

Abstract
The growth factor-like lipid mediator, lysophosphatidic acid (LPA), is a potent signaling molecule that influences numerous physiologic and pathologic processes. Manipulation of LPA signaling is of growing pharmacotherapeutic interest, especially because LPA resembles compounds with drug-like features. The action of LPA is mediated through activation of multiple types of molecular targets, including six G protein-coupled receptors that are clear targets for drug development. However, the LPA signaling has been linked to pathological responses that include promotion of fibrosis, atherogenesis, tumorigenesis, and metastasis. Thus, a question arises: Can we harness, in an LPA-like drug, the many beneficial activities of this lipid without eliciting its dreadful actions? We developed octadecyl thiophosphate (OTP; subsequently licensed as Rx100), an LPA mimic with higher stability in vivo than LPA. This article highlights progress made toward developing analogs like OTP and exploring prosurvival and regenerative LPA signaling. We determined that LPA prevents cell death triggered by various cellular stresses, including genotoxic stressors, and rescues cells condemned to apoptosis. LPA2 agonists provide a new treatment option for secretory diarrhea and reduce gastric erosion caused by nonsteroidal anti-inflammatory drugs. The potential uses of LPA2 agonists like OTP and sulfamoyl benzoic acid-based radioprotectins must be further explored for therapeutic uses.
Keywords: autotaxin, Rx100, apoptosis, radiation mitigator, secretory diarrhea, gastric erosion, deoxyribonucleic acid damage repair
Graphical Abstract
While working with Xenopus oocytes in the late 1980s, we stumbled across an unexpected finding; rabbit serum elicited oscillatory chloride currents in the oocytes at dilutions higher than 1,000,000-fold (1). We became puzzled by the extreme potency of the putative serum factor that activated IP3 production and Ca2+ release causing the opening of Ca-activated chloride channels in the plasma membrane. Activation of the IP3-Ca2+ signaling pathway was already a known feature of G protein-coupled receptor (GPCR) activation. The serum factor was active in the sera of all vertebrate species tested; whereas, plasma showed minimal activity. Because of its unusually high potency and its presumed GPCR-mediated action, we embarked on a project that has led to the purification of lysophosphatidic acid (LPA) and several of its structural variants (2).
Structure-activity relationship (SAR) studies showed that LPA GPCRs had a preference for agonists that contained fatty acyl groups with one or two double bonds over LPA with saturated fatty acids. Molecular cloning identified the endothelial differentiation gene (EDG) family encoding eight GPCRs, three specific for LPA (LPA1/2/3) and five specific for sphingosine-1-phosphate (S1P) (S1P1/2/3/4/5) (3). These eight GPCRs showed highly similar sequences and it soon became possible to express them individually in yeast and mammalian cell types lacking endogenous Ca2+ responses, allowing for the examination of the SAR of the individual LPA receptor subtypes (4–7). In addition to the three EDG family LPA GPCRs, a second subfamily with three distinct receptors (designated LPA4/5/6) within the purinergic GPCR cluster has been identified (3). In addition to specific GPCRs, LPA activates multiple molecular targets via direct and specific binding interaction (Table 1). These LPA targets activate distinct cellular signals that mediate cellular responses unique to the target. This article is not intended as a comprehensive overview of the growing field of LPA receptors/targets and biological actions, and the reader should look up the references and review the articles listed in Table 1 for additional information about LPA biology and signaling.
TABLE 1.
Molecular targets and biological responses of LPA
| Molecular Target | Signaling Mechanism | Biological Response | Reference |
| LPA1 GPCR | Gq/11, Gi, G12/13, PDZ | Cell migration, bone development, cell proliferation, cortical development, carcinoma cell invasion, metastasis | (3, 57) |
| LPA2 GPCR | Gq/11, Gi, G12/13, PDZ, LIM | Cell survival, cell proliferation, cell motility, invasion and metastasis | (3, 57) |
| LPA3 GPCR | Gq/11, Gi, Gs | Embryo implantation, cell motility, | (3, 57) |
| LPA4 GPCR | Gs, Gq/11, Gi, G12/13, | Cell motility, lymphangiogenesis | (3, 57) |
| LPA5 GPCR | Gq/11, G12/13, | Inhibition of T cell receptor, B cell receptor, platelet aggregation | (3, 57–60) |
| LPA6 GPCR | Gs, Gi, G12/13, | Hair growth, vascular development | (3, 57) |
| PPARγ | Genes with PPRE elements | Regulation of lipid uptake, metabolism, arterial wall remodeling and neointima | (61–66) |
| TRPV1 channel | Ca2+ influx | Nociception and itch | (39, 40, 67, 68) |
| TRPA1 channel | Ca2+ influx | Itch sensation | (68, 69) |
| M-type K+ channel | K+ influx | Increased cell excitability | (69, 70) |
| Villin | LPA binding, carrier, sequestration | Epithelial cell migration, lamellipodia formation | (71) |
| Gelsolin | LPA binding, carrier, sequestration | Regulation of actin polymerization and inhibition of inflammation | (72–80) |
| Fragmin and adseverin | Inhibition of PI 4,5-P2 binding and enhanced phosphorylation by c-src | Inhibition of actin severing | (75) |
| Gintonin latex-like protein | LPA-binding carrier | LPA delivery to LPA GPCR | (72) |
COMPUTATIONAL MODELING OF LPA RECOGNITION BY LPA GPCRs
We developed homology models of the EDG family S1P and LPA GPCRs (8–11). The models were validated with extensive site-directed mutagenesis that revealed several previously unknown features of the EDG family of GPCRs. One such striking discovery was the identification of a single amino acid residue, glutamate 3.29, in the third putative transmembrane domain of S1P1 that, when mutated to glutamine, converted the specificity of the GPCRs from S1P to LPA (10). Whereas all S1P receptors have a conserved glutamate that is predicted to ion-pair with the amine moiety of S1P, all LPA-specific members contain a conserved glutamine predicted to hydrogen bond with the hydroxyl group of LPA. Furthermore, computational predictions from the models identified two additional conserved positively charged amino acids, arginine 3.28 and lysine/arginine 7.34, in every EDG GPCR, that were required for activation by the cognate ligand (6, 10, 11).
The computational modeling, SAR, and mutagenesis all pointed out that the glycerol backbone was not a required structural component of a minimal LPA ligand (12). Based on this hypothesis, we explored the ligand properties of fatty alcohol phosphates as ligands of LPA GPCRs. The choice of the fatty alcohol phosphate scaffold was based on the importance of the glycerol backbone required for substrate recognition by phospholipases, to which this scaffold is resistant (13). To further enhance the stability of the ligands, we inserted a thiophosphate in place of the phosphate group, as thiophosphates also show increased resistance to cleavage by many lipases. Octadecyl thiophosphate (OTP; licensed under the trade name Rx100) showed full agonist activity at LPA2 and LPA5, with reasonable submicromolar potency at all LPA GPCRs (Table 2) (14). Resistance to degradation by phospholipases and lipid phosphatases is the likely cause for the long ∼10.5 h plasma half-life of OTP in Rhesus monkey plasma (15). OTP has an oral bioavailability of approximately 80%, is highly protein bound (>99%), and is metabolically (S9 fractions and human liver microsomes) and chemically (37°C) stable. The majority of our efficacy studies with OTP have been performed using the oral or subcutaneous route of administration. Intraperitoneal administration is also another route of administration that is widely used in exploratory and proof-of-concept studies. Continued development includes optimizing dosing schedules and clinically relevant routes of administration. The improved in vivo stability over LPA combined with the drug-like properties of OTP prompted us to explore the biological actions of this LPA mimic in vitro and in vivo. As a result of these improvements, OTP was licensed by RxBio Inc. and named Rx100. OTP/Rx100 is the most advanced LPA mimic and is nearing filing of an Investigational New Drug application.
TABLE 2.
1. Rx100 has favorable drug profile including drug substance stability ≥5 years at refrigerated temperatures
| Chemical formula | C18H37O3SP |
| Molecular weight | 364.52 |
| Salt form | L-Lysine |
| Aqueous solubility | >250 mg/ml |
| EC50 at human LPA1/2/3/4/5 (nM) in Ca2+ mobilization assay | 850, 23, 120, 950, 3 |
| Pharmacokineticsa | T1/2 10.5 h |
In Rhesus macaques.
WHAT ARE THE STRUCTURAL REQUIREMENTS OF THE ANTI-APOPTOTIC RESPONSE MEDIATED BY LPA2?
Many reports have established that LPA has a major action in promoting cell survival by preventing programmed cell death elicited by different cellular stresses ranging from serum withdrawal to radiation-induced DNA damage (16–19). A unique feature of the anti-apoptotic effect of LPA was that it not only prevented programmed cell death when administered prior to triggering apoptosis, but it was also capable of rescuing cells that were otherwise condemned to apoptosis (16). OTP treatment also arrested the progression of the apoptotic cascade in cultured cells (16). In addition, OTP prevented death in mice and Rhesus macaques when exposed to lethal levels of ionizing radiation (see below) (Fig. 1A, B) (20, 21). Experiments have pointed to the unique anti-apoptotic action of the LPA2 GPCR subtype coupled to G protein-mediated and non-G protein-mediated pathways (12, 17–19, 22).
Fig. 1.

All animal experiments shown in the figure were reviewed and approved by the IACUC of the University of Tennessee Health Science Center and the California National Primate Research Center. A: Kaplan-Meier survival curve from a study conducted under Good Laboratory Practice standards using partial body irradiation (PBI) and dosing of either vehicle or OTP (3 mg/kg sc) beginning 24 h after 15.62 Gy exposure in female C57/BL6 mice >12 weeks (P < 0.0001, log-rank test). Median survival time was 17 days for OTP-treated animals and 7 days for vehicle-treated animals. Taken from study report: BARDA Contract HHSO100201100036C. B: Kaplan-Meier survival curve from a study with male Rhesus macaques at the California National Primate Research Center under RC-2 grant 1RC2AI087550. This model used 11.5 Gy PBI with 5% bone marrow shielding (PBI-BM5) and treatment with vehicle or OTP (0.1 mg/kg sc) beginning 4 h after radiation (P = 0.02, log-rank test). C: Ki67 immunohistological staining of a jejunum section taken on postirradiation day from a vehicle-treated Rhesus macaque after PBI-BM5 (11.5 Gy). Note the lack of crypts and uneven villus lengths in this section. D: A jejunum section from an OTP-treated Rhesus macaque (0.1 mg/kg for 5 days starting at 4 h after irradiation) from the same study as shown in C. Note the robust labeling of regenerating crypts uniformly lining the gut wall. E: Hematoxylin-eosin stained duodenum section from a vehicle-treated Rhesus macaque exposed to 11.5 Gy irradiation as in C and D taken 8 days after radiation exposure. Note the atrophic Brunner gland with extensive fibrosis. E: Hematoxylin-eosin stained duodenum section from a Rhesus macaque treated with OTP 0.3 mg/kg for 5 days starting at 24 h postirradiation and euthanized on day 8. Note that the Bunner glands show normal cellularity and no sign of fibrosis. Taken from the final grant report to NIAID on award AI 87550.
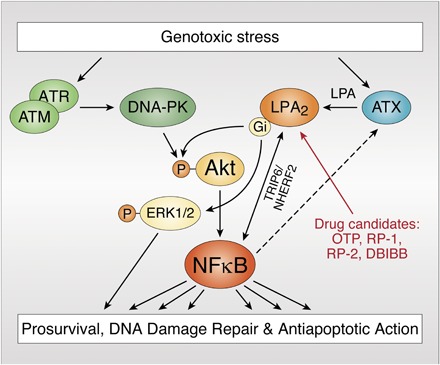
Truncation and site-directed mutagenesis studies conducted with the EDG receptors that otherwise share ∼80% sequence identity in their transmembrane regions suggested that the unique signaling actions of LPA2 are tied to the C terminus of this receptor subtype that shows less than 25% homology with the other two EDG family LPA GPCRs (19, 22). Only LPA2 contains a C-terminal -S-x-L/V sequence PDZ-protein-recognition motif and a -C311x-x-C- LIM protein binding motif near its C-terminal tail (Fig. 2) (22). The LPA2 PDZ-recognition motif has been shown to mediate the interaction with Na-H exchange response factor (NHERF)1, NHERF2 (23), membrane associated guanylate kinase WW and PDZ domain containing protein (MAGI)-2, MAGI-3, neurabin, PDZ-RhoGEF, and LARG proteins (22). These PDZ-dependent interactions mediate and enhance signals to targets like the cystic fibrosis transmembrane regulator (CFTR), phospholipase C-β3, protein kinase B (AKT), ERK1, ERK2, and the small GTPase, RhoA (22). The LIM-domain-mediated macromolecular complexes include the pro-apoptotic protein, Siva-1, and thyroid receptor-interacting protein 6 (TRIP6). These latter two scaffold proteins interact with Bcl-XL, ERK1/2, AKT, c-SRC, protein tyrosine phosphatase L1, NFkB, and the MDM2-P53 tumor suppressor axis (24). Based on these potential interactions mediated by the PDZ and LIM motifs of the LPA2 C terminus, we hypothesized that these two motifs play important roles in arresting the progression of apoptosis via the Siva-1, NFkB, MDM2-P53, and Bcl-XL pathways and promote cell regeneration via the enhanced activation of the ERK1/2 and AKT prosurvival kinases. This hypothesis has been tested using a series of C-terminal site-specific mutants of the different EDG-family LPA GPCRs. We used cell lines lacking endogenous LPA receptors for transfection with LPA2 receptor mutants (19, 22, 23), as well as LPA1, LPA2, and LPA1/2 KO mice exposed to LPA and apoptosis-inducing treatments, including γ-irradiation (12). Using a series of C-terminal site-specific mutants of the different EDG-family LPA GPCRs, we have established that LPA2 via its -C311-x-x-C- half Zn-finger LIM domain makes a macromolecular complex with the pro-apoptotic immediate-early gene product, Siva-1, that is a transcriptional target of p53 and E2F1 (25). Siva-1, when expressed after DNA damage, binds to the anti-apoptotic Bcl-XL protein in the mitochondrial outer membrane. Bcl-XL sequestered by Siva-1 no longer stabilizes the mitochondrial membrane and initiates the apoptotic cascade. Subsequent to ligand activation of LPA2, Siva-1 binds to the LIM domain and triggers the polyubiquitination and proteasomal degradation of the complex (19). The proteasomal elimination of Siva-1 effectively halts the activation of the mitochondrial apoptosis cascade by preventing the sequestration of the anti-apoptotic Bcl-XL. This LPA2-coupled macromolecular signaling mechanism appears to be responsible for the attenuation and arrest of the apoptotic cascade.
Fig. 2.

A: Alignment of the C-terminal sequence of the human EDG and P2Y family of LPA GPCRs. Note the high degree of sequence diversity in the C termini of these receptors even within the EDG and P2Y subgroups. The C311-x-x-C motif is only found in LPA2, although some other LPA receptors have PDZ-interaction motifs. B: Schematic summary of LPA2 signaling pertinent to protection against apoptosis-inducing stress. See the text for details.
LPA2 can also bind to TRIP6 via the same -C311-x-x-C- motif. However, TRIP6, which is a PDZ-motif-binding protein itself, makes yet another interaction with the NHERF2 dimer that is docked to the C-terminal -DSTL PDZ-motif. Thus, a ternary macromolecular complex is assembled that consists of LPA2 and the homodimer of NHERF2, which on one hand binds to the PDZ-motif of TRIP6 and the C-terminal PDZ-motif of LPA2. Once this ligand-activated macromolecular complex is assembled, it enhances the duration and the magnitude of prosurvival signals mediated via AKT and ERK1/2 kinases (19, 26, 27).
Studies using WT, LPA1 KO, and LPA2 KO mice showed that exposure of these animals to lethal levels (>10 Gy) of γ-irradiation led to the development of acute hematopoietic (HEM) and gastrointestinal radiation syndrome (GI-ARS). GI-ARS is characterized by radiation-induced loss of intestinal crypt stem cells and disruption of the intestinal barrier. In the bone marrow, there is loss of HEM stem and progenitor cells leading to death. Using the GI-ARS mouse model, we found that LPA and OTP treatment attenuated crypt loss, enhanced the intestinal barrier, and increased survival in WT and LPA1 KO mice, but was completely ineffective in LPA2 KO mice. Vehicle-treated LPA2 KO mice showed more extensive damage in their crypt counts and a higher number of apoptotic cells compared with WT or LPA1 KO mice. We also found that LPA2 KO mice showed delayed resolution of DNA double-strand breaks (28). This suggests that LPA2 is important in the natural protection against radiation, mediated in part by augmenting the DNA damage repair (DDR) response. Furthermore, we found that mouse embryonic fibroblasts (MEFs) derived from LPA1/2 double KO mice (DKO-MEFs) are highly sensitive to apoptosis-inducing chemotherapeutics and γ-irradiation (19, 26, 29). However, when DKO-MEFs were reconstituted with the human LPA2, treatment with LPA or OTP protected these cells from a number of apoptosis-inducing stressors, including γ-irradiation (22, 30). In summary, macromolecular LPA2 signalosomes provide enhanced protection to cells against a wide range of apoptosis-inducing environmental stresses, as we have been able to demonstrate in murine and nonhuman primate models of radiation-induced genotoxic injury (Fig. 1A, B). Does treatment of radiation injury with LPA and its analogs promote malignant transformation as has been shown for multiple LPA GPCRs, including LPA2 (31)? We have tested this hypothesis using LPA2-transduced MEF cells exposed to 15 Gy γ-irradiation in soft-agar colony growth assays with and without treatment with the LPA2-selective agonist, GRI977143 (30). These studies showed that cultures treated with the LPA2 agonist showed no statistically significant increase in colony numbers compared with vehicle-treated irradiated cultures and unirradiated control cultures. We have not yet been able to collect data for tumor incidence from a statistically relevant number of irradiated and treated animals that were kept beyond the ∼35 day endpoint that is considered representative for the recovery from the GI-ARS. Such experiments will be necessary to decide any pro-tumorigenic action of the LPA2 agonists. However, one must also take into consideration that LPA2 is a natural component of the DDR pathway and accelerates the resolution of DNA double-strand breaks indicated by the reduction of γH2AX staining in irradiated WT mice versus LPA2 KO mice (28). The role of LPA2 in DDR combined with the relatively short 3–6 day treatment of irradiated mice could provide an explanation for the lack of increased tumorigenesis following selective stimulation of LPA2 signaling. It is also important to recognize that LPA levels increase significantly during physiological conditions like pregnancy, brain development, and wound healing without any evidence of the causing or promotion of malignant transformation. RxBio, Inc. licensed OTP/Rx100 for development and has made significant progress toward investigational new drug (IND) filing.
VIRTUAL HIGH-THROUGHPUT SCREENING-GUIDED DRUG DESIGN OF LPA2 SPECIFIC AGONISTS
Studies concerning the anti-apoptotic preventive and mitigative (i.e., poststress) actions of LPA revealed the unique role of the LPA2-coupled macromolecular signals. LPA and its mimic, OTP, are not specific agonists of the LPA2 subtype. Thus, a drug discovery effort aimed at the development of LPA2 GPCR-specific agonists was launched. Subsequent to the computational modeling of the EDG family GPCRs (6, 7, 9, 10, 32), several crystal structures of LPA and S1P GPCRs have been solved (24, 33–35). The validated computational models and the crystal structures now provide a robust modeling platform for virtual high-throughput-guided LPA GPCR ligand discovery. First, using our LPA2 model, we conducted a virtual high-throughput screening for ligands using the Genome Research Institute’s chemical library (36). Our virtual screen yielded compounds NSC12404, GRI977143, H2L55-47924, and H2L5828102, each a novel nonlipid LPA2 agonist (36). From this set of compounds, we selected the GRI977143 compound for preclinical efficacy testing (30). The GRI977143 compound has a modest potency of EC50 3.3 μM at LPA2 and has a weak LPA3 antagonist activity (IC50 ∼6.6 μM). In radiation- and chemotherapy-induced apoptosis assays, GRI977143 showed dose-dependent anti-apoptotic actions by blocking caspase 3/7/8/9 activation and DNA fragmentation, preventing BAX depletion and PARP cleavage (30). The compound was without any anti-apoptotic effect in DKO-MEFs lacking LPA2 expression. When tested in mice using a total body irradiation model, GRI977143 administered 24 h after radiation exposure significantly increased mean survival time and reduced mortality (30). Subsequently, we conducted medicinal chemistry studies to improve the potency of GRI977143 and dial out its antagonist effect at LPA3. The synthetic improvements using the sulfamoyl benzoic acid (SBA) scaffold have identified multiple new compounds with subnanomolar agonist activity specific to the LPA2 without any effect at the other LPA receptors (Fig. 3A) (14, 37). We chose one of the SBA analogs {2-[4-(1,3-dioxo-1H,3H-benzoisoquinolin-2-yl) butylsulfamoyl]benzoic acid (DBIBB)} for further testing. These experiments showed that DBIBB was a highly effective mitigator of radiation-induced cell death in vitro and in vivo (Fig. 3B) (21). DBIBB had therapeutic efficacy against both the HEM and gastrointestinal radiation injuries, and it was effective when drug treatment was delayed to 72 h postirradiation. In cultured human CD34 HEM progenitor cells, after a 4 Gy irradiation, DBIBB significantly enhanced cell survival and the differentiation of the myeloid cell lineage (21).
Fig. 3.

A. Structural formulas of LPA, OTP, and the LPA2-specific agonists DBIBB, RP-1, and RP-2. B: Kaplan-Meier survival plots for groups of 15 C57BL/6 mice exposed to 15.69 Gy of 137Cs γ-irradiation using a partial body 5% bone marrow shielding model. DBIBB was dissolved in 1% ethanol/2% propanediol in PBS (vehicle) and was administered starting at +26 ± 2 h postirradiation for 3 days as a single daily subcutaneous injection. DBIBB increased survival during the GI-ARS phase (days 8–12) and also the HEM-ARS phase (days 15–20) [see Patil et al. (21) for details]. C: Effect of RP-1 on the survival of small intestinal enteroids after 5 Gy γ-irradiation. Enteroid cultures were established for 6 days before treatment with 1 μM RP-1 or vehicle. On day 7, the cultures were exposed to 5 Gy γ-irradiation and allowed to grow for 2 more days before the surviving enteroids were counted. Data points are the means of triplicate wells and the results are representative of at least three biological replicates (*P = 0.05, **P ≤ 0.01, ***P < 0.001). Panel C is taken from Kuo et al. (14).
Enteroid cultures derived from the small intestine recapitulate the growth and maturation of the stem cell-derived enterocytes into functionally differentiated enterocytes found in the villus. The gut has two distinct intestinal stem cell (ISC) populations that are distinguished by the expression of the Bmi1 and the LGR-5 markers. We used murine enteroid cultures established from mice that expressed enhanced green fluorescent protein driven by the LGR-5 promoter and also the red cherry tomato protein from a tamoxifen-inducible LGR-5 promoter. Double labeling of the LGR-5-positive ISC allowed us not only to identify this stem cell population but also enabled us to induce the expression of the red protein, which would only be synthesized in surviving cells capable of protein synthesis. These enteroid cultures were irradiated and treated with the most potent SBA analog, designated radioprotectin (RP)-1 (14). RP-1 has a 25 nM EC50 at the murine LPA2 and a 5 pM EC50 at the human LPA2 (14, 37). RP-1 significantly enhanced enteroid growth and survival of LGR-5-positive ISCs after exposure to 5 Gy γ-irradiation (Fig. 3C). Furthermore, RP-1 decreased mortality in murine models of the HEM-ARS and the GI-ARS after a 16 Gy γ-irradiation dose (14). In summary, the follow-on new generation SBA analogs are specific LPA2 agonists and effectively mitigate γ-irradiation-induced apoptosis and enhance the regeneration of LGR-5-positive ISCs, indicating that the LPA2 expressed in this ISC subset represents the molecular target of SBA drugs.
LPA is a unique water-soluble lipid generated from highly hydrophobic precursors that are insoluble in aqueous solutions without a proper carrier protein. Mother Nature uses albumin primarily to overcome the insolubility of LPA in the presence of divalent cations present at millimolar concentrations in biological fluids. Because of the amphipathic properties of LPA, it has been a challenge to identify synthetic nonlipid-like compounds that fit the ligand recognition pockets of the LPA target. Conceptually, the cleavage of water insoluble lysophosphatidylcholine to LPA by the lysophospholipase D enzyme, autotaxin (ATX), increases its hydrophilicity by unmasking the negatively charged phosphate moiety of the molecule. The phosphate group in LPA or S1P is negatively charged at physiological pH and plays an essential role in binding to the lysophospholipid GPCRs (9, 10, 24, 38). Modeling studies and crystal structures indicate that an interaction surface made up of polar residues is present in the ligand-binding pockets that LPA targets. Such polar molecular surfaces have also been identified in ion channel targets of LPA (39, 40) and ATX too (41, 42). Molecular modeling of the nonlipid LPA receptor agonists suggests that these compounds engage the hydrophilic surface of the receptor’s ligand binding pocket or the catalytic pocket of ATX (12, 36, 37, 43, 44). Although most man-made LPA GPCR ligands are highly hydrophobic, they all contain a charged group, which is required for biological activity (45). We anticipate that the search for small molecule modulators of LPA targets with the availability of high-throughput screening methods designed to recognize allosteric modulators of GPCRs should identify more hydrophilic ligands that engage polar surfaces on the receptors.
IS THE LPA2-ATX AXIS A NATURAL DEFENSE AGAINST GENOTOXIC STRESS?
LPA2 KO mice showed a significantly higher number of apoptotic cells and fewer surviving crypts after irradiation (12). This observation has led us to hypothesize that the endogenous expression of LPA2 conveys resistance against radiation-induced cell death (Fig. 2B). Radiation-induced DNA double-strand breaks are detected by ATM and ATR kinases, which activate DDR, cell survival, and stress response pathways (46, 47). The decision between survival and apoptosis of a cell exposed to a genotoxic insult also depends on inputs from the cell microenvironment. LPA derived from stromal ATX can affect the decision about the cell’s fate (28, 48). The repair of DNA double-strand breaks can be monitored by the resolution of phosphorylated histone 2AX (γ-H2AX). We have found that DBIBB accelerated γ-H2AX resolution in DKO-MEFs reconstituted with the human LPA2 (LPA2 DKO-MEFs), but not in vector control DKO-MEF cells (21). We compared γ-H2AX resolution in the guts of WT and LPA2 KO mice. The resolution of γ-H2AX-positive gut cells was protracted in gut sections of LPA2 KO compared with WT mice (28). In LPA2 DKO-MEFs, the duration of AKT and ERK1/2 kinase activation protracted beyond 8 h after LPA stimulation (28). LPA2 activation-dependent enhancement of γ-H2AX resolution, AKT, and ERK1/2 kinase activation was deficient in DKO-MEFs reconstituted with CA-CA-LA mutant LPA2, in which LIM and PDZ-binding motifs were mutated (28).
We have also investigated the effect of γ-irradiation on the expression of LPA GPCR subtype and ATX. These experiments showed that exposure to ionizing radiation dose-dependently increases the mRNA levels of LPA2 in nontransformed IEC-6 intestinal crypt-like cells. LPA2 upregulation at the transcript level was maintained for 24 h and was accompanied by a higher efficacy/responsiveness of irradiated cells stimulated with RP-1. ATX transcript and plasma LPA levels were also increased in irradiated mice (28).
The endogenous dosimeter-like properties of LPA2 expression intrigued us to question whether this receptor is an integral part of the DDR response and whether it is coupled to the activation of ATM and ATR kinases in the genotoxic stress response pathway. The ATM/ATR kinase inhibitor, CGK-733, dose-dependently reduced radiation-induced upregulation of LPA2. Deletion of the NFkB recognition site in the LPA2 promoter abolished radiation-induced upregulation of LPA2, establishing an additional link between LPA2 expression in the DDR pathway. Corroborating evidence for the radiation-dependent transcriptional upregulation of LPA2 was found in murine enteroid cultures (14).
The requirement of the NFkB site in the LPA2 promoter has led us to hypothesize that direct and indirect links might govern DDR and LPA-induced regenerative response. First, the ATM/ATR kinases directly phosphorylate AKT, which in turn can activate the canonical NFkB pathway driving LPA2 and ATX expression. Second, ionizing radiation causes a surge in IL-6 and TNFα production, which in turn activates NFkB and stimulates expression of LPA2 and ATX. Third, LPA2 activation led to a prolonged AKT and ERK phosphorylation extending beyond 12 h (28). This long-lasting activation of AKT signaling represents yet another mechanism that could contribute to sustained NFkB activation and upregulation of LPA2 and ATX. Fourth, radiation-induced increase in ATX expression and LPA production is important for anti-apoptotic and prosurvival signaling (28). LPA2-TRIP6 physically interacts with NFkB and promotes its nuclear translocation and expression of NFkB target genes. Lai et al. (27) have shown that TRIP6 binds to Fas and interferes with the recruitment of FADD to Fas, which in turn antagonizes Fas-induced apoptosis and enhances the anti-apoptotic action of LPA. We demonstrated the anti-apoptotic action of the LPA2-TRIP6-NHERF2 complex against radiation- and chemotherapy-induced apoptosis (22). The macromolecular complex consisting of LPA2-TRIP6-NHERF2-NFkB is a noncanonical activation mechanism of NFkB-dependent prosurvival signals. This signaling pathway represents a feed forward protective mechanism against apoptosis that is centered around multiple mechanisms, each driving LPA2 expression. Taken together, transcriptional LPA2 expression via the canonical and noncanonical activation of NFkB appears to be central to the cell’s response to genotoxic stress. Because many chemotherapeutics and radiation kill neoplastic cells by inducing genotoxic-stress, the role of the LPA-ATX axis in cancer resistance is an important mechanism that offers therapeutic utility for the use of ATX inhibitors and LPA2 antagonists (29, 49–51). In a recent report, Seo et al. (51) demonstrated the importance of the ATX-LPA-AKT signaling pathway in the growth of ovarian cancer stem-like cells. Taken together, the mechanisms detailed above justify the need for more research concerning the role of LPA in cancer progression and metastasis (50). In this context, the lack of specific high-potency LPA2 antagonists/modulators is becoming a limitation to the field.
LPA promotes the proliferation, invasion, and therapy resistance of tumor cells (49, 52, 53). Therefore, there is concern that the use of LPA mimics for therapeutic purposes might have grave consequences by activating dormant carcinoma cells in the body. At the present time, other than the physiologically elevated LPA concentrations during pregnancy and wound healing, no specific knowledge is available in the literature about the effects of selective and long-term stimulation of LPA targets. Therefore, one can only speculate that specific stimulation or inhibition of a single LPA target when applied for a limited period of time might be therapeutically beneficial without activating the potentially dire side effects mediated by one or another LPA target.
WHAT ARE OTHER MEDICAL INDICATIONS FOR THERAPEUTIC USE OF LPA2-SPECIFIC AGONISTS?
The LPA2 C-terminal PDZ binding motif via NHERF2 makes physical interaction with CFTR. In the apical membrane of the enterocyte, this complex brings the heterotrimeric Gi protein to a microdomain in the immediate vicinity of adenylyl cyclase and inhibits local cAMP production, thereby reducing the opening of the CFTR Cl− channel (23). CFTR-dependent Cl− flux into the intestinal lumen generates an osmotic gradient that is responsible for paracellular Na+ and water movement, leading to a disease designated as secretory diarrhea (SED). SED is caused by bacterial toxins, like cholera toxin, that activate adenylyl cyclase, leading to CFTR-mediated secretion responsible for fluid discharge. SED causes over 2.5 million deaths annually in the developing world and often spreads as an epidemic in postdisaster conditions. Currently, oral rehydration therapy (ORT) with salt solutions is the standard of care of SED. However, due to limited availability of clean water, as has been the case and also the cause of many cholera epidemics, ORT is a symptomatic rather than causative treatment of SED. Using open- and closed-intestinal-loop SED models, we have shown that LPA and OTP effectively reduce cholera toxin-induced Cl− secretion into the intestinal lumen (Fig. 4A, B) (23, 54). Disruption of the macromolecular complex between LPA2 and CFTR abrogates the effect of LPA on Cl− secretion (23). To model conditions that accompany infection with toxin-producing bacteria, we used a Citrobacter rodentium infection-induced SED model. We have determined that 80% of OTP administered orally is bioavailable, and for this reason we delivered OTP via oral gavage to mice infected with C. rodentium. In this infection model, OTP significantly reduced fecal Cl− secretion and weight loss of mice (54). Because patients with SED also vomit, they often cannot keep down the massive amounts of liquid used as ORT. A self-injectable syringe or a pill containing OTP or other LPA2 agonist represents a novel therapeutic approach that targets the root core of the disease.
Fig. 4.

The animal experiments shown in the figure were reviewed and approved by the IACUC of the University of Tennessee Health Science Center and the University of Tokushima, Japan. A: OTP prevents infection-induced weight loss. C57BL/6 mice were infected on day 0 with C. rodentium orally. Mice received placebo or OTP (0.1 or 1 mg/kg sc) on days 1–14 (#P < 0.05; *P < 0.01). OTP significantly reduced weight loss in animals infected with C. rodentium compared with infected animals not receiving OTP. Error bars represent SEM; control groups n = 4, other groups n = 5–6. B: Effect of delayed administration of OTP on the fluid accumulation ratio in the open loop cholera toxin (CTX) model. CTX was administered at 0 h and OTP (1 mg/kg po) was administered with delays between 0 and 3 h after CTX administration. Samples were collected 6 h after CTX administration. CTX significantly increases fluid accumulation compared with vehicle (**P < 0.005). Delaying administration by up to 2 h after CTX administration results in significantly reduced fluid accumulation than that in animals receiving CTX and vehicle (*P < 0.001). Error bars represent SEM; n = 6 per group. C: LPA2-specific agonists, RP-2 and DBIBB, reduce aspirin-induced stomach erosions in BALB/C mice (n = 5 per group). Aspirin (300 mg/kg body weight) was made up in 3% w/v carboxymethyl cellulose, and 200 μl of this suspension was delivered into the stomach via oral gavage. For details of the model see (56). The drugs (1 mM stock) were made up in PBS containing 0.1% mouse serum albumin in 3% carboxymethyl cellulose, and 200 μl was administered 15 min after the aspirin suspension via oral gavage. Mice were euthanized 2.5 h after the first treatment and stomachs were dissected and photographed. The length of erosions was measured using the ImageJ program (The National Institutes of Health, Bethesda, MD) and compared with the vehicle group using Student’s t-test. Note that RP-2 and DBIBB significantly reduced the total length of stomach erosions. D: Representative images of stomachs from mice that were treated with aspirin, LPA (1 mM), and OTP (1 mM). Note that the bloody erosions are almost completely absent in the OTP-treated stomach. Panels A and B are taken with permission and edits from Thompson et al. (54).
Nonsteroidal anti-inflammatory drugs (NSAIDs), such as aspirin, indomethacin, and many others in this drug family, are the most used over-the-counter drugs worldwide. However, NSAIDs often cause erosions in the stomach mucosa and, in severe cases, can lead to gastric bleeding and hematemesis. Tanaka and colleagues have demonstrated that delivery of LPA into the stomach via LPA2 reduces gastric erosions elicited by different NSAIDs (55, 56). These authors demonstrated that LPA2-activated prostaglandin E production is a key step in the protective mechanism against gastric erosion (55). In collaboration with the Tanaka group, we have obtained proof of concept that, similarly to LPA, the SBA LPA2 agonists and OTP delivered before, together, or after intragastric gavage of indomethacin prevent and reduce gastric erosions (Fig. 4C, D). Based on these observations, coformulation of LPA2 agonists with NSAIDs offers an intriguing possibility that could benefit large patient populations who depend on NSAIDs for the treatment of arthritis and other chronic inflammatory conditions.
Although this perspective focused only on the chemical biology of drug-like ligands of the LPA2 GPCRs, similar drug discovery and chemical biology efforts will have to explore the other LPA GPCR subtypes to gain full benefits to harnessing the therapeutic potential of this system.
Acknowledgments
The studies reviewed in this paper have been generated by a terrific group of coworkers whom we had the privilege of working with over the decades. Instead of listing approximately 35 coauthors in this paper, We have cited the pertinent publications that our coworkers contributed to, recognizing their contributions, their dedicated and creative work without which we would not have been able to advance the field toward the development of an LPA-based human medicine.
Footnotes
Abbreviations:
- AKT
- protein kinase B
- ATX
- autotaxin
- CFTR
- cystic fibrosis transmembrane regulator
- DBIBB
- 2-[4-(1,3-dioxo-1H,3H-benzoisoquinolin-2-yl) butylsulfamoyl]benzoic acid
- DDR
- DNA damage repair
- DKO
- double KO
- DKO-MEF
- mouse embryonic fibroblast derived from lysophosphatidic acid 1 and 2 double KO mice
- EDG
- endothelial differentiation gene
- GI-ARS
- gastrointestinal radiation syndrome
- GPCR
- G protein-coupled receptor
- γ-H2AX
- phosphorylated histone 2AX
- HEM
- hematopoietic
- ISC
- intestinal stem cell
- LPA
- lysophosphatidic acid
- MEF
- mouse embryonic fibroblast
- NHERF
- Na-H exchange response factor
- NSAID
- nonsteroidal anti-inflammatory drug
- ORT
- oral rehydration therapy
- OTP
- octadecyl thiophosphate (Rx100)
- RP
- radioprotectin
- SAR
- structure-activity relationship
- SBA
- sulfamoyl benzoic acid
- SED
- secretory diarrhea
- S1P
- sphingosine-1-phosphate
- TRIP6
- thyroid receptor-interacting protein 6
This work was supported by National Institutes of Health Grants CA092160, AI1RC2AI087550, and U01AI080405; U.S. Department of Veterans Affairs Grant 1101BX001187; the Van Vleet Endowment; Biomedical Advanced Research and Development Authority Grant HHSO100201100036C; and RxBio Inc. (1R43DK105719-01A1). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. G.J.T., L.R.J., and W.S.M. are founders of RxBio Inc.; G.J.T. and L.R.J. are paid consultants at RxBio Inc.; and G.J.T., L.R.J., W.S.M., K.T., and A.B. are stockholders in RxBio Inc.
REFERENCES
- 1.Tigyi G., Henschen A., and Miledi R.. 1991. A factor that activates oscillatory chloride currents in Xenopus oocytes copurifies with a subfraction of serum albumin. J. Biol. Chem. 266: 20602–20609. [PubMed] [Google Scholar]
- 2.Tigyi G., and Miledi R.. 1992. Lysophosphatidates bound to serum albumin activate membrane currents in Xenopus oocytes and neurite retraction in PC12 pheochromocytoma cells. J. Biol. Chem. 267: 21360–21367. [PubMed] [Google Scholar]
- 3.Chun J., Blaho V., Frantz A., Hla T., Jones D., Kihara Y., Mizuno H., Moolenaar W., Mpamhanga C., Spiegel S., and Yung Y. C.. 2018. Lysophospholipid (LPA) receptors. In IUPHAR/BPS Guide to PHARMACOLOGY. Accessed February 6, 2019, at http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=36.
- 4.Erickson J. R., Wu J. J., Goddard G., Kawanishi K., Tigyi G., Tomei L. D., and Kiefer M. C.. 1998. The putative lysophosphatidic acid receptor Edg-2/Vzg-1 functionally couples to the yeast response pathway. J. Biol. Chem. 273: 1506–1510. [DOI] [PubMed] [Google Scholar]
- 5.Fischer D. J., Liliom K., Guo Z., Nusser N., Virag T., Murakami-Murofushi K., Kobayashi S., Erickson J. R., Sun G., Miller D. D., et al. 1998. Naturally occurring analogs of lysophosphatidic acid elicit different cellular responses through selective activation of multiple receptor subtypes. Mol. Pharmacol. 54: 979–988. [DOI] [PubMed] [Google Scholar]
- 6.Fujiwara Y., Osborne D. A., Walker M. D., Wang D. A., Bautista D. A., Liliom K., Van Brocklyn J. R., Parrill A. L., and Tigyi G.. 2007. Identification of the hydrophobic ligand binding pocket of the S1P1 receptor. J. Biol. Chem. 282: 2374–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujiwara Y., Sardar V., Tokumura A., Baker D., Murakami-Murofushi K., Parrill A., and Tigyi G.. 2005. Identification of residues responsible for ligand recognition and regioisomeric selectivity of lysophosphatidic acid receptors expressed in mammalian cells. J. Biol. Chem. 280: 35038–35050. [DOI] [PubMed] [Google Scholar]
- 8.Bautista D. L., Baker D. L., Wang D., Fischer D. J., Van Brocklyn J., Spiegel S., Tigyi G., and Parrill A. L.. 2000. Dynamic modeling of EDG1 receptor structural changes induced by site-directed mutations. Theochem. 529: 219–224. [Google Scholar]
- 9.Parrill A. L., Wang D-A., Bautista D. L., Van Brocklyn J. R., Lorincz Z., Fischer D. J., Baker D. L., Liliom K., Spiegel S., and Tigyi G.. 2000. Identification of Edg1 receptor residues that recognize sphingosine 1-phosphate. J. Biol. Chem. 275: 39379–39384. [DOI] [PubMed] [Google Scholar]
- 10.Wang D. A., Lorincz Z., Bautista D. L., Liliom K., Tigyi G., and Parrill A. L.. 2001. A single amino acid determines ligand specificity of the S1P1 (EDG1) and LPA1 (EDG2) phospholipid growth factor receptors. J. Biol. Chem. 276: 49213–49220. [DOI] [PubMed] [Google Scholar]
- 11.Sardar V. M., Bautista D. L., Fischer D. J., Yokoyama K., Nusser N., Virag T., Wang D., Baker D. L., Tigyi G., and Parrill A. L.. 2002. Molecular basis for lysophosphatidic acid receptor antagonist selectivity. Biochim. Biophys. Acta. 1582: 309–317. [DOI] [PubMed] [Google Scholar]
- 12.Deng W., Shuyu E., Tsukahara R., Valentine W. J., Durgam G., Gududuru V., Balazs L., Manickam V., Arsura M., VanMiddlesworth L., et al. 2007. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology. 132: 1834–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Virag T., Elrod D. B., Liliom K., Sardar V. M., Parrill A. L., Yokoyama K., Durgam G., Deng W., Miller D. D., and Tigyi G.. 2003. Fatty alcohol phosphates are subtype-selective agonists and antagonists of lysophosphatidic acid receptors. Mol. Pharmacol. 63: 1032–1042. [DOI] [PubMed] [Google Scholar]
- 14.Kuo B., Szabó E., Lee S. L., Balogh A., Norman D. D., Inoue A., Ono Y., Aoki J., and Tigyi G. J.. 2018. The LPA2 receptor agonist radioprotectin-1 spares LGR-5 positive intestinal stem cells from radiation injury in murine enteroids. Cell. Signal. 51: 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosanam H., Ma F., He H., Ramagiri S., Gududuru V., Tigyi G. J., Van Rompay K., Miller D. D., and Yates C. R.. 2010. Development of an LC-MS/MS assay to determine plasma pharmacokinetics of the radioprotectant octadecenyl thiophosphate (OTP) in monkeys. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 878: 2379–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng W., Balazs L., Wang D. A., Van Middlesworth L., Tigyi G., and Johnson L. R.. 2002. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology. 123: 206–216. [DOI] [PubMed] [Google Scholar]
- 17.Deng W., Poppleton H., Yasuda S., Makarova N., Shinozuka Y., Wang D. A., Johnson L. R., Patel T. B., and Tigyi G.. 2004. Optimal lysophosphatidic acid-induced DNA synthesis and cell migration but not survival require intact autophosphorylation sites of the epidermal growth factor receptor. J. Biol. Chem. 279: 47871–47880. [DOI] [PubMed] [Google Scholar]
- 18.Deng W., Wang D. A., Gosmanova E., Johnson L. R., and Tigyi G.. 2003. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 284: G821–G829. [DOI] [PubMed] [Google Scholar]
- 19.Lin F. T., Lai Y. J., Makarova N., Tigyi G., and Lin W. C.. 2007. The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J. Biol. Chem. 282: 37759–37769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng W., Kimura Y., Gududuru V., Wu W., Balogh A., Szabo E., Thompson K. E., Yates C. R., Balazs L., Johnson L. R., et al. 2015. Mitigation of the hematopoietic and gastrointestinal acute radiation syndrome by octadecenyl thiophosphate, a small molecule mimic of lysophosphatidic acid. Radiat. Res. 183: 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patil R., Szabo E., Fells J. I., Balogh A., Lim K. G., Fujiwara Y., Norman D. D., Lee S. C., Balazs L., Thomas F., et al. 2015. Combined mitigation of the gastrointestinal and hematopoietic acute radiation syndromes by an LPA2 receptor-specific nonlipid agonist. Chem. Biol. 22: 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shuyu E., Lai Y. J., Tsukahara R., Chen C. S., Fujiwara Y., Yue J., Yu J. H., Guo H., Kihara A., Tigyi G., et al. 2009. Lysophosphatidic acid 2 receptor-mediated supramolecular complex formation regulates its antiapoptotic effect. J. Biol. Chem. 284: 14558–14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C., Dandridge K. S., Di A., Marrs K. L., Harris E. L., Roy K., Jackson J. S., Makarova N. V., Fujiwara Y., Farrar P. L., et al. 2005. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J. Exp. Med. 202: 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parrill A. L., and Tigyi G.. 2013. Integrating the puzzle pieces: the current atomistic picture of phospholipid-G protein coupled receptor interactions. Biochim. Biophys. Acta. 1831: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fortin A., MacLaurin J. G., Arbour N., Cregan S. P., Kushwaha N., Callaghan S. M., Park D. S., Albert P. R., and Slack R. S.. 2004. The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1. J. Biol. Chem. 279: 28706–28714. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H., Xu X., Gajewiak J., Tsukahara R., Fujiwara Y., Liu J., Fells J. I., Perygin D., Parrill A. L., Tigyi G., et al. 2009. Dual activity lysophosphatidic acid receptor pan-antagonist/autotaxin inhibitor reduces breast cancer cell migration in vitro and causes tumor regression in vivo. Cancer Res. 69: 5441–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai Y. J., Lin V. T., Zheng Y., Benveniste E. N., and Lin F. T.. 2010. The adaptor protein TRIP6 antagonizes Fas-induced apoptosis but promotes its effect on cell migration. Mol. Cell. Biol. 30: 5582–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balogh A., Shimizu Y., Lee S. C., Norman D. D., Gangwar R., Bavaria M., Moon C., Shukla P., Rao R., Ray R., et al. 2015. The autotaxin-LPA2 GPCR axis is modulated by gamma-irradiation and facilitates DNA damage repair. Cell. Signal. 27: 1751–1762. [Erratum. 2015. Cell. Signal. 27: 2137.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brindley D. N., Lin F. T., and Tigyi G. J.. 2013. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim. Biophys. Acta. 1831: 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiss G. N., Lee S. C., Fells J. I., Liu J., Valentine W. J., Fujiwara Y., Thompson K. E., Yates C. R., Sumegi B., and Tigyi G.. 2013. Mitigation of radiation injury by selective stimulation of the LPA(2) receptor. Biochim. Biophys. Acta. 1831: 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin S., Lee S. J., Shim H., Chun J., and Yun C. C.. 2010. The absence of LPA receptor 2 reduces the tumorigenesis by ApcMin mutation in the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 299: G1128–G1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valentine W. J., Fells J. I., Perygin D. H., Mujahid S., Yokoyama K., Fujiwara Y., Tsukahara R., Van Brocklyn J. R., Parrill A. L., and Tigyi G.. 2008. Subtype-specific residues involved in ligand activation of the endothelial differentiation gene family lysophosphatidic acid receptors. J. Biol. Chem. 283: 12175–12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanson M. A., Roth C. B., Jo E., Griffith M. T., Scott F. L., Reinhart G., Desale H., Clemons B., Cahalan S. M., Schuerer S. C., et al. 2012. Crystal structure of a lipid G protein-coupled receptor. Science. 335: 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chrencik J. E., Roth C. B., Terakado M., Kurata H., Omi R., Kihara Y., Warshaviak D., Nakade S., Asmar-Rovira G., Mileni M., et al. 2015. Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell. 161: 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taniguchi R., Inoue A., Sayama M., Uwamizu A., Yamashita K., Hirata K., Yoshida M., Tanaka Y., Kato H. E., Nakada-Nakura Y., et al. 2017. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature. 548: 356–360. [DOI] [PubMed] [Google Scholar]
- 36.Kiss G. N., Fells J. I., Gupte R., Lee S. C., Liu J., Nusser N., Lim K. G., Ray R. M., Lin F. T., Parrill A. L., et al. 2012. Virtual screening for LPA2-specific agonists identifies a nonlipid compound with antiapoptotic actions. Mol. Pharmacol. 82: 1162–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patil R., Fells J. I., Szabo E., Lim K. G., Norman D. D., Balogh A., Patil S., Strobos J., Miller D. D., and Tigyi G. J.. 2014. Design and synthesis of sulfamoyl benzoic acid analogues with subnanomolar agonist activity specific to the LPA2 receptor. J. Med. Chem. 57: 7136–7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naor M. M., Walker M. D., Van Brocklyn J. R., Tigyi G., and Parrill A. L.. 2007. Sphingosine 1-phosphate pKa and binding constants: intramolecular and intermolecular influences. J. Mol. Graph. Model. 26: 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morales-Lázaro S. L., Serrano-Flores B., Llorente I., Hernandez-Garcia E., Gonzalez-Ramirez R., Banerjee S., Miller D., Gududuru V., Fells J., Norman D., et al. 2014. Structural determinants of the transient receptor potential 1 (TRPV1) channel activation by phospholipid analogs. J. Biol. Chem. 289: 24079–24090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nieto-Posadas A., Picazo-Juarez G., Llorente I., Jara-Oseguera A., Morales-Lazaro S., Escalante-Alcalde D., Islas L. D., and Rosenbaum T.. 2011. Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nat. Chem. Biol. 8: 78–85. [Erratum. 2012. Nat. Chem. Biol. 8: 737.] [DOI] [PubMed] [Google Scholar]
- 41.Kawaguchi M., Okabe T., Okudaira S., Nishimasu H., Ishitani R., Kojima H., Nureki O., Aoki J., and Nagano T.. 2013. Screening and X-ray crystal structure-based optimization of autotaxin (ENPP2) inhibitors, using a newly developed fluorescence probe. ACS Chem. Biol. 8: 1713–1721. [DOI] [PubMed] [Google Scholar]
- 42.Nishimasu H., Okudaira S., Hama K., Mihara E., Dohmae N., Inoue A., Ishitani R., Takagi J., Aoki J., and Nureki O.. 2011. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 18: 205–212. [DOI] [PubMed] [Google Scholar]
- 43.Fells J. I., Lee S. C., Fujiwara Y., Norman D. D., Lim K. G., Tsukahara R., Liu J., Patil R., Miller D. D., Kirby R. J., et al. 2013. Hits of a high-throughput screen identify the hydrophobic pocket of autotaxin/lysophospholipase D as an inhibitory surface. Mol. Pharmacol. 84: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fells J. I., Lee S. C., Norman D. D., Tsukahara R., Kirby J. R., Nelson S., Seibel W., Papoian R., Patil R., Miller D. D., et al. 2014. Targeting the hydrophobic pocket of autotaxin with virtual screening of inhibitors identifies a common aromatic sulfonamide structural motif. FEBS J. 281: 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Llona-Minguez S., Ghassemian A., and Helleday T.. 2015. Lysophosphatidic acid receptor (LPAR) modulators: The current pharmacological toolbox. Prog. Lipid Res. 58: 51–75. [DOI] [PubMed] [Google Scholar]
- 46.Bhatti S., Kozlov S., Farooqi A. A., Naqi A., Lavin M., and Khanna K. K.. 2011. ATM protein kinase: the linchpin of cellular defenses to stress. Cell. Mol. Life Sci. 68: 2977–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith J., Tho L. M., Xu N., and Gillespie D. A.. 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108: 73–112. [DOI] [PubMed] [Google Scholar]
- 48.Tentner A. R., Lee M. J., Ostheimer G. J., Samson L. D., Lauffenburger D. A., and Yaffe M. B.. 2012. Combined experimental and computational analysis of DNA damage signaling reveals context-dependent roles for Erk in apoptosis and G1/S arrest after genotoxic stress. Mol. Syst. Biol. 8: 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banerjee S., Norman D. D., Lee S. C., Parrill A. L., Baker D. L., Tigyi G. J., and Miller D. D.. 2017. Highly potent non-carboxylic acid autotaxin inhibitors reduce melanoma metastasis and chemotherapeutic resistance of breast cancer stem cells. J. Med. Chem. 60: 1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee S. C., Fujiwara Y., and Tigyi G. J.. 2015. Uncovering unique roles of LPA receptors in the tumor microenvironment. Receptors Clin. Investig. 2: e440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seo E. J., Kwon Y. W., Jang I. H., Kim D. K., Lee S. I., Choi E. J., Kim K. H., Suh D. S., Lee J. H., Choi K. U., et al. 2016. Autotaxin regulates maintenance of ovarian cancer stem cells through lysophosphatidic acid-mediated autocrine mechanism. Stem Cells. 34: 551–564. [DOI] [PubMed] [Google Scholar]
- 52.Gotoh M., Fujiwara Y., Yue J., Liu J., Lee S., Fells J., Uchiyama A., Murakami-Murofushi K., Kennel S., Wall J., et al. 2012. Controlling cancer through the autotaxin-lysophosphatidic acid receptor axis. Biochem. Soc. Trans. 40: 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gupte R., Patil R., Liu J., Wang Y., Lee S. C., Fujiwara Y., Fells J., Bolen A. L., Emmons-Thompson K., Yates C. R., et al. 2011. Benzyl and naphthalene methylphosphonic acid inhibitors of autotaxin with anti-invasive and anti-metastatic activity. ChemMedChem. 6: 922–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson K. E., Ray R. M., Alli S., Ge W., Boler A., Shannon McCool W., Meena A. S., Shukla P. K., Rao R., Johnson L. R., et al. 2018. Prevention and treatment of secretory diarrhea by the lysophosphatidic acid analog Rx100. Exp. Biol. Med. (Maywood). 243: 1056–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Afroz S., Yagi A., Fujikawa K., Rahman M. M., Morito K., Fukuta T., Watanabe S., Kiyokage E., Toida K., Shimizu T., et al. 2018. Lysophosphatidic acid in medicinal herbs enhances prostaglandin E2 and protects against indomethacin-induced gastric cell damage in vivo and in vitro. Prostaglandins Other Lipid Mediat. 135: 36–44. [DOI] [PubMed] [Google Scholar]
- 56.Tanaka T., Morito K., Kinoshita M., Ohmoto M., Urikura M., Satouchi K., and Tokumura A.. 2013. Orally administered phosphatidic acids and lysophosphatidic acids ameliorate aspirin-induced stomach mucosal injury in mice. Dig. Dis. Sci. 58: 950–958. [DOI] [PubMed] [Google Scholar]
- 57.Kihara Y., Mizuno H., and Chun J.. 2015. Lysophospholipid receptors in drug discovery. Exp. Cell Res. 333: 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu J., Oda S. K., Shotts K., Donovan E. E., Strauch P., Pujanauski L. M., Victorino F., Al-Shami A., Fujiwara Y., Tigyi G., et al. 2014. Lysophosphatidic acid receptor 5 inhibits B cell antigen receptor signaling and antibody response. J. Immunol. 193: 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oda S. K., Strauch P., Fujiwara Y., Al-Shami A., Oravecz T., Tigyi G., Pelanda R., and Torres R. M.. 2013. Lysophosphatidic acid inhibits CD8 T cell activation and control of tumor progression. Cancer Immunol. Res. 1: 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khandoga A. L., Fujiwara Y., Goyal P., Pandey D., Tsukahara R., Bolen A., Guo H., Wilke N., Liu J., Valentine W. J., et al. 2008. Lysophosphatidic acid-induced platelet shape change revealed through LPA(1–5) receptor-selective probes and albumin. Platelets. 19: 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng Y., Makarova N., Tsukahara R., Guo H., Shuyu E., Farrar P., Balazs L., Zhang C., and Tigyi G.. 2009. Lysophosphatidic acid-induced arterial wall remodeling: requirement of PPARgamma but not LPA1 or LPA2 GPCR. Cell. Signal. 21: 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McIntyre T. M., Pontsler A. V., Silva A. R., St Hilaire A., Xu Y., Hinshaw J. C., Zimmerman G. A., Hama K., Aoki J., Arai H., et al. 2003. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARgamma agonist. Proc. Natl. Acad. Sci. USA. 100: 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsukahara T. 2012. The role of PPARgamma in the transcriptional control by agonists and antagonists. PPAR Res. 2012: 362361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsukahara T., Matsuda Y., and Haniu H.. 2017. Lysophospholipid-related diseases and PPARgamma signaling pathway. Int. J. Mol. Sci. 18: E2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tsukahara T., Tsukahara R., Yasuda S., Makarova N., Valentine W. J., Allison P., Yuan H., Baker D. L., Li Z., Bittman R., et al. 2006. Different residues mediate recognition of 1-O-oleyllysophosphatidic acid and rosiglitazone in the ligand binding domain of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 281: 3398–3407. [DOI] [PubMed] [Google Scholar]
- 66.Zhang C., Baker D. L., Yasuda S., Makarova N., Balazs L., Johnson L. R., Marathe G. K., McIntyre T. M., Xu Y., Prestwich G. D., et al. 2004. Lysophosphatidic acid induces neointima formation through PPARgamma activation. J. Exp. Med. 199: 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chemin J., Patel A., Duprat F., Zanzouri M., Lazdunski M., and Honore E.. 2005. Lysophosphatidic acid-operated K+ channels. J. Biol. Chem. 280: 4415–4421. [DOI] [PubMed] [Google Scholar]
- 68.Kittaka H., Uchida K., Fukuta N., and Tominaga M.. 2017. Lysophosphatidic acid-induced itch is mediated by signalling of LPA5 receptor, phospholipase D and TRPA1/TRPV1. J. Physiol. 595: 2681–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hernández-Araiza I., Morales-Lazaro S. L., Canul-Sanchez J. A., Islas L. D., and Rosenbaum T.. 2018. Role of lysophosphatidic acid in ion channel function and disease. J. Neurophysiol. 120: 1198–1211. [DOI] [PubMed] [Google Scholar]
- 70.Telezhkin V., Reilly J. M., Thomas A. M., Tinker A., and Brown D. A.. 2012. Structural requirements of membrane phospholipids for M-type potassium channel activation and binding. J. Biol. Chem. 287: 10001–10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khurana S., Tomar A., George S. P., Wang Y., Siddiqui M. R., Guo H., Tigyi G., and Mathew S.. 2008. Autotaxin and lysophosphatidic acid stimulate intestinal cell motility by redistribution of the actin modifying protein villin to the developing lamellipodia. Exp. Cell Res. 314: 530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arora P. D., Janmey P. A., and McCulloch C. A.. 1999. A role for gelsolin in stress fiber-dependent cell contraction. Exp. Cell Res. 250: 155–167. [DOI] [PubMed] [Google Scholar]
- 73.Goetzl E. J., Lee H., Azuma T., Stossel T. P., Turck C. W., and Karliner J. S.. 2000. Gelsolin binding and cellular presentation of lysophosphatidic acid. J. Biol. Chem. 275: 14573–14578. [DOI] [PubMed] [Google Scholar]
- 74.Karliner J. S., Honbo N., Summers K., Gray M. O., and Goetzl E. J.. 2001. The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 33: 1713–1717. [DOI] [PubMed] [Google Scholar]
- 75.Meerschaert K., De Corte V., De Ville Y., Vandekerckhove J., and Gettemans J.. 1998. Gelsolin and functionally similar actin-binding proteins are regulated by lysophosphatidic acid. EMBO J. 17: 5923–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mintzer E., Sargsyan H., and Bittman R.. 2006. Lysophosphatidic acid and lipopolysaccharide bind to the PIP2-binding domain of gelsolin. Biochim. Biophys. Acta. 1758: 85–89. [DOI] [PubMed] [Google Scholar]
- 77.Osborn T. M., Dahlgren C., Hartwig J. H., and Stossel T. P.. 2007. Modifications of cellular responses to lysophosphatidic acid and platelet-activating factor by plasma gelsolin. Am. J. Physiol. Cell Physiol. 292: C1323–C1330. [DOI] [PubMed] [Google Scholar]
- 78.Christofidou-Solomidou M., Scherpereel A., Solomides C. C., Christie J. D., Stossel T. P., Goelz S., and DiNubile M. J.. 2002. Recombinant plasma gelsolin diminishes the acute inflammatory response to hyperoxia in mice. J. Investig. Med. 50: 54–60. [DOI] [PubMed] [Google Scholar]
- 79.Dinubile M. J. 2007. Plasma gelsolin: in search of its raison d’etre. Focus on “Modifications of cellular responses to lysophosphatidic acid and platelet-activating factor by plasma gelsolin”. Am. J. Physiol. Cell Physiol. 292: C1240–C1242. [DOI] [PubMed] [Google Scholar]
- 80.Jordan J. R., Moore E. E., Damle S. S., Eckels P., Johnson J. L., Roach J. P., Redzic J. S., Hansen K. C., and Banerjee A.. 2007. Gelsolin is depleted in post-shock mesenteric lymph. J. Surg. Res. 143: 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]