

Abstract
Sphingosine phosphate lyase (SPL) is the final enzyme in the sphingolipid degradative pathway, catalyzing the irreversible cleavage of long-chain base phosphates (LCBPs) to yield a long-chain aldehyde and ethanolamine phosphate (EP). SPL guards the sole exit point of sphingolipid metabolism. Its inactivation causes product depletion and accumulation of upstream sphingolipid intermediates. The main substrate of the reaction, sphingosine-1-phosphate (S1P), is a bioactive lipid that controls immune-cell trafficking, angiogenesis, cell transformation, and other fundamental processes. The products of the SPL reaction contribute to phospholipid biosynthesis and programmed cell-death activation. The main features of SPL enzyme activity were first described in detail by Stoffel et al. in 1969. The first SPL-encoding gene was cloned from budding yeast in 1997. Reverse and forward genetic strategies led to the rapid identification of other genes in the pathway and their homologs in other species. Genetic manipulation of SPL-encoding genes in model organisms has revealed the contribution of sphingolipid metabolism to development, physiology, and host-pathogen interactions. In 2017, recessive mutations in the human SPL gene SGPL1 were identified as the cause of a novel inborn error of metabolism associated with nephrosis, endocrine defects, immunodeficiency, acanthosis, and neurological problems. We refer to this condition as SPL insufficiency syndrome (SPLIS). Here, we share our perspective on the 50-year history of SPL from discovery to disease, focusing on insights provided by model organisms regarding the pathophysiology of SPLIS and how SPLIS raises the possibility of a hidden role for sphingolipids in other disease conditions.
Keywords: sphingolipids, sphingosine-1-phosphate, SGPL1, sphingosine phosphate lyase insufficiency syndrome
Graphical Abstract
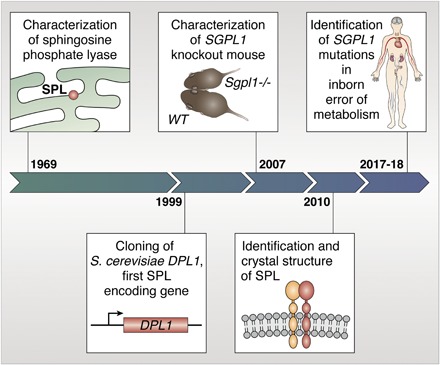
SPHINGOSINE PHOSPHATE LYASE GUARDS THE EXIT OF THE SPHINGOLIPID DEGRADATIVE PATHWAY
Sphingolipids represent a diverse and ubiquitous class of lipids discovered in 1884 by Thudichum, who named them for the functional enigma they posed (1). By the 1960s, sphingolipids were recognized as normal constituents of the plasma membrane, myelin sheath, and plasma and known to accumulate in patients with sphingolipidoses, storage disorders caused by inborn errors of sphingolipid metabolism.
Sphingolipid structure is centered on a long-chain base (LCB) backbone. In mammalian cells, the most prevalent LCB is sphingosine. The LCB anchor can be modified by the addition of polar headgroups at the C1 position and acylation of the free amino group, generating a vast array of sphingolipid species. In 1969–1970, Stoffel and colleagues published a series of reports mapping the final metabolic fate of sphingolipids in the yeast Hansenula ciferrii (2, 3). Using radioactive precursors, they showed that all sphingolipids are ultimately degraded to LCBs. Degradation of LCBs involved an initial phosphorylation (suggested by an ATP requirement). We now know that this step is catalyzed by sphingosine kinases and yields LCB phosphates (LCBPs), including the prolific bioactive lipid, sphingosine-1-phosphate (S1P), which ligates a family of G protein-coupled receptors [S1P receptors (S1PRs)] that play key roles in development, physiology, and immunology. In the final metabolic step, LCBPs are cleaved, yielding ethanolamine phosphate (EP) and a corresponding long-chain aldehyde. Both products are further metabolized to carbon dioxide or reutilized in the synthesis of phospholipids (2). In particular, the aldehyde product is reutilized in the formation of plasmalogens (2).
SPHINGOSINE PHOSPHATE LYASE IS A PYRIDOXAL 5′-PHOSPHATE-DEPENDENT ENZYME
Stoffel et al. demonstrated that the enzyme responsible for the final cleavage reaction was enriched in the endoplasmic reticulum (ER) (2). Its topology in the ER membrane with its catalytic site facing the cytosol was later described (4). The enzyme was recently crystallized and shown to function as a homodimer (5). Stoffel et al. named the enzyme “dihydrosphingosine-1-phosphate aldolase (sphinganine-1-phosphate alkanal-lyase)” (2). Based on the similarity of the reaction to that catalyzed by pyridoxal 5′-phosphate (PLP)-dependent threonine and allothreonine aldolases, he hypothesized and proved that the lyase reaction requires PLP. Stoffel et al. also showed that the cleavage of LCBPs was an irreversible step (2). Thus, sphingosine phosphate lyase (SPL) guards the sole exit point of sphingolipid metabolism.
CLONING OF DPL1 PROVIDES INSIGHT AND A GENETIC SCREEN TO IDENTIFY SPL-ENCODING GENES
Evolutionary conservation of sphingolipids allowed the cloning of the first SPL gene, DPL1, from Saccharomyces cerevisiae using a genetic screen for sphingosine resistance (6). Disruption of DPL1 conferred profound sensitivity to LCBs, creating a facile screen to identify other SPL genes and isolate suppressors (Fig. 1). The identification and genetic manipulation of SPL-encoding genes that followed revealed many biological functions of SPL, which appears to be encoded by a single gene in every genome in which it has been identified.
Fig. 1.

Effects of 1 mM d-erythro-sphingosine on BST1/DPL1 overexpression and bst1/dpl1Δ yeast strains. Dilutions across are 0, 1:2, 1:4, 1:40, 1:400, and 1:4,000. Lanes 1–6 represent six different bst1/dpl1Δ strains. Lane 7 represents the BST1/DPL1 overexpression strain JS14. Lane 8 represents WT SGP3. Reproduced with permission from ref. 6.
In addition to exhibiting a profound sensitivity to LCBs and calcium, dpl1Δ accumulated LCBPs and was resistant to heat shock and nutrient deprivation, suggesting a protective role for LCBPs (7, 8). However, when combined with mutations in S1P phosphatase—identified as a high-copy dpl1Δ suppressor (9)—dpl1Δ was lethal, accumulating massive amounts of LCBPs and exhibiting disrupted calcium homeostasis (8, 10). Thus, inhibition of SPL and LCBP accumulation may have positive consequences. However, there seems to be a sweet spot beyond which toxicity occurs. The latter could be due to toxic effects of very high levels of LCBPs or upstream sphingolipid intermediates. Alternatively, lethality could be due to an indirect effect on global sphingolipid homeostatic regulatory networks. Sphingolipid biosynthesis is tightly controlled by ORM proteins, which inhibit the rate-limiting sphingolipid biosynthetic enzyme serine palmitoyltransferase (SPT) (11). Complete inhibition of sphingolipid degradation could result in feedback signals that prevent de novo biosynthesis of essential sphingolipids needed for membrane stability and organization. The recognition of SPL inhibition as a double-edged sword reappears as a consistent theme from yeast to man.
INVERTEBRATE MODELS REVEAL SPL TO BE ESSENTIAL FOR LIFE
In Caenorhabditis elegans, silencing the SPL gene spl-1 results in semilethality, delayed development, and reproductive defects. Worms lacking SPL are sluggish, starved, and pump food poorly, characteristics of aging in nematodes and consistent with the identification of Spl-1 in a C. elegans screen for genes that prevent accelerated aging (12).
The Sply gene encodes fruit fly SPL (13). Sply-null larvae exhibit semilethality; the rare larvae that survive to adulthood exhibit reproductive defects and are flightless due to abnormal flight muscle patterning and neuromuscular junction defects (14). Interestingly, Sply mutant phenotypes are attenuated when sphingolipid biosynthesis is suppressed (13). This suggests that Sply mutants represent a sphingolipidosis that can be ameliorated by substrate depletion strategies.
Sgpl1 REPORTER AND KO MICE ILLUSTRATE SPL’S ROLES IN MAMMALIAN PHYSIOLOGY
Sgpl1 LacZ reporter mice reveal the ubiquitous expression of mammalian SPL (15) (Fig. 2). Specific patterns, such as rich expression in the thymic reticular network, gut epithelium, sebaceous glands, corticomedullary region and pelvis of the kidney, discrete olfactory bulb ganglia, spinal cord gray matter, and cranial nerves, intimate myriad undiscovered roles for SPL in mammalian physiology.
Fig. 2.

Whole-mount survey of β-Gal-stained adult SPL reporter mouse organs. A: Whole brain. B: Olfactory bulb, showing staining of specific olfactory ganglia. C: Forebrain. D: Midbrain. E: Cerebellum and brainstem. F: Hindbrain. G: Basicranium, showing positively stained trigeminal nerve ganglia and unstained pituitary. H: Spinal cord, showing gray matter staining. I: Thymus and heart. Thymus is intensely stained. The pin is within the ventricular wall of the bisected heart, and the view shows the interior of the ventricle. J: Kidney, showing strong staining in the renal pelvis. K: Bladder, showing epithelial staining. L: Female gonads, including ovaries, fallopian tubes, and uterus. M: Foot. N: Skin. O: Preputial gland. P: Ribcage. Q: Liver. R: Stomach. S: Jejunum, with a Peyer’s patch in the center and showing less staining than the surrounding jejunal tissue. T: Ileum. U: Brown adipose tissue. V: Harderian gland and eye. W: Adrenal gland. X: Tail. Data are representative of results obtained from four animals (two male and two female). Reproduced with permission from ref. 15.
Global Sgpl1 disruption causes runting and early death (16) (Fig. 3). Sgpl1 KO (SPL KO) mice accumulate sphingolipids, triglycerides, and cholesteryl esters (17). They exhibit lymphopenia, thymic atrophy, neutrophilia, anemia, nephrosis (excessive protein loss in the urine), increased cortical bone thickness, and rare developmental phenotypes often involving vascular structures (16–20). [SPL is a target of the platelet-derived growth factor pathway, which controls vascular integrity, skeletal patterning, and glomeruli development (16).]
Fig. 3.

Gene trap disruption of the Sgpl1 gene causes S1P lyase deficiency. A: Sphingolipid metabolic pathway and linkage to triacylglycerol and PtdE synthesis. B: Appearance and life span of Sgpl1−/−, Sgpl1+/−, and Sgpl1+/+ mice. Photo shows knockout mouse on the left and wild type mouse on the right. Reproduced with permission from ref. 17.
Conditional SPL KO mice live longer and have, thus, allowed deeper investigation of SPL’s functions. S1P-mediated activation of S1PR1 on mature lymphocytes mediates their egress from the thymus (21). An S1P chemotactic gradient between tissues where S1P is low and blood where it is high is needed to enable T-cell egress, exemplified by the gradient disruption and lymphopenia in SPL KO mice (22). Dendritic cell-specific SPL KO mice recapitulate the lymphopenia of global KO mice. This reveals the importance of SPL metabolic activity within dendritic cells (a tiny population of resident thymic cells located at the site of egress) in generating the S1P gradient (23). SPL is influenced by stress conditions, raising the possibility that, as dendritic cells patrol the body in search of danger-associated antigens, modulation of their SPL activity could regulate lymphocyte egress in response to environmental conditions.
Neural-specific SPL KO mice accumulate LCBs/LCBPs in the brain and exhibit deficits of spatial learning, memory, and motor function (24). These defects are accompanied by presynaptic pathology and reduced synaptic plasticity associated with a calcium-dependent induction of the unfolded protein response. Consistent with these findings, S1P is known to exhibit neurotoxic activity (25). However, S1P is also needed for synaptic transmission (26). Tight regulation of sphingolipid metabolism in precise tissue compartments may be critical to support healthy neuronal function.
SPL produces EP, which can be incorporated into phosphatidylethanolamine (PtdE). PtdE plays a key role in autophagy, which neurons use to clear protein aggregates. Cultured SPL KO neurons exhibit a block in autophagic flux that is alleviated by exogenous PtdE (10, 27). Thus, neuronal SPL may facilitate autophagic clearance of misfolded proteins and protein aggregates associated with aging and age-related neurodegeneration.
SPL is also essential for kidney, skin, and platelet functions, as was shown by the nephrosis, podocyte effacement, skin irritation, and increased platelet activation in an inducible SPL KO mouse (28).
DISCOVERY OF SPLIS, A NEW INBORN ERROR OF SPHINGOLIPID METABOLISM
In 2017–2018, a novel human syndrome associated with inactivating SGPL1 mutations was discovered by diagnostic genome sequencing (14, 29–33). We refer to this condition as SPL insufficiency syndrome (SPLIS). The most severely affected suffered from hydrops fetalis and/or perinatal death. Others presented with congenital nephrotic syndrome, adrenal insufficiency, and/or neurological symptoms (developmental delay or regression, ataxia, cranial nerve defects, seizures, and peripheral neuropathy) (Fig. 4). Some exhibited hypothyroidism, ichthiosis, immunodeficiency, cranial nerve palsies, bony abnormalities, hypocalcemia, gonadal defects, and other birth defects. Another case of primary adrenal insufficiency caused by SGPL1 mutations was recently reported (34). We are also aware of five additional unreported cases of SPLIS (unpublished observations).
Fig. 4.

A: Homozygosity mapping and whole-exon sequencing reveal SGPL1 mutations as causing a human syndrome with nephrosis, ichthyosis, adrenal insufficiency, or neurologic defects and immunodeficiency. Some features of SPLIS include the following. B: Ptosis. C: Skin image showing brownish black desquamation on sebostatic skin with multipleradial papules with a blueish/black erythema and central calcinosis. D, E: Median and ulnar nerve paralysis. F: H&E-stained epidermal section showing acanthosis/orthokeratotic hyperkeratosis (black arrowhead) and calcinosis (white arrowhead). G: Renal histology (silver staining) showing focal segmental glomerulosclerosis. Scale bars: 100 μm. Reproduced with permission from ref. 29.
SPLIS mutations were varied and included nonsense mutations, splicing defects, and missense mutations. All were associated with low to undetectable SPL abundance and activity. Mutations were distributed throughout the polypeptide sequence. Cellular findings suggested that many SPLIS missense mutations induce protein instability and rapid clearance due to misfolding, a common defect of inborn errors of metabolism (35).
SPLIS patient blood and fibroblast cultures accumulated sphingolipids (14, 29–33). S1P and sphingosine were elevated, whereas ceramides and other sphingolipid levels were variably affected. Changes in the level and composition of ceramides may be relevant to SPLIS skin manifestations, because specific ceramide species contribute to the integument’s barrier function (36). The ratio of sphingosine to dihydrosphingosine is increased in SPLIS plasma, suggesting that de novo biosynthesis is inhibited (14). Cholesterol and triglyceride levels were also elevated in some SPLIS patients, which could be related to nephrotic syndrome or a direct impact of SPL deficiency on global lipid metabolism.
Phenotypes observed in murine and invertebrate SPL KO models had signaled the devastating consequences of SPL insufficiency in man. The fetal and perinatal deaths of the most severely affected SPLIS patients and the pleiotropic disease features provided a stark and unequivocal confirmation of the essential role SPL plays in human metabolism.
MECHANISM OF SPLIS PATHOLOGIES
Silencing Sply in fruit fly neurons triggers axonal degeneration and neuromuscular junction developmental defects (14). Drosophila Sply mutant nephrocytes, functional equivalents to human podocytes, exhibit reduced foot process density and impaired albumin uptake (29). SPL is highly expressed in murine glomerular podocytes and mesangial cells, with little expression in endothelium. Silencing SGPL1 in mesangial cells impairs migration via inactivation of small Rho GTPase signaling downstream of S1PR1/3. Although silencing SPL by siRNA in human podocytes had no appreciable effect, SGPL1 disruption causes changes that suggest injury (unpublished observations). SPL KO mouse adrenal glands revealed disrupted adrenocortical zonation and reduced expression of steroidogenic enzymes, indicating a role for SPL in adrenal gland development (30).
Although an immune-mediated mechanism in SPLIS would be plausible, to date, there is no evidence to support this. Adrenal calcifications and glomerular dysfunction could be caused by vascular compromise and hemorrhage, consistent with S1P’s role in vascular maturation, permeability, and shear stress (37). Whether S1P-mediated effects on calcium homeostasis, autophagy, epigenetic regulation, and key proinflammatory transcriptional hubs contribute to SPLIS remains to be determined (38–41). Dysregulation of actin cytoskeletal organization by aberrant S1PR signaling through Rho-GTPases could compromise mesangial cells, podocytes, and neurons, all of which require migration or maintenance of morphological structures, such as foot processes, dendrites, and axons for proper function. SPL product deficiency may contribute to cellular pathologies in SPLIS. In addition to EP’s role in autophagic flux, hexadecenal/BAX and S1P/BAK cooperative interactions trigger mitochondrial outer-membrane permeability changes in response to apoptotic signals (42). Finally, the impact of SPL insufficiency on aging, carcinogenesis, and metabolic disease remain unknown. Long-term consequences that might be expected in SPLIS patients in light of S1P’s role in cell transformation and cancer, the role of ceramides in diabetes, and the identification of aging phenotypes in SPL mutants may be obscured by the immediate consequences of organ failure.
SPLIS TREATMENT STRATEGIES: RECEPTORS, REDUCTION, REPLACEMENT, REPAIR, AND REVIVAL
One can envision different strategies for treating SPLIS patients. Targeting S1PRs could correct disease manifestations caused by aberrant S1PR signaling. A Food and Drug Administration-approved S1PR antagonist (fingolimod) is available, with others in development (43). Substrate reduction has a role in sphingolipidosis treatment (44). The attenuation of Sply mutant phenotypes in response to SPT inhibition is encouraging in that regard (13). Sphingosine kinase inhibitors could mitigate S1P-dependent toxicities, but might worsen consequences of LCB and ceramide accumulation. SPL replacement via bone-marrow transplantation, gene therapy, or enzyme replacement could be considered. Delivery of soluble SPL had positive effects in a preclinical model of SPL deficiency (45). Gene editing could repair mutant SGPL1. Misfolded SPL proteins might be revived with chaperones. Importantly, PLP can act as a chaperone for some PLP-dependent enzymes (46).
SPLIS REVEALS A POTENTIAL ROLE FOR SPHINGOLIPIDS IN OTHER DISEASE CONDITIONS
Although a rare disease, SPLIS may serve as a harbinger of a broader role for sphingolipid metabolism in more common medical conditions, such as nephrosis associated with metabolic or vascular conditions, neurodegenerative diseases (spinal muscular atrophy, vascular dementia, and Alzheimer’s disease), primary immunodeficiency, adrenal insufficiency, and other endocrine defects. Much work remains to elucidate how and to what extent sphingolipid metabolism may contribute to the pathophysiology of these sporadic diseases.
In addition to the role of SPL in human biology revealed by SPLIS, recent studies have shown that S1P-dependent host-pathogen interactions can be modulated by both the host’s SPL and by pathogen-derived SPL homologs, further expanding the relevance of SPL to human health (47, 48).
SUMMARY: 50 YEARS OF LYASE AND A MOMENT OF TRUTH
Fifty years ago, Stoffel characterized the enzyme that guards the exit of sphingolipid metabolism. Since then, nearly every gene of sphingolipid metabolism has been cloned and its human ortholog identified. Genetic manipulation of the pathway has revealed myriad functions of sphingolipids in nature. SPL’s essential role in humans should come as no surprise, considering SPL’s fundamental roles in model organism development, aging, reproduction, and cellular homeostasis. Every biochemical consequence of SPL deficiency—cytotoxic sphingolipid accumulation, aberrant S1P signaling, and product depletion—likely contributes to the pathophysiology of SPLIS. Further analysis of SPLIS pathologies, patient cell lines, and model systems should elucidate the biochemical and cellular basis of the disease and may point to therapeutic strategies for this novel sphingolipidosis and more common conditions in which sphingolipids may play a role.
Footnotes
Abbreviations:
- EP
- ethanolamine phosphate
- LCB
- long-chain base
- LCBP
- long-chain base phosphate
- PtdE
- phosphatidylethanolamine
- PLP
- pyridoxal 5′-phosphate
- S1P
- sphingosine-1-phosphate
- S1PR
- sphingosine-1-phosphate receptor
- SPL
- sphingosine phosphate lyase
- SPLIS
- sphingosine phosphate lyase insufficiency syndrome
This work was supported by National Institutes of Health Public Health Service Grant DK115669; a gift from Ultragenyx Pharmaceutical; and funds from the Swim Across America Foundation (J.D.S.). The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health. The author declares no conflict of interest.
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Thudichum J. L. W. (1884) A Treatise on the Chemical Constitution of Brain. Bailliere, Tindall, and Cox, London. [Google Scholar]
- 2.Stoffel W., LeKim D., and Sticht G.. 1969. Distribution and properties of dihydrosphingosine-1-phosphate aldolase (sphinganine-1-phosphate alkanal-lyase). Hoppe Seylers Z. Physiol. Chem. 350: 1233–1241. [DOI] [PubMed] [Google Scholar]
- 3.Stoffel W. 1970. Studies on the biosynthesis and degradation of sphingosine bases. Chem. Phys. Lipids. 5: 139–158. [DOI] [PubMed] [Google Scholar]
- 4.Ikeda M., Kihara A., and Igarashi Y.. 2004. Sphingosine-1-phosphate lyase (SPL) is an endoplasmic reticulum-resident, integral membrane protein with the pyridoxal 5′-phosphate binding domain exposed to the cytosol. Biochem. Biophys. Res. Commun. 325: 338–343. [DOI] [PubMed] [Google Scholar]
- 5.Bourquin F., Riezman H., Capitani G., and Grutter M. G.. 2010. Structure and function of sphingosine-1-phosphate lyase, a key enzyme of sphingolipid metabolism. Structure. 18: 1054–1065. [DOI] [PubMed] [Google Scholar]
- 6.Saba J. D., Nara F., Bielawska A., Garrett S., and Hannun Y. A.. 1997. The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. J. Biol. Chem. 272: 26087–26090. [DOI] [PubMed] [Google Scholar]
- 7.Gottlieb D., Heideman W., Zhou J., Oskouian B., and Saba J.. 1999. The DPL1 gene is involved in mediating the response to nutrient deprivation in Saccharomyces cerevisiae. Mol. Cell Biol. Res. Commun. 1: 66–71. [DOI] [PubMed] [Google Scholar]
- 8.Birchwood C. J., Saba J. D., Dickson R. C., and Cunningham K. W.. 2001. Calcium influx and signaling in yeast stimulated by intracellular sphingosine 1-phosphate accumulation. J. Biol. Chem. 276: 11712–11718. [DOI] [PubMed] [Google Scholar]
- 9.Mao C., Wadleigh M., Jenkins G. M., Hannun Y. A., and Obeid L. M.. 1997. Identification and characterization of Saccharomyces cerevisiae dihydrosphingosine-1-phosphate phosphatase. J. Biol. Chem. 272: 28690–28694. [DOI] [PubMed] [Google Scholar]
- 10.Kim S., Fyrst H., and Saba J.. 2000. Accumulation of phosphorylated sphingoid long chain bases results in cell growth inhibition in Saccharomyces cerevisiae. Genetics. 156: 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Breslow D. K., Collins S. R., Bodenmiller B., Aebersold R., Simons K., Shevchenko A., Ejsing C. S., and Weissman J. S.. 2010. Orm family proteins mediate sphingolipid homeostasis. Nature. 463: 1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuelson A. V., Carr C. E., and Ruvkun G.. 2007. Gene activities that mediate increased life span of C. elegans insulin-like signaling mutants. Genes Dev. 21: 2976–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herr D. R., Fyrst H., Phan V., Heinecke K., Georges R., Harris G. L., and Saba J. D.. 2003. Sply regulation of sphingolipid signaling molecules is essential for Drosophila development. Development. 130: 2443–2453. [DOI] [PubMed] [Google Scholar]
- 14.Atkinson D., Nikodinovic Glumac J., Asselbergh B., Ermanoska B., Blocquel D., Steiner R., Estrada-Cuzcano A., Peeters K., Ooms T., De Vriendt E., et al. 2017. Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy. Neurology. 88: 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borowsky A. D., Bandhuvula P., Kumar A., Yoshinaga Y., Nefedov M., Fong L. G., Zhang M., Baridon B., Dillard L., de Jong P., et al. 2012. Sphingosine-1-phosphate lyase expression in embryonic and adult murine tissues. J. Lipid Res. 53: 1920–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmahl J., Raymond C. S., and Soriano P.. 2007. PDGF signaling specificity is mediated through multiple immediate early genes. Nat. Genet. 39: 52–60. [DOI] [PubMed] [Google Scholar]
- 17.Bektas M., Allende M. L., Lee B. G., Chen W., Amar M. J., Remaley A. T., Saba J. D., and Proia R. L.. 2010. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J. Biol. Chem. 285: 10880–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weber C., Krueger A., Munk A., Bode C., Van Veldhoven P. P., and Graler M. H.. 2009. Discontinued postnatal thymocyte development in sphingosine 1-phosphate-lyase-deficient mice. J. Immunol. 183: 4292–4301. [DOI] [PubMed] [Google Scholar]
- 19.Allende M. L., Bektas M., Lee B. G., Bonifacino E., Kang J., Tuymetova G., Chen W., Saba J. D., and Proia R. L.. 2011. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 286: 7348–7358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weske S., Vaidya M., Reese A., von Wnuck Lipinski K., Keul P., Bayer J. K., Fischer J. W., Flogel U., Nelsen J., Epple M., et al. 2018. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat. Med. 24: 667–678. [DOI] [PubMed] [Google Scholar]
- 21.Matloubian M., Lo C. G., Cinamon G., Lesneski M. J., Xu Y., Brinkmann V., Allende M. L., Proia R. L., and Cyster J. G.. 2004. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 427: 355–360. [DOI] [PubMed] [Google Scholar]
- 22.Schwab S. R., Pereira J. P., Matloubian M., Xu Y., Huang Y., and Cyster J. G.. 2005. Lymphocyte sequestration through S1P lyase inhibition an disruption of S1P gradients. Science. 309: 1735–1739. [DOI] [PubMed] [Google Scholar]
- 23.Zamora-Pineda J., Kumar A., Suh J. H., Zhang M., and Saba J. D.. 2016. Dendritic cell sphingosine-1-phosphate lyase regulates thymic egress. J. Exp. Med. 213: 2773–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitroi D. N., Deutschmann A. U., Raucamp M., Karunakaran I., Glebov K., Hans M., Walter J., Saba J., Gräler M., Ehninger D., et al. 2016. Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci. Rep. 6: 37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagen N., Van Veldhoven P. P., Proia R. L., Park H., Merrill A. H. Jr., and Van Echten-Deckert G.. 2009. Subcellular origin of sphingosine-1-phosphate is essential for its toxic effect in lyase deficient neurons. J. Biol. Chem. 284: 11346–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan J. P., Hu Z., and Sieburth D.. 2012. Recruitment of sphingosine kinase to presynaptic terminals by a conserved muscarinic signaling pathway promotes neurotransmitter release. Genes Dev. 26: 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitroi D. N., Karunakaran I., Graler M., Saba J. D., Ehninger D., Ledesma M. D., and van Echten-Deckert G.. 2017. SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy. 13: 885–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schümann J., Grevot A., Ledieu D., Wolf A., Schubart A., Piaia A., Sutter E., Côté S., Beerli C., Pognan F., et al. 2015. Reduced activity of sphingosine-1-phosphate lyase induces podocyte-related glomerular proteinuria, skin irritation, and platelet activation. Toxicol. Pathol. 43: 694–703. [DOI] [PubMed] [Google Scholar]
- 29.Lovric S., Goncalves S., Gee H. Y., Oskouian B., Srinivas H., Choi W. I., Shril S., Ashraf S., Tan W., Rao J., et al. 2017. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Invest. 127: 912–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prasad R., Hadjidemetriou I., Maharaj A., Meimaridou E., Buonocore F., Saleem M., Hurcombe J., Bierzynska A., Barbagelata E., Bergada I., et al. 2017. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J. Clin. Invest. 127: 942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Janecke A. R., Xu R., Steichen-Gersdorf E., Waldegger S., Entenmann A., Giner T., Krainer I., Huber L. A., Hess M. W., Frishberg Y., et al. 2017. Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum. Mutat. 38: 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linhares N. D., Arantes R. R., Araujo S. A., and Pena S. D. J.. 2018. Nephrotic syndrome and adrenal insufficiency caused by a variant in SGPL1. Clin. Kidney J. 11: 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bamborschke D., Pergande M., Becker K., Koerber F., Dotsch J., Vierzig A., Weber L. T., and Cirak S.. 2018. A novel mutation in sphingosine-1-phosphate lyase causing congenital brain malformation. Brain Dev. 40: 480–483. [DOI] [PubMed] [Google Scholar]
- 34.Settas N., Persky R., Faucz F. R., Sheanon N., Voutetakis A., Lodish M., Metherell L. A., and Stratakis C. A.. SGPL1 deficiency: a rare cause of primary adrenal insufficiency. J. Clin. Endocrinol. Metab. Epub ahead of print. December 3, 2018;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bross P., Corydon T. J., Andresen B. S., Jorgensen M. M., Bolund L., and Gregersen N.. 1999. Protein misfolding and degradation in genetic diseases. Hum. Mutat. 14: 186–198. [DOI] [PubMed] [Google Scholar]
- 36.Behne M., Uchida Y., Seki T., de Montellano P. O., Elias P. M., and Holleran W. M.. 2000. Omega-hydroxyceramides are required for corneocyte lipid envelope (CLE) formation and normal epidermal permeability barrier function. J. Invest. Dermatol. 114: 185–192. [DOI] [PubMed] [Google Scholar]
- 37.Yanagida K., and Hla T.. 2017. Vascular and immunobiology of the circulatory sphingosine 1-phosphate gradient. Annu. Rev. Physiol. 79: 67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H., Deng J., Kujawski M., Yang C., Liu Y., Herrmann A., Kortylewski M., Horne D., Somlo G., Forman S., et al. 2010. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 16: 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihlefeld K., Claas R. F., Koch A., Pfeilschifter J. M., and Meyer Zu Heringdorf D.. 2012. Evidence for a link between histone deacetylation and Ca2+ homoeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts. Biochem. J. 447: 457–464. [DOI] [PubMed] [Google Scholar]
- 40.Hait N. C., Allegood J., Maceyka M., Strub G. M., Harikumar K. B., Singh S. K., Luo C., Marmorstein R., Kordula T., Milstien S., et al. 2009. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 325: 1254–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tommasino C., Marconi M., Ciarlo L., Matarrese P., and Malorni W.. 2015. Autophagic flux and autophagosome morphogenesis require the participation of sphingolipids. Apoptosis. 20: 645–657. [DOI] [PubMed] [Google Scholar]
- 42.Chipuk J. E., McStay G. P., Bharti A., Kuwana T., Clarke C. J., Siskind L. J., Obeid L. M., and Green D. R.. 2012. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 148: 988–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chew W. S., Wang W., and Herr D. R.. 2016. To fingolimod and beyond: The rich pipeline of drug candidates that target S1P signaling. Pharmacol. Res. 113: 521–532. [DOI] [PubMed] [Google Scholar]
- 44.Arenz C. 2017. Recent advances and novel treatments for sphingolipidoses. Future Med. Chem. 9: 1687–1700. [DOI] [PubMed] [Google Scholar]
- 45.Huwiler A., Bourquin F., Kotelevets N., Pastukhov O., Capitani G., Grutter M. G., and Zangemeister-Wittke U.. 2011. A prokaryotic S1P lyase degrades extracellular S1P in vitro and in vivo: implication for treating hyperproliferative disorders. PLoS One. 6: e22436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lorenz E. C., Lieske J. C., Seide B. M., Meek A. M., Olson J. B., Bergstralh E. J., and Milliner D. S.. 2014. Sustained pyridoxine response in primary hyperoxaluria type 1 recipients of kidney alone transplant. Am. J. Transplant. 14: 1433–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo Y. J., Blake C., Alexander S., and Hahm B.. 2010. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J. Virol. 84: 8124–8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Custódio R., McLean C. J., Scott A. E., Lowther J., Kennedy A., Clarke D. J., Campopiano D. J., Sarkar-Tyson M., and Brown A. R.. 2016. Characterization of secreted sphingosine-1-phosphate lyases required for virulence and intracellular survival of Burkholderia pseudomallei. Mol. Microbiol. 102: 1004–1019. [DOI] [PubMed] [Google Scholar]