

Abstract
Diet, hormones, gene transcription, and posttranslational modifications control the hepatic metabolism of FAs; metabolic dysregulation causes chronic diseases, including cardiovascular disease, and warrants exploration into the mechanisms directing FA and triacylglycerol (TAG) synthesis and degradation. Long-chain FA metabolism begins by formation of an acyl-CoA by a member of the acyl-CoA synthetase (ACSL) family. Subsequently, TAG synthesis begins with acyl-CoA esterification to glycerol-3-phosphate by a member of the glycerol-3-phosphate acyltransferase (GPAT) family. Our studies of the isoforms ACSL1 and GPAT1 strongly suggest that these proteins are members of larger protein assemblies (interactomes). ACSL1 targeted to the ER interacts with peroxisomal, lipid droplet, and tethering proteins, uncovering a dynamic role for ACSL1 in organelle and lipid droplet interactions. On the outer mitochondrial membrane (OMM), PPARα upregulates ACSL1, which interacts with proteins believed to tether lipid droplets to the OMM. In contrast, GPAT1 is upregulated nutritionally by carbohydrate and insulin in a coordinated sequence of enzyme reactions, from saturated FA formation via de novo lipogenesis to FA esterification by GPAT1 and entry into the TAG biosynthesis pathway. We propose that involved enzymes form a dynamic protein interactome that facilitates esterification and that other lipid-metabolizing pathways will exist in similar physiologically regulated interactomes.
Keywords: acyl-coenzyme A, lipid droplets, de novo lipogenesis, beta oxidation
Graphical Abstract
The storage and degradation of long-chain FA underpin eukaryotic energy metabolism. The two extremes of dysregulated energy metabolism, obesity and cachexia, are linked to major noninfectious chronic disorders, including diabetes, cardiovascular disease, and cancer. Thus, for the past 60 years, scientists and clinicians have worked to understand FAs and triacylglycerol (TAG) in terms of their pathways of synthesis, regulation, and physiological effects in liver, skeletal muscle, heart, and adipose tissue.
Although we have long been aware that cellular compartments, including the cytosol, are packed tightly with proteins, textbook descriptions of biochemical pathways suggest that substrates and products of sequential enzyme steps wander around randomly before encountering the next enzymatic active site. Textbook drawings are similarly deceptive in showing sequential enzymes isolated from the myriad of other soluble and membrane-associated proteins and lipids. These misleading portrayals are particularly problematic for the initial steps in lipid metabolism, long-chain acyl-CoA synthetase (ACSL) and glycerol-3-phosphate acyltransferase (GPAT). As products of ACSL and as substrates for GPAT, long-chain acyl-CoAs are not uniformly available within the cytosol.
In theory, sequestration of acyl-CoAs should not be possible: acyl-CoAs are water-soluble and amphipathic, and should therefore be able to move freely within the cytosol and within membrane monolayers. However, genetic information, biochemical data, and studies of knockout mice challenge this idea.
The initiation of glycerolipid biosynthesis begins with ACSL-mediated thioesterification of FAs to produce long-chain acyl-CoAs. GPAT then esterifies these acyl-CoAs to form lysophosphatidic acid (LPA). Subsequent esterification steps and the action of phosphatidic acid (PA) phosphohydrolase result in the synthesis of TAG. The PA and diacylglycerol (DAG) intermediates in this pathway are also precursors of all the glycerophospholipids. Although this series of five biochemical steps was fully elucidated by 1960 (1, 2), we have since learned that each step in the pathway of TAG synthesis is catalyzed by at least two, and as many as 13 independent proteins, each encoded by a separate gene (Fig. 1) (3). Why are so many isoenzymes required to catalyze each step? Does each isoform have a different function or are the isoforms redundant? If each isoform has a different function, what mechanism ensures this? Differences in tissue expression may underlie some specific biochemical or metabolic phenotypes. The current data suggest an underlying complexity of lipid metabolism that strongly indicates that lipids are channeled within cells, both functionally and mechanistically.
Fig. 1.

Synthesis of glycerolipids from long-chain FAs. Members of the family of long-chain ACSLs (ACSL, ACSVL, ACSBg) thioesterify long-chain FAs to form acyl-CoAs. These may be esterified to the sn-1 position of glycerol-3-phosphate by one of four GPATs to form LPA. One of several 1-acylglycerol-3-phosphate acyltransferases (AGPATs; also known as LPA acyltransferases) uses an acyl-CoA to form PA. After one of three PA phosphohydrolases (PAPases/lipins) cleaves the phosphate, the remaining DAG product is esterified by one of two DGATs to form TAG. The TAG may remain in the cytosol within a lipid droplet or, in liver, be secreted as part of a VLDL. The LPA, PA, and DAG intermediates may initiate signaling cascades, and PA and DAG are also precursors of all the glycerophospholipids: phosphatidylinositol (PI), phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and cardiolipin (CL). Acyl-CoAs can also be converted to acyl-carnitines by carnitine palmitoyltransferase (CPT1) to enter the mitochondria for β-oxidation.
CHANNELING IN FA METABOLISM
Enzymes that control metabolic pathways are frequently regulated by multiple mechanisms. We propose that a complex network of interacting proteins constitutes an unexplored mechanism for FA regulatory channeling. Data from studies of GPAT and ACSL isoforms have led us to conclude that assemblies of interacting proteins must facilitate the channeling of FAs and acyl-CoAs into specific downstream pathways.
EVIDENCE FOR COMPARTMENTALIZATION OF ACYL-CoAs
Long-chain FAs must be converted to acyl-CoAs by one of 13 long-chain ACSL isoforms before they can enter most synthetic or degradative pathways (Fig. 1). In highly oxidative tissues, such as skeletal muscle, brown adipose, and heart, ACSL1 is the major isoform and is primarily located on the outer mitochondrial membrane (OMM) (4) where it interacts with CPT1 (5) and directs FAs toward mitochondrial β-oxidation (6–8). Mice lacking ACSL1 in skeletal muscle are able to run only half as far as controls despite having muscle content of long-chain acyl-CoA that is twice as high, indicating that acyl-CoAs synthesized by other ACSL isoforms are unavailable for β-oxidation. Similarly, in adipose tissue devoid of ACSL1, FA oxidation is markedly impaired while TAG synthesis remains unaffected (6). These studies strongly suggest that acyl-CoAs are compartmentalized within the cytosol.
ACSL1 INTERACTOMES
In adipose tissue, skeletal muscle, and heart, ACSL1 is primarily located on the OMM and channels FAs specifically into the mitochondria for β-oxidation (6–9). In liver, however, the ACSL1 located on the OMM appears to direct FAs toward β-oxidation, but the fate of the acyl-CoAs synthesized by the remaining 50% of ACSL1 protein on the ER remained unclear (10).
Differential FA partitioning is likely to require ACSL1 to interact with other specific proteins. In order to understand which protein interactions were unique to ACSL1 in its two locations, we used the unbiased protein interaction discovery technique, BioID (11). This method detects interacting proteins, including those that may have weak or transient interactions with the bait protein. As a fusion protein with the Escherichia coli biotin ligase, BirA*, ACSL1 was targeted to either the ER or the OMM of Hepa1-6 (mouse hepatoma) cells (12). Proteomic analysis identified 98 proteins that specifically interacted with ACSL1 at the ER, 55 at the OMM and 43 proteins common to both subcellular locations. Cohorts of peroxisomal and lipid droplet proteins, tethering proteins, and vesicle proteins uncovered a dynamic role for ACSL1 in organelle and lipid droplet interactions.
Using primary mouse hepatocytes from both male and female mice, we confirmed by coimmunoprecipitation that ACSL1 interacts with specific networks of proteins that enable its acyl-CoA product to be directed into the mitochondria for β-oxidation or into niche pathways at the ER related to ceramide and branched-chain FA metabolism (12). Proteins that interacted with ACSL1 targeted to the OMM included a group of proteins believed to tether lipid droplets to the OMM; these include SNAP23, Stx7, and VAMP2, -4, and -5 (12). These results confirmed that the intracellular location of ACSL1 allows it to interact with independent networks of proteins.
GLUCOSE ALTERS PROTEIN INTERACTIONS WITH ACSL1
Primary hepatocytes can be used to investigate weak or transient interactions because they respond well to physiological stimuli. Thus, specific interactions of ACSL1 with SNAP23 and VAMP4 were abrogated in the presence of 25 mM of glucose (12), suggesting that the link between ACSL1 and FAs released from lipid droplets depends on the cell’s nutrient status (Fig. 2). Supporting this interpretation, incubation in the absence of glucose enhanced FA oxidation, whereas incubation with glucose enhanced incorporation of FA into complex lipids (12). In contrast to the modulation of SNAP23 and VAMP4 interactions, the interaction of ACSL1 with the OMM protein, CPT1, did not change. These data show that the transient interactions are specific and relevant to the disposition of acyl-CoAs during fasting and feeding.
Fig. 2.

Altered ACSL1 interactome at the OMM in the absence or presence of glucose. Primary hepatocytes from male and female mice were incubated with or without 25 mM of glucose plus 1 mM of pyruvate for 16 h before immunoprecipitation of Ad-ACSL1-Flag (12). Carnitine palmitoyltransferase-1 (CPT) coimmunoprecipitated with ACSL1 under both conditions, but the OMM-lipid droplet tethering proteins, VAMP4 and SNAP23, coimmunoprecipitated only when glucose was absent.
EVIDENCE FOR COMPARTMENTALIZATION OF GPATs
Of the three liver GPAT isoenzymes, only GPAT1 is an integral member of the pathway that converts excess dietary carbohydrate into TAG, a principal source of diet-related hepatic steatosis (13). GPAT1 is a target of the insulin- and nutrient-activated transcription factors, SREBP-1c and ChREBP (14–19). In the presence of high dietary carbohydrate and insulin, these transcription factors upregulate the export of mitochondrial citrate (20) and the enzymes that use citrate for de novo lipogenesis (DNL) (Fig. 3) (16). Although both GPAT1 and -4 use exogenously derived palmitate, only GPAT1 initiates TAG synthesis from FA synthesized de novo from acetate (21), thereby linking nutrient excess to hepatic DNL and TAG synthesis (3). When GPAT1 is absent, acetate incorporation into TAG is almost totally blocked, and its incorporation into phospholipids is diminished by 60–80% (21). Because FAS releases free palmitate, a specific ACSL must activate this FA and direct it to GPAT1, which, unlike GPAT3/4, has a twofold preference for saturated FAs (3). Although none of the 13 ACSLs (22) is a known target of SREBP-1c or ChREBP, both ACSL3 and ACSL5 are upregulated by refeeding a high carbohydrate diet after a 24 h fast (unpublished observations), suggesting that these isoforms might link DNL FAs to TAG synthesis. Although the “handoff” of LPA to a specific AGPAT or to a further downstream DAG acyltransferase (DGAT) isoenzyme would seem reasonable, no AGPAT, PAPase/lipin, or DGAT isoforms are known to be upregulated by SREBP-1c.
Fig. 3.

Suggested interaction of enzymes involved in DNL and TAG synthesis via GPAT1. The citrate carrier (CiC) transports citrate out of the mitochondria and into the cytosol where it is cleaved by ATP citrate lyase to form oxaloacetate (OAA) and acetyl-CoA. Malate dehydrogenase (DH) and malic enzyme convert the OAA successively to malate and pyruvate together with the production of NADPH. Acetyl-CoA carboxylase (ACC) converts the acetyl-CoA to malonyl-CoA, which is subsequently converted primarily to palmitate by FAS. After activation to palmitoyl-CoA by an ACSL, GPAT1 esterifies it to glycerol-3-phosphate to form LPA. Subsequent steps described in Fig. 1 convert the LPA to TAG or glycerophospholipids (PL) in the ER. Some of these steps may occur on structures variously termed mitochondria-associated membranes (MAM) and mitochondria-associated vesicles (MAV) (48).
In the absence of GPAT1, newly synthesized FAs are oxidized in the mitochondria, demonstrating the importance of the DNL-GPAT1 liaison in avoiding a futile cycle by preventing the immediate degradation of newly synthesized FAs (Fig. 1) (23). Because each of the GPAT isoforms uses the same substrates, the most likely mechanism by which long-chain acyl-CoAs can be channeled into specific downstream pathways is via close interactions between the pathway enzymes to traffic newly synthesized FAs into TAG synthesis. Thus, GPAT1 and at least some of the enzymes indicated in yellow and green (Fig. 3) must interact, even though their locations are in separate cellular areas (cytosol, OMM, and ER) (3). Furthermore, because TAG synthesis occurs primarily at the ER (24), it also follows that additional downstream enzymes may be integrated with this pathway. Thus, excess oxidation of DNL FAs in the absence of GPAT1 protects against diet-induced insulin resistance (25), decreases hepatic steatosis and VLDL secretion in chow-fed and high fat-fed mice (26), and reverses preexisting hepatic steatosis in ob/ob mice (27).
ADDITIONAL FEATURES OF THE GPAT ISOFORMS SUGGEST INDEPENDENT FUNCTIONS
During fasting, when insulin is low, GPAT1 mRNA, protein, and activity decrease, so that FAs that enter the liver from lipolyzed adipose TAG are either oxidized or esterified, presumably by GPAT3 and -4. Despite its importance in initiating TAG synthesis, little is known about the acute regulation of GPAT1 or other enzymes in the TAG synthetic pathway. AMP-activated kinase inhibits (28) and casein kinase-II stimulates (29) hepatic GPAT1 activity, but no functional consequences are known. Does inhibiting or inducing lipolysis with insulin, glucose deprivation, or AMPK activation alter the ability of GPAT1 to use de novo synthesized FAs, cause impaired insulin signaling, or modify the binding of specific proteins to GPAT1?
Supporting the idea that the different GPATs are not functionally equivalent and that cytosolic FA pools do not mix is the fact that markedly different phenotypes are observed in Gpat1-, Gpat3-, and Gpat4-null mice (21, 23, 26, 30–33). Each of these studies was carried out in whole-body knockout models, so information from isolated hepatocytes may be more relevant than the liver phenotype itself. Although GPAT3 and -4 have been less well studied, GPAT4 may contribute primarily to phospholipid synthesis (34, 35).
Differences are also observed in hepatic signaling via mTORC2; both GPAT1 and GPAT4, but not GPAT3, initiate the synthesis of DAG and PA, the intermediates that inhibit mTORC2 phosphorylation of Akt and impair insulin signaling (36, 37). Differential signaling is consistent with the presence of compartmentalized lipid intermediates.
CHANNELING AND DYNAMICS OF PATHWAY INTEGRATION AND FLUX
To reconcile the experimental data, we propose that the ACSLs and GPATs are part of compartmentalized pathways that are organized by multi-enzyme assemblies; the metabolites would be channeled to enzymes in a sequential manner without equilibrating with the cytosolic aqueous phase or nearby membrane monolayers. Channeling involves what Paul Srere defined as a metabolon, a supramolecular complex of sequential metabolic enzymes and cellular structural elements (38, 39). In addition to intracellular organelles, the cytosol also appears to be structurally organized, as observed in centrifuged Neurospora and Euglena in which cell contents were layered with a final top “cytosolic” layer, surprisingly devoid of macromolecules (40, 41).
Substrates might be channeled within metabolons by movement along protein surfaces, by tunneling within associated proteins (42), or by probabilistic channeling within a large group of clustered proteins (Fig. 4). The clustered proteins might interact directly or via scaffolding to regional structural or membrane proteins. It has been proposed that surface movements could occur via electrostatic interactions with the substrate (43), which would be possible with acyl-CoAs, but perhaps not with their downstream glycerolipid intermediates. More likely is the idea of clustered scaffolded proteins like glycogen granules that contain metabolic enzymes and regulatory kinases and phosphatases (44).
Fig. 4.

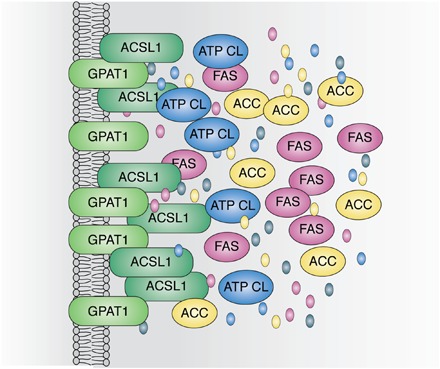
Potential organization of a protein assembly that links enzymes of DNL and GPAT1. ATP citrate lyase (ATP CL), acetyl-coA carboxylase (ACC), FAS, and ACSL ultimately produce the palmitoyl-CoA that GPAT1 esterifies to glycerol-3-phosphate. Although thought of as cytosolic, the enzymes of DNL may interact with membrane and organellar proteins, and both GPAT1 and its associated ACSL are membrane-bound proteins. Interactions at specific sites could help to organize the metabolon. The substrates and products, oxaloacetate, acetyl-CoA, malonyl-CoA, and palmitate, are depicted as colored ovals integrated within the hypothesized protein assembly.
LIPID CHANNELING
The textbook concept of enzyme pathways underlies traditional analyses of enzyme kinetics. Thus, isolated proteins are evaluated for substrate affinity, substrate preference, and Vmax. This is particularly problematic with enzymes that metabolize lipids. In such cases, Kornberg’s “Commandment IV”, “Do not waste clean thinking on dirty enzymes,” (Ref. 45; p. 3614) must give way to his subsequent thought that for some analyses, the marked dilution of proteins in solution must be restored to the more normal crowded molecular state (“Commandment VII”) (45). Molecular dilution is even more of a problem for membrane-associated enzymes in which substrates are likely to be highly concentrated within the membrane mono- or bilayer. For example, the acyl-CoA “concentration” within a cell is a meaningless number unless one considers the amount near the GPAT1 active site at the membrane-cytosol interface. Even cations and molecules such as Na, K, ATP, amino acids, and glucose are probably not distributed uniformly within the cytosol (38).
BENEFITS ARISE FROM CHANNELING OR COMPARTMENTALIZATION OF ENZYME PATHWAYS
As multi-enzyme assemblies, pathway efficiency should be enhanced because substrates and intermediates are not diluted into the bulk phase, but instead, remain near potential subsequent proteins where they can interact productively with active sites. This process allows for better regulation of the steady state flux and ensures associations that can enhance the stability of intermediates and avoid interference by other cellular constituents. The interactome can increase reaction rates by increasing local substrate concentrations and by restricting intermediates from entering competing reactions. Moreover, this concept does not preclude “leakiness” that permits substrates to enter branch-point pathways.
We propose that protein interactomes constitute novel and unexplored regulatory mechanisms that facilitate FA and acyl-CoA channeling and metabolism. The interactions of multi-enzyme assemblies might be direct via surface binding or via structural proteins that form a scaffold for multiple members of the pathway interactome. These interactions might be transient, as observed with purinosomes that form in the cytosol to enhance purine synthesis (46) or with the insulin signaling pathway that forms and disassembles depending on the interaction of insulin with its receptor (47). In addition to the described interactomes for ACSL1 and GPAT1, we predict that other pathways that metabolize lipids will prove to exist in similar physiologically regulated protein interactomes.
Acknowledgments
The author is grateful to Dr. Florencia Pascual for her insightful comments.
Footnotes
Abbreviations:
- ACSL
- acyl-CoA synthetase
- DAG
- diacylglycerol
- DGAT
- diacylglycerol acyltransferase
- DNL
- de novo lipogenesis
- GPAT
- glycerol-3-phosphate acyltransferase
- LPA
- lysophosphatidic acid
- OMM
- outer mitochondrial membrane
- PA
- phosphatidic acid
- TAG
- triacylglycerol
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK56598 and DK59935. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health. The author declares no conflicts of interest.
REFERENCES
- 1.Kennedy E. P. 1961. Biosynthesis of complex lipids. Fed. Proc. 20: 934–940. [PubMed] [Google Scholar]
- 2.Kornberg A., and Pricer W. E. Jr.. 1953. Enzymatic synthesis of the conenzyme A derivatives of long-chain fatty acids. J. Biol. Chem. 204: 329–343. [PubMed] [Google Scholar]
- 3.Coleman R. A., and Mashek D. G.. 2011. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev. 111: 6359–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper D. E., Young P. A., Klett E. L., and Coleman R. A.. 2015. Physiological consequences of compartmentalized acyl-CoA metabolism. J. Biol. Chem. 290: 20023–20031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee K., Kerner J., and Hoppel C. L.. 2011. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 286: 25655–25662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellis J. M., Li L. O., Wu P. C., Koves T. R., Ilkayeva O., Stevens R. D., Watkins S. M., Muoio D. M., and Coleman R. A.. 2010. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab. 12: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis J. M., Mentock S. M., Depetrillo M. A., Koves T. R., Sen S., Watkins S. M., Muoio D. M., Cline G. W., Taegtmeyer H., Shulman G. I., et al. 2011. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol. Cell. Biol. 31: 1252–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L. O., Grevengoed T. J., Paul D. S., Ilkayeva O., Koves T. R., Pascual F., Newgard C. B., Muoio D. M., and Coleman R. A.. 2015. Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes. 64: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grevengoed T. J., Cooper D. E., Young P. A., Ellis J. M., and Coleman R. A.. 2015. Loss of long-chain acyl-CoA synthetase isoform 1 impairs cardiac autophagy and mitochondrial structure through mechanistic target of rapamycin complex 1 activation. FASEB J. 29: 4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L. O., Ellis J. M., Paich H. A., Wang S., Gong N., Altshuller G., Thresher R. J., Koves T. R., Watkins S. M., Muoio D. M., et al. 2009. Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. J. Biol. Chem. 284: 27816–27826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roux K. J., Kim D. I., Raida M., and Burke B.. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Young P. A., Senkal C. E., Suchanek A. L., Grevengoed T. J., Lin D. D., Zhao L., Crunk A. E., Klett E. L., Fullekrug J., Obeid L. M., et al. 2018. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem. 293: 16724–16740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lambert J. E., Ramos-Roman M. A., Browning J. D., and Parks E. J.. 2014. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 146: 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimano H. 2001. Sterol regulatory element-binding proteins (SREBPs): transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 40: 439–452. [DOI] [PubMed] [Google Scholar]
- 15.Shimano H. 2009. SREBPs: physiology and pathophysiology of the SREBP family. FEBS J. 276: 616–621. [DOI] [PubMed] [Google Scholar]
- 16.Horton J. D., Goldstein J. L., and Brown M. S.. 2002. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109: 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linden A. G., Li S., Choi H. Y., Fang F., Fukasawa M., Uyeda K., Hammer R. E., Horton J. D., Engelking L. J., and Liang G.. 2018. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J. Lipid Res. 59: 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ericsson J., Jackson S. M., Kim J. B., Spiegelman B. M., and Edwards P. A.. 1997. Identification of glycerol-3-phosphate acyltransferase as an adipocyte determination and differentiation factor 1-and sterol regulatory element-binding protein-responsive gene. J. Biol. Chem. 272: 7298–7305. [DOI] [PubMed] [Google Scholar]
- 19.Huo M., Zang H. L., Zhang D. J., Wang B., Wu J., Zhang X. Y., Chen L. H., Li J., Yang J. C., and Guan Y. F.. 2009. Role of increased activity of carbohydrate response element binding protein in excessive lipid accumulation in the liver of type 2 diabetic db/db mouse. [Article in Chinese] Beijing Da Xue Xue Bao Yi Xue Ban. 41: 307–312. [PubMed] [Google Scholar]
- 20.Giudetti A. M., Stanca E., Siculella L., Gnoni G. V., and Damiano F.. 2016. Nutritional and hormonal regulation of citrate and carnitine/acylcarnitine transporters: two mitochondrial carriers Involved in fatty acid metabolism. Int. J. Mol. Sci. 17: E817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wendel A. A., Cooper D. E., Ilkayeva O. R., Muoio D. M., and Coleman R. A.. 2013. Glycerol-3-phosphate acyltransferase (GPAT)-1, but not GPAT4, incorporates newly synthesized fatty acids into triacylglycerol and diminishes fatty acid oxidation. J. Biol. Chem. 288: 27299–27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watkins P. A., Maiguel D., Jia Z., and Pevsner J.. 2007. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 48: 2736–2750. [DOI] [PubMed] [Google Scholar]
- 23.Hammond L. E., Neschen S., Romanelli A. J., Cline G. W., Ilkayeva O. R., Shulman G. I., Muoio D. M., and Coleman R. A.. 2005. Mitochondrial glycerol-3-phosphate acyltransferase-1 is essential in liver for the metabolism of excess acyl-CoAs. J. Biol. Chem. 280: 25629–25636. [DOI] [PubMed] [Google Scholar]
- 24.Poppelreuther M., Sander S., Minden F., Dietz M. S., Exner T., Du C., Zhang I., Ehehalt F., Knuppel L., Domschke S., et al. 2018. The metabolic capacity of lipid droplet localized acyl-CoA synthetase 3 is not sufficient to support local triglyceride synthesis independent of the endoplasmic reticulum in A431 cells. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids. 1863: 614–624. [DOI] [PubMed] [Google Scholar]
- 25.Neschen S., Morino K., Hammond L. E., Zhang D., Liu Z. X., Romanelli A. J., Cline G. W., Pongratz R. L., Zhang X. M., Choi C. S., et al. 2005. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knock out mice. Cell Metab. 2: 55–65. [DOI] [PubMed] [Google Scholar]
- 26.Hammond L. E., Gallagher P. A., Wang S., Hiller S., Kluckman K. D., Posey-Marcos E. L., Maeda N., and Coleman R. A.. 2002. Mitochondrial glycerol-3-phosphate acyltransferase-deficient mice have reduced weight and liver triacylglycerol content and altered glycerolipid fatty acid composition. Mol. Cell. Biol. 22: 8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendel A. A., Li L. O., Li Y., Cline G. W., Shulman G. I., and Coleman R. A.. 2010. Glycerol-3-phosphate acyltransferase 1 deficiency in ob/ob mice diminishes hepatic steatosis but does not protect against insulin resistance or obesity. Diabetes. 59: 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muoio D. M., Seefeld K., Witters L. A., and Coleman R. A.. 1999. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 338: 783–791. [PMC free article] [PubMed] [Google Scholar]
- 29.Onorato T. M., Chakraborty S., and Haldar D.. 2005. Phosphorylation of rat liver mitochondrial glycerol-3-phosphate acyltransferase by casein kinase 2. J. Biol. Chem. 280: 19527–19534. [DOI] [PubMed] [Google Scholar]
- 30.Vergnes L., Beigneux A. P., Davis R. G., Watkins S. M., Young S. G., and Reue K.. 2006. Agpat6 deficiency causes subdermal lipodystrophy and resistance to obesity. J. Lipid Res. 47: 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beigneux A. P., Vergnes L., Qiao X., Quatela S., Davis R., Watkins S. M., Coleman R. A., Walzem R. L., Philips M., Reue K., et al. 2006. Agpat6–a novel lipid biosynthetic gene required for triacylglycerol production in mammary epithelium. J. Lipid Res. 47: 734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao J., Li J. L., Li D., Tobin J. F., and Gimeno R. E.. 2006. Molecular identification of microsomal acyl-CoA:glycerol-3-phosphate acyltransferase, a key enzyme in de novo triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA. 103: 19695–19700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper D. E., Grevengoed T. J., Klett E. L., and Coleman R. A.. 2015. Glycerol-3-phosphate acyltransferase isoform-4 (GPAT4) limits oxidation of exogenous fatty acids in brown adipocytes. J. Biol. Chem. 290: 15112–15120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagle C. A., Vergnes L., Dejong H., Wang S., Lewin T. M., Reue K., and Coleman R. A.. 2008. Identification of a novel sn-glycerol-3-phosphate acyltransferase isoform, GPAT4, as the enzyme deficient in Agpat6−/− mice. J. Lipid Res. 49: 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y. Q., Kuo M. S., Li S., Bui H. H., Peake D. A., Sanders P. E., Thibodeaux S. J., Chu S., Qian Y. W., Zhao Y., et al. 2008. AGPAT6 is a novel microsomal glycerol-3-phosphate acyltransferase (GPAT). J. Biol. Chem. 283: 10048–10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C., Cooper D. E., Grevengoed T. J., Li L. O., Klett E. L., Eaton J. M., Harris T. E., and Coleman R. A.. 2014. Glycerol-3-phosphate acyltransferase-4-deficient mice are protected from diet-induced insulin resistance by the enhanced association of mTOR and rictor. Am. J. Physiol. Endocrinol. Metab. 307: E305–E315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C., Wendel A. A., Keogh M. R., Harris T. E., Chen J., and Coleman R. A.. 2012. Glycerolipid signals alter mTOR complex 2 (mTORC2) to diminish insulin signaling. Proc. Natl. Acad. Sci. USA. 109: 1667–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srere P. A. 1987. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 56: 89–124. [DOI] [PubMed] [Google Scholar]
- 39.Ovádi J., and Srere P. A.. 2000. Macromolecular compartmentation and channeling. Int. Rev. Cytol. 192: 255–280. [DOI] [PubMed] [Google Scholar]
- 40.Kempner E. S., and Miller J. H.. 1968. The molecular biology of Euglena gracilis. IV. Cellular stratification by centrifuging. Exp. Cell Res. 51: 141–149. [DOI] [PubMed] [Google Scholar]
- 41.Zalokar M. 1960. Cytochemistry of centrifuged hyphae of Neurospora. Exp. Cell Res. 19: 114–132. [DOI] [PubMed] [Google Scholar]
- 42.Hyde C. C., Ahmed S. A., Padlan E. A., Miles E. W., and Davies D. R.. 1988. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 263: 17857–17871. [PubMed] [Google Scholar]
- 43.Sweetlove L. J., and Fernie A. R.. 2018. The role of dynamic enzyme assemblies and substrate channelling in metabolic regulation. Nat. Commun. 9: 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roach P. J., Depaoli-Roach A. A., Hurley T. D., and Tagliabracci V. S.. 2012. Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441: 763–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kornberg A. 2000. Ten commandments: lessons from the enzymology of DNA replication. J. Bacteriol. 182: 3613–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pedley A. M., and Benkovic S. J.. 2017. A new view into the regulation of purine metabolism: the purinosome. Trends Biochem. Sci. 42: 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo S. 2014. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J. Endocrinol. 220: T1–T23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pellon-Maison M., Montanaro M. A., Coleman R. A., and Gonzalez-Baro M. R.. 2007. Mitochondrial glycerol-3-P acyltransferase 1 is most active in outer mitochondrial membrane but not in mitochondrial associated vesicles (MAV). Biochim. Biophys. Acta. 1771: 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]