rodent Diet CE-2, CLEA Japan). Male mice of 2-4 month old were used for experiments unless otherwise stated. Only male mice were used to avoid sex-related confounding effects. The T0901317 (J-Star Research, South Plainfield, NJ) treatment studies were carried out as described previously 27. For dietary studies, a rodent diet high in fructose (69 kcal% fructose, 10 kcal% fat, 20 kcal% protein; #D08040107, Research Diets, Inc.) was used. For olive-oil gavage studies, 16-hour fasted mice were given oral gavage of olive oil (300 μl/body), after the pretreatment with intraperitoneal injection of normal saline or Triton WR-1339 (500 mg/kg-body weight) 28. For all the animal studies, blood was drawn from the retro-orbital sinus with heparinized capillary tubes, and plasma was separated immediately for analysis. Animal experiments described in this manuscript have been approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee, and the Animal Care Committees of the University of Tokyo.
Biochemical Analyses
Plasma levels of cholesterol and triglycerides (TG) were measured enzymatically as described previously using kits 29. Plasma levels of insulin were assayed with the mouse insulin enzyme-linked immuno-sorbent assay (ELISA) kits (Ultra Sensitive Rat Insulin ELISA Kit from Crystal Chem Inc.; Morinaga, Tokyo, Japan) as described 29.
Fast Performance Liquid Chromatography
For fast performance liquid chromatography (FPLC) in Figure 1CD and Figure 5E, pooled plasma (1.7 ml) of each group of mice in Figure 1AB and Figure 5D, respectively, was subjected to ultracentrifugation at a density (d) of 1.215 g/ml. Plasma of all mice in the same group was pooled. The resulting lipoprotein fractions (d < 1.215) were adjusted to a final volume of 3 ml with a buffer containing 0.15 M sodium chloride, 0.01% (wt/vol) sodium EDTA, and 0.02% (wt/vol) sodium azide at pH 7.2, after which 2 ml was subjected to FPLC using a Superose 6B column. Fractions (2 ml) were collected, and 0.1 ml from each fraction was used to determine the content of total cholesterol and triglycerides 27,30.
Figure 1. Plasma lipids and lipoproteins in wild-type and Apoa5−/− mice fed a chow diet with or without LXR agonist (T0901317).

(A and B) Plasma lipid levels in wild-type (WT) and Apoa5−/− mice (2-5 months old, male) fed an ad libitum chow diet or a diet supplemented with 0.015% T0901317 (T). Aliquots of blood were obtained by retro-orbital bleeding at the indicated time after the start of the diet and used for the measurement of plasma triglycerides (A) and cholesterol (B) levels. Each value represents the mean ± SEM of data from 5-7 mice. (C and D) FPLC profiles of plasma lipoproteins from WT (C) and Apoa5−/− mice (D) fed a chow diet with (filled circles) or without (open circles) 0.015% T0901317 for 6 days. Plasma from 5-7 mice of each group was pooled and subjected to ultracentrifugation at d = 1.215 g/ml. The lipoprotein fractions (d < 1.215 g/ml) were subjected to gel filtration by FPLC, and the content of triglycerides in each fraction was measured as described in Methods. FPLC, fast performance liquid chromatography. (E-G) Electron microscopy of negatively stained plasma VLDL (d < 1.006 g/ml) from WT and Apoa5−/− mice fed a chow diet with or without 0.015% T0901317 (T) for 6 days. Plasma samples from 4 mice of each group were pooled, and VLDL was isolated for viewing by electron microscopy (E) as described in Methods. Magnification × 100,000. Size distribution of VLDL particles from wild-type (F) and Apoa5−/− (G) fed a chow diet with (filled bars) or without (open bars) 0.015% T0901317 for 6 days. The diameters of more than 300 VLDL particles from each group were measured, and the percentages of VLDL particles of different size were shown. Statistical significance was determined by two-way ANOVA with post hoc test. *P < 0.05, between genotypes. See also Figure I in the online-only Data Supplement.
Figure 5. Effects of olive oil gavage and aging on plasma lipids and lipoproteins in wild-type (WT), Apoa5−/−, and Apoa5−/−;Srebp-1c−/− mice.

(A) Wild-type and Apoa5−/− littermates (10-14 weeks old, male, n = 4-7) (upper) or Apoa5−/− and Apoa5−/−;Srebp-1c−/− littermates (13-14 weeks old, male, n = 4) (lower) were pretreated with saline (left) or Triton WR-1339 (500 mg/kg-body weight) (right), followed by gavage with olive oil (300 μl/body). Aliquots of blood were obtained by retro-orbital bleeding at the indicated time points and used to determine the plasma levels of triglycerides. Each value represents the mean ± SEM of data from 4 mice. (B and C), Apoa5−/− (8~17 weeks old (n = 25, male), 18~27 weeks old (n = 30, male), 28~ weeks old (n = 23, male)) were fed an ad libitum chow diet, and aliquots of blood were obtained by retro-orbital bleeding. Plasma levels of triglycerides (B) and cholesterol (C) were determined. Data are presented as the mean ± SEM. (D and E), Wild-type (WT) (n = 10, black), Apoa5−/− (n = 7, red), and Apoa5−/−;Srebp-1c−/− littermates (n = 7, blue) (20-30 weeks old, male) were fed an ad libitum chow diet, and aliquots of blood were obtained by retro-orbital bleeding. Plasma levels of triglycerides were determined (D). Data are presented as the mean ± SEM. Pooled plasma from each group was subjected to FPLC analysis, and the content of triglycerides in each fraction was measured (E) as described in Methods. FPLC, fast performance liquid chromatography. Statistical significance was determined by two-way ANOVA with post hoc test (for A), Kruskal-Wallis test with post hoc test (B), and one-way ANOVA with post hoc test (for C and D). *P < 0.05 versus wild-type mice (for A) or versus all other groups (for B-D). See also Figure I in the online-only Data Supplement.
High Performance Liquid Chromatography
The size distribution of plasma lipoproteins was analyzed by gel-permeation high-performance liquid chromatography (GP-HPLC) system as described previously 31 (LipoSEARCH®, Skylight Biotech, Inc., Akita, Japan). Briefly, plasma lipoproteins were separated with tandemly connected gel permeation columns, and TG levels in the column effluent were measured and expressed in mV. Lipoproteins were classified on the basis of their particle sizes by component peak analyses with the Gaussian curve fitting technique. The ranges of eluting positions for major lipoproteins are as follows: CM or large VLDL (>80 nm), 15~19.1 ± 1.0 min; VLDL (30~80 nm), 19.1~22.7 ± 1.0 min; LDL (16~30 nm), 22.7~25.1 ± 1.0 min; HDL (8~16 nm), 25.1~28.0 ± 1.0 min. The lipoprotein fraction of VLDL size (30~80 nm) was further classified into 3 subclasses on the basis of their particle sizes as shown in the inset of the figure.
Apolipoprotein analyses of plasma VLDL
Plasma VLDL (d < 1.006 g/ml) was isolated from the pooled plasma of each group of mice by ultracentrifugation. The protein concentrations in VLDL were measured as described previously 27. VLDLs (6 μg protein) from each group were delipidated with ethanol/ether (1:1), precipitated, and dissolved in SDS loading buffer. Equal amounts of VLDL protein (7.5 ng per lane) were subjected to 4-15% SDS gradient gels and immunoblot analyses using primary antibodies at a dilution of 1:1000 (anti-apoB and apoE antibody, #K23300R, Biodesign International, Saco, Maine, USA).
Electron Microscopy of Plasma VLDL
Plasma VLDL (d < 1.006 g/ml) was isolated from the pooled plasma of each group of mice by ultracentrifugation. VLDL fractions were negatively stained and visualized by electron microscopy. The diameters of VLDL particles were measured by ImageJ 1.39u software (NIH) as described previously 27.
Quantitative Real-Time PCR Analysis
Total RNA was prepared from mouse livers using a TRIzol reagent (Invitrogen Corp., Tokyo, Japan) and subjected to quantitative real-time PCR as previously described 24,29. The relative amounts of mRNAs were calculated using the comparative CT method. Mouse cyclophilin mRNA was used as the invariant control. Most of the primers for real-time PCR analysis were described previously 24,27. The primers used for the measurement of individual mRNAs are available on request.
Recombinant Adenoviruses
Recombinant adenoviruses that express β-galactosidase (lacZ) or APOA5 under the control of the cytomegalovirus promoter were constructed using the pAd/cytomegalovirus/V5-DEST Gateway system (Invitrogen Corp.) as described previously 32. The recombinant adenoviruses were expanded in HEK293 cells, purified by cesium chloride ultracentrifugation, and stored in 10% (v/v) glycerol in phosphate-buffered saline at −80 °C. A total of 0.5 × 1011 particles of lacZ virus (Ad-lacZ) or APOA5 virus (Ad-APOA5) were injected into the jugular vein of mice anesthetized with 60 mg/kg of sodium pentobarbital.
Hepatic VLDL-TG Secretion
Secretion rates of VLDL-TG in vivo were estimated by the intravenous administration of Triton WR-1339 as described previously 28. Briefly, mice were fasted overnight (16 hours) prior to intravenous injection of Triton WR-1339 (500 mg/kg-body weight). Plasma was obtained by retro-orbital bleeding at the indicated time points after injection. Plasma TG levels were measured as described above.
The Effect of Heparin on CM Clearance
Apoa5−/− mice (11-16 weeks old, male) were fasted for 16 hours, followed by gavage with olive oil (10 μl/g-body weight). Four hours after the gavage, mice were randomly assigned to a group injected with normal saline (100 μl/body) or a group injected with heparin (50 IU/100 μl/body). Aliquots of blood were obtained by retro-orbital bleeding at the indicated time points (0, 3, 15, 30, 120 min) and used to determine the plasma levels of triglycerides.
Measurement of Plasma Lipase Activities
Activities of Lipoprotein lipase (LPL) and hepatic lipase (HL) in post-heparin plasma were determined as described 33,34. Post-heparin plasma was collected after injection of heparin (50 IU/100 μl/body) via the jugular vein at the indicated time points 35. To eliminate the possible contamination of plasma VLDL-TG into the lipase assay reaction, plasma was ultracentrifuged at 48,000 rpm (204,782 g) for 16 hours to remove the VLDL fraction (top). An aliquot of the bottom fraction (equivalent to 5 μl of plasma) was then diluted in a final 100 μl normal saline solution and used as an enzymatic source in the lipase activity assay 34. LPL activity was calculated by subtracting the HL activity (assayed in the presence of 1M NaCl) from the total lipase activity (assayed in the absence of 1M NaCl) as described 34.
Statistical Analyses
All values are given as mean ± SEM. Statistical analyses were performed using PRISM 8 for Mac OS X (GraphPad Software, Inc., La Jolla, CA). Data were tested for normality and equal variance to confirm the appropriateness of parametric tests. Data that followed a normal distribution were analyzed by Student t test, Welch t test, one-way ANOVA with Tukey post hoc test, or two-way ANOVA with Tukey or Sidak post hoc tests, where appropriate. Data that failed normal distribution were analyzed by Mann-Whitney U test or by Kruskal-Wallis test with Dunn post hoc test. A P value of less than 0.05 was considered significant.
Results
LXR-agonist induce severe HTG accompanying the accumulation of large VLDL particles in Apoa5−/− Mice
We first characterized the lipid and lipoprotein profiles of Apoa5−/− mice under chow-fed conditions as well as in the pharmacological conditions where the production of large VLDL particles are stimulated by LXR agonist (T0901317). Production of large-sized VLDLs has been suggested as a prominent feature of environment-induced HTG8,9, and we and others have previously demonstrated that the activation of SREBP1a30, LXR36, or LXR-SREBP-1c pathway27, produces large-sized VLDL particles of chylomicron (CM) size (>80 nm), which massively accumulates in the plasma of Ldlr−/− mice. The large-sized VLDL particles produced by LXR agonist has been shown to be of hepatic origin by primary hepatocyte study27. Immunoblot analyses of apoB-48 and apoB-100 in VLDL proteins (Figure IA in the online-only Data Supplement) were compatible with the notion that LXR-induced large VLDL particles are mainly of hepatic origin, as the apoB-48 to apoB-100 ratio of VLDL particles were not increased, but decreased by T0901317 treatment.
On chow-fed condition, these mice exhibited a moderate increase in plasma TG (wild-type;chow, 201 ± 14 mg/dl; Apoa5−/−;chow, 479 ± 62 mg/dl, P < 0.01) (Figure 1A, chow) and a slight non-significant increase in plasma cholesterol (wild-type;chow, 129 ± 11 mg/dl vs. Apoa5−/−;chow, 154 ± 13 mg/dl) (Figure 1B, chow), which is consistent with the previous report 11. Elevated levels of plasma TGs are confined to VLDL fraction as revealed by fast performance liquid chromatography (FPLC) (Figure 1, C and D, chow). Notably, the median size of plasma VLDL particles (d < 1.006) in Apoa5−/− (39.2 nm) is slightly larger than in wild-type mice (36.5 nm) as determined by negative stain electron microscopy (Figure 1, E-G, chow), which is consistent with previous reports 16,37.
Treatment of mice with T0901317 resulted in a robust increase in the mRNA expression of SREBP-1c and its target genes in both wild-type and Apoa5−/− mice, which is consistent with previous reports 27(data not shown). When Apoa5−/− mice were treated with T0901317, they exhibited a massive increase in plasma TGs, reaching over 4,000 mg/dl (Figure 1A), accompanying a modest increase in plasma cholesterol levels (Figure 1B). In clear contrast, in the presence of apoA-V, wild-type mice treated with T0901317 exhibited an only modest increase in plasma TG (Figure 1A) and a mild increase in plasma cholesterol (Figure 1B). FPLC analysis revealed that the elevated TGs in Apoa5−/− were contained in VLDL particles (Figure 1D). T0901317 increased the size of VLDL particles in wild-type mice, where the median size of VLDL particles increased from 36.5 to 49.6 nm (Figure 1, E and F)27,36, which is more pronounced in Apoa5−/− (39.2 to 58.2 nm) (Figure 1, E and G).
The percentage of the large-sized VLDL particles of CM size (>80 nm) was increased in wild-type (1.4% to 12.8%) (Figure 1F), which is again more pronounced in Apoa5−/− mice (2.8% to 17.5%) (Figure 1G).
These results suggest that large-sized VLDL particles are removed from the plasma in the presence of apoA-V, but not efficiently in the absence of apoA-V.
The Large-Sized VLDL Particles Are Cleared by ApoA-V
To determine if apoA-V clears the large-sized VLDL particles from plasma, we overexpressed apoA-V in several models of large VLDL accumulation by using recombinant adenovirus carrying human APOA5 (Ad-APOA5). We first confirmed the TG-lowering effect of Ad-APOA5 in Apoa5−/− mice. Severe HTG was induced by 6 days of T0901317 treatment in Apoa5−/− mice (694 ± 122 mg/dl (chow) vs. 5540 ± 823 mg/dl (T0901317)), which was reversed 3 days after the injection of Ad-APOA5 but not after the control virus injection (Ad-lacZ, 4849 ± 977 mg/dl vs. Ad-APOA5, 485 ± 122 mg/dl, P < 0.05 by Welch t test). We next tested another model of large VLDL accumulation (Ldlr−/−;T0901317) 27. As shown in Figure 2, the overexpression of human APOA5 by recombinant adenovirus resulted in a rapid reduction of plasma TG levels (Figure 2A) accompanying a delayed reduction in plasma TC levels (Figure 2B) in Ldlr−/−;T0901317 mice. The slower reduction of the plasma levels of cholesterol than triglycerides suggests the role of apoA-V in TG hydrolysis. Measurement of the VLDL size revealed a reduction in the percentage of large VLDL particles (>80 nm) by APOA5 overexpression (Ldlr−/−;chow;Ad-lacZ, 0.2%; Ldlr−/−;T0901317;Ad-lacZ, 12.6%; Ldlr−/−;T0901317;Ad-APOA5, 2.4%) (Figure 2, C and D). Together with additional supportive evidence in Ldlr−/−;Vldlr−/− mice (Figure 2E) and in MX1-Cre+;Lrp1fl/fl;Ldlr−/− mice (Figure 2F), where LXR-agonist induced HTG was rescued by Ad-APOA5 with closely similar time courses as in Ldlr−/− mice (Figure 2, A and B), these results demonstrate that apoA-V enhances the clearance of large VLDL particles produced by LXR agonist.
Figure 2. Effects of APOA5 overexpression on plasma lipids and VLDL size in Ldlr−/−, Vldlr−/−;Ldlr−/−, and MX1-Cre+;Lrp1fl/fl;Ldlr−/− mice treated with T0901317.

(A and B) Male Ldlr−/− mice (12-20 weeks old) were fed with a chow diet (black, open circles), or a chow diet containing 0.015% T0901317 (T) (filled circles). Six days after the start of the T0901317 treatment, mice were randomly allocated to two groups and injected via the jugular vein with 0.5 × 1011 particles per mouse of recombinant adenoviruses encoding lacZ (Ad-lacZ, black, filled circles) or human APOA5 (Ad-APOA5, red, filled circles). Blood was obtained from retro-orbital plexus at the indicated time and used for measuring the plasma levels of triglycerides (A) and cholesterol (B). Each value represents the mean ± SEM of data from 4 mice. (C) Electron microscopy of negatively stained plasma VLDL (d < 1.006 g/ml). Plasma samples from 4 mice of each group were pooled, and VLDL was isolated for viewing by electron microscopy as described in Methods. Magnification x100,000. (D) The percentages of large VLDL particles (>80 nm). The diameters of more than 300 VLDL particles from each group were measured, and the percentages of large VLDL particles (>80 nm) were shown. (E and F) Male Vldlr−/−;Ldlr−/− mice (8-16 weeks old) (E), or male MX1-Cre+;Lrp1fl/fl;Ldlr−/− mice (12-20 weeks old) (F) were fed with a chow diet (black, open circles), or a chow diet containing 0.015% T0901317 (T) (filled circles). Six days after the start of the T0901317 treatment, mice were randomly allocated to two groups and injected via the jugular vein with 0.5 × 1011 particles per mouse of recombinant adenoviruses encoding lacZ (Ad-lacZ, black, filled circles) or human APOA5 (Ad-APOA5, red, filled circles). Blood was obtained from retro-orbital plexus at the indicated time and used for measuring the plasma levels of triglycerides (upper) and cholesterol (lower). Each value represents the mean ± SEM of data from 4 mice. Statistical significance was determined by Mann-Whitney U test. * P < 0.05, versus control virus.
Rescue of LXR Agonist-Induced HTG by SREBP-1c Deficiency
To define the molecular mechanisms underlying the LXR-agonist induced accumulation of large VLDL particles in Apoa5−/− mice, we asked if the deletion of SREBP-1c could rescue this response. We crossbred Srebp-1c−/− mice with Apoa5−/− mice to generate doubly mutant mice (Apoa5−/−;Srebp-1c−/−). We confirmed that the hepatic mRNA expression of SREBP-1c was abolished and those of SREBP-1c target genes were largely blunted in Apoa5−/−;Srebp-1c−/− mice (Table I in the online-only Data Supplement, chow). On a chow diet, the deletion of SREBP-1c almost completely normalized the plasma TG levels of Apoa5−/− mice to the levels of wild-type mice (wild-type;chow, 146 ± 17 mg/dl; Apoa5−/−;chow, 638 ± 173 mg/dl; Apoa5−/−;Srebp-1c−/−;chow, 144 ± 16 mg/dl) (Figure 3A). Strikingly, the LXR-agonist induced HTG in Apoa5−/− was nearly completely rescued in Apoa5−/−;Srebp-1c−/− mice (wild-type;T0901317, 158 ± 33 mg/dl; Apoa5−/−;T0901317, 2780 ± 836 mg/dl; Apoa5−/−;Srebp-1c−/−;T0901317, 200 ± 51 mg/dl) (Figure 3A). The size of plasma VLDL was increased by LXR agonist in the presence of SREBP-1c (wild-type and Apoa5−/−), but not in the absence of SREBP-1c (Apoa5−/−;Srebp-1c−/−) (Figure 3, B and C). The percentage of large VLDL particles (>80 nm) of chylomicron size was increased by LXR agonist in the presence of SREBP-1c (wild-type, 0.7% vs. 3.7% (chow vs. T0901317); Apoa5−/−, 2.8% vs. 19.2% (chow vs. T0901317)), but not in the absence of SREBP-1c (Apoa5−/−;Srebp-1c−/−, 2.0% vs. 0.4% (chow vs. T0901317)) (Figure 3, B and C). These results demonstrate the essential role of the SREBP-1c large-VLDL pathway in severe HTG induced by LXR agonist in apoA-V deficiency.
Figure 3. Effects of treatment with LXR agonist (T0901317) on plasma lipids and lipoproteins in wild-type (WT), Apoa5−/−, and Apoa5−/−;Srebp-1c−/− mice.

(A) Male mice with the indicated genotype were fed a chow diet with (filled symbols) or without (open symbols) 0.015% T0901317 (T). Aliquots of blood were obtained by retro-orbital bleeding at the indicated time (0, 2, or 6 days) after the start of the diet and used to determine the plasma levels of triglycerides. Each value represents the mean ± SEM of data from 4 mice. (B) Electron microscopy of negatively stained plasma VLDL (d < 1.006 g/ml). Plasma samples from 4 mice of each group were pooled, and VLDL was isolated for viewing by electron microscopy as described in Methods. Magnification x100,000. (C) Size distribution of VLDL particles from wild-type (WT) (black, top), Apoa5−/− (red, middle), and Apoa5−/−;Srebp-1c−/− (blue, bottom) fed a chow diet with (filled bars) or without (open bars) 0.015% T0901317 (T) for 6 days. The diameters of more than 300 VLDL particles from each group were measured, and the percentages of VLDL particles of different size were shown. Statistical significance was determined by two-way ANOVA with post hoc test. *P < 0.05 versus all other groups.
Carbohydrate-Induced HTG and Its Rescue by SREBP-1c Deficiency
The important unanswered question here is if the SREBP-1c large-VLDL pathway plays a dominant role in the development of severe HTG in pathophysiological conditions. To this end, we thoroughly tested for environmental factors that induce severe HTG in human APOA5 deficient patients 22,23 (Figures 4 and 5). Figure 4 shows an experiment comparing the responses of wild-type, Apoa5−/−, and Apoa5−/−;Srebp-1c−/− mice to high-fructose feeding over a 14 days period. When wild-type mice were fed a high-fructose diet (69 kcal% fructose), they showed a marginal increase in plasma TG as well as plasma cholesterol (Figure 4, A and B). In marked contrast, we discovered that Apoa5−/− mice treated with a high-fructose diet exhibited a massive increase in plasma TGs, reaching ~3,000 mg/dl at day 14, accompanying a mild increase in plasma cholesterol (Figure 4A, P < 0.001, two-way ANOVA; Figure 4B, P < 0.001, two-way ANOVA).
Figure 4. Effects of high-fructose feeding on plasma lipids and lipoproteins in wild-type, Apoa5−/−, and Apoa5−/−;Srebp-1c−/− mice.

(A-C) Wild-type (WT, black) and Apoa5−/− (red) littermates (9-13 months old, male) (A and B), or Apoa5−/− (red) and Apoa5−/−;Srebp-1c−/− (blue) littermates (9-17 months old, male) (C) were fed a chow diet (open circles) or a high-fructose diet containing 69 kcal% fructose (filled circles). Aliquots of blood were obtained by retro-orbital bleeding at the indicated time and used to determine the plasma levels of triglycerides and cholesterol. Each value represents the mean ± SEM of data from 4-5 mice. (D) Pooled plasma from each group of mice was subjected to gel-permeation high-performance liquid chromatography (GP-HPLC) analysis, and the content of triglycerides in each fraction was measured as described in Methods. Bold dashed lines and bold lines represent chow and high-fructose feeding groups, respectively, and thin lines represent ± SEM of data from 4-5 mice per each group. The inset shows the content of triglycerides in each fraction of lipoproteins: Apoa5−/− (SKO, red) and Apoa5−/−;Srebp-1c−/− (DKO, blue) fed a chow diet (open circles) or a high-fructose diet containing 69% fructose (filled circles). Component peak analyses with the Gaussian curve fitting technique were used to estimate the size of lipoproteins as described in Methods. Fraction #1 contains lipoproteins larger than 80 nm in diameter; #2, #3, #4 contains lipoproteins with diameters in the range of 30~80 nm. The fraction of lipoproteins contains larger particles in the numerical order (#1 (>80 nm) > #2 > #3 > #4). Data are presented as the mean ± SEM. Statistical significance was determined by two-way ANOVA with post hoc test (for A-C) and one-way ANOVA with post hoc test (for the inset of D). *P < 0.05 versus wild-type mice (for A and B) or versus all other groups (for C and D). See also Figure SI and Table I in the online-only Data Supplement.
Next, we tested the role of SREBP-1c in this fructose-induced HTG of apoA-V deficiency. Previous work demonstrated that high-fructose feeding induces hepatic lipogenesis partially via SREBP-1c 38–41. Consistently, fructose-feeding increased the mRNA levels of all fatty acid and triglyceride biosynthetic genes in Apoa5−/− mice, which were partially suppressed in Apoa5−/−;Srebp-1c−/− mice (Table I in the online-only Data Supplement). Consequently, the high-fructose feeding increased the TG content in livers of Apoa5−/− mice (7.2 ± 0.8 vs. 43.3 ± 6.4 mg/g-liver, chow vs. high-fructose, P < 0.001), which was again partially suppressed in Apoa5−/−;Srebp-1c−/− mice (4.1 ± 0.3 vs. 26.9 ± 1.9 mg/g-liver, chow vs. high-fructose, P < 0.001) (Table 1).
Table 1.
Effects of high-fructose feeding in wild-type, Apoa5−/−, and Apoa5−/−;Srebp-1c−/− mice.
| Apoa5−/− | Apoa5−/−;Srebp-1c−/− | |||
|---|---|---|---|---|
| Chow n=5 |
High fructose n=5 |
Chow n=4 |
High fructose n=4 |
|
| Body weight (g) | 29.9 ± 0.3 | 29.9 ± 1.2 | 28.4 ± 1.3 | 30.3 ± 1.6 |
| Food intake (g/day) | 4.6 ± 0.1 | 4.5 ± 0.1 | 4.8 ± 0.1 | 4.5 ± 0.2 |
| Liver weight/body weight (%) | 5.3 ± 0.1 | 6.5 ± 0.2 a | 5.2 ± 0.1 | 5.9 ± 0.1 a |
| Liver triglyceride content (mg/g) | 7.2 ± 0.8 | 43.3 ± 6.4 a | 4.1 ± 0.3 | 26.9 ± 1.9 a,b |
| Liver cholesterol content (mg/g) | 12.2 ± 0.5 | 14.8 ± 1.2 | 12.6 ± 0.6 | 17.5 ± 3.5 |
| Plasma glucose (mg/dl) | 162.2 ± 4.8 | 179.0 ± 20.2 | 169.5 ± 8.7 | 180.5 ± 21.6 |
Mice were fed an ad libitum chow diet or a high-fructose diet for 14 days. Each value represents the mean ± SEM of 4-5 values. One-way ANOVA with Tukey post hoc test was used to evaluate statistical significance
between dietary treatments within the same genotype (P < 0.05) and
between genotypes within the same dietary treatment (P < 0.05).
The deletion of SREBP-1c produced the more pronounced effect on the plasma TG levels: SREBP-1c deficiency nearly completely rescued the fructose-induced HTG in Apoa5−/− mice (Figure 4C, P < 0.0001, two-way ANOVA). The complete rescue of the HTG phenotype by SREBP-1c deficiency can be explained if SREBP-1c is essential for the production of large VLDL particles not only by LXR agonist treatment (Figure 3) 27 but also by high-fructose feeding. Indeed, lipoprotein analyses by HPLC methods demonstrated that the high-fructose feeding induced the accumulation of large VLDL particles (>80 nm) in Apoa5−/− mice, which is almost completely rescued in Apoa5−/−;Srebp-1c−/− mice (Figure 4D). Furthermore, the component peak analyses with the Gaussian curve fitting technique revealed that VLDLs of larger size were preferentially accumulated by high-fructose feeding in Apoa5−/− mice (Figure 4D, Inset), which was nearly completely abrogated in Apoa5−/−;Srebp-1c−/− mice.
Importantly, the rate of VLDL secretion was not different between Apoa5−/− and Apoa5−/−;Srebp-1c−/− mice on a chow diet (Figure IB in the online-only Data Supplement), suggesting that the basal rate of VLDL secretion is not controlled by SREBP-1c at least on a chow-fed condition.
In the experiments not shown, we tested the effect of high-glucose and high-sucrose feeding and found no increase in plasma TG levels in Apoa5−/−;Srebp-1c−/− mice by any of these high-carbohydrate diets (data not shown).
Collectively, these results reveal the essential role of the SREBP-1c large-VLDL pathway in severe HTG induced by various carbohydrates.
SREBP-1c Deficiency Does Not Affect CM Accumulation in Apoa5−/− Mice
Another diet that induces severe HTG in human APOA5 deficiency is a lipid-rich diet. As shown in Figure 5A (top, left), olive oil gavage in wild-type mice resulted in a transient increase in plasma TG levels that returned to baseline after 8 hours, reflecting the production of CM particles followed by rapid clearance. In marked contrast, olive-oil gavage in Apoa5−/− mice resulted in a massive and persistent increase in the plasma TG levels, reaching over 5,000 mg/dl after 8 hours (Figure 5A, top, left panel, P < 0.0001 by 2-way ANOVA). When these mice were pretreated with Triton WR-1339, an inhibitor of LPL-mediated TG lipolysis, olive oil gavage induced CM accumulation both in wild-type and Apoa5−/− mice at similar rates to similar levels, demonstrating the role of apoA-V in LPL-mediated CM-TG clearance (Figure 5A, top, right panel). Previous work suggested that apoA-V stimulates LPL pathway by enhancing the interaction between TRL and LPL via its dual binding affinity for TRL and GPIHBP1/HSPGs that tether LPL on endothelial cell surfaces 12–14. If this is the case, one would expect that heparin injection would rescue TRL accumulation in apoA-V deficiency by releasing LPL from endothelial cell surfaces into circulation and restoring the interaction between LPL and TRL. As expected, the administration of heparin to olive-oil fed Apoa5−/− mice rapidly cleared pre-accumulated CM-TG (Figure IC in the online-only Data Supplement, P < 0.0001, two-way ANOVA). Heparin injection also cleared pre-accumulated VLDL-TG in Apoa5−/− mice (Figure ID in the online-only Data Supplement). Importantly, heparin-releasable LPL activity was not decreased in Apoa5−/− compared to WT mice (Figure IE in the online-only Data Supplement). These results support the notion that apoA-V deficiency decreases the substrate (TRL) -enzyme (LPL) interaction but not the amount of enzyme (the heparin-releasable activity of LPL protein). We next tested the role of SREBP-1c in CM metabolism of Apoa5−/− mice. In marked contrast to the rescue of VLDL accumulation in Apoa5−/− by SREBP-1c deficiency (Figures 3 and 4), the rates of CM accumulation were not affected by SREBP-1c deletion either in the presence or absence of Triton WR-1339 (Figure 5A, lower panel), indicating that SREBP-1c deficiency affects neither clearance nor production of CMs after a fat load. These results suggest that SREBP-1c controls VLDL levels at least partially by regulating the size of VLDL particles, without directly affecting CM production or the CM/VLDL removal system.
Age-Related HTG in Apoa5−/− Mice and Its Rescue by the SREBP-1c Deletion
Lastly, we tested whether aging aggravates HTG in Apoa5−/− mice as is often observed in human APOA5 deficiency 22,23, and, if so, whether the deletion of Srebp-1c could rescue the response. As shown, the age-related severe HTG phenotype in human APOA5 deficiency (~ 6,000 mg/dl) was successfully reconstituted in Apoa5−/− mice (Figure 5B). In Apoa5−/− mice, aging severely increased the plasma TG levels (8~17 weeks old, 959 ± 118 mg/dl; 18~27 weeks old, 2019 ± 193 mg/dl; 28~ weeks old, 3362 ± 388 mg/dl, Spearman r = 0.6715, P < 0.0001) (Figure 5B) and moderately increased the plasma TC levels (8~17 weeks old, 231 ± 13 mg/dl; 18~27 weeks old, 328 ± 16 mg/dl; 28~ weeks old, 410 ± 29 mg/dl, Spearman r = 0.6059, P < 0.0001) (Figure 5C). The plasma insulin levels also increased along with age (Figure IF in the online-only Data Supplement) (8~17 weeks old, 0.89 ± 0.12 ng/ml; 18~27 weeks old, 2.63 ± 0.43 ng/ml; 28~ weeks old, 3.65 ± 0.82 ng/ml, Spearman r = 0.6806, P < 0.0001). Notably, the plasma levels of TG correlate well with those of insulin in Apoa5−/− mice (Figure IH in the online-only Data Supplement; Spearman r = 0.7121, P < 0.0001). Inasmuch as age-associated hyperinsulinemia (Figure IF in the online-only Data Supplement) activates SREBP-1c 42,43, the age-related HTG in apoA-V deficiency would be mediated by SREBP-1c. Indeed, the deletion of SREBP-1c nearly completely abolished the age-related accumulation of TG in VLDL fraction in apoA-V deficiency (WT, 162 ± 11 mg/dl, Apoa5−/−, 2225 ± 303 mg/dl, Apoa5−/−;Srebp-1c−/−, 286 ± 29 mg/dl, P < 0.0001) (Figure 5, D and E). We isolated plasma VLDL from each group of aged mice in Figure 5DE, and measured the size of VLDL particles. As in younger mice (Figure 1 and 3), plasma VLDL particles of Apoa5−/− mice (median size, 39.9 nm; percentage of VLDL > 80 nm, 4.0%) was larger than that of WT mice (36.5 nm and 1.6%, respectively), and this phenotype was abolished in Apoa5−/−;Srebp-1c−/− mice (30.3 nm and 0.4%, respectively), suggesting that the SREBP-1c large-VLDL pathway may at least in part play a role in the rescue of age-related HTG by SREBP-1c deficiency.
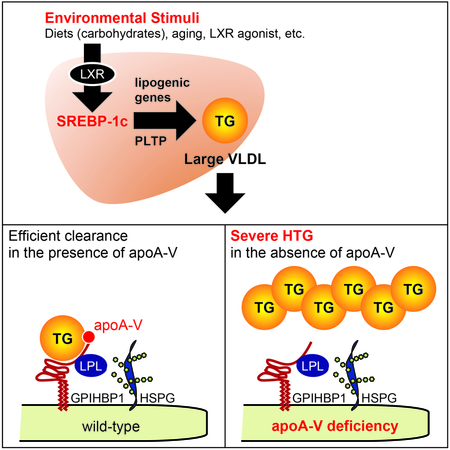
Together, these works clearly demonstrate the pivotal role of the SREBP-1c large-VLDL pathway in environment-induced severe HTG of apoA-V deficiency (Figure 6).
Figure 6. Schematic representation of the pivotal role of the SREBP-1c large-VLDL pathway in environment-induced HTG.

(A-C) ApoA-V is required for the clearance of large TRL particles. By biding both TRLs and GPIHBP1/HSPG that tethers LPL, apoA-V facilitates the interaction between TRLs and LPL and stimulates LPL-mediated TRL-TG clearance. Under a normal non-stressed situation, TRL particles are scarce, resulting in only moderate increases in plasma TGs in apoA-V deficiency (A). Under metabolic stresses that increase the production of TRLs (B, C), these particles accumulate in the absence of apoA-V. SREBP-1c is essential for the accumulation of large VLDLs (B), but not CMs (C), via its essential role in the production of large VLDL particles. LPL, lipoprotein lipase; GPIHBP1, glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1; HSPG, heparan sulfate proteoglycan.
Discussion
In this report, we identify the pivotal role of the SREBP-1c large-VLDL pathway in the environment-induced HTG using a disease mouse model. We found that SREBP-1c is required for the accumulation of VLDLs, but not CMs, via its specific role in large VLDL formation. Our main findings are summarized in Figure 6. Without environmental stress, VLDLs and CMs are scarce (Figure 6A). Under conditions where SREBP-1c is activated (LXR activation, high-carbohydrate diets, hyperinsulinemia, etc.), large VLDL particles are increasingly produced via SREBP-1c. As large TRL particles (large VLDL, CM) require apoA-V for efficient clearance (Figures 1–5 and Figure I in the online-only Data Supplement), these large VLDLs massively accumulate in Apoa5−/− (Figure 6B). Without SREBP-1c, large VLDL particles are not produced, resulting in clear rescue of severe HTG (Figure 6B). On the other hand, SREBP-1c does not affect CM metabolism (Figure 6C).
A number of genes have been identified as causative genes of severe HTG (LPL, APOC2, GPIHBP1, LMF1, and APOA5) 5. However, little is known about the genes that mediate environmental effects. The major drawback to investigate the gene-environment interactions has been the lack of suitable animal models. Although several different genetic mouse models of severe HTG have been described previously (e.g., Lpl−/−44, Lmf1 mutant mice 45, Gpihbp1−/−46, and Apoc2 hypomorphic mice 47), these mice manifest severe HTG without environmental stress 46,47 or even neonatal death due to severe HTG at birth 44,45,47. As a suitable experimental model to study gene-environment interactions of severe HTG, we focused on apoA-V deficient mice, which are unique among other mouse models in that they are neither lethal nor severely hypertriglyceridemic at a younger age 11. By using Apoa5−/− mice, we successfully reconstituted the severe type V HTG phenotype of human APOA5 deficiency in response to environmental stimuli, which enables us to delineate the underlying molecular mechanisms.
Severe HTG is still an orphan disease with limited therapeutic options 5. Newly developing approaches aim to target LPL pathway genes by inhibition (e.g., antisense or monoclonal antibody against APOC3 or ANGPTL4) or overexpression (e.g., adeno-associated virus (AAV) for LPL). However, these modalities are costly and still under development before wide and safe clinical use. Targeting apoA-V is promising: APOA5 variants are strongly associated with HTG as well as CAD risks 48,49 and multiple studies in mice have demonstrated the potent TG-lowering and athero-protective effects of apoA-V, albeit by overexpression methods (e.g., recombinant proteins, adenoviruses, AAV or transgenic animals) 18,50 (Figure 2). Our results offer a novel approach to target apoA-V pathway by inhibiting gene(s) that mediate the environmental effect (Figure 6). Current therapy for environment-induced HTG is only aimed at controlling environmental factors, which is often difficult to attain (strict fat/carbohydrate restriction, adequate control of diabetes mellitus) or impossible to manipulate (aging). Inhibition of SREBP-1c large-VLDL pathway provides a translatable approach to alleviate these environmental effects. Theoretically, reduction of VLDL by SREBP-1c inhibition may, in turn, decrease the level of CM, inasmuch as VLDL and CM compete for a common, saturable, plasma TG removal system of LPL pathway 6.
Our studies may have relevance not only to severe HTG but also to more common types of HTG such as postprandial or atherogenic dyslipidemia. Previous studies in humans have shown the association of APOA5 variants with postprandial HTG after a high-fat meal 51,52. The indispensable role of apoA-V in postprandial TG clearance is clearly demonstrated in our data (Figures 4, 5, and S1). Variants of APOA5 has also been shown to be associated with atherogenic dyslipidemia, which is characterized by a triad of HTG, low HDL-C, and a predominance of small, dense LDL 53,54. Our data suggest that the accumulation of large VLDLs in the face of apoA-V dysfunction (Figures 1–4) may increase small, dense LDLs, inasmuch as large VLDLs are the source of small, dense LDLs 9. The well-known association between metabolic syndrome and small, dense LDLs 55 may be explained by the activation of SREBP-1c in metabolic syndrome 42,43. Inhibition of SREBP-1c has therapeutic potential for these common, atherogenic type of dyslipidemia.
The current studies advance our understanding of the molecular pathophysiology of environment-induced severe HTG. In vivo kinetics studies in humans have indicated that the overproduction of large VLDL is a prominent feature of environment-induced severe HTG8,9. Molecular studies have identified several genes that control large VLDL production including LXR, SREBP-1a, and SREBP-1c27,30,36. However, whether these molecular pathways control HTG in physiological conditions has not been tested. Our data for the first time demonstrate that a single gene (SREBP-1c) plays an essential role in environment-induced severe HTG using a disease model of apoA-V deficiency.
Inhibitors of SREBP is currently under clinical development5. CAT-2003 and CAT-2054 are conjugates of eicosapentaenoic acid and niacin and inhibit the maturation of both SREBP-1 and SREBP-256. These compounds are now tested for its efficacy and safety for treating hypercholesterolemia as well as HTG. More specific inhibitors of SREBP-1c might be more effective in treating severe HTG, which deserves further study.
Our study may answer another long-standing enigma of apoA-V: how can an apolipoprotein with much lower plasma concentration than VLDL plays a dominant role in VLDL metabolism 57. The plasma concentration of apoA-V (~100-400 ng/ml) 58,59 is estimated as only 4% of that of total VLDL particles in plasma 57. This enigma can be solved, providing that apoA-V is required primarily for the clearance of large VLDL particles of chylomicron size (> 80 nm) (Figures 1–4). As large VLDL particles (> 80 nm) constitute only a proportion of total VLDL particles (estimated as < 3% of total VLDLs in our data) (Figures 1–3), the concentrations of apoA-V (~4% of total VLDLs) and large VLDLs (~3% of total VLDLs) are within the same range. As a molar basis, Merkel et al. estimated that about 1 apoA-V molecule is present in 24 VLDL particles. Given that large VLDLs constitute ~3% of total VLDLs (Figures 1–3), 1 apoA-V molecule is present in ~1 large VLDL particle.
As to the mode of action of apoA-V, our results are compatible with the notion that apoA-V enhances LPL action by facilitating the interaction between TGRL and LPL. However, our results do not preclude the possibility that apoA-V enhances whole particle uptake of TGRL13,16,17, or inhibits VLDL secretion via its intracellular role to promote lipid droplet formation 18,19.
Our study may have several limitations. First, the results presented were performed in male mice, and further validation in female mice is required to assess the possible effect of sex-related confounding factors that affect lipid metabolism. Second some studies used relatively small numbers of mice (n=4-5) per each group, although we repeated experiments to confirm the results. Third, the underlying molecular mechanisms on the clear rescue of age-related HTG by SREBP-1c deficiency may involve not only large VLDL formation but also other factors such as VLDL secretion rate, which deserves further study. Fourth, our data are compatible with the role of SREBP-1c in large VLDL formation but do not preclude the possibility that SREBP-1c may play a role in CM formation as well. Further study in tissue-specific mouse models is required to rule out this possibility. Fifth, although our data are consistent with the model whereby apoA-V clears large VLDL of chylomicron size, the direct proof by in vitro biochemical approaches requires further study.
In conclusion, our studies uncover a pivotal role of the SREBP-1c large-VLDL pathway in environment-induced severe HTG of type V HLP. The molecular dissection of gene (apoA-V)-environment (SREBP-1c-mediated large VLDL production) interactions not only expands our understanding of plasma TG homeostasis and HTG pathogenesis but also offers a brand-new approach to treat severe HTG by inhibiting genes that mediate environmental effects.
Supplementary Material
Highlights:
Environment-induced HTG in human APOA5 deficiency was successfully reconstituted in Apoa5−/− mice.
The deletion of SREBP-1c in Apoa5−/− mice nearly completely rescued severe HTG induced by carbohydrates or aging.
In response to environmental stimuli, SREBP-1c is activated to produce large VLDL particles, which are cleared from plasma in the presence of apoA-V but massively accumulate in apoA-V deficiency.
Targeting a single gene (SREBP-1c) that mediates environmental effects may be an effective approach to treat severe HTG.
Acknowledgments
We thank Dr. Alyssa H. Hasty for her critical reading of the manuscript; Drs. Michael S. Brown and Joseph L. Goldstein for continuous help and support.
Sources of Funding
This work was supported in part by NIH grant HL-20948 (G.L.); Banyu Life Science Foundation International (H.O.); Health, Labour and Welfare Sciences Research Grant for Research on Rare and Intractable Diseases (H.O.); and Japan Society for the Promotion of Science (JSPS) KAKENHI, Grant Numbers JP17K09858 (Grant-in-Aid for Scientific Research (C) to H.O.) and JP23890039 (Grant-in-Aid for Research Activity start-up to H.O.).
Nonstandard Abbreviations and Acronyms
- SREBP
sterol regulatory element-binding protein
- VLDL
very low density lipoprotein
- HDL
high-density lipoprotein
- apoA-V
apolipoprotein A-V
- HTG
hypertriglyceridemia
- TG
triglyceride
- TRL
triglyceride-rich lipoprotein
- CM
chylomicron
- LPL
lipoprotein lipase
- CVD
cardiovascular disease
- GPIHBP1
glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1
- LMF1
LPL chaperone lipase maturation factor 1
- LXR
liver X receptor
Footnotes
Disclosures
None.
References
- 1.Havel RJ, Kane JP. Chapter 114: Introduction: Structure and Metabolism of Plasma Lipoproteins In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson M, Mitchell G, eds. The Online Metabolic & Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001. [Google Scholar]
- 2.Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circulation Research. 2016;118:547–563. doi: 10.1161/CIRCRESAHA.115.306249. [DOI] [PubMed] [Google Scholar]
- 3.Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med. 2017;377:211–221. doi: 10.1056/NEJMoa1612790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. doi: 10.1038/nrg.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brahm AJ, Hegele RA. Chylomicronaemia--current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11:352–362. doi: 10.1038/nrendo.2015.26. [DOI] [PubMed] [Google Scholar]
- 6.Brunzell JD, Hazzard WR, Porte D, Bierman EL. Evidence for a common, saturable, triglyceride removal mechanism for chylomicrons and very low density lipoproteins in man. J Clin Invest. 1973;52:1578–1585. doi: 10.1172/JCI107334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kesaniemi YA, Grundy SM. Dual defect in metabolism of very-low-density lipoprotein triglycerides. Patients with type 5 hyperlipoproteinemia. JAMA. 1984;251:2542–2547. [PubMed] [Google Scholar]
- 8.Melish J, Le NA, Ginsberg H, Steinberg D, Brown WV. Dissociation of apoprotein B and triglyceride production in very-low-density lipoproteins. Am J Physiol. 1980;239:E354–362. doi: 10.1152/ajpendo.1980.239.5.E354. [DOI] [PubMed] [Google Scholar]
- 9.Adiels M, Olofsson S-O, Taskinen M-R, Borén J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192. [DOI] [PubMed] [Google Scholar]
- 10.van der Vliet HN, Sammels MG, Leegwater AC, Levels JH, Reitsma PH, Boers W, Chamuleau RA. Apolipoprotein A-V: a novel apolipoprotein associated with an early phase of liver regeneration. J Biol Chem. 2001;276:44512–44520. doi: 10.1074/jbc.M106888200. [DOI] [PubMed] [Google Scholar]
- 11.Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, Krauss RM, Rubin EM. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 12.Merkel M, Loeffler B, Kluger M, Fabig N, Geppert G, Pennacchio LA, Laatsch A, Heeren J. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. 2005;280:21553–21560. doi: 10.1074/jbc.M411412200. [DOI] [PubMed] [Google Scholar]
- 13.Lookene A, Beckstead JA, Nilsson S, Olivecrona G, Ryan RO. Apolipoprotein A-V-heparin interactions: implications for plasma lipoprotein metabolism. J Biol Chem. 2005;280:25383–25387. doi: 10.1074/jbc.M501589200. [DOI] [PubMed] [Google Scholar]
- 14.Gin P, Yin L, Davies BSJ, Weinstein MM, Ryan RO, Bensadoun A, Fong LG, Young SG, Beigneux AP. The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J Biol Chem. 2008;283:29554–29562. doi: 10.1074/jbc.M802579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shu X, Nelbach L, Weinstein MM, Burgess BL, Beckstead JA, Young SG, Ryan RO, Forte TM. Intravenous injection of apolipoprotein A-V reconstituted high-density lipoprotein decreases hypertriglyceridemia in apoav−/− mice and requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1. Arterioscler Thromb Vasc Biol. 2010;30:2504–2509. doi: 10.1161/ATVBAHA.110.210815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosskopf I, Baroukh N, Lee S-J, Kamari Y, Harats D, Rubin EM, Pennacchio LA, Cooper AD. Apolipoprotein A-V deficiency results in marked hypertriglyceridemia attributable to decreased lipolysis of triglyceride-rich lipoproteins and removal of their remnants. Arterioscler Thromb Vasc Biol. 2005;25:2573–2579. doi: 10.1161/01.ATV.0000186189.26141.12. [DOI] [PubMed] [Google Scholar]
- 17.Gonzales JC, Gordts PLSM, Foley EM, Esko JD. Apolipoproteins E and AV mediate lipoprotein clearance by hepatic proteoglycans. J Clin Invest. 2013;123:2742–2751. doi: 10.1172/JCI67398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forte TM, Sharma V, Ryan RO. Apolipoprotein A-V gene therapy for disease prevention / treatment: a critical analysis. J Biomed Res. 2016;30:88–93. doi: 10.7555/JBR.30.20150059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blade AM, Fabritius MA, Hou L, Weinberg RB, Shelness GS. Biogenesis of apolipoprotein A-V and its impact on VLDL triglyceride secretion. J Lipid Res. 2011;52:237–244. doi: 10.1194/jlr.M010793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Priore Oliva C, Pisciotta L, Li Volti G, Sambataro MP, Cantafora A, Bellocchio A, Catapano A, Tarugi P, Bertolini S, Calandra S. Inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2005;25:411–417. doi: 10.1161/01.ATV.0000153087.36428.dd. [DOI] [PubMed] [Google Scholar]
- 21.Marçais C, Verges B, Charrière S, Pruneta V, Merlin M, Billon S, Perrot L, Drai J, Sassolas A, Pennacchio LA, Fruchart-Najib J, Fruchart J-C, Durlach V, Moulin P. Apoa5 Q139X truncation predisposes to late-onset hyperchylomicronemia due to lipoprotein lipase impairment. J Clin Invest. 2005;115:2862–2869. doi: 10.1172/JCI24471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talmud PJ. Rare APOA5 mutations--clinical consequences, metabolic and functional effects: an ENID review. Atherosclerosis. 2007;194:287–292. doi: 10.1016/j.atherosclerosis.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 23.Albers K, Schlein C, Wenner K, Lohse P, Bartelt A, Heeren J, Santer R, Merkel M. Homozygosity for a partial deletion of apoprotein A-V signal peptide results in intracellular missorting of the protein and chylomicronemia in a breast-fed infant. Atherosclerosis. 2014;233:97–103. doi: 10.1016/j.atherosclerosis.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 24.Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem. 2002;277:9520–9528. doi: 10.1074/jbc.M111421200. [DOI] [PubMed] [Google Scholar]
- 25.Frykman PK, Brown MS, Yamamoto T, Goldstein JL, Herz J. Normal plasma lipoproteins and fertility in gene-targeted mice homozygous for a disruption in the gene encoding very low density lipoprotein receptor. Proc Natl Acad Sci USA. 1995;92:8453–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rohlmann A, Gotthardt M, Hammer RE, Herz J. Inducible inactivation of hepatic LRP gene by cre-mediated recombination confirms role of LRP in clearance of chylomicron remnants. J Clin Invest. 1998;101:689–695. doi: 10.1172/JCI1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okazaki H, Goldstein JL, Brown MS, Liang G. LXR-SREBP-1c-phospholipid transfer protein axis controls very low density lipoprotein (VLDL) particle size. J Biol Chem. 2010;285:6801–6810. doi: 10.1074/jbc.M109.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okazaki H, Tazoe F, Okazaki S, et al. Increased cholesterol biosynthesis and hypercholesterolemia in mice overexpressing squalene synthase in the liver. J Lipid Res. 2006;47:1950–1958. doi: 10.1194/jlr.M600224-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Takanashi M, Taira Y, Okazaki S, et al. Role of Hormone-sensitive Lipase in Leptin-Promoted Fat Loss and Glucose Lowering. J Atheroscler Thromb. 2017;24:1105–1116. doi: 10.5551/jat.39552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton JD, Shimano H, Hamilton RL, Brown MS, Goldstein JL. Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J Clin Invest. 1999;103:1067–1076. doi: 10.1172/JCI6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okazaki M, Usui S, Ishigami M, Sakai N, Nakamura T, Matsuzawa Y, Yamashita S. Identification of unique lipoprotein subclasses for visceral obesity by component analysis of cholesterol profile in high-performance liquid chromatography. Arterioscler Thromb Vasc Biol. 2005;25:578–584. doi: 10.1161/01.ATV.0000155017.60171.88. [DOI] [PubMed] [Google Scholar]
- 32.Okazaki H, Igarashi M, Nishi M, et al. Identification of neutral cholesterol ester hydrolase, a key enzyme removing cholesterol from macrophages. J Biol Chem. 2008;283:33357–33364. doi: 10.1074/jbc.M802686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nilsson-Ehle P, Schotz MC. A stable, radioactive substrate emulsion for assay of lipoprotein lipase. J Lipid Res. 1976;17:536–541. [PubMed] [Google Scholar]
- 34.Gotoda T, Yamada N, Kawamura M, Kozaki K, Mori N, Ishibashi S, Shimano H, Takaku F, Yazaki Y, Furuichi Y. Heterogeneous mutations in the human lipoprotein lipase gene in patients with familial lipoprotein lipase deficiency. J Clin Invest. 1991;88:1856–1864. doi: 10.1172/JCI115507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinstein MM, Yin L, Beigneux AP, Davies BSJ, Gin P, Estrada K, Melford K, Bishop JR, Esko JD, Dallinga-Thie GM, Fong LG, Bensadoun A, Young SG. Abnormal patterns of lipoprotein lipase release into the plasma in GPIHBP1-deficient mice. J Biol Chem. 2008;283:34511–34518. doi: 10.1074/jbc.M806067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grefhorst A, Elzinga BM, Voshol PJ, Plösch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, Kuipers F. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- 37.Nelbach L, Shu X, Konrad RJ, Ryan RO, Forte TM. Effect of apolipoprotein A-V on plasma triglyceride, lipoprotein size, and composition in genetically engineered mice. J Lipid Res. 2008;49:572–580. doi: 10.1194/jlr.M700281-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaka T, Shimano H, Yahagi N, et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes. 2004;53:560–569. [DOI] [PubMed] [Google Scholar]
- 39.Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K. Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. American Journal of Physiology-Endocrinology and Metabolism. 2010;299:E685–E694. doi: 10.1152/ajpendo.00283.2010. [DOI] [PubMed] [Google Scholar]
- 40.Haas JT, Miao J, Chanda D, Wang Y, Zhao E, Haas ME, Hirschey M, Vaitheesvaran B, Farese RV, Kurland IJ, Graham M, Crooke R, Foufelle F, Biddinger SB. Hepatic insulin signaling is required for obesity-dependent expression of SREBP-1c mRNA but not for feeding-dependent expression. Cell Metab. 2012;15:873–884. doi: 10.1016/j.cmet.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linden AG, Li S, Choi HY, Fang F, Fukasawa M, Uyeda K, Hammer RE, Horton JD, Engelking LJ, Liang G. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J Lipid Res. 2018;59:475–487. doi: 10.1194/jlr.M081836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferré P, Foretz M, Azzout-Marniche D, Bécard D, Foufelle F. Sterol-regulatory-element-binding protein 1c mediates insulin action on hepatic gene expression. Biochem Soc Trans. 2001;29:547–552. [DOI] [PubMed] [Google Scholar]
- 43.Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 2008;7:95–96. doi: 10.1016/j.cmet.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Weinstock PH, Bisgaier CL, Aalto-Setälä K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, Zechner R, Breslow JL. Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J Clin Invest. 1995;96:2555–2568. doi: 10.1172/JCI118319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Péterfy M, Ben-Zeev O, Mao HZ, Weissglas-Volkov D, Aouizerat BE, Pullinger CR, Frost PH, Kane JP, Malloy MJ, Reue K, Pajukanta P, Doolittle MH. Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat Genet. 2007;39:1483–1487. doi: 10.1038/ng.2007.24. [DOI] [PubMed] [Google Scholar]
- 46.Beigneux AP, Davies BSJ, Gin P, et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. doi: 10.1016/j.cmet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sakurai T, Sakurai A, Vaisman BL, Amar MJ, Liu C, Gordon SM, Drake SK, Pryor M, Sampson ML, Yang L, Freeman LA, Remaley AT. Creation of Apolipoprotein C-II (ApoC-II) Mutant Mice and Correction of Their Hypertriglyceridemia with an ApoC-II Mimetic Peptide. J Pharmacol Exp Ther. 2016;356:341–353. doi: 10.1124/jpet.115.229740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sarwar N, Sandhu MS, Ricketts SL, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–1639. doi: 10.1016/S0140-6736(10)60545-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Do R, Stitziel NO, Won H-H, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–106. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mansouri RM, Baugé E, Gervois P, Fruchart-Najib J, Fiévet C, Staels B, Fruchart J-CC. Atheroprotective effect of human apolipoprotein A5 in a mouse model of mixed dyslipidemia. Circ Res. 2008;103:450–453. doi: 10.1161/CIRCRESAHA.108.179861. [DOI] [PubMed] [Google Scholar]
- 51.Kim JY, Kim OY, Koh SJ, Jang Y, Yun S-S, Ordovas JM, Lee JH. Comparison of low-fat meal and high-fat meal on postprandial lipemic response in non-obese men according to the −1131T>C polymorphism of the apolipoprotein A5 (APOA5) gene (randomized cross-over design). J Am Coll Nutr. 2006;25:340–347. [DOI] [PubMed] [Google Scholar]
- 52.Moreno-Luna R, Perez-Jimenez F, Marin C, Perez-Martinez P, Gomez P, Jimenez-Gomez Y, Delgado-Lista J, Moreno JA, Tanaka T, Ordovas JM, Lopez-Miranda J. Two independent apolipoprotein A5 haplotypes modulate postprandial lipoprotein metabolism in a healthy Caucasian population. J Clin Endocrinol Metab. 2007;92:2280–2285. doi: 10.1210/jc.2006-1802. [DOI] [PubMed] [Google Scholar]
- 53.Austin MA, Talmud PJ, Farin FM, Nickerson DA, Edwards KL, Leonetti D, McNeely MJ, Viernes H-M, Humphries SE, Fujimoto WY. Association of apolipoprotein A5 variants with LDL particle size and triglyceride in Japanese Americans. Biochim Biophys Acta. 2004;1688:1–9. [DOI] [PubMed] [Google Scholar]
- 54.Mar R, Pajukanta P, Allayee H, Groenendijk M, Dallinga-Thie G, Krauss RM, Sinsheimer JS, Cantor RM, de Bruin TWA, Lusis AJ. Association of the APOLIPOPROTEIN A1/C3/A4/A5 gene cluster with triglyceride levels and LDL particle size in familial combined hyperlipidemia. Circ Res. 2004;94:993–999. doi: 10.1161/01.RES.0000124922.61830.F0. [DOI] [PubMed] [Google Scholar]
- 55.Reaven GM, Chen YD, Jeppesen J, Maheux P, Krauss RM. Insulin resistance and hyperinsulinemia in individuals with small, dense low density lipoprotein particles. J Clin Invest. 1993;92:141–146. doi: 10.1172/JCI116541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zimmer M, Bista P, Benson EL, et al. CAT-2003: A novel sterol regulatory element-binding protein inhibitor that reduces steatohepatitis, plasma lipids, and atherosclerosis in apolipoprotein E*3-Leiden mice. Hepatol Commun. 2017;1:311–325. doi: 10.1002/hep4.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Merkel M, Heeren J. Give me A5 for lipoprotein hydrolysis! J Clin Invest. 2005;115:2694–2696. doi: 10.1172/JCI26712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishihara M, Kujiraoka T, Iwasaki T, Nagano M, Takano M, Ishii J, Tsuji M, Ide H, Miller IP, Miller NE, Hattori H. A sandwich enzyme-linked immunosorbent assay for human plasma apolipoprotein A-V concentration. J Lipid Res. 2005;46:2015–2022. doi: 10.1194/jlr.D500018-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.O’Brien PJ, Alborn WE, Sloan JH, Ulmer M, Boodhoo A, Knierman MD, Schultze AE, Konrad RJ. The novel apolipoprotein A5 is present in human serum, is associated with VLDL, HDL, and chylomicrons, and circulates at very low concentrations compared with other apolipoproteins. Clin Chem. 2005;51:351–359. doi: 10.1373/clinchem.2004.040824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.