

Abstract
Chaperone-mediated autophagy (CMA) was the first studied process that indicated that degradation of intracellular components by the lysosome can be selective — a concept that is now well accepted for other forms of autophagy. Lysosomes can degrade cellular cytosol in a nonspecific manner but can also discriminate what to target for degradation with the involvement of a degradation tag, a chaperone and a sophisticated mechanism to make the selected proteins cross the lysosomal membrane through a dedicated translocation complex. Recent studies modulating CMA activity in vivo using transgenic mouse models have demonstrated that selectivity confers on CMA the ability to participate in the regulation of multiple cellular functions. Timely degradation of specific cellular proteins by CMA modulates, for example, glucose and lipid metabolism, DNA repair, cellular reprograming and the cellular response to stress. These findings expand the physiological relevance of CMA beyond its originally identified role in protein quality control and reveal that CMA failure with age may aggravate diseases, such as ageing-associated neurodegeneration and cancer.
Last year, Yoshimori Ohsumi received the Nobel Prize in Medicine or Physiology for his molecular discoveries in autophagy. Work from many groups since the discovery of lysosomes has paved the way to our current understanding of the autophagic mechanisms that mediate the delivery of cytosolic proteins and organelles to lysosomes for degradation1,2. These new findings have proved that autophagy can be very selective in what it targets for degradation (cargo)3, an idea that originally encountered considerable resistance.
It was during this early resistance that the discovery of chaperone-mediated autophagy (CMA) occurred. At that time, the best-characterized form of autophagy was macroautophagy, a process during which cargo that is sequestered in double-membrane vesicles (autophagosomes) is delivered to lysosomes through vesicular fusion4 (Fig. 1). This apparent in bulk trapping of cytoplasm inside autophagosomes was not compatible with the idea of selective degradation. A similar in bulk principle applied to microautophagy, whereby invaginations at the lysosomal membrane (or the vacuole in yeast) internalize cytosolic cargo into small vesicles that then detach into the lumen for degradation5 (Fig. 1).
Fig. 1. Autophagic pathways in mammals.

a Macroautophagy sequesters cytosolic cargo by a delimiting membrane that forms through conjugation of specific proteins among themselves and with lipids in a complex multistep process. The membrane then seals into an autophagosome that is trafficked by microtubules. Fusion of autophagosomes with lysosomes mediates degradation of the trapped cargo. Macroautophagy can be in bulk or selective depending on the cargo sequestered. b Microautophagy entraps cytosolic cargo in small vesicles formed by invagination of the lysosomal membrane either in bulk or selectively via recognition and targeting by heat shock cognate 71 kDa protein (HSC70; also known as HSPA8) and cochaperones that are yet to be determined. c Chaperone-mediated autophagy (CMA) involves the selective degradation of KFERQ-like motif-bearing proteins delivered to the lysosomes via chaperone HSC70 and cochaperones, such as carboxyl terminus of HSC70-interacting protein (CHIP), heat shock protein 40 (HSP40; also known as DNABJ1) and HSP70–HSP90 organizing protein (HOP), and their internalization in lysosomes via the receptor lysosome-associated membrane protein type 2A (LAMP2A). Bottom: evolutionary conservation of each autophagy pathway. CASA, chaperone-assisted selective autophagy; GFAP, glial fibrillary acidic protein; lys-HSC70, lysosomal HSC70; Ub, ubiquitin.
Macroautophagy and microautophagy are now known to be capable of exquisite cargo selectivity. Macroautophagy uses cytosolic receptor proteins (for example, sequestosome 1 (p62)), next to BRCA1 gene 1 protein (NBR1) and NIP3-like protein X (NIX; also known as BNIP3L), that bring cargo and the autophagy machinery together3. This spatial coincidence allows assembly of the autophagy proteins to form a limiting membrane around the cargo. This membrane grows and seals, giving rise to an autophagosome with the cargo and receptors inside. Selective organelle degradation by microautophagy occurs by interaction of organelle proteins with surface proteins at the vacuole, lysosomes or late endosomes6,7, whereas a cytosolic chaperone, the heat shock cognate 71 kDa protein (HSC70; also known as HSPA8), mediates selective protein degradation by micro-autophagy (known as endosomal microautophagy in mammals)8 (Fig. 1).
Selectivity has been associated with CMA since its discovery because of the unique mechanism that underlies lysosomal cargo delivery. Proteins, the only cargo degraded by this pathway, cross the lysosomal membrane one by one. Not all proteins can undergo degradation via CMA. To be CMA substrates, proteins must contain a specific targeting motif in their amino acid sequence9. This motif binds to a cytosolic chaperone (HSC70), which brings the substrate protein to the lysosomal surface for internalization and rapid intralysosomal degradation.
The focus of CMA studies has shifted through the years. Early studies optimized systems to reconstitute CMA in vitro, which helped in understanding chaperone–cargo recognition and substrate lysosomal translocation10,11. Molecular dissection of CMA has been slow because model systems, such as yeast, worms or flies, that accelerated discovery in macroautophagy are of no use in the study of CMA as one of the essential CMA components is absent in these species and, to date, has been described only in birds and mammals12. The introduction of mammalian RNAi technology facilitated the identification of new components and regulators of the CMA pathway13–16. The ability to genetically modulate CMA has been key, in recent years, to linking CMA malfunctioning to human diseases, such as neurodegeneration and cancer17,18. Current studies on CMA at the cellular and organism level are providing a more comprehensive view of its physiological relevance by discovering previously unknown CMA functions19–21. For example, genetic blockage of CMA in mouse liver has revealed a key role for CMA in the regulation of glucose and lipid metabolism, whereas mouse models with compromised CMA in T cells support the requirement for a fully functional CMA to reach maximal T cell activation.
In this Review, we describe recent advances in our understanding of the mechanisms of CMA and the finely tuned orchestration of CMA activity as part of the proteostasis networks. We also discuss new findings on the pathophysiological relevance of CMA, particularly in the context of ageing and two age-related disorders (neurodegeneration and cancer).
Mechanisms of CMA
Biochemical and genetic approaches have helped dissect the steps and molecular machinery involved in CMA. These studies have confirmed the unique characteristics of CMA regarding substrate identification and delivery inside lysosomes when compared to other types of autophagy.
The basis for cargo selectivity
The concept of selective lysosomal degradation by CMA originated from the finding that not all cytosolic proteins could undergo lysosomal degradation. Using ribonuclease A, which is one of the proteins that can be degraded, the late American biochemist and cell biologist Fred Dice and his team identified an 11-amino-acid region in the protein, later narrowed down to the pentapeptide KFERQ, which is necessary and sufficient to target proteins for lysosomal degradation22.
The properties of the residues that constitute the motif, rather than the specific amino acids, determine whether the CMA-targeting chaperone HSC70 can bind to this region9. The motif is always flanked by a glutamine on one of the sides (as the pentapeptide functions as a targeting sequence in both directions) and contains one or two of the positive residues K and R, one or two of the hydrophobic residues F, L, Ior V andone of the negatively charged E or D residues. Approximately 40% of proteins in the mammalian proteome contain a canonical KFERQ-like motif. In addition, in some substrate proteins, the same targeting motif can be generated through post-translational modifications, thus expanding the number of potential CMA substrates. Phosphorylation of S, T or Y present in a motif containing only four of the canonical residues and missing the negatively charged one can complete the motif and convert the protein to a CMA substrate23–26. In some instances, Q can be replaced by K, which upon acetylation acquires properties similar to Q, thus completing the motif 27,28. Ubiquitylation or acetylation of the same K could, in theory, become a switch between proteasomal and lysosomal degradation. Characteristics of canonical or putative CMA motifs and recommendations for motif validation are described in Box 1.
Box 1. Validation of KFerQ-like motifs for cMa targeting.
Despite the dynamic nature that post-translational modifications have conferred to the KFerQ-like motif, it is important to notice that the specific requirements for a motif to function in chaperone-mediated autophagy (CMa) targeting have been established and validated experimentally. several published studies have proposed as CMa-targeting motifs what appear to be randomly chosen sequences. in order to maintain consistency and avoid confusion in the field, it is important that any new proposed motif is validated experimentally according to the following original criteria: elimination of the motif abolishes lysosomal degradation of the protein independent of macroautophagy; insertion of the proposed motif in a well-characterized non-CMa substrate makes it amenable to CMa degradation; and elimination or mutation of residues in the motif suppresses binding of the protein to heat shock cognate 71 kDa protein (HsC70; also known as HsPa8), or if there is still HsC70 binding, it can no longer compete with a KFerQ-containing peptide.
The figure summarizes the types of post-translational modifications to take into consideration as possible enablers or disruptors of HsC70 bind ing to substrate proteins through the KFerQ-like motif (details in the main text). examples of canonical motifs and how post-translational modifications can complete a motif are shown in the left panel, whereas the right panel illustrates how post-translational modifications in a protein outside the motif can change the protein conformation and unmask the KFerQ-like motif.
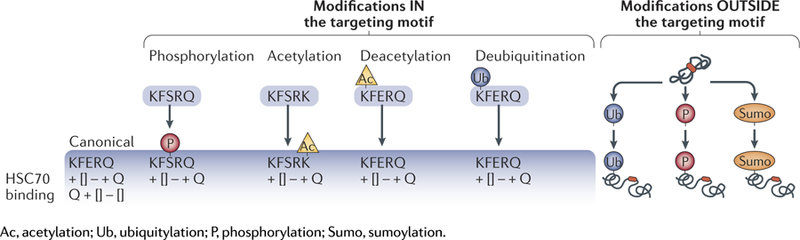
Post-translational modifications outside the motif can also modulate CMA targeting of proteins bearing canonical motifs by facilitating conformational changes that expose or mask the motif. For example, CMA-dependent degradation of hypoxia-inducible factor 1α (HiF1α), a validated CMA substrate, takes place only when it is ubiquitylated on Lys63 by the E3 ubiquitin-protein ligase STUB1 (REF.29). By contrast, acetylation of mammalian Ste20-like kinase 1 (MST1; also known as STK4) in a residue far from the canonical CMA motif prevents its lysosomal degradation, and only upon deacetylation can HSC70 bind the canonical motif 30.
HSC70 involvement in CMA
The KFERQ-like motif provided the ‘bait’ to identify HSC70 as the cytosolic chaperone that, upon binding that region, targeted proteins for lysosomal degradation31. Although cochaperones, such as carboxyl terminus of HSC70-interacting protein (CHIP), heat shock protein 40 (HSP40; also known as DNABJ1) and HSP70–HSP90 organizing protein HOP modulate substrate targeting to lysosomes in an HSC70-dependent manner32,33, to date, HSC70 remains the only chaperone proved to directly bind the KFERQ-like motif. HSC70 participates in multiple cellular functions by facilitating folding of unfolded or misfolded proteins. HSC70 binds hydrophobic regions to assist in protein folding. In contrast, HSC70 binds KFERQ-like motifs to target proteins for degradation via CMA.
The relationship of cytosolic HSC70 with autophagy has expanded beyond CMA. In cooperation with the cochaperones BAG family molecular chaperone regulator 1 (BAG1) and BAG3, HSC70 participates in the selective degradation of ubiquitin-positive protein aggregates by a type of macroautophagy known as chaperone-assisted selective autophagy (CASA)34 (Fig. 1). The search for a process in mammalian cells that is homologous to yeast microautophagy led to the discovery of endosomal microautophagy, in which cytosolic proteins enter endosomal compartments inside vesicles generated at the surface of late endosomes through the assembly of endosomal sorting complexes required for transport (ESCRT)8. Although cytosolic proteins are trapped in a nonselective manner as these vesicles form, a subset of cytosolic proteins are selectively targeted there by HSC70 (REF.8) (Fig. 1). As in CMA, HSC70 binds endosomal microautophagy cargo proteins through a KFERQ-like motif. HSC70 is thus at the centre of protein triage between three different types of autophagy in mammals.
Discovery of selective endosomal microautophagy8 has necessitated revising the criteria for bona fide CMA substrates. Dependence on HSC70 for lysosomal degradation, for many years a distinctive characteristic of CMA substrates, is now insufficient to attribute degradation to this pathway. Dependence on the lysosomal membrane receptor lysosome-associated membrane protein type 2A (LAMP2A, discussed below), is the best criteria to determine whether protein degradation occurs via CMA, as LAMP2A is not required for CASA34 or endosomal-microautophagy8. Whereas HSC70 requires LAMP2A for lysosomal docking in CMA, the chaperone binds directly to lipids at the endosomal membrane in endosomal microautophagy8.
A fraction of cellular HSC70 that is constitutively present in lysosomes, both at their membrane cytosolic side and in the lysosomal lumen, also participates in CMA15,35 (Fig. 2). Membrane-bound HSC70 in a complex with the cochaperones HSP90, HSP40, HSP70-interacting protein (HIP) and HOP has been proposed to participate in substrate unfolding33, which is required before lysosomal translocation36. Membrane HSC70 is also required to sustain continuous cycles of CMA because after the substrate is internalized, HSC70 facilitates dissociation of the receptor protein from the CMA translocation complex to allow for binding of new substrates15.
Fig. 2. Lysosomal effectors and regulators of CMA.

Steps in selective degradation of proteins by chaperone-mediated autophagy (CMA): recognition of the KFERQ-like motif in the substrate by heat shock cognate 71 kDa protein (HSC70; also known as HSPA8) (step 1); binding of the substrate–chaperone complex to lysosome-associated membrane protein type 2A (LAMP2A) (step 2); unfolding of the substrate by the chaperone complex (step 3); formation of the CMA translocation complex (step 4); substrate translocation mediated by lysosomal HSC70 (lys-HSC70) (step 5); substrate degradation by lysosomal proteases (step 6); and dissociation of LAMP2A from the translocation complex (step 7). CMA regulation via cytosolic and lysosomal signalling events is shown. Turnover of LAMP2A occurs in lipid microdomains by the dual action of cathepsin A and a metalloproteinase. Top right: activators and inhibitors of CMA. EF1α, elongation factor 1-α; GFAP, glial fibrillary acidic protein; HSP90, heat shock protein 90; mb-HSC70, membrane-associated HSC70; NFAT1, nuclear factor of activated T cells 1; PHLPP1, PH domain leucine-rich repeat-containing protein phosphatase 1; RAR, retinoic acid receptor; TORC2, TOR complex 2.
The second form of lysosomal HSC70 is located in the lumen35,37 (Fig. 2). The stability of HSC70 in this acidic environment with high concentrations of proteases relies on specific properties of the luminal HSC70. Although both cytosolic and lysosomal HSC70 originate from the same gene, it was shown that luminal HSC70 has a highly acidic isoelectric point, which is very different from the neutral isoelectric point of the cytosolic chaperone37. Luminal HSC70 becomes unstable with small changes in lysosomal acidification, which are physiologically used to modulate the percentage of lysosomes competent for CMA at a given time35. As luminal HSC70 is necessary to complete substrate translocation, lysosomes lacking this chaperone cannot perform CMA. An unresolved question is how HSC70 gains access to the lysosomal lumen. Blockage of macro-autophagy or CMA does not reduce the levels of luminal HSC70 (REF.13,38), thus discarding the possibility that it might be internalized through these pathways. It is possible that HSC70 internalization from the cytosol occurs into late endosomes through microautophagy and that lysosome–endosome fusion facilitates HSC70 transfer to the lysosomal lumen.
Diverse functions of the lysosomal receptor LAMP2A
LAMP2A was the first identified lysosomal component required for CMA39. Participation of a receptor protein in CMA was inferred because substrate internalization in lysosomes was saturable and trypsinization of the surface of lysosomes (to remove protein components) ablated substrate binding and internalization35,40. LAMP2A is one of the three splice variants of a single gene, LAMP212. LAMP2A, LAMP2B and LAMP2C have identical luminal regions but different transmembrane and cytosolic regions. LAMP2A is the only one of the three isoforms required for CMA39,41 (Fig. 2). Although LAMP2A may share with LAMP2B and LAMP2C some of the functions attributed to the luminal region that is identical in the three LAMP2 variants42, it is not the most abundant of the three proteins, and its blockage remains, to date, the most specific way to inhibit CMA. LAMP2A contributes to different steps of CMA.
Substrate binding.
The 12-amino-acid cytosolic tail of LAMP2A is required for lysosomal docking of HSC70– substrate complexes39,41. HSC70 and the substrate can bind this tail at the same time, suggesting that substrate recognition and targeting are coupled processes39,41,43; however, the KFERQ-like motif in the substrate is not required for LAMP2A binding39. Three positive residues in the cytosolic tail of LAMP2A are essential to complete CMA of substrates41, leaving open the possibility that transferring of the unfolding substrate requires interaction only with negatively charged residues in the substrate protein. Studies using an unfoldable artificial substrate demonstrated that unfolding is not required for lysosomal surface binding but is necessary for its translocation36.
LAMP2A assembly.
Multimerization of LAMP2A into a 700 kDa multimeric protein complex is essential for substrate translocation into the lumen14,15 (Fig. 2). Assembly of this translocation complex occurs step-wise and includes the formation of an intermediate LAMP2A homotrimer44 that increases substrate affinity and probably prevents its aggregation as the substrate unfolds44. Cytosolic HSC70 associates with LAMP2A monomers and homotrimers, but it is released before the fully functional translocation complex is assembled15. A form of HSP90 associated at the luminal part of the lysosomal membrane interacts with and stabilizes LAMP2A during this transition15 (Fig. 2). HSP90 may cover protease-sensitive regions of LAMP2A that are otherwise exposed during its conformational arrangement for multimeric assembly15.
Regulation of lysosomal levels of LAMP2A.
Luminal HSC70 is required for CMA, but LAMP2A is the rate-limiting component of this pathway. Changes in levels and dynamics of LAMP2A at the lysosomal membrane regulate CMA flux (that is, the rate at which cargo is degraded). De novo synthesis of LAMP2A occurs during mild oxidative stress21,45, genotoxic damage24 or hypoxia32,46 ‒ all conditions that induce CMA activation. This transcriptional upregulation of LAMP2A is still poorly understood. The transcription factor nuclear factor of activated T cells (NFAT1) has been shown to directly promote the transcriptional upregulation of LAMP2A during T cell activation21 (Fig. 3). Transcription of several CMA components, including LAMP2A, have been shown to be under the negative control of the nuclear retinoic acid receptor-α47. Although most lysosomal proteins, including other LAMPs, are part of the programme of the transcription factor EB (TFEB), LAMP2 transcription is not regulated, at least directly, by this factor48.
Fig. 3. Main physiological roles of CMA.

In most cells, chaperone-mediated autophagy (CMA) participates in protein quality control by degrading oxidized and damaged proteins under stress conditions and also contributes amino acids through degradation of proteins at advanced times of starvation. In addition, depending on the protein substrate degraded, CMA has a modulatory role in multiple cellular pathways. This CMA-mediated selective remodelling of the proteome has recently demonstrated a role for CMA in modulation of carbohydrate and lipid metabolism, transcriptional programmes, immune responses and the cell cycle. The selective CMA substrates linked to those pathways are shown in blue boxes. GFAP, glial fibrillary acidic protein; HSC70, heat shock cognate 71 kDa protein; HSP90, heat shock protein 90; LAMP2A, lysosome-associated membrane protein type 2A; lys-HSC70, lysosomal HSC70; TCA, tricarboxylic acid.
The increase in LAMP2A levels in other conditions that activate CMA, such as starvation, does not involve de novo synthesis of LAMP2A and is instead mediated through changes in LAMP2A degradation at the lysosomal membrane, which is a finely controlled process43,49 (Fig. 2). LAMP2A is usually well protected from the luminal proteases by its abundant glycosylation and unique spread conformation against the inner leaflet of the membrane50. Degradation of LAMP2A requires its mobilization to lipid microdomains49,51, where it undergoes a dual cleavage by cathepsin A and a yet unidentified metalloproteinase to be released into the lumen for rapid degradation51 (Fig. 2). Changes in the lipid composition of the lysosomal membrane with age or upon high-lipid dietary challenges accelerate this regulated degradation of LAMP2A because it becomes trapped in the expanded lipid microdomains52,53. The half-life of LAMP2A in lysosomes is approximately 36 h, but it becomes more than 72 h in hepatocytes and in fibroblasts in culture during prolonged starvation51. This reduced degradation leads to a net increase in LAMP2A levels in lysosomes. Mobilization of a fraction of LAMP2A resident in the lysosomal lumen (likely in micelles) to the lysosomal membrane is responsible for the further increase in LAMP2A levels at the lysosomal membrane observed when starvation persists beyond 48 h (REF.43).
There is no evidence yet that Golgi complex-to-lysosome trafficking of LAMP2A modulates CMA activity under physiological conditions. However, mis-targeting of LAMP2A has been identified as the basis of reduced CMA observed in the lysosomal storage disorder cystinosis54. Defective trafficking seems specific for LAMP2A, as other LAMPs reach lysosomes normally in this disease, thus suggesting the existence of LAMP2A-dedicated Golgi-to-lysosomal transport machinery. The small GTPase Ras-related protein Rab- 11A (RAB11) and the RAB7 effector Rab-interacting lysosomal protein (RILP) are likely part of this transport machinery, as correction of the lower levels observed for both proteins in patients is sufficient to correct LAMP2A mistargeting in disease cells55. Small molecules that activate CMA by releasing the inhibitory effect of retinoic acid receptor-α (RARα) on several CMA components47 also correct LAMP2A trafficking55. Altered endosome-to-Golgi retrieval of LAMPs has been found in cells bearing mutations in the retromer component vacuolar protein sorting-associated protein 35 (VPS35). These mutations have been identified in patients with Parkinson disease (PD)56, which was the first neurodegenerative disorder associated with defective CMA activity17. However, contrary to the selectivity for LAMP2A observed in cystinosis, VPS35 mutations alter trafficking of multiple lysosomal proteins, suggesting a more general lysosomal malfunction in these conditions56.
Dynamics of LAMP2A assembly.
Changes in rates of LAMP2A assembly and disassembly from the translocation complex impact overall CMA activity (Fig. 2). A pair of proteins, the monomeric form of the intermediate filament protein glial fibrillary acidic protein (GFAP) and its GTP-binding partner elongation factor 1-α (EF1α) modulate the stability of multimeric LAMP2A in a GTP-dependent manner14. Part of GFAP at the lysosomal membrane is phosphorylated and bound to EF1α, and part is unmodified and bound to the LAMP2A multimeric complex, conferring stability14. In the presence of GTP, EF1α dissociates from phospho-GFAP, thus promoting mobilization of GFAP from the translocation complex to phospho-GFAP and the subsequent disassembly of LAMP2A14.
Conditions that change the lipid composition of the lysosomal membrane impact CMA. High-lipid-content diets change the lysosomal membrane lipidome, which compromises both LAMP2A stability and its ability to multimerize52. Interestingly, changes in the lysosomal membrane lipidome with age have comparable effects on LAMP2A stability and dynamics52. It is plausible that interventions to modify lipid membrane composition could be utilized to modulate CMA activity under pathological conditions.
Physiological regulation of CMA
CMA was initially described as part of the cellular response to stress, but basal CMA activity is detectable in almost all mammalian cells57. The tools and assays currently available to study CMA are summarized in TABLES 1,2. Basal levels of CMA differ widely among cell types and tissues, suggesting possible differences in their dependence on CMA. CMA is upregulated in response to a large variety of stressors, such as starvation, oxidative stress, genotoxic insults, lipid challenges, hypoxia and radiation24,29,40,45,58,59. Integration of CMA with the rest of the cellular stress response requires fine-tuning of CMA activity. Although much is yet to be discovered about physiological regulators of CMA, cues from cytosolic signalling pathways and signalling events at the lysosomal membrane have been shown to regulate CMA (Fig. 2).
Table 1.
The CMA toolkit: overview of the approaches that can be used to measure CMA activity
| assay | Principle and description | Key features |
|---|---|---|
| CMA reportera | Uses photoconvertible and photoactivable fluorescent reporters that upon conversion change colour or upon activation start to fluoresce. CMA activation is detected as a change in the fluorescence pattern, from cytosolic diffuse to lysosomal punctate pattern, and is quantified as number of puncta per cell | • Pulse-chase experiments • CMA activity measured in intact cells in culture • Test sample size is not a limitation |
| Lysosomal uptakea | Intact isolated lysosomes pretreated or not with protease inhibitors are incubated with a known CMA substrate, and centrifugation after incubation and immunoblot is used to measure the amount of substrate associated with lysosomes | • In vitro assay • Direct measure of CMA activity • Binding and uptake can be separately measured |
| Lysosomal protein degradationa | Intact isolated lysosomes are incubated with radiolabelled protein substrates | • In vitro assay • Direct measure of CMA activity • Recapitulates binding, uptake and degradation |
| Dynamics of CMA translocation complex | Isolated lysosomes are subjected to blue-native electrophoresis (immunoblot) to assess the amount of lysosomal LAMP2A present in the 700 kDa multimeric translocation complex | • In vitro assay • Provides information on CMA dynamics • Useful to determine possible failures in this CMA step |
| Protein degradation by CMA | Metabolic labelling of intact cells and measurement of the conversion of labelled protein to free-labelled amino acids in the presence or absence of lysosomal inhibitors (to block all forms of autophagy) and of macroautophagy inhibitors | • CMA activity measured in intact cells • Possible compensatory changes • Assesses other forms of autophagy in the same experiment |
| Immunofluorescence | CMA-active lysosomes are labelled by costaining with antibodies against LAMP2A and HSC70. | • Indirect measurement of CMA • Measures number of lysosomes performing CMA • Useful to determine changes in CMA-related compartments |
For comprehensive details on assays and methodology to measure CMA, see REF.117. CMA, chaperone-mediated autophagy; HSC70, heat shock cognate 71 kDa protein; LAMP2A, lysosome-associated membrane protein type 2A.
Gold-standard assays in the field.
Table 2.
The CMA toolkit: overview of the approaches that can be used to identify CMA substrates
| Substrate feature | Description of the assay | Additional notes and features |
|---|---|---|
| Presence of KFERQ motif | • Scan for the motif in the amino acid sequence of the protein of interest • Incomplete motifs could be converted to a complete motif by post-translational modifications (Box 1) |
Also present in eMI substrates |
| Association with lysosomes | • Colocalization with CMA-active lysosomes (HSC70+) in intact cells • Immunoblot in isolated lysosomes to demonstrate preferential association with CMA-active lysosomes |
Proteins can associate with lysosomes to be functional |
| Degradation in lysosomes | Increased association of the protein of interest with CMA-active lysosomes upon lysosomal inhibition assayed by either immunofluorescence in intact cells or immunoblot of isolated lysosomes | Levels of substrates for other autophagic pathways also increase |
| Change in protein levels and degradation when CMA activity is modified | • Increased levels of the protein of interest or reduced degradation rates upon blockage of CMA (knockdown or knockout for LAMP2A) • Reduced levels of the protein of interest or increased degradation rates upon activation of CMA (overexpression of LAMP2A or AR7 compound) |
Compensatory upregulation of other forms of autophagy might mask this effect |
| Interaction with CMA components | Coimmunoprecipitation of the protein of interest with HSC70 in cytosolic fractions or LAMP2A in lysosomal fractions | Binding to HSC70 is not a sufficient criterion as it can occur because of unfolding or targeting to other pathways |
| Lysosomal uptake | In vitro assay with intact isolated lysosomes pretreated or not with protease inhibitors incubated with the protein of interest | Most conclusive proof of being a CMA substrate |
For comprehensive details on assays and methodology to validate a protein as a CMA substrate, see REF.117. CMA, chaperone-mediated autophagy; eMI, endosomal microautophagy; HSC70, heat shock cognate 71 kDa protein (also known as HSPA8); LAMP2A, lysosome-associated membrane protein type 2A.
NFAT and calcium signalling
The first signalling pathway identified in the activation of CMA was the calcineurin–NFAT pathway, required for CMA activation in T cells21, which also provided novel insights on CMA activation in response to oxidative stress45. The transcription factor NFAT1 directly binds the lamp2 proximal promoter region, which contains several putative NFAT1-binding sites. Generation of reactive oxygen species (ROS) during T cell activation promotes the nuclear translocation of NFAT1 and the subsequent increase in lamp2a expression. Both calcineurin inhibition by cyclosporine A or blocking ROS production ablate CMA activation in these cells21. T cell receptor (TCR)-induced CMA activation favours CMA degradation of TCR signalling inhibitors such as E3 ubiquitin-protein ligase Itchy homolog (ITCH) and regulator of calcineurin 1 (RCAN1) — a requirement for maintaining T cell activation in these cells21.
RARα signaling
Signalling through RARα, but not through other abundant members of this nuclear receptor family (such as RARβ, RARγ or RXR), inhibits CMA47. Genetic and chemical blockage of RARα demonstrate that not only LAMP2A47 but also proteins that facilitate LAMP2A trafficking to lysosomes, such as RAB11 or RILP, are also under the negative regulation of this receptor55. It is thus likely that RARα controls a discrete CMA-related transcriptional programme. In fact, structure-based chemical design has made possible generating synthetic derivatives of all-trans retinoic acid that block only the inhibitory effect of RARα on CMA without affecting the RARα-dependent transcriptional programme47. Chemical upregulation of CMA with these compounds rendered cellular protection from oxidative stress and proteotoxicity47. Given the ubiquitous expression of RARα, these atypical RARα inhibitors hold great promise in the systemic restoration of impaired CMA in ageing.
The TORC2–AKT1–PHLPP1 axis
Physiological regulation of CMA activity also occurs directly at the lysosomal membrane. As previously mentioned, dynamics of the CMA translocation complex depend on GFAP phosphorylation status14, which determines its oligomeric state. Lysosomal GFAP phosphorylation is performed by a form of AKT1 that resides at the lysosomal membrane16. AKT1 activity is closely monitored by the phosphatase PH domain leucine-rich repeat-containing protein phosphatase 1 (PHLPP1) and the kinase TOR complex 2 (TORC2)16 at the lysosomal membrane (Fig. 2). Lysosomal TORC2 phosphorylates AKT1, which, by phosphorylating GFAP, puts a break on CMA activity. Contrary to TORC1, TORC2 does not shuttle and is always present at the membrane of CMA-active lysosomes16. TORC2 and AKT1 exert sustained inhibition on CMA as chemical TORC2 inhibitors such as torin (at concentrations that do not affect TORC1), knockdown of the TORC2 component rapamycin-insensitive companion of mTOR (RICTOR), or chemical or genetic blockage of AKT1 all lead to CMA induction16. The TORC2–AKT1-axis inhibition on CMA is physiologically released through RAC1-dependent recruitment of the phosphatase PHLPP1 to the lysosomal membrane whenever CMA activation is needed16. PHLPP1 dephosphorylates AKT1, and inactivation of this kinase makes dephosphorylated GFAP available to accelerate the rounds of LAMP2A assembly and disassembly16 (Fig. 2). Confining these signalling events to the lysosomal membrane may allow fine-tuning of CMA to rapidly adjust to the cellular needs. Interestingly, several of the CMA regulators at the lysosomal membrane (that is, GFAP, EF1α, RICTOR and AKT1) contain CMA-targeting motifs and undergo some, albeit discrete, lysosomal degradation14,16. It is tempting to speculate that the targeting motif may be used not only for degradation but, in some instances, for delivery of CMA regulators to the lysosomal membrane and that degradation of these regulators by CMA may be a way to terminate their stimulatory or inhibitory effect on CMA.
Physiological roles of CMA
The diverse nature of already identified CMA substrates and the additional potential CMA substrates that can be generated by post-translational modification justifies the variety of cellular processes impacted by CMA (Fig. 3). Early studies emphasized the role of CMA in protein quality control and considered abnormally synthesized or post-translationally damaged KFERQ-containing proteins as main CMA substrates45. However, the finding that during starvation most KFERQ-bearing proteins can undergo CMA degradation revealed that protein damage is not a requirement for CMA targeting. There is evidence that properly folded, fully functional proteins can become CMA substrates in specific cellular scenarios and that their timely selective degradation by CMA contributes to the regulation of the cellular pathways in which these proteins participate. Experimental approaches to designate a protein as a CMA substrate are described in TABLE 2.
CMA and protein quality control
CMA activation is often triggered by stressors that cause protein damage, such as mild oxidative stress45, hypoxic stress32,58 or protein denaturation60, and in fact, CMA-incompetent cells and organs accumulate oxidized and aggregated proteins13,20 (Fig. 3). CMA cannot degrade proteins once they aggregate, but it is part of the first line of defence against protein aggregation by mediating degradation of single proteins upon damage or partial unfolding17,61. Interestingly, in tissues such as the liver, loss of proteostasis is not imminent upon CMA blockage because compensatory upregulation of macroautophagy and the proteasome can handle the load of CMA substrates19. However, ageing or additional stressors (that is, oxidative stress or lipid challenge) increase protein damage and aggregation in CMA-incompetent tissues, where the compensatory systems become insufficient to maintain proteostasis20. In further support of the role for CMA in protein quality control, both genetic and chemical upregulation of CMA in vitro and in vivo reduce levels of oxidized and aggregated proteins and improve cellular resistance against proteotoxicity47,62.
CMA in the response to starvation
Lack of nutrients activates CMA in most organs and cell types tested22,40,59. Considering the role of autophagy in adaptation to starvation and the timing of CMA activation — after at least 8 h of starvation13,63, when macroautophagy has already peaked — it was suggested that starvation-induced CMA provides cells with free amino acids for the generation of energy and for the synthesis of crucial proteins required for cell function (Fig. 3). In organs such as the liver, proteolysis through macroautophagy lasts for about 8 h of starvation and then switches to preferential degradation of lipids64. In the face of nutrient deprivation beyond 10 h, the pool of intracellular amino acids may be mostly replenished through CMA, which is upregulated and sustained at this high level for up to three days in rodents40. CMA generates amino acids that sustain protein synthesis but some amino acids, such as alanine and glutamine, also provide the carbon skeletons for gluconeogenesis. Interestingly, tissues with high gluconeogenesis rates, such as the liver and kidney, are very active for CMA. In support of the contribution of CMA to cellular energetics, in vitro and in vivo models with impaired CMA exhibit reduced levels of ATP during nutrient deprivation13,18,19, while genetic restoration of CMA in aged mice normalizes their ATP levels62. The ability of CMA to generate free amino acids through degradation has been proposed to be utilized by pathogens, such as Salmonella, which recruits CMA components to gain host-derived peptides for its growth65.
The signals that activate CMA during starvation are still for the most part unknown. Ketone bodies, the choice fuel in prolonged starvation, activate CMA59, and hydrolysis of amino acids derived from CMA generate α-ketoacids themselves, thus establishing a positive feedback loop between CMA and energy production. Intriguingly, CMA activation by ketone bodies occurs through generation of ROS59, suggesting that similar mechanisms to those described during T cell activation are in play.
An interesting combination of CMA-dependent quality control and cellular energetics has been recently described to occur through the CMA degradation of oxidatively damaged nonfunctional mitochondrial enzyme protein/nucleic acid deglycase DJ-1 (PARK7)66. By efficiently degrading this mitochondrial protein, CMA helps preserve mitochondrial function and should have a positive impact on cellular energetics66.
Recent studies in mice with CMA-incompetent livers have revealed a new level of complexity in the interplay between CMA and cellular energetics whereby, as described in the next section, CMA regulates the rates of metabolic pathways through timely selective degradation of key enzymes in these pathways19,67.
Selective proteome remodelling by CMA
Protein degradation is extensively utilized by cells to downregulate cellular processes in which these proteins participate. The selectivity of CMA makes it suitable for this type of regulatory degradation, which underlies the basis of some of the recent novel physiological functions attributed to CMA (Fig. 3)
Regulation of metabolic pathways.
CMA contributes to the regulation of glucose and lipid metabolism through selective degradation of key enzymes in these pathways (Fig. 3). Studies in mice have revealed that basal degradation of many metabolic enzymes via CMA in the liver is accelerated several-fold during fasting19,40. Timely CMA degradation of glycolytic enzymes contributes, for example, to shutting off hepatic glycolysis during starvation, and failure to do so leads to abnormally elevated rates of hepatic glycolysis and the subsequent energy deficiency in peripheral organs19. Comparative proteomic analysis of lysosomes from CMA-defective mouse livers and from control littermates confirmed that the majority of the glycolytic enzymes and some of the enzymes in the tricarboxylic acid cycle undergo CMA degradation during starvation19. Abnormal upregulation of CMA activity, such as that observed in patients with tripeptidyl peptidase II (TPPII) mutations68 can also become deleterious for cellular energetics. In this case, CMA activation is secondary to the lysosomal expansion that occurs upon the loss of TPPII in an attempt to maintain the pool of free amino acids normally provided by this peptidase. The caveat, however, is that general lysosomal upregulation increases degradation via CMA of hexokinase 2, a key glycolytic enzyme, which further impairs cellular energetics in these patients68.
CMA also contributes to regulate lipid metabolism (Fig. 3). Proteins involved in lipid metabolism — lipogenesis enzymes, lipid carriers and lipid droplet coat proteins — have been identified as bona fide CMA substrates19,67. Control of both lipogenesis and lipolysis by CMA underlies the basis of the marked derangements in lipid usage and pronounced hepatosteatosis observed in CMA-incompetent mouse liver19. CMA facilitates lipolysis by selective degradation of lipid droplet coat proteins, such as perilipins 2 and 3 (PLIN2 and PLIN3)67. Selective removal of these proteins from the surface of the lipid droplets by CMA is required to facilitate access of cytosolic lipolytic enzymes and the macroautophagy machinery to the lipids stored in the core of the droplets to initiate both lipolysis and lipophagy67. These findings place CMA upstream of both lipolytic pathways. Timely removal of lipid droplet coat proteins by CMA during starvation is triggered in part via 5’-AMP-activated protein kinase (AMPK)-dependent phosphorylation of PLIN2 (REF.23). CMA contributes to keeping intracellular lipid levels in check by reducing lipogenesis and increasing lipolysis19. In fact, cells upregulate CMA in response to acute lipid challenges, and failure to do so results in cellular toxicity52.
Transcriptional regulation by CMA.
Additional regulatory roles for CMA, beyond the control of metabolic pathways, have been recently identified (Fig. 3). CMA regulates transcriptional programmes mediated by nuclear factor-κB (NF-κB) by degrading its inhibitor NF-κ-B inhibitor-α (IκBα) during starvation69. Timely CMA degradation of paired-box protein PAX2, a transcription factor important for cell proliferation and differentiation70, maintains renal size and growth, whereas CMA protects neurons against stressors through selective degradation of the transcription factors myocyte-specific enhancer factor 2A (MEF2A) and MEF2D, which are required for neuronal survival71,72.
CMA and the immune response.
CMA is important for CD4+ T cell activation because it selectively degrades two negative regulators of TCR signalling, namely, ITCH and RCAN1 (REF.21) (Fig. 3). Persistent high levels of these two factors in CMA-incompetent T cells are responsible for their reduced cell proliferation and cytokine secretion following activation. These observations unveil an important link between CMA activity and the immune response, further supported by the fact that mice with defective T cell CMA are compromised in their responses to immunization and to infection by pathogens such as Listeria monocytogenes21. Recent studies have revealed contribution of CMA to the regulation of innate immunity through degradation of stimulator of interferon genes protein (STING), a signalling protein in the response against cytosolic nucleic acids73. The trigger for STING degradation is desumoylation, which unmasks its KFERQ-like motif during the late phase of viral infection to shut off the innate immune response73.
Control of cell cycle.
Following genotoxic insults, CMA is required to initiate cell cycle progression after DNA repair has been completed24. This effect is mediated in part by degrada tion of phosphorylated serine/ threonine-protein kinase CHK1 (also known as cell cycle checkpoint kinase), which in the absence of CMA, stays in the nucleus, interfering with DNA repair24 (Fig. 3). CMA also degrades HIF1α32,46, another regulator of cell cycle progression, thus adjusting its levels to the different phases of the cell cycle under conditions of hypoxia. This close regulation is possible through the opposite effects of a pair of cyclin-dependent kinases, whereby CDK1 blocks HIF1α lysosomal degradation, and CDK2 promotes it46. K63 ubiquitylation appears to be the trigger for degradation of HIF1α via CMA29.
Anti-ageing functions of CMA
Age-dependent decrease in CMA activity occurs in almost all cell types and tissues in rodents and in humans21,22,62,74,75. Lower stability and therefore reduced levels of LAMP2A at the lysosomal membrane is one of the main causes of decline in CMA activity53. Changes in the lipid composition of the lysosomal membrane with age are responsible for the loss of LAMP2A stability49,52,53. Recently generated, tissue-specific AMP2A-knockout mice19 have begun to expand our understanding of the organism’s reaction to CMA malfunctioning and the consequences of reduced function of this pathway with age. Studies in cultured cells have previously demonstrated crosstalk between macroautophagy and CMA, whereby cells respond to blockage of one of these pathways by upregulating the other13,38. Similar positive compensation by macroautophagy has also been found in vivo in mouse models with selective blockage of CMA in liver or in T cells19,21. However, studies in mouse retina suggest that compensation for CMA failure is not universal75. While the age-related decline in retinal macroautophagy coincides with pronounced upregulation of CMA, blockage of CMA does not elicit a compensatory activation of macroautophagy in this organ75, thus explaining its higher sensitivity to stress upon CMA blockage75. In general, although macroautophagy and CMA can compensate for some of each other’s functions, they are not redundant, and their loss of function, even when the other pathway is upregulated, becomes evident. For example, CMA-incompetent livers do not accumulate damaged or aggregated proteins because increases in macroautophagy and the proteasome take over some of the CMA protein quality control functions. However, owing to the different timing of activation and substrate-targeting mechanisms of these two pathways, they cannot compensate for CMA regulatory functions and problems with metabolic flux, DNA repair, and so on, which become evident in these animal livers19. Furthermore, upregulation of macroautophagy and the proteasome in CMA-incompetent livers is rapidly lost when animals are subjected to stressors (that is, oxidative stress and lipid challenges) and as the animals age20. Compensation failure with age can explain the gradual loss of protein homeostasis and higher sensitivity to stress of aged AMP2A-knockout mice livers.
Genetic interventions to restore CMA function in old mouse liver have shown remarkable improvement in age-related changes in this organ. Expressing an inducible exogenous copy of Lamp2a protected aged livers from stressors and led to overall improvement in proteostasis and organ function62. Preventing the systemic decline of CMA with age may prove an attractive strategy against organism functional loss and age-associated disorders.
The contribution of CMA failure to human disease
A better understanding of CMA dynamics, the identification of novel CMA molecular effectors and regulators and the development of tools to track CMA (TABLES 1,2) have all contributed to linking CMA malfunctioning to a growing number of human diseases. We have selected two types of pathologies with defective CMA to illustrate examples in which diminished or increased CMA activity contributes to disease progression. A more comprehensive description of other CMA-related diseases, summarized in TABLE 3, can be found in recent reviews76,77.
Table 3.
Human diseases associated with defects in CMA
| Disease subtype | CMA substrate | CMA activity | Observations | Refs |
|---|---|---|---|---|
| Cancer | ||||
| Malignant transformation | CIP2A | Reduced | CMA regulates levels of MYC oncogene | 114 |
| TCTP (acetylated) | Reduced | CMA regulates levels of TCTP | 28 | |
| MDM2 | Reduced | CMA degrades the oncogene MDM2 | 115 | |
| Unknown | Reduced | CMA is required for efficient exposure of ecto-CRT (eat me signal) in immunogenic cell death | 116 | |
| Tumour survival and/or growth | Unknown | Increased | CMA required for Warburg effect | 18 |
| PEA-15 | n.d. | CMA degrades the anti-oncogenic variant | 25 | |
| PKM2 | Increased | CMA required for Warburg effect | 27 | |
| BBC3a | Increased | CMA degrades this pro-apoptotic protein, reducing cancer cell apoptosis | 110 | |
| Breast cancer | MST1 (de-acetylated) | Increased | CMA favours degradation of tumour suppressor MST1 | 30 |
| Oxidized proteins | Increased | CMA provides cancer cells chemoresistance and defence against ROS | 101 | |
| HSD17B4 (acetylated) | Increased | Failure to acetylate makes the protein resistant to CMA; favours cancer | 102 | |
| Colorectal cancer | P300 | Increased | CMA of acetyltransferase p300 confers 5-fluorouracil resistance | 103 |
| Gastric cancer | RND3 | Increased | CMA degradation of RND3 sustains proliferation | 26 |
| Hepatocellular carcinoma | Unknown | Increased | LAMP2A blockage reduces cancer cell viability | 104 |
| HMGB1 | n.d. | CMA degrades HMGB1 to downregulate p53, and this confers resistance to irradiation-induced apoptosis | 105 | |
| Cyclin D1? | Increased | Macrophage-secreted IL-17 increases LAMP2A — proposed mechanism for oxaliplatin resistance | 107b | |
| Unknown | Increased | Increased LAMP2A levels proposed to promote cancer cell survival in the cirrhotic liver | 106b | |
| Non-small-cell lung cancer | MCL1 | n.d. | CMA stabilizes the prosurvival protein MCL1 | 111 |
| NCOR | n.d. | CMA degrades misfolded NCOR reducing ER stress | 108 | |
| Myeloid leukaemia | AF1Qa | n.d. | CMA degrades the poor prognosis biomarker AF1Q | 118 |
| Hexokinase II | n.d. | CMA depletes hexokinase and induces metabolic catastrophe upon blockage of macroautophagy | 112 | |
| Neurodegenerative disorders | ||||
| General neuronal degeneration | MEF2A | n.d. | CMA degradation of oxidized MEF2A protects primary neurons from oxidative stress | 72 |
| MEF2Da | n.d. | CMA degradation of oxidized MEF2D protects primary neurons from oxidative stress | 119 | |
| Unknown | Reduced | Blocking CMA leads to dopaminergic neurodegeneration | 90 | |
| Unknown | n.d. | Inverse correlation between LAMP2A levels in brain regions and their susceptibility to protein aggregation | 79b | |
| Parkinson disease | α-Synuclein | Reduced | α-Synuclein is degraded by CMA but pathogenic α-synuclein blocks CMA | 17,61,78,89 |
| α-Synuclein | Reduced | Blockage of LAMP2A in substantia nigra in rats leads to neurodegeneration | 90 | |
| LRRK2 | Reduced | LRRK2 is degraded by CMA but pathogenic LRRK2 blocks CMA | 81 | |
| PARK7 (oxidized) | n.d. | CMA protects mitochondrial function by degrading nonfunctional PARK7 | 66 | |
| UCHL1 | Reduced | Pathogenic UCHL1 blocks CMA | 82,83 | |
| VPS35 mutant | Reduced | Pathogenic VPS35 impairs endosome-to-Golgi complex retrieval of LAMP2A and accelerates LAMP2A degradation | 56 | |
| GBA1 mutant | Reduced | GBA1 mutants prevent autophagy lysosomal reformation, affecting all forms of autophagy | 93 | |
| α-Synuclein mutant | n.d. | Nrf2 expression in astrocytes slows down CMA decline in PD mice | 92 | |
| Unknown | Increased | LAMP2A upregulation (AAV) protects PD neurons | 91b | |
| Unknown | Reduced | Reduced levels of LAMP2A in PD patient brains | 94b | |
| Unknown | Reduced | Reduced LAMP2A and HSC70 transcripts in PD brains due to increase of six miRNAs that control LAMP2A expression | 97b | |
| α-Synuclein | Reduced | miRNA against HSC70 (miR-320a) reduces α-synuclein degradation | 120b | |
| α-Synuclein | Reduced | Reduced LAMP2A protein in early stage PD brains (even before α-synuclein accumulation). CMA dysfunction is an early event | 95b | |
| Unknown | Reduced | GBA-affected brain regions have reduced LAMP2A levels | 95b | |
| Unknown | Reduced | Reduced LAMP2A or HSC70 mRNA and LAMP2A and HSC70 protein levels in leukocytes from PD patients | 121b,122b | |
| Unknown | n.d. | Sequence variants and SNPs in the LAMP2 promoter in PD patients | 96b | |
| Prion diseases | Prion protein | Increased | Upregulation of LAMP2A and HSC70 (protective) | 123b |
| Alzheimer disease | Tau | Decreased | Inefficient CMA | 84 |
| APP c-term | n.d. | Eliminating KFERQ from APP leads to C-terminal fragment accumulation | 98 | |
| Frontotemporal dementia | TDP-43 | n.d. | CMA contributes to degradation of TDP-43 | 85 |
| Huntington disease | Huntingtin | n.d. | CMA contributes to degradation of huntingtin | 86–88 |
| Heart diseases | ||||
| Heart failure | Ryanodine R2 (oxidized) | Reduced | CMA removes oxidized ryanodine receptor type 2; inability to do so may lead to heart failure (hypothesis) | 124 |
| Danon disease | Unknown | Reduced | No LAMP2A, undegraded proteins attributed to low CMA | 125b |
| Eye diseases | ||||
| Leber congenital amaurosis | Unknown | Increased | Upregulation of total LAMP2 expression suggested to protect against early apoptotic events | 126b |
| Liver diseases | ||||
| Acute liver failure | Unknown | Reduced | Reduced LAMP2A levels | 127b |
| NASH | Unknown | Reduced | Increased LAMP2A expression (compensatory) | 128b |
| Hepatosteatosis | Unknown | Reduced | Reduced LAMP2A levels | 129b |
| Alcoholic fatty liver | Unknown | Reduced | Reduced LAMP2A levels | 130b |
| Lung diseases | ||||
| Emphysema (cigarette smoke) | Unknown | Increased | CMA upregulated, protective against apoptosis | 131 |
| Kidney diseases | ||||
| Diabetes | PAX2 | Reduced | Reduced PAX2 CMA leads to kidney hypertrophy | 132 |
| Muscle diseases | ||||
| Sporadic inclusion-body myositis | Unknown | Increased | LAMP2A mRNA and protein upregulated (likely in response to macroautophagy and proteasome decrease) | 133b |
| Neuronal diseases | ||||
| Spinal cord injury | ROS damaged proteins? | Increased | HDAC6 upregulates CMA (protective) | 134 |
| Brain ischaemia | No specific | Increased | CMA upregulation (protective) | 58 |
| Traumatic brain injury | Damaged proteins | Increased | LAMP2A levels increase during the recovery time (protective) | 135b |
| Immune system | ||||
| Immunosenescence | ITCH, RCAN1 | Reduced | Failure to activate T cells because of reduced CMA; LAMP2A expression restores T cell response | 21 |
| Innate antiviral response | STING (desumoylated) | n.d. | CMA degrades STING in the late phase of infection to shut down the antiviral response | 73 |
| Salmonella | Unknown | Increased? | Recruits LAMP2A and HSC70 (CMA for nutrients?) | 65 |
| Lupus | Unknown | Increased | Increased LAMP2A expression | 136b |
| Lysosomal storage disorders | ||||
| Galactosialidosis | Unknown | Increased | Higher CMA by stabilizing LAMP2A | 51 |
| Cystinosis | Unknown | Reduced | Compromised LAMP2A trafficking to lysosomes | 54,55 |
| Mucolipidosis | Unknown | Reduced | Patient cells have lower CMA activity | 137 |
APP, amyloid precursor protein; AAV, adeno-associated virus; BBC3, Bcl-2-binding component 3 (also known as PUMA); CIP2A, protein CIP2A (also known as KIAA1524); CMA, chaperone-mediated autophagy; ecto-CRT, surface-exposed calreticulin; ER, endoplasmic reticulum; GBA, glucocerebrosidase; HDAC6, histone deacetylase 6; HMGB1, high mobility group protein B1; HSC70, heat shock cognate 71 kDa protein (also known as HSPA8); HSD17B4, peroxisomal multifunctional enzyme type 2; IL-17, interleukin-17; ITCH, E3 ubiquitin-protein ligase Itchy homolog; LAMP2A, lysosome-associated membrane protein type 2A; LRRK2, leucine-rich repeat serine/threonine-protein kinase 2; MEF, myocyte-specific enhancer factor; miRNA, microRNA; MST1, mammalian Ste20-like kinase 1 (also known as STK4); MYC, MYC proto-oncogene protein; NASH, nonalcoholic steatohepatitis; n.d., not determined; NCOR, nuclear receptor corepressor; PD, Parkinson disease; PEA-15, 15 kDa phosphoprotein enriched in astrocytes (also known as PED); PARK7, protein/nucleic acid deglycase DJ-1; RCAN1, regulator of calcineurin 1; RND3, Rho-related GTP-binding protein RhoE; ROS, reactive oxygen species; SNPs, single nucleotide polymorphisms; STING, stimulator of interferon genes protein; Tau, tau protein; TCTP, translationally-controlled tumour protein; TDP-43, TAR DNA-binding protein 43; UCHL1, ubiquitin carboxyl-terminal hydrolase isozyme L1; VPS35, vacuolar protein sorting-associated protein 35.
Indicates no bona-fide KFERQ-like motif has been identified upon sequence analysis.
Indicates studies in which changes in CMA are proposed on the basis of LAMP2A level measurements but CMA activity has not yet been analysed.
CMA failure in neurodegenerative disorders
The first connection between CMA malfunctioning and a human disease was with PD17. Since then, CMA failure has been linked to the pathogenesis of a growing number of neurodegenerative disorders (TABLE 3). CMA contributes to the degradation of several proteins that have a propensity to aggregate and, consequently, a decline in CMA activity leads to the accumulation of toxic aggregates.
The pathogenic variants of several CMA substrates are involved in PD, Alzheimer disease, frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurodegeneration-related proteins validated as CMA substrates include α-synuclein17,61,78–80, PARK7 (REF.66), leucine-rich repeat serine/threonine-protein kinase 2 (LRRK2)81 and ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1)82,83 (in PD), Tau protein84, TAR DNA-binding protein 43 (TDP-43)85 (in tauopathies such as Alzheimer disease or frontotemporal dementia) and huntingtin86–88 (in Huntington disease). However, it is possible that a larger number of proteins that are associated with neurodegeneration are CMA substrates, as many other proteins in these diseases contain the KFERQ-targeting motif.
A common characteristic of these neurodegeneration-related proteins is that, in most cases, the unmodified protein behaves as a conventional CMA substrate, whereas the pathogenic variants are targeted to lysosomes but fail to degrade and inhibit CMA (Fig. 4). In PD, pathogenic forms of α-synuclein17,61,89, UCHL1 (REFS82,83) and LRRK2 (REF.81) bind cytosolic HSC70 but, unlike their wild-type counterparts, interact abnormally with CMA components at the lysosomal membrane. Their obliterated entry into the lysosomal lumen often favours their organization into oligomeric complexes at the lysosomal surface, which negatively impacts CMA-mediated degradation of other substrates and further disrupts neuronal proteostasis17,61,78,80,82,85,90. Failed assembly of LAMP2A into the CMA translocation complex upon binding of PD proteins to this receptor has been demonstrated as the basis of their toxic effect on CMA81. Accordingly, in vitro and in vivo upregulation of CMA with small molecules47 or by viral-mediated LAMP2A expression91 has been shown to decrease α-synuclein levels and to protect against neurodegeneration91. Similarly, Nrf2 (also known as Nfe2l2) expression in astrocytes slows down CMA decline in PD mice by as-yet unidentified mechanisms and has a similar neuroprotective effect92. CMA malfunction is becoming a common feature of most PD types, although CMA failure may originate through different mechanisms. Besides the direct toxic effect on CMA described for pathogenic α-synuclein, UCHL1 or LRRK2, PD-related proteins, such as mutant VPS35 (REF.56) or GBA1 (REF.93), may reduce CMA activity by affecting lysosomal biogenesis. Although direct measurement of CMA in PD patient brains is still not feasible, correlative evidence supporting CMA malfunctioning in PD patients has populated the literature in recent years (TABLE 3). Reduced LAMP2A levels have been reported in both familial and idiopathic PD patients’ brains94, especially in the areas more vulnerable to disease95. PD-linked sequence variants in the LAMP2 promoter96 and increased levels of specific microRNA in PD patients97 seem responsible, at least in part, for the changes in LAMP2A in PD brains. Early stage PD brains show reduced LAMP2A levels, even before α-synuclein accumulation, suggesting that CMA dysfunction is an early event in PD pathogenesis95.
Fig. 4. CMA in neurodegeneration and cancer.

Chaperone-mediated autophagy (CMA) malfunctioning has been observed in multiple human disorders. Depicted here are two conditions, neurodegeneration and cancer, for which stronger experimental evidence of CMA involvement has been obtained. In neurodegeneration, pathogenic proteins can exert a toxic effect on CMA directly by disrupting the dynamics of the CMA translocation systems or indirectly by affecting levels of CMA effectors or lysosomal biogenesis. Most studies support that CMA has an anti-oncogenic role in normal untransformed cells and prevents malignant transformation, at least in part, through the mechanisms depicted here. In contrast, CMA has a protumorigenic effect in cancer cells by favouring their replication and growth and protecting them from extracellular insults. GFAP, glial fibrillary acidic protein; HSC70, heat shock cognate 71 kDa protein (also known as HSPA8); HSP90, heat shock protein 90; LAMP2A, lysosome-associated membrane protein type 2A; lys-HSC70, lysosomal HSC70.
Several of the proteins associated with frontotemporal dementias and tauopathies, including Alzheimer disease, have also been validated as bona fide CMA substrates whose degradation through this pathway is reduced in the diseased brains84,85,98,99. Similar to PD-related proteins, mutant forms of tau protein are targeted to lysosomes but fail to reach the lysosomal lumen84. In this case, the mutant proteins do not disrupt LAMP2A multimerization, but their translocation through this complex is halted halfway into the lysosomal lumen84 (Fig. 4). Incomplete translocation of mutant tau leads to its partial cleavage into highly amyloidogenic peptides that form irreversible oligomers at the lysosomal membrane, thus inhibiting CMA84. CMA also contributes to the degradation of other neurodegeneration-related proteins such as RCAN199, ubiquilin 1 (REF.100) and TDP-43 (REF.85), but less information is available about the CMA of their pathogenic variants or their impact on CMA. An unexpected relation has been suggested between CMA and the processing of amyloid precursor protein (APP), a protein closely associated with Alzheimer disease98. APP is a single-span membrane protein and, as such, is not a candidate for CMA degradation. However, APP undergoes physiological cleavage to release a short cytosolic stub where, coincidentally, the KFERQ motif is located. Although direct evidence for its CMA degradation is still missing, mutation of this motif led to cytosolic accumulation of this small, highly toxic peptide98.
Despite most studies pointing towards faulty functioning of CMA in neurodegenerative disorders, in Huntington disease, CMA activity is upregulated as a compensatory response to macroautophagy failure87. In fact, CMA can degrade both wild-type and mutant huntingtin, the protein that aggregates in the affected neurons, and artificial CMA targeting of mutant huntingtin has proved effective in reducing neurotoxicity in experimental mouse models86,88. However, the therapeutic applicability of mutant huntingtin re-routing towards CMA may be limited by the fact that the normal age-related decline in CMA activity is accelerated in the Huntington disease brain87.
Associations between CMA and other neuropathogenic proteins, such as prion proteins, and changes in CMA components in conditions, such as brain ischaemia or traumatic brain and spinal cord injury, have begun to appear (TABLE 3).
Accumulating evidence points towards a failure in CMA in several neurodegenerative diseases. A common theme that has also emerged is that the mechanism of this CMA inhibition varies on a case-by-case basis. Studying the pathological protein along with the wild-type version and delineating the exact step affected in CMA would be key in providing specific therapeutic interventions.
Complex role of CMA in cancer biology
Despite connections between CMA and cancer having been first established little more than 6 years ago18, some common key features of this interplay have since emerged. CMA activity is markedly upregulated in most cancer cell lines, and levels of CMA components, mainly LAMP2A, are elevated in a large array of human tumours18,30,101 (TABLE 3). Blocking CMA in cancer cells adversely affects cell survival and decreases their tumorigenicity, whereas blocking CMA in preformed xenografts causes tumour shrinkage and reduces metastasis18. The bases for the pro-oncogenic effect of CMA are probably multiple and depend on the type and stage of cancer (Fig. 4). The contribution of CMA to protein quality control explains why upregulating CMA provides the cancer cells several survival benefits, for example, resistance to hypoxia, oxidative stress and DNA damage — all key elements of a growing tumour microenvironment and of most anti-oncogenic interventions26,101–108. The role of CMA in tumour growth goes beyond quality control of damaged proteins and has been linked to selective degradation of specific regulatory proteins. Thus, CMA activity is required in some cancers to sustain the Warburg effect — aerobic glycolysis important for tumour progression18,27. CMA degradation of some glycolytic enzymes, such as acetylated pyruvate kinase PKM2, results in increased levels of glycolytic intermediates, which promote growth and proliferation27. Additionally, CMA indirectly reduces p53 levels, thus releasing its transcriptional inhibition on key glycolytic enzymes and resulting in higher glycolytic rates18. CMA also sustains tumour growth by degrading antiproliferation proteins (such as Rho-related GTP-binding protein RhoE (RND3)26), tumour suppressor proteins (such as MST1 (REF.30), 15 kDa phosphoprotein enriched in astrocytes (PEA-15; also known as PED)25 and mutant p53 (REF.109)) or pro-apoptotic proteins (such as Bcl-2-binding component 3 (BBC3; also known as PUMA110)), and by indirectly contributing to stabilize prosurvival proteins such as the induced myeloid leukaemia cell differentiation protein MCL1 (REF.111). Accordingly, blockage of CMA in cancer cells causes oxidative stress and higher susceptibility to chemotherapeutic agents and apoptosis101, and is being pursued as a possible antitumorigenic intervention. Upregulation of CMA — beyond that already occurring in the tumour — after blocking macroautophagy and the proteasome promotes cell death in specific tumours through metabolic collapse due to rapid degradation of glycolytic enzymes via CMA112,113.
Despite this protumorigenic role of CMA in cancer cells, it is important to note that the role of CMA in non-transformed cells is remarkably the opposite and that all evidence supports an anti-oncogenic role for CMA under physiological conditions (Fig. 4). CMA plays a tumour suppressive role in nontumorigenic cells by accelerating MYC’s proteasomal degradation and thus abolishing its oncogenic activity114 by directly degrading oncoproteins such as E3 ubiquitin-protein ligase MDM2 (REF.115), and by perhaps facilitating immunogenic cell death116. CMA may also protect against malignant transformation by assuring genome stability through its role in efficient DNA repair24. Progressive decrease in CMA with age may thus contribute to the higher risk of malignant transformation in ageing. In fact, mice with hepatic blockage of CMA develop spontaneous tumours20. Therefore, interventions aiming to preserve or restore CMA activity in ageing individuals may have potential value in cancer prevention.
Conclusions and perspectives
Research on CMA has a long way to go to reach the wealth of knowledge accumulated on other forms of autophagy during the past 2 decades. However, the identification of the essential CMA molecular players, the better understanding of the unique dynamics of CMA substrate degradation and the in vivo validation of the physiological relevance of CMA through the study of transgenic mouse models have all contributed to the coming of age of CMA.
Discovery of the regulated LAMP2A multimerization at the lysosomal membrane was key in understanding how CMA is regulated at the mechanistic level, but in the cellular context, the number of signalling pathways known in CMA regulation is still discrete, especially when considering the variety of CMA-activating stimuli present. Whether endosomal microautophagy is the CMA alternative in species where LAMP2A is not present or whether there could be yet another CMA-equivalent pathway in these species needs exploration. The growing evidence in support of a close interplay between CMA and macroautophagy makes it a priority to understand the molecular players involved in this crosstalk. Ongoing studies using high-resolution technologies on CMA reconstituted translocation units should provide new insights on the energetic requirements, driving forces and roles of cochaperones and chaperones at each side of the lysosomal membrane.
CMA bears functions common to all cell types (that is, protein quality control) and also more cell-type- specific regulatory functions that stem from its ability to selectively degrade single still-functional proteins. The introduction of mouse models with increased or reduced CMA and the future expansion of the tissue-specific conditional models will help us better understand these CMA regulatory functions and how tissue-specific changes in CMA impact whole organism function. Similarly, despite the already impressive list of connections between CMA malfunctioning and disease, a large number of them are still correlative, and experimental evidence of changes in CMA activity, the CMA step affected and the molecular mechanisms by which CMA contributes to pathogenesis is still missing. Is the toxic effect of α-synuclein or tau protein on CMA reversible? What are the more vulnerable CMA steps and/or components? Is CMA upregulation in these disease conditions always possible, or is there a ‘point of no return’? Multiple questions also surround the dual role of CMA as an anti-oncogenic mechanism in normal cells but a protumorigenic one in transformed cells. What mediates the switch from low to high CMA during transformation? How do oncogenic proteins protect themselves from CMA degradation in the cancer cell? As our knowledge of the contribution of CMA malfunction to disease grows, the need for good chemical modulators of CMA will increase. Despite recent success in the development of selective CMA activators47, the field still lacks selective inhibitors of CMA beyond those that inhibit lysosomal proteolysis and thus disrupt all forms of autophagy. Research on CMA has reached a mature stage, and the recent advances in understanding its cellular functions and uncovering new links between CMA failure and human diseases open new avenues for basic and clinical research.
Chaperone
A protein that assists during folding of other proteins until they reach their functional conformations. Chaperone substrates include both de novo synthesized native proteins and previously folded proteins that undergo partial unfolding.
Proteostasis networks
Intracellular components, mainly chaperones and proteolytic systems, that control each of the processes that occur from protein synthesis to degradation to prevent protein aggregation and ensure maintenance of a stable proteome.
HIF1α
The hypoxia-inducible factor 1α is a transcription factor that modulates the cellular response to hypoxia. HiF1α orchestrates the transcription of a large set of genes involved in cell proliferation, cell survival, and glucose and iron metabolism.
E3 ubiquitin-protein ligase
Intracellular enzyme that participates in the covalent attachment of ubiquitin moieties to cargo proteins. This tagging is commonly used for the targeting of the protein to the proteasome system for degradation.
MST1
The mammalian STE20-like protein kinase 1 is a protein component of the Hippo signalling pathway that modulates cell proliferation and differentiation.
Cochaperones
Proteins that assist chaperones in their functions by modulating their ATP hydrolytic capability.
HSP90
Heat shock protein 90 is a chaperone that stabilizes proteins in transient conformations and facilitates their final folding. HSP90 often acts in conjunction with other intracellular chaperones such as HSC70.
Lysosome-associated membrane protein (LAMP).
Single-span membrane proteins at the lysosomal membrane with a short C-terminus (12 amino acids) exposed to the cytosol while the rest of the protein is in the lysosomal lumen.
Isoelectric point
The pH at which the net charge on a protein is zero.
Nuclear retinoic acid receptor-α
Nuclear receptor activated by retinoic acid that activates or represses gene expression. its wide range of targets include genes involved in development, apoptosis, differentiation, autophagy and circadian regulation.
Lipid microdomains
Cholesterol and/or glycosphin-golipid-rich regions in membranes that present higher order and density than the surrounding membrane. They can be transient and highly dynamic in terms of resident proteins and lipids and in their size.
Cathepsin
Protease located in the lysosomal lumen that is maximally active at acidic pH.
Cystinosis
Lysosomal storage disorder characterized by the abnormal accumulation of cysteine inside lysosomes owing to a defect in its normal export from this organelle.
Retromer
Multiprotein complex made up of membrane-associated sorting nexin and vacuolar-protein-sorting proteins that recycle transmembrane proteins from endosomes to the golgi complex and the plasma membrane.
AKT1
Intracellular kinase that participates in signalling pathways that regulate a wide array of intracellular processes, including proliferation, cell survival, metabolism, growth and angiogenesis.
TOR complex 2 (TORC2).
One of the two functional multiprotein complexes containing the nutrient sensing kinase ToR. ToRC2 plays regulatory roles in actin cytoskeleton dynamics, proliferation, growth and metabolism.
Perilipins
Proteins that cover the surface of lipid droplets and shield the hydrophobic lipid core from the aqueous cytosol. Perilipins also regulate the rate of consumption of the lipids (lipolysis) in the lipid droplet.
Warburg effect
Unique metabolic characteristic of many cancer cells whereby they sustain very high levels of glycolysis followed by lactic acid fermentation rather than the common usage of oxidation of pyruvate in mitochondria that follows glycolysis in most cells.
Immunogenic cell death
Functionally peculiar variant of regulated cell death that — in immunocompetent syngeneic hosts — is sufficient to activate an adaptive immune response against dead cell-associated antigens.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Green DR & Levine B To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L et al. Molecular definitions of autophagy and related processes. EMBO J 36, 1811–1836 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stolz A, Ernst A & Dikic I Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol 16, 495–501 (2014). [DOI] [PubMed] [Google Scholar]
- 4.De Duve C & Wattiaux R Functions of lysosomes. Annu. Rev. Physiol 28, 435–492 (1966). [DOI] [PubMed] [Google Scholar]
- 5.Marzella L, Ahlberg J & Glaumann H Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Archiv B Cell Pathol. Incl. Mol. Pathol 36, 219–234 (1981). [DOI] [PubMed] [Google Scholar]
- 6.Roberts P et al. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol. Biol. Cell 14, 129–141 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakai Y, Koller A, Rangell L, Keller G & Subramani S Peroxisome degradation by microautophagy in Pichia pastoris. Identification of specific steps and morphological intermediates. J. Cell Biol 141, 625–636 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahu R et al. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; First report of endosomal microautophagy.
- 9.Dice JF Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci 15, 305–309 (1990). [DOI] [PubMed] [Google Scholar]; First characterization of the properties of the KFERQ-like signal.
- 10.Terlecky SR, Chiang H-L, Olson TS & Dice JF Protein and peptide binding and stimulation of in vitro lysosomal proteolysis by the 73-kDa heat shock cognate protein. J. Biol. Chem 267, 9202–9209 (1992). [PubMed] [Google Scholar]
- 11.Cuervo AM, Terlecky SR, Dice JF & Knecht E Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J. Biol. Chem 269, 26374–26380 (1994). [PubMed] [Google Scholar]
- 12.Eskelinen EL et al. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic 6, 1058–1061 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Massey AC, Kaushik S, Sovak G, Kiffin R & Cuervo AM Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl Acad. Sci. USA 103, 5805–5810 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R & Cuervo AM Identification of regulators of chaperone-mediated autophagy. Mol. Cell 39, 535–547 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bandyopadhyay U, Kaushik S, Varticovski L & Cuervo AM The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol 28, 5747–5763 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of the CMA translocation complex at the lysosomal membrane.
- 16.Arias E et al. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol. Cell 59, 270–284 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT & Sulzer D Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004). [DOI] [PubMed] [Google Scholar]; First connection of CMA malfunction with a human disorder (neurodegeneration).
- 18.Kon M et al. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl Med 3, 109ra117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider JL, Suh Y & Cuervo AM Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20, 417–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; First mouse model with tissue-specific CMA blockage in vivo.
- 20.Schneider JL et al. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 14, 249–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valdor R et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol 15, 1046–1054 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dice JF Altered degradation of proteins microinjected into senescent human fibroblasts. J. Biol. Chem 257, 14624–14627 (1982). [PubMed] [Google Scholar]
- 23.Kaushik S & Cuervo AM AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 12, 432–438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park C, Suh Y & Cuervo AM Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun 6, 6823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quintavalle C et al. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J. Cell. Physiol 229, 1359–1368 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou J et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy 12, 515–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv L et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. Cell 42, 719–730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonhoure A et al. Acetylation of translationally controlled tumor protein promotes its degradation through chaperone-mediated autophagy. Eur. J. Cell Biol 96, 83–98 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Ferreira JV, Soares AR, Ramalho JS, Pereira P & Girao H K63 linked ubiquitin chain formation is a signal for HIF1A degradation by chaperone-mediated autophagy. Sci. Rep 5, 10210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L et al. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene 35, 4048–4057 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Chiang H, Terlecky S, Plant C & Dice JF A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385 (1989). [DOI] [PubMed] [Google Scholar]; Identification of HSC70 as the chaperone for CMA.
- 32.Ferreira JV et al. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9, 1349–1366 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Agarraberes F & Dice JF A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci 114, 2491–2499 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Arndt V et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol 20, 143–148 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Cuervo AM, Dice JF & Knecht E A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J. Biol. Chem 272, 5606–5615 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Salvador N, Aguado C, Horst M & Knecht E Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem 275, 27447–27456 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Agarraberes F, Terlecky S & Dice J An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol 137, 825–834 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaushik S, Massey A, Mizushima N & Cuervo AM Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 19, 2179–2192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuervo AM & Dice JF A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 (1996). [DOI] [PubMed] [Google Scholar]; Identification of the lysosomal membrane receptor for CMA.
- 40.Cuervo AM, Knecht E, Terlecky SR & Dice JF Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol 269, C1200–C1208 (1995). [DOI] [PubMed] [Google Scholar]
- 41.Cuervo AM & Dice JF Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell Sci 113, 4441–4450 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Li J & Pfeffer SR Lysosomal membrane glycoproteins bind cholesterol and contribute to lysosomal cholesterol export. eLife 5, e21635 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuervo AM & Dice JF Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–583 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Rout AK, Strub MP, Piszczek G & Tjandra N Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J. Biol. Chem 289, 35111–35123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiffin R, Christian C, Knecht E & Cuervo AM Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 15, 4829–4840 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hubbi ME et al. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1alpha to promote cell-cycle progression. Proc. Natl Acad. Sci. USA 111, E3325–E3334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anguiano J et al. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol 9, 374–382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sardiello M et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Kaushik S, Massey AC & Cuervo AM Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J 25, 3921–3933 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of the mechanisms that determine LAMP2A stability in lysosomes.
- 50.Wilke S, Krausze J & Bussow K Crystal structure of the conserved domain of the DC lysosomal associated membrane protein: implications for the lysosomal glycocalyx. BMC Biol 10, 62 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cuervo AM, Mann L, Bonten EJ, d’Azzo A & Dice JF Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J 22, 12–19 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez-Navarro JA et al. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl Acad. Sci. USA 109, E705–E714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kiffin R et al. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci 120, 782–791 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Napolitano G et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med 7, 158–174 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J. Biol. Chem 292, 10328–10346 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang FL et al. VPS35 in dopamine neurons is required for endosome-to-Golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci 35, 10613–10628 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koga H, Martinez-Vicente M, Macian F, Verkhusha VV & Cuervo AM A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun 2, 386 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dohi E et al. Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int 60, 431–442 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Finn PF & Dice JF Ketone bodies stimulate chaperone-mediated autophagy. J. Biol. Chem 280, 25864–25870 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Cuervo AM, Hildebrand H, Bomhard EM & Dice JF Direct lysosomal uptake of alpha 2-microglobulin contributes to chemically induced nephropathy. Kidney Int 55, 529–545 (1999). [DOI] [PubMed] [Google Scholar]
- 61.Martinez-Vicente M et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest 118, 777–788 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang C & Cuervo AM Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med 14, 959–965 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mizushima N, Yamamoto A, Matsui M, Yoshimori T & Ohsumi Y In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101–1111 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh R et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Singh V et al. Salmonella co-opts host cell chaperone-mediated autophagy for intracellular growth. J. Biol. Chem 292, 1847–1864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang B et al. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy 12, 1215–1228 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaushik S & Cuervo AM Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol 17, 759–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; Identification of the regulatory role of CMA in lipid metabolism.
- 68.Lu W et al. Dual proteolytic pathways govern glycolysis and immune competence. Cell 159, 1578–1590 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuervo AM, Hu W, Lim B & Dice JF IkappaB is a substrate for a selective pathway of lysosomal proteolysis. Mol. Biol. Cell 9, 1995–2010 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Franch HA, Sooparb S, Du J & Brown NS A mechanism regulating proteolysis of specific proteins during renal tubular cell growth. J. Biol. Chem 276, 19126–19131 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Yang Q et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323, 124–127 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang L et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 10, 1015–1035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hu MM et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45, 555–569 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Cuervo AM & Dice JF Age-related decline in chaperone-mediated autophagy. J. Biol. Chem 275, 31505–31513 (2000). [DOI] [PubMed] [Google Scholar]
- 75.Rodriguez-Muela N et al. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell 12, 478–488 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cai Z et al. Chaperone-mediated autophagy: roles in neuroprotection. Neurosci. Bull 31, 452–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cuervo AM & Wong E Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24, 92–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM & Di Monte DA Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem 285, 13621–13629 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Malkus KA & Ischiropoulos H Regional deficiencies in chaperone-mediated autophagy underlie alpha-synuclein aggregation and neurodegeneration. Neurobiol. Dis 46, 732–744 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vogiatzi T, Xilouri M, Vekrellis K & Stefanis L Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem 283, 23542–23556 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orenstein SJ et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci 16, 394–406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kabuta T, Furuta A, Aoki S, Furuta K & Wada K Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem 283, 23731–23738 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andersson FI et al. The effect of Parkinson’s-disease- associated mutations on the deubiquitinating enzyme UCH-L1. J. Mol. Biol 407, 261–272 (2011). [DOI] [PubMed] [Google Scholar]
- 84.Wang Y et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet 18, 4153–4170 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang CC et al. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci 127, 3024–3038 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Bauer PO et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol 28, 256–263 (2010). [DOI] [PubMed] [Google Scholar]
- 87.Koga H et al. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci 31, 18492–18505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qi L et al. The role of chaperone-mediated autophagy in huntingtin degradation. PLoS ONE 7, e46834 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xilouri M, Vogiatzi T, Vekrellis K, Park D & Stefanis L Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 4, e5515 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xilouri M et al. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 12, 2230–2247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xilouri M et al. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 136, 2130–2146 (2013). [DOI] [PubMed] [Google Scholar]; First evidence of antidegenerative effect of genetic enhancement of CMA.
- 92.Gan L, Vargas MR, Johnson DA & Johnson JA Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci 32, 17775–17787 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Magalhaes J et al. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum. Mol. Genet 25, 3432–3445 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alvarez-Erviti L et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol 67, 1464–1472 (2010). [DOI] [PubMed] [Google Scholar]
- 95.Murphy KE et al. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 137, 834–848 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pang S, Chen D, Zhang A, Qin X & Yan B Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson’s disease. Neurosci. Lett 526, 63–67 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Alvarez-Erviti L et al. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis 4, e545 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Park JS, Kim DH & Yoon SY Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep 49, 337–342 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu H, Wang P, Song W & Sun X Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J 23, 3383–3392 (2009). [DOI] [PubMed] [Google Scholar]
- 100.Rothenberg C et al. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet 19, 3219–3232 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Saha T LAMP2A overexpression in breast tumors promotes cancer cell survival via chaperone-mediated autophagy. Autophagy 8, 1643–1656 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Y et al. Acetylation targets HSD17B4 for degradation via the CMA pathway in response to estrone. Autophagy 13, 538–553 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Du C et al. 5-Fluorouracil targets histone acetyltransferases p300/CBP in the treatment of colorectal cancer. Cancer Lett 400, 183–193 (2017). [DOI] [PubMed] [Google Scholar]
- 104.Ding ZB et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol 49, 2367–2376 (2016). [DOI] [PubMed] [Google Scholar]
- 105.Wu JH et al. CMA down-regulates p53 expression through degradation of HMGB1 protein to inhibit irradiation-triggered apoptosis in hepatocellular carcinoma. World J. Gastroenterol 23, 2308–2317 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chava S et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 8, 40019–40036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guo B et al. M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 8, 44465–44476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ali AB, Nin DS, Tam J & Khan M Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS ONE 6, e25268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vakifahmetoglu-Norberg H et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev 27, 1718–1730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xie W et al. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy 11, 1623–1635 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Suzuki J, Nakajima W, Suzuki H, Asano Y & Tanaka N Chaperone-mediated autophagy promotes lung cancer cell survival through selective stabilization of the pro-survival protein, MCL1. Biochem. Biophys. Res. Commun 482, 1334–1340 (2017). [DOI] [PubMed] [Google Scholar]
- 112.Xia HG et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J. Cell Biol 210, 705–716 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Galan-Acosta L, Xia H, Yuan J & Vakifahmetoglu-Norberg H Activation of chaperone-mediated autophagy as a potential anticancer therapy. Autophagy 11, 2370–2371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gomes LR, Menck CFM & Cuervo AM Chaperone-mediated autophagy prevents cellular transformation by regulating MYC proteasomal degradation. Autophagy 13, 928–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lu TL et al. Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem. Biophys. Res. Commun 398, 26–31 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Garg AD, Dudek AM & Agostinis P Calreticulin surface exposure is abrogated in cells lacking, chaperone-mediated autophagy-essential gene, LAMP2A. Cell Death Dis 4, e826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaushik S & Cuervo AM Methods to monitor chaperone-mediated autophagy. Methods Enzymol 452, 297–324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li P et al. Degradation of AF1Q by chaperone-mediated autophagy. Exp. Cell Res 327, 48–56 (2014). [DOI] [PubMed] [Google Scholar]
- 119.Gao L et al. Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxid. Redox Signal 20, 2936–2948 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li G et al. Targeted suppression of chaperone-mediated autophagy by miR-320a promotes alpha-synuclein aggregation. Int. J. Mol. Sci 15, 15845–15857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu G et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res 1394, 105–111 (2011). [DOI] [PubMed] [Google Scholar]
- 122.Sala G et al. Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson’s disease. Brain Res 1546, 46–52 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Wang H et al. Overexpression of PLK3 mediates the degradation of abnormal prion proteins dependent on chaperone-mediated autophagy. Mol. Neurobiol 54, 4401–4413 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Pedrozo Z et al. Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc. Res 98, 277–285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fidzianska A, Walczak E & Walski M Abnormal chaperone-mediated autophagy (CMA) in cardiomyocytes of a boy with Danon disease. Folia Neuropathol 45, 133–139 (2007). [PubMed] [Google Scholar]
- 126.Metrailler S, Schorderet DF & Cottet S Early apoptosis of rod photoreceptors in Rpe65(−/−) mice is associated with the upregulated expression of lysosomal-mediated autophagic genes. Exp. Eye Res 96, 70–81 (2012). [DOI] [PubMed] [Google Scholar]
- 127.Li Y, Lu L, Luo N, Wang YQ & Gao HM Inhibition of PI3K/AKt/mTOR signaling pathway protects against d-galactosamine/ lipopolysaccharide-induced acute liver failure by chaperone-mediated autophagy in rats. Biomed. Pharmacother 92, 544–553 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Das S et al. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol 305, G950–G963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sharma S, Mells JE, Fu PP, Saxena NK & Anania FA GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS ONE 6, e25269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cai Y et al. The detrimental role played by lipocalin-2 in alcoholic fatty liver in mice. Am. J. Pathol 186, 2417–2428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lee CH, Lee KH, Jang AH & Yoo CG The impact of autophagy on the cigarette smoke extract-induced apoptosis of bronchial epithelial cells. Tuberc. Respir. Dis 80, 83–89 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sooparb S, Price SR, Shaoguang J & Franch HA Suppression of chaperone-mediated autophagy in the renal cortex during acute diabetes mellitus. Kidney Int 65, 2135–2144 (2004). [DOI] [PubMed] [Google Scholar]
- 133.Cacciottolo M, Nogalska A, D’Agostino C, Engel WK & Askanas V Chaperone-mediated autophagy components are upregulated in sporadic inclusion-body myositis muscle fibres. Neuropathol. Appl. Neurobiol 39, 750–761 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Su M et al. HDAC6 regulates the chaperone-mediated autophagy to prevent oxidative damage in injured neurons after experimental spinal cord injury. Oxid. Med. Cell Longev 2016, 7263736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Park Y et al. Chaperone-mediated autophagy after traumatic brain injury. J. Neurotrauma 32, 1449–1457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Macri C et al. Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 11, 472–486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Venugopal B et al. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J. Cell. Physiol 219, 344–353 (2009). [DOI] [PubMed] [Google Scholar]