

Abstract
Sleep is a fundamental conserved physiological state in animals and humans. It may serve multiple functions, ranging from energy conservation to higher brain operation. Understanding sleep functions and the underlying mechanisms requires the study of sleeplessness and its consequences. The traditional approach to remove sleep is sleep deprivation (SD) by sensory stimulation. However, stimulation‐induced SD can be stressful and can cause non‐specific side effects. An emerging alternative method is “genetic SD”, which removes sleep using genetics or optogenetics. Sleep requires sleep‐active neurons and their regulators. Thus, genetic impairment of sleep circuits might lead to more specific and comprehensive sleep loss. Here, I discuss the advantages and limits of genetic SD in key genetic sleep model animals: rodents, zebrafish, fruit flies and roundworms, and how the study of genetic SD alters our view of sleep functions. Genetic SD typically causes less severe phenotypes compared with stimulation‐induced SD, suggesting that sensory stimulation‐induced SD may have overestimated the role of sleep, calling for a re‐investigation of sleep functions.
Keywords: genetics, model organism, optogenetics, sleep, sleep deprivation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Molecular Biology of Disease; Neuroscience
Glossary
- ALA
name of a specific Caenorhabditis elegans interneuron/mechanosensory neuron
- AMPA
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid
- CNO
clozapine‐N‐oxide
- dFB
dorsal fan‐shaped body
- EEG
electroencephalogram
- EGF
epidermal growth factor
- GABA
γ‐aminobutyric acid
- GPCR
G protein‐coupled receptor
- HPA
hypothalamic–pituitary–adrenal axis
- MS
medial septum
- PB
parabrachial nuclei
- PI
pars intercerebralis
- PZ
parafacial zone
- REM
rapid eye movement
- RIS
ring interneuron s, name of a specific C. elegans interneuron
- ROS
reactive oxygen species
- SD
sleep deprivation
- SIK3
salt‐inducible kinase 3
- VLPO
ventrolateral preoptic nucleus
Introduction
We spend about one‐third of our lives asleep. Sleep curtailment is detrimental to human health and is associated with an increased risk of infection, depression, cardiovascular disease, obesity, type 2 diabetes, and cancer, illustrating the importance of this time investment. The high prevalence of insomnia and insufficient sleep quality in modern societies thus presents a massive unmet health problem 1, 2, 3, 4. Sleep appears to affect virtually all aspects of animal physiology, and numerous processes have been proposed to depend on the regular occurrence of sleep. While the broad detrimental consequences of sleep loss are obvious, there still is no consensus as to what the direct consequences of sleep loss are, and how sleep carries out its functions at the molecular level.
Sleep research first focused on humans. Seminal work using EEG recordings showed that humans have two types of sleep, REM sleep and non‐REM sleep, which are also called active and quiet sleep, respectively. During REM sleep, the brain shows high asynchronous activity similar to wake, concomitant with paralysis of striated muscles, with a few exceptions including the musculature controlling eye movement or breathing. During non‐REM sleep, both muscles and neurons show reduced activity with highly synchronous brain activity 5, 6. Using the electrophysiological characteristics of human sleep, it has been possible to detect both types of sleep in a wide range of mammals and birds 7, 8. Even reptiles possess two different states of sleep that resemble non‐REM and REM sleep, whereas amphibians appear to only show quiet sleep 9. This led to the conclusion that sleep diverged at the base of the amniotes into non‐REM and REM sleep.
Behavioral quiescence has long been observed across species, including invertebrates. However, defining sleep using EEG correlates for REM and non‐REM sleep often is not possible due to brain anatomical differences. Nevertheless, quiescence can also be identified as sleep by using four key behavioral criteria 10. (i) A typical posture is assumed that is compatible with reduced muscle activity. (ii) Sleep reduces responsiveness to sensory stimulation, indicating a global neural dampening that extends to sensory systems, and which contrasts sleep to quiet wakefulness. (iii) Sleep is rapidly reversible, meaning that the human or animal can be readily awoken, separating sleep from coma or paralysis. (iv) Sleep is under homeostatic regulation, implying that mechanisms exist that ensure that this state takes place, underscoring its importance 10. By applying these behavioral criteria, sleep has been identified in all animals that have a nervous system, with cnidaria presenting the most basal phylum in which sleep has been detected 11. Thus, sleep is much more widespread among animals than initially thought 12. Particularly important was the identification of sleep in three key non‐mammalian animal models, zebrafish, Drosophila, and Caenorhabditis elegans, as these models facilitate a molecular and mechanistic dissection of sleep 13, 14, 15, 16, 17.
Compelling evidence for the existence of a species that has a nervous system but never sleeps is lacking, but the amount of sleep is highly plastic and some animals can get away with little sleep. Environmental conditions impact sleep need, and the time spent in sleep differs substantially across species. Under extreme conditions, temporary sleep restriction or even complete loss appears to exist and confers a selective advantage. For example, migrating and mating birds appear to be able to suspend or reduce the need to sleep for at least several days 18, 19. Also, some species, such as large herbivores or cave‐dwelling fish, manage to live with sleeping only little, and even 3 h per day can be sufficient 20, 21. On the other extreme, some animals such as bats sleep up to 20 h per day 21. This suggests that the amount of sleep is adapted to, and depends on ecological constraints, perhaps to regulate behavior and to preserve energy 22. Because animals appear to be asleep for at least 10% of their time, a lower limit of how little sleep is required for survival seems to exist (Fig 1).
Figure 1. Sleep time fraction varies greatly but does not drop below 10%.
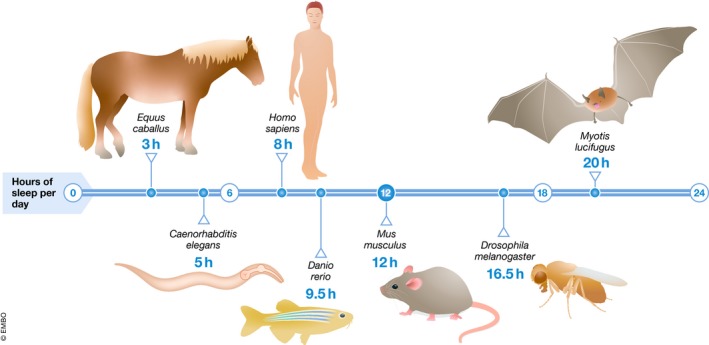
Sleep time fraction varies between 3–20 h/24 h with large herbivores sleeping little and bats sleeping a lot 21. Model organisms fall within the range of wild species 38, 85, 103, 124.
Functions and molecular underpinnings of sleep
The physiological state of sleep has been proposed to play multiple roles that can be coarsely sorted into three groups that are overlapping and not mutually exclusive. (i) The first group of sleep function theories posits that sleep plays a role in optimizing behavior and the conservation or allocation of energy. (ii) The second group states that sleep may regulate core molecular and cellular processes. (iii) And the third group suggests that sleep serves higher brain functions 12, 23 (Fig 2).
Figure 2. Summary of some of the hypothesized functions of sleep.
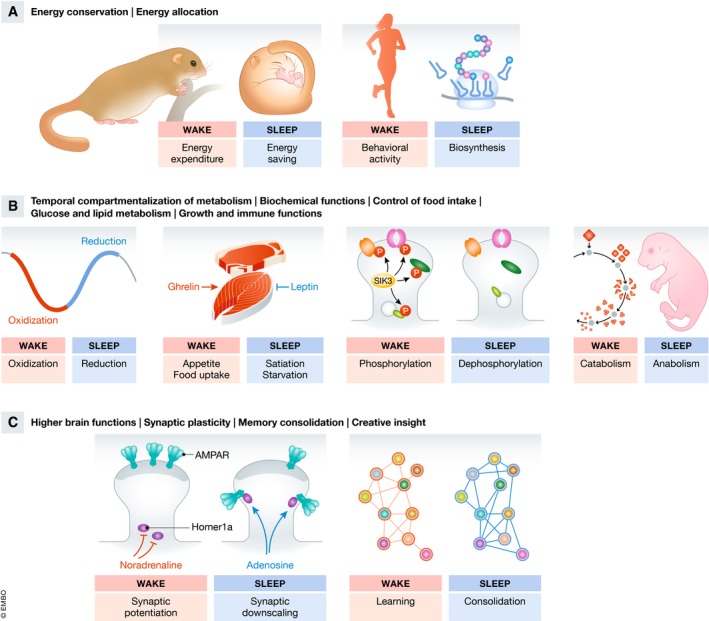
Various ideas exist as to the functions of sleep and molecular changes underlying sleep, and some hypotheses are depicted here. (A) In its most simple form, sleep may save energy when activity is not adaptive. It would thus serve a similar function as hibernation 22. Energy may not only be saved for later use but could rather be allocated for other processes such as anabolic reactions including protein synthesis 25. (B) Sleep may become adaptive by compartmentalizing processes such as conflicting metabolic reactions which would make these processes more efficient 36. Sleep controls hormones, food intake, and metabolism (including lipid and sugar metabolism) 3, 4. Sleep controls cyclic biochemical reactions. Wakefulness, for example, is associated with the phosphorylation of synaptic proteins and sleep is associated with dephosphorylation 37. Various other ideas as to sleep homeostasis exist, including accumulation of extracellular adenosine 144. Sleep is important for growth and immune functions 32, 33, 34. (C) Sleep controls higher brain functions such as synaptic plasticity including learning and memory. Synaptic changes during sleep include a downscaling of weak synapses, a process that appears to be promoted by Homer1a. Strong synapses are preserved 45, 47, 145. Sleep may support systems memory consolidation by re‐activating and re‐distributing memory across brain areas and circuits 49. These brain re‐arrangements may even facilitate novel insight and creativity in humans 50. Note that these ideas are overlapping. Most evidence in support of these theories stems from sleep deprivation by sensory stimulation.
An adaptive value of sleep could be understood by viewing sleep as an inactive state. At times when wakefulness is not advantageous, the organism would enter an inactive state and thus save energy. A strong argument that energetic and ecological constraints play a role in determining sleep is the large variation in sleep amount and intensity seen across species 22. Sleep would thus share an energy‐saving function with torpor, a metabolically and behaviorally inactive phase found in mammals and birds that is characterized by a massive drop in body temperature, for instance during hibernation. Both the transitions from wakefulness to torpor as well as the exit from torpor into wakefulness involve a phase of non‐REM sleep, suggesting that they are related 22, 24, 25. Sleep and torpor differ behaviorally as sleep is defined as a readily reversible state, whereas torpor generally is not rapidly reversible. A main functional difference of torpor and sleep is that sleep need does not appear to dissipate during torpor 26, 27. Thus, sleep seems to serve benefits that cannot be simply explained by an energy conservation function alone. According to the energy allocation theory of sleep, energy is not primarily conserved for later use but is diverted to restorative processes such as anabolic biosynthetic reactions 25, 28.
It has been proposed that sleep becomes regenerative by allowing or facilitating key molecular and cellular housekeeping functions. This view has been supported by biochemical and transcriptomic studies that found that sleep is associated with an increase in the expression of genes required for biosynthesis and transport 29, 30, 31. Anabolic metabolism during sleep could, for example, facilitate growth, increase stress resistance, and support the immune system 32, 33, 34, 35. Sleep may control metabolism, at least in part, by regulating the rhythmic timing of food intake. For instance, sleep restriction in humans increases the concentration of the appetite‐stimulating hormone ghrelin, whereas it reduces the concentration of the appetite‐inhibiting hormone leptin, and sleep restriction is associated with metabolic syndrome, obesity, and type 2 diabetes 3, 4. Sleep may itself present a metabolic cycle, which provides a temporal compartmentalization of processes that are difficult to reconcile or that are more energetically favorable if carried out subsequently 36. An example of a cycling biochemical reaction is phosphorylation of a substantial fraction of synaptic proteins, which is globally increased during wakefulness, but decreased during sleep 37. The key kinase responsible for this phosphorylation cycle is SIK3, and a gain‐of‐function mutation of SIK3 called sleepy causes excessive sleep duration and intensity 38. SIK3 is a known regulator of lipid and sugar metabolism, suggesting a molecular link between sleep and cyclic metabolic activity 39, 40. Completing the picture of cellular housekeeping, it has been observed that sleep in mice is also a period in which potentially toxic molecules such as protein aggregates are removed from the brain. This removal may involve neuronal shrinkage increasing the flux of interstitial fluid 41.
Seminal experiments showed that a good night's sleep is important for learning and memory. Memory formation requires synaptic and cellular changes across neural circuits and brain regions that encode this memory. Transcriptome analysis of sleeping brains has found that an increased expression of genes required for plasticity and protein synthesis during sleep is required for memory formation, suggesting that sleep serves the expression of plasticity genes to support learning 42, 43, 44. Plasticity involves alterations in the size and composition of synapses. For new memories to form, specific synapses need to strengthen and new synapses need to form whereas other synapses need to weaken or disappear. It has been proposed that wakefulness leads to a net increase in synapse size and that sleep causes a subsequent net synaptic downscaling, which mostly affects weak synapses and leaves strong synapses intact 45. The weakening of synapses involves a phosphorylation and subsequent removal of AMPA receptors, a process that is supported by Homer1a 46. According to the working model, Homer1a is kept out of the synapse during wakefulness by noradrenergic signaling and enters the synapse during sleep. This recruitment of Homer1a to the synapse is promoted by adenosine, a somnogenic (sleep‐promoting) factor that is thought to accumulate as a function of wakefulness and that promotes homeostatic sleep drive 47, 48. Besides these cell biological changes of synapse size and composition, the process of memory consolidation occurs at the systems level involving recurrent reactivation of memories and its redistribution and integration into existing circuits, allowing the updating of knowledge. Disconnecting neural circuits from sensory input may facilitate the massive restructuring of brain circuits as memories mature 49. Thus, sleep may even allow novel associations and creative insights into problems that are hard to solve during wakefulness 50. REM sleep may help consolidate certain types of memories, a process that, at least in part, is mediated by rhythmic activity in the hippocampus, though the underlying mechanisms are not well understood 6, 49.
Sleep is induced by sleep‐active neurons
The nervous system plays a crucial role in sleep induction. Research on the neural substrates of sleep control started with work on human patients who had suffered from sleep loss as a consequence of infection‐induced neural injury. Lesions in a particular brain area, the anterior hypothalamus, led to a reduction of sleep, demonstrating that dedicated centers exist in the mammalian brain that control sleep 51. This work motivated mechanistic studies of neuronal sleep control centers, mostly by using mammals such as cats, rats, and mice. Central to sleep induction are sleep‐active sleep‐promoting neurons that express inhibitory neurotransmitters, GABA, and neuropeptides. Sleep‐active neurons depolarize specifically at the onset of sleep to inhibit wake‐promoting circuits and thus to promote sleep. These neurons can be inhibited by sensory stimulation and arousal to allow quick reversibility. They are over‐activated in the process of sleep homeostasis and confer increased sleep drive. Sleep‐active neurons thus present the motor of sleep, which in turn is regulated by upstream driver mechanisms that determine when and how much the sleep motor is active 52, 53.
Sleep deprivation reveals sleep functions
Most of the theories regarding the functions of sleep are based on observations of processes that correlate with sleep, and causality is established by studying the consequences of sleep deprivation. Sleep is under the control of wakefulness‐promoting and sleep‐promoting circuits, which oppose each other to generate discrete states 54. SD is typically induced by sensory stimulation, i.e., by increasing the activity of the wake‐promoting arousal system leading to an inhibition of the sleep‐promoting system. Stimulation‐induced SD accounts for virtually all the causal testing of the theories summarized above. Acute complete SD has been used to study the essential functions of sleep. Complete SD in rodents caused weight loss, skin ulceration, sepsis, and ultimately death in experimental animals 55. To prevent lethality, SD can be applied partially to shorten sleep and then is often called sleep restriction, which often is imposed chronically to study sleep functions. Chronic sleep restriction in animal models has been important to understand the effects of chronic sleep curtailment on human health. For example, sleep restriction in rodents leads to neuronal injury and reduced vigilance 56. However, it has been difficult to attribute the detrimental consequences of complete or partial SD to sleep loss rather than to stress. The pleiotropic consequences of complete SD have also made it impossible to clearly deduce the more immediate consequences of sleep loss. Sleep, arousal, and stress are intimately linked across species, and hyperarousal caused by mental stress is the main cause of insomnia in humans 2. In mammals, hyperarousal activates the HPA axis and thus sets off a physiological stress response, which maintains arousal and suppresses sleep, perpetuating a vicious cycle 57, 58. Gentler protocols are standard today and aim to arouse by motivating instead of stressing. Nevertheless, SD still is achieved by an over stimulation of sensory and arousal pathways (Fig 3) 59. A second confounding factor for studying sleep functions after SD is the interference of homeostatic sleep rebound with wake functions. SD leads to homeostatic increases in sleep pressure that can even lead to “lapses” or “microsleep” bouts that can disturb wake functions. SD in humans causes deficits in attention, working memory, and information processing 60. While it is important to study the consequences of SD on brain performance, it is difficult to understand whether the observed defects are directly caused by sleep loss or whether they are caused by homeostatic rebound mechanisms.
Figure 3. Classic SD suppresses sleep by increasing arousal, whereas genetic SD impairs the sleep‐inducing system.
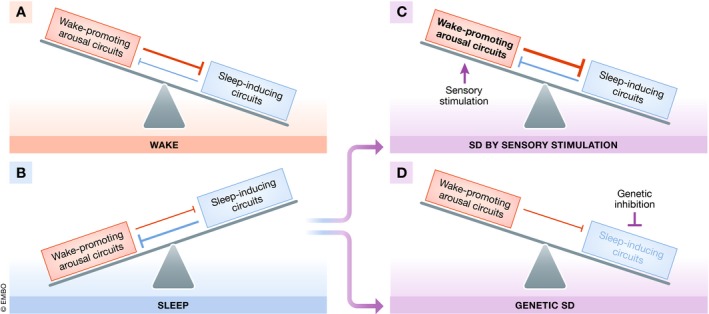
According to the flip‐flop switch model, sleep and wake are under the control of two antagonizing systems, a wake‐inducing arousal system and a sleep‐inducing system 52. (A) During wake, the arousal system dominates and suppresses sleep. (B) During sleep, the sleep‐inducing system dominates and suppresses wake. (C) Sensory stimulation during sleep increases the activity of the arousal system, suppressing sleep despite increased sleep drive. (D) Genetically impairing the sleep‐inducing system permits wakefulness by disinhibition. Sleep‐active neurons might also contribute to arousal dampening as part of the normal waking behavior and thus their ablation might cause some level of hyperarousal. However, this arousing effect likely is smaller than the level of hyperactivity caused by sensory stimulation‐induced SD, and genetic manipulations can remove sleep without causing massive hyperactivity. Both SD approaches change the organism by fundamentally different means and are thus complementary. Both approaches should be pursued for establishing a causal link between sleep and phenotypes observed after sleep deprivation.
Genetic sleep deprivation
An alternative strategy to SD by sensory stimulation is to render model animals sleepless by impairing the sleep‐inducing system. In this paradigm, the organism specifically lacks sleep induction, not requiring additional stimulation. The increase in arousal following sleep neuron inhibition should be attributable to a disinhibition of the wake‐promoting system (Fig 3). How can the sleep‐inducing system be impaired? While it is possible to ablate brain parts using neurosurgical methods, a more specific way to impair sleep‐inducing brain centers is through genetic targeting. Here, I thus call the use of genetics to remove sleep “genetic SD”. Genetic SD may be achieved by the deletion of sleep genes or by genetic ablation of neurons that are required for sleep induction. Complete genetic SD likely results in lethality in many systems requiring either conditional or partial approaches. Conditional genetic SD could be generated by optogenetic or chemogenetic inhibition of sleep‐active neurons as well as by inducible knockouts to create a genetic analog of SD by sensory stimulation. Alternatively, genetic SD could be induced only partially by using hypomorphic mutations to generate genetic analogs of chronic sleep restriction. In systems in which sleep loss is not imminently lethal, chronic complete SD might be a good choice to generate strong phenotypes. As an alternative to targeting sleep‐active neurons directly, manipulating neurons that are upstream or downstream of sleep‐active neurons could be employed for removing sleep. This could be achieved, for instance, by activating neurons that inhibit sleep‐active neurons or by preventing activity reduction of wake neurons that are normally inhibited by sleep‐active neurons. To complement genetic SD studies, gain‐of‐function experiments can be devised that activate the sleep‐inducing system and cause increased sleep, or “genetic sleep gain”.
Specificity of the sleep mutant phenotype is essential to link sleep loss to its consequences. However, many mutations affect sleep indirectly. For example, circadian rhythms control global physiology, and their abrogation can also result in sleep loss 61, 62. In mutants that confer a strong circadian phenotype, it will be difficult to attribute physiological phenotypes to sleep loss. Similarly, sleep loss can be caused by mutations leading to hyperactivity. However, hyperactivity also strongly affects wake behavior and causes the same problems as SD by sensory stimulation 63. The most specific sleep loss would probably be obtained by mutating genes that are specifically required for sleep induction, i.e., sleep‐active neurons and their circuits. Because sleep‐active neurons inhibit wake circuits, the removal of the sleep‐active neurons should lead to an increase in arousal. Assuming that sleep‐active neurons play only a minor role in limiting wakefulness activity but rather a prominent role in inducing sleep, their ablation may result in moderate arousal but should not result in severe hyperarousal during normal wakefulness. Consistent with this idea, mutants exist that reduce sleep without causing hyperactivity (see below). It is possible that sleep genes and neurons play roles also in other processes and that thus complete specificity of genetic SD will be difficult or impossible in some or even all systems. However, it is likely that a high degree of specificity can be achieved in most systems, which should be sufficient for studying sleep functions.
Chronic sleep restriction in humans is associated with long‐term health consequences, and model animals that genetically reduce sleep will be important tools to study the mechanisms underlying chronic sleep restriction. For studying the functions of sleep in model organisms, it may be favorable if the degree of sleep removal is high, perhaps even complete. Homeostatic compensatory processes exist that can compensate for sleep loss. For example, reduction of sleep amount in experimental models can lead to increased sleep depth during the remaining sleep time, which, at least in part, ameliorates the consequences of sleep loss. Some animals can live with little sleep, suggesting that relatively small amounts of sleep can be sufficient to fulfill sleep's essential functions 21, 52. Thus, some sleep functions may not be detectable as long as residual sleep is present and it would be advantageous to be able to ablate sleep bound. Because sleep homeostasis induces rebound sleep through over‐activation of sleep‐active neurons, the targeting of these neurons should not only allow the control of baseline sleep, but also rebound sleep 54, 64.
Genetically removing sleep in model systems: rodents
Seminal discoveries on sleep were made using a variety of mammalian models including mice, rats, cats, and monkeys. These model animals have been pivotal in studying both non‐REM and REM sleep. The brain structures controlling sleep in mammals have turned out to be highly conserved. Its molecular amenability has made the mouse the most intensively used species for genetic sleep studies in mammals 23, 65, 66. SD by sensory stimulation has been the main method by which sleep functions have been investigated in mammals. Genetic SD is partially possible in rodent models for both REM sleep and non‐REM sleep. Forward genetic screening for sleep mutants identified a mouse mutant called Dreamless, a dominant mutation in a gene that encodes an ion channel required to control neural excitability, leading to a strong reduction of REM sleep but also causing defects in other rhythmic processes 38. REM sleep is induced from non‐REM sleep by GABAergic neurons in the ventral medulla of the brain stem. Inhibition of these neurons reduces REM sleep, and it has also been possible to induce REM sleep by optogenetically depolarizing these neurons 67. Thus, the Dreamless mutant and optogenetic induction of REM sleep present tools to investigate REM sleep functions, but such studies have not yet been published. Proving causality for REM sleep functions has been a challenge because manipulating REM sleep typically also affects non‐REM sleep 6. REM sleep is thought to be involved in specific types of memory formation and consolidation through brain activity characterized by high‐amplitude theta waves in the hippocampal EEG. To study the effects of hippocampal theta activity on memory, the activity of GABAergic MS neurons, which are required for theta activity during REM sleep but not for REM sleep itself, was optogenetically silenced during REM sleep. Silencing GABAergic MS neurons specifically during REM sleep caused defects in specific types of memory formation, providing a causal link between hippocampal theta activity during REM sleep and memory formation 68. This example shows how optogenetics can be employed for functional studies of REM sleep 6.
Mutants that specifically and completely remove non‐REM sleep in mammals have not yet been described, and the known mutants that show reduced sleep all display only partial sleep loss and often are not very specific but also confer additional phenotypes and are thus not ideal for genetic SD 62, 69. However, manipulations of specific brain areas can lead to substantial sleep loss or gain (Fig 4). There are two principal approaches for triggering sleep loss via manipulations of brain areas that have been successfully applied in rodents. (i) The activity of wake‐promoting areas can be increased and (ii) sleep‐inducing centers can be impaired. (i) An important wake‐promoting area is the PB, which causes arousal in many brain areas and which can be activated chemogenetically to extend wakefulness and restrict sleep for several days without causing hyperarousal 70. Alternatively to activating the PB, wakefulness can also be extended by activating other arousal centers of the brain including supramammillary glutamatergic neurons 71. (ii) Sleep‐active neurons were first found in the VLPO and lesioning this area in rodents reduced sleep by approximately 50% without causing stress, hyperarousal, or strong circadian effects 72, 73. VLPO sleep‐active neurons can also be controlled using optogenetics 74. Sleep‐promoting VLPO neurons can not only be silenced directly but also indirectly, for instance though chemogenetic activation of inhibitors of sleep‐inducing centers, such as GABAergic neurons of the ventral lateral hypothalamus or basal forebrain 75, 76. Other brain areas such as the basal forebrain, the lateral hypothalamus, brain stem, and cortex also contain sleep‐active neurons 66. For example, GABAergic neurons of the PZ of the medulla of the brainstem present an important sleep‐inducing brain region in mammals. These neurons were shown to be sleep‐active, ablation of this region led to a reduction of sleep by about 40%, and chemogenetic activation of this region led to an increase in sleep (Fig 5) 77, 78, 79. The occurrence of multiple populations of sleep‐active neurons in mammalian brains may explain why ablation of subsets of sleep‐active neurons only caused a partial removal of sleep. It would be fascinating to completely remove sleep from mice by ablating all or at least the key populations of sleep‐active neurons. A reasonable next step could be to combine genetic ablations of GABAergic sleep‐active neurons of the VLPO and PZ to see whether this would lead to a stronger or even complete sleep loss.
Figure 4. Genetic SD across the four key genetic animal models.
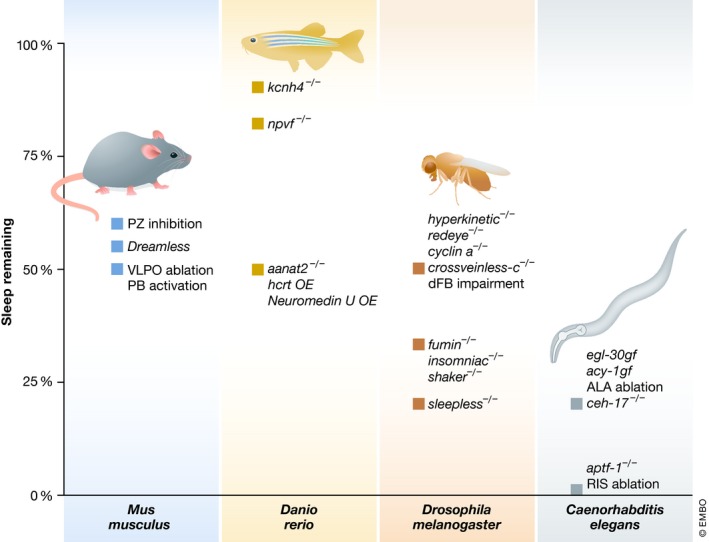
Shown are examples of genetic and neural ablations that result in reduction of sleep, some of which have low specificity, for instance manipulations leading to generally increased activity levels or impairment of circadian rhythms do not present specific manipulations. Inhibition of the PZ reduces non‐REM sleep by about 40% 77. Dreamless reduces REM sleep by 44% 38. Chemogenetic activation of the PB and ablation of the VLPO reduce sleep by about 50% (estimated from 70 for the first day of CNO application) 72. In zebrafish, mutation of the sleep‐promoting npvf gene reduces sleep by about 10% 95, voltage‐gated potassium channel gene kcnh4a knockout caused a 15% reduction 94, melatonin receptor mutation aanat2 −/− led to approximately 50% reduction 96, and overexpression of wake‐promoting factors such as hcrt and neuromedin u genes led to a variable reduction of sleep of around half 90, 91. In Drosophila, inhibition of SIFamide receptor‐expressing PI neurons reduces sleep by about 30%(estimated from 110, 146) and interference with the mushroom body by about 45% without causing hyperactivity during wake 111. Mutation of redeye 99, cyclin A RNAi 100, deletion of hyperkinetic 101, or interference with sleep‐promoting neurons of the dFB (activating cAMP signaling 115 or crossveinless‐c RNAi 113) reduce sleep by about half. insomniac 103, fumin 104, or shaker led to approximately 2/3 reduction of sleep 102. One of the strongest mutations in Drosophila is sleepless with > 80% sleep reduction 105. Caenorhabditis elegans physiological sleep during lethargus is reduced by about 80% in hyperactive mutants (egl‐30gf 127 or acy‐1gf 128) as well as in ALA mutant ceh‐17(−) (locomotion quiescence 20 min after heat shock) 35 and is virtually abolished across several physiological conditions (reduction here displayed as 99%) by aptf‐1 −/− or ablation of the sleep‐active RIS neuron 124, 134, 139.
Figure 5. Chemogenetics and optogenetics allow specific gain‐of‐function experiments for sleep.
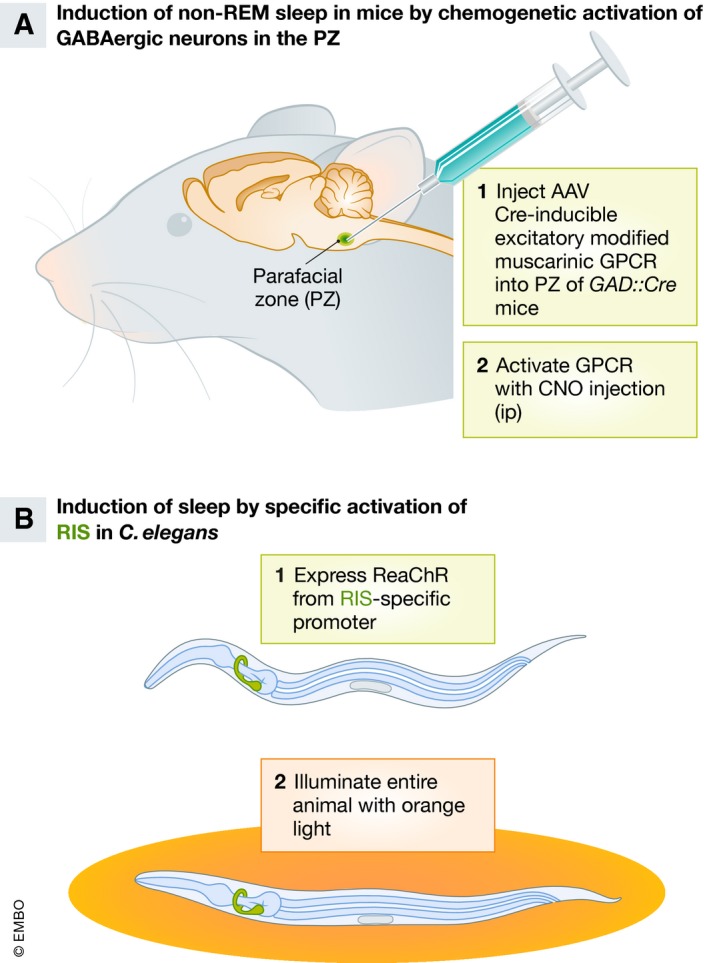
Shown are examples from mouse and Caenorhabditis elegans, but chemogenetic and optogenetic sleep control is also applicable to other models such as Drosophila and zebrafish. (A) Non‐REM sleep can be triggered in mice by chemogenetic activation of GABAergic neurons in the PZ. To achieve specific activation of GABAergic neurons within a specific brain locus, a transgenic mouse is taken that expresses Cre recombinase from the GABA‐specific GAD2 promoter. A Cre‐inducible excitatory muscarinic modified G protein‐coupled receptor is expressed using an adeno‐associated virus construct, which is injected locally into the PZ and transforms only the neurons in the vicinity of the injections. Intraperitoneal injection of CNO, an agonist of the excitatory muscarinic modified G protein‐coupled receptor, then leads to an increased activity of GABAergic PZ neurons, leading to the induction of non‐REM sleep. Mice with increased non‐REM sleep can then be analyzed for phenotypes such as learning and memory 78. (B) Sleep can be induced optogenetically in Caenorhabditis elegans by depolarizing the GABAergic and peptidergic sleep‐active RIS neuron 134. Transgenic animals are generated that express Channelrhodopsin (here the red‐light‐activated variant ReaChR) specifically in RIS, which is achieved by using a specific promoter. Illuminating the entire animal, which is transparent, with red light leads to the depolarization of RIS and sleep induction. The phenotypes caused by increased sleep can then be studied.
Restriction of sleep by VLPO lesion in rats has been used to study the role of sleep in metabolism. While ghrelin was increased and leptin was reduced, a decreased body weight and no signs of metabolic syndrome or obesity were found in this model 73. Restricting sleep by activating the PB in mice led to a modest increase in blood glucose levels and decreased leptin, but body weight was reduced similar to the results obtained by VLPO lesion 70. Interestingly, in these sleep restriction models, the metabolic consequences were thus either minor or contrary to what has been found after sleep restriction in humans 3, 4. Together these results suggest that either there are different responses of humans and rodents to sleep restriction or that the consequences of sleep restriction observed in humans may not be caused directly by sleep loss but by other factors such as stress or circadian effects, underscoring the importance to re‐evaluate sleep function theories using genetic SD models.
Genetically removing sleep in model systems: zebrafish
The zebrafish Danio rerio presents an important vertebrate sleep model system between rodent and invertebrate models. Like humans and unlike rodents, zebrafish sleep mostly during the night. Zebrafish appear to have a quiet sleep state but evidence for a sleep state that resembles REM is lacking. While one study could not find evidence for rapid eye movement during sleep, this result does not exclude the possibility that other components of REM sleep are present in zebrafish 80. Major advantages of zebrafish as a sleep model are the high level of conservation of genes involved in sleep control, such as neuropeptide systems, a high level of conservation of key brain anatomical structures within a transparent brain, the possibility to model neuropsychiatric disorders as well as the possibility to scale up genetic and pharmacological screens 13, 14, 81, 82, 83, 84. Several physical methods exist for SD in zebrafish. For instance, electrical shocks and physical shaking have been used but are quite harsh and can even injure the animal 83, 85. Light potently suppresses sleep in fish leading to a 90% reduction of sleep 85. This level of sleep deprivation is impressive but sleep deprivation by light still might cause unspecific effects through sensory stimulation and alternations of the circadian clock. Perhaps the gentlest method for physical SD in zebrafish is through constant water flow 86. Physical SD in zebrafish has been mostly used to study sleep reversibility and homeostasis, but some studies have also started to address the effects of SD on cognitive functions and learning 87, 88, 89.
Through genetic screening multiple mutants with reduced sleep have been identified. For example, knockout of the sleep‐promoting neuropeptides QRFP and prokineticin 2 reduce sleep. However, these mutants produce only small effects because these factors control the relatively small amount of sleep that occurs during the day. Overexpression of wake‐promoting genes such as hcrt or neuromedin U causes hyperactivity and suppresses sleep. The effects of transient overexpression are quite variable but can suppress about half of the sleep time 90, 91. Chemogenetic or optogenetic activation or inhibition of hcrt neurons can be used to decrease or increase sleep, respectively 92, 93. Consistent with these findings, the kcnh4a potassium channel genes act in hcrt neurons to regulate their activity, with kcnh4a knockout resulting in a 15% sleep reduction 94. Loss of function of the npvf neuropeptide gene also causes hyperactivity and reduces sleep by 10% 95. Mutation of the melatonin receptor gene aanat2 in zebrafish reduces night sleep in the presence of light–dark cycles by about 50%. In free‐running conditions (i.e., constant darkness), the increase of sleep during the subjective night is almost completely eliminated. These results suggest that melatonin is the major factor for circadian regulation of sleep in zebrafish 96 (Fig 4). Reports on sleep functions based on genetic SD are still lacking in the literature. While sleep‐active neurons have not yet been reported in zebrafish, they likely exist and their ablation should provide a valuable model for studying the consequences of sleep loss.
Genetically removing sleep in model systems: Drosophila
Drosophila melanogaster has emerged as a leading model system to study the molecular basis of sleep. Its main advantages are genetic amenability and a clear coupling of sleep to the circadian rhythm. Like humans and zebrafish, Drosophila sleep mostly during the dark phase and also have a period of behavioral inactivity during the middle of the light phase that is called a siesta. Thus, behavioral activity in fruit flies occurs mostly during both the morning and the evening hours. Drosophila has been instrumental in solving the molecular underpinnings of circadian rhythms and thus presents a prime system to study the control of sleep and its regulation by the circadian clock 15, 97, 98. Genetic accessibility has motivated multiple large‐scale screens for mutations that alter sleep behavior. Mutations and neural manipulations in Drosophila can severely reduce sleep. For instance, mutation of the nicotinic acetylcholine receptor α subunit gene redeye, the potassium channel regulator hyperkinetic, or RNAi of cyclin A or its regulator reduced sleep by about half 99, 100, 101. Mutation of the shaker potassium channel, the ubiquitin ligase adapter complex gene insomniac, and the dopamine transporter gene fumin reduced sleep by about two‐thirds 102, 103, 104. Among the strongest mutations that reduce sleep is the sleepless mutation with about 80% of sleep reduction. sleepless encodes a neurotoxin that regulates shaker 105, 106 (Fig 4). However, several of these mutants are severely hyperactive. Thus, results regarding sleep functions based on hyperactive mutants should be interpreted with caution 101, 104, 105, 107.
Fly brains possess several centers that contain wake‐promoting or sleep‐promoting neurons. Wake‐promoting centers are, for example, cyclin A‐expressing neurons of the pars lateralis 108. Important sleep‐promoting centers are formed by sub‐populations of neurons in the mushroom body, dorsal paired medial neurons, and peptidergic neurons in the PI 109, 110, 111. As another example, sleep‐promoting neurons of the dFB can actively induce sleep and confer homeostatic sleep drive stemming from R2 neurons of the ellipsoid body and are thus similar to mammalian sleep‐promoting neurons 112, 113, 114. Interference with the function of dFB neurons, for instance by RNAi of crossveinless‐c, a Rho GTPase‐activating gene, reduced sleep by about half. Importantly, mutation of crossveinless‐c decreases sleep without causing signs of hyperactivity 113, 115. This supports the hypothesis that genetic SD without hyperactivity is possible in Drosophila (Fig 4). Thus, specific interference of dFB neurons and crossveinless‐c mutants present specific, albeit partial, genetic SD in Drosophila and should, along with other mutants, provide useful models for studying the effects of sleep restriction in fruit flies. Similar to mammals, several populations of sleep‐promoting neurons exist and the ablation of individual populations causes partial sleep loss. It is not well understood how the various sleep centers in Drosophila interact to cause sleep, but they likely act, at least in part, in parallel pathways. It might be possible to combine mutations that target different sleep‐promoting areas and test whether this would result in near‐complete sleep loss. This would not only shed light on how the different sleep centers interact but might also generate stronger models of genetic SD. It will be interesting to see whether near‐complete genetic SD will be possible and whether and how it would result in lethality.
Sensory stimulation‐induced SD leads to hyperarousal, the activation of cellular stress responses in Drosophila, and is detrimental 116. Genetic sleep reduction has been associated with reduced lifespan in many but not all Drosophila sleep mutants. For instance, loss of the sleepless gene causes both a shortening of sleep and lifespan, while neuronal knockdown of insomniac leads to sleep reduction without a shortening of longevity 102, 103, 105, 117. Also, knockout of fumin did not cause a shortening of lifespan but a reduction of brood size 104, 118. Also, defects in memory have been observed in sleep mutants 101. Genetic sleep reduction by neuronal knockdown of insomniac did not demonstrate a role for sleep in survival of infection or starvation. The short‐sleeping mutant did, however, exhibit a sensitivity to survive oxidative stress. Several other short‐sleeping mutants showed oxidative stress sensitivity as well, suggesting that the sensitivity was probably not conferred by pleiotropic side effects caused by the mutation but rather is broadly associated with sleep loss. Consistent with this finding, increasing sleep genetically or pharmacologically conferred greater resistance to oxidative stress 107. These experiments not only identified resistance to oxidative stress as a potential core function of sleep in Drosophila, but also illustrate how the use of multiple sleep mutants distills a sleep phenotype from potentially pleiotropic mutations.
Genetically removing sleep in model systems: C. elegans
Caenorhabditis elegans is the genetic animal model with the smallest nervous system, as it has only about 0.3% the number of neurons of an adult Drosophila or zebrafish embryo brain. The connectome of the 302 neurons of the hermaphrodite has been mapped, providing an entry point for circuit studies 119. Sleep in C. elegans is attractive to study due to its genetic amenability and the invariant number of neurons allowing straightforward genetic SD. Caenorhabditis elegans shows sleeping behavior across many life stages and conditions. In the developing larva, sleep is linked to the molting cycle, and sleep bouts occur during a phase called lethargus prior to the molt 120, 121, 122. This developmentally controlled sleep does not seem to be coupled to the day–night cycle, but its timing still is controlled by the circadian period homolog lin‐42 123. If hatched in the absence of food, larvae arrest development and during this phase alternate between sleep and wake cycles 124. In the presence of adverse conditions, worms develop into an enduring alternative larval stage called the “dauer”, which spends much of its time sleeping 121, 124. Adult worms sleep both in the presence and in the absence of food, with food amount and quality determining the amount of sleep 124, 125, 126. Finally, C. elegans sleep following severe cellular stress 35.
As in other species, hyperactive mutations can reduce sleep in C. elegans; however, they do not present specific manipulations 127, 128. Caenorhabditis elegans possess two major individual neurons that have been implicated in the induction of sleep. Cellular stress causes the secretion of EGF, which activates EGF receptor signaling in a neuron called ALA 35, 129, 130. EGF activation leads to the secretion of multiple neuropeptides from ALA, which have both overlapping and distinct inhibitory functions on behavioral activity by binding to downstream receptors, likely involving a diffusional mechanism 131, 132, 133. It is not yet clear whether ALA presents a sleep‐active neuron in the sense that it depolarizes specifically during a sleep bout or whether it promotes sleep by a different mechanism. ALA can be easily ablated physically or genetically. Loss of function of the homeobox transcription factor genes ceh‐17 or ceh‐14 renders ALA dysfunctional and thus strongly impairs sleep following cellular stress 129 (Fig 4).
The second major known sleep‐promoting neuron of C. elegans is called RIS. This neuron is sleep‐active as it depolarizes at the onset of sleep bouts and its optogenetic depolarization acutely induces sleep 134, 135, 136 (Fig 5). Similar to ALA, RIS can be easily ablated physically or genetically. A mutation in the AP2 transcription factor gene aptf‐1 renders RIS inactive, because AP2 is required for the expression of sleep‐inducing neuropeptides 134. Interestingly, AP2 transcription factors are conserved regulators of sleep also in Drosophila and humans 137, 138. Sleep bouts become undetectable in these “RIS mutants” during many life stages and physiological conditions. aptf‐1 mutant worms show no severe hyperactivity during wake, indicating that they are not strongly hyperaroused following sleep loss and that sleep loss is likely not a consequence of increased arousal 124, 134, 135, 139. Thus, during many physiological conditions, RIS inactivation in C. elegans presents both a virtually complete as well as a highly specific model for sleeplessness (Fig 4). It has been proposed that ALA and RIS present mostly parallel systems that act during un‐physiological and physiological conditions, respectively, and whether and how these neurons interact is not known 140. Together, ALA and RIS ablation present valuable tools for studying the functions of sleep in different conditions.
Loss of ALA function is viable during physiological conditions but impairs survival upon cellular stress, demonstrating the importance of sleep in recuperating from cellular insult. The need to sleep after cellular stress is plastic and is reduced if the general stress resistance is increased, suggesting that sleep is part of a stress resistance program 35, 129, 130, 141. RIS‐ablated C. elegans are viable and display much less severe consequences compared with SD by sensory stimulation, which can even be lethal 134, 139, 142, 143. It is possible that sensory stimulation causes non‐specific side effects or that long‐term genetic SD is compensated for by development or other homeostatic processes. Caenorhabditis elegans lives a boom‐and‐bust lifestyle and alternates between short periods of superfluous food and long periods of starvation. Consistent with these ecological constraints, sleep becomes essential during larval starvation‐induced developmental arrest. It was shown that genetic SD shortens the survival of starved larvae by half. Rather than merely conserving energy, sleep appears to slow down the progression of aging phenotypes 124. RIS‐ablated worms have a normal lifespan under ideal laboratory conditions 124, but sleep during old age has not been examined. Caenorhabditis elegans has a short generation time and life span and may thus be uniquely able to survive sleep loss under ideal laboratory conditions. In summary, similar to Drosophila, essential sleep functions following genetic SD in C. elegans are most obvious under challenging conditions.
Conclusion
Recent advances in the understanding of how sleep is regulated in genetically accessible model organisms have made it possible to genetically remove sleep to a high degree and specificity. Acute SD by sensory deprivation and chronic genetic SD are obviously different experiments and can lead to different conclusions as to the functions of sleep. In future studies, it will be important to understand the basis of these differences. It is as of now unclear whether SD by sensory stimulation overestimates the role of sleep because it causes non‐specific side effects or whether genetic SD underestimates the role of sleep because of compensation processes. Genetic SD models can be used to study the consequences of sleep restriction or loss. Until now, specific phenotypes from genetic SD are scarce. However, sleepless model animals are increasingly employed for studies aiming to understand the consequences of sleep loss and will likely be key to comprehend why animals and humans need to sleep. Initial results indicate that much of the phenotypes observed after SD may not be a direct consequence of the lost sleep. For instance, the metabolic consequences of sleep loss in humans have been challenged by more specific surgical or genetic SD in rodents. Similarly, genetic SD in Drosophila and C. elegans produces smaller phenotypes compared with stimulation‐induced SD. Work from Drosophila and C. elegans suggests that sleep becomes especially important for survival during challenging conditions. Improving the genetic sleep loss models by increasing the degree and specificity of sleeplessness as well as fine‐tuning the amount and timing of lost or gained sleep will be important next steps in facilitating the study of sleep functions in animals. Analyzing phenotypes of genetic SD models will help define core functions of sleep and support our endeavor to understand how sleep becomes vital.
Conflict of interest
The author declares that he has no conflict of interest.
In need of answers.
-
What are the vital functions of sleep?
The functions of sleep have been studied for decades, mostly by either correlation or SD induced by sensory stimulation. Genetic SD is an emerging alternative to remove sleep but typically produces weaker phenotypes compared with stimulation‐induced SD. It could be that constitutive genetic SD leads to compensatory changes, whereas acute SD cannot be easily compensated for. However, the power of constitutive genetic SD lies in the potential accumulation of the consequences of sleep loss over time. Also, transgenerational effects of sleep loss should be studied for long‐term effects of sleep loss. Thus, a thorough analysis of the different SD methods and a re‐evaluation of the previously proposed roles of sleep will be necessary to understand sleep functions.
-
Can sleep be removed specifically and completely using genetic SD?
A prerequisite for genetic SD is specificity of the manipulation as well as a high degree of deprivation. However, it is yet unclear what level of specificity can be achieved. Genes and neurons that control sleep may have functions that overlap with other processes. Also, complete genetic SD likely is lethal in many systems such as mammals. Thus, partial or conditional genetic SD will be the methods of choice for studying sleep functions in this case.
-
How did sleep evolve and how conserved are sleep functions?
Molecular analysis has suggested that there is a high level of conservation of sleep regulation but it is less clear how conserved molecular sleep functions are. Also, it is not clear for which initial functions sleep has been selected for. Speculatively, sleep emerged in evolution to save energy or serve basic needs and was co‐opted later to also serve higher brain functions. Studying sleep functions across models should shed light on these questions. Evolutionary studies would be aided by studying sleep and sleeplessness in additional models beyond the widely used animals described herein.
-
How does sleep exert its functions?
While many ideas exist as to the potential functions of sleep, little is known about the underlying mechanisms. For example, it is not known what molecules are preserved, how resources are allocated, and how cellular processes are aided. It is unclear how basic molecular and cellular functions of sleep support a healthy physiology and how sleep is linked to aging. Also, how sleep aids higher brain functions is not clear. To answer these questions, the consequences of sleep loss need to be studied using multiple molecular and systems approaches across animals.
Acknowledgements
I would like to thank Mimi Shirasu‐Hiza, Mary Osborn, Jason Rihel, and David Prober for comments on the manuscript. This work was supported by the Max Planck Society (Max Planck Research Group) and the European Research Council (Horizon 2020 Starting Grant, agreement no. 637860, SLEEPCONTROL).
EMBO Reports (2019) 20: e46807
See the Glossary for abbreviations used in this article.
References
- 1. Irwin MR (2015) Why sleep is important for health: a psychoneuroimmunology perspective. Annu Rev Psychol 66: 143–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drake CL, Roehrs T, Roth T (2003) Insomnia causes, consequences, and therapeutics: an overview. Depress Anxiety 18: 163–176 [DOI] [PubMed] [Google Scholar]
- 3. Van Cauter E, Spiegel K, Tasali E, Leproult R (2008) Metabolic consequences of sleep and sleep loss. Sleep Med 9(Suppl 1): S23–S28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knutson KL, Spiegel K, Penev P, Van Cauter E (2007) The metabolic consequences of sleep deprivation. Sleep Med Rev 11: 163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aserinsky E, Kleitman N (1953) Regularly occurring periods of eye motility, and concomitant phenomena, during sleep. Science 118: 273–274 [DOI] [PubMed] [Google Scholar]
- 6. Peever J, Fuller PM (2017) The biology of REM sleep. Curr Biol 27: R1237–R1248 [DOI] [PubMed] [Google Scholar]
- 7. Beckers GJ, Rattenborg NC (2015) An in depth view of avian sleep. Neurosci Biobehav Rev 50: 120–127 [DOI] [PubMed] [Google Scholar]
- 8. Vorster AP, Born J (2015) Sleep and memory in mammals, birds and invertebrates. Neurosci Biobehav Rev 50: 103–119 [DOI] [PubMed] [Google Scholar]
- 9. Shein‐Idelson M, Ondracek JM, Liaw HP, Reiter S, Laurent G (2016) Slow waves, sharp waves, ripples, and REM in sleeping dragons. Science 352: 590–595 [DOI] [PubMed] [Google Scholar]
- 10. Campbell SS, Tobler I (1984) Animal sleep: a review of sleep duration across phylogeny. Neurosci Biobehav Rev 8: 269–300 [DOI] [PubMed] [Google Scholar]
- 11. Nath RD, Bedbrook CN, Abrams MJ, Basinger T, Bois JS, Prober DA, Sternberg PW, Gradinaru V, Goentoro L (2017) The jellyfish cassiopea exhibits a sleep‐like state. Curr Biol 27: 2984–2990.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cirelli C, Tononi G (2008) Is sleep essential? PLoS Biol 6: e216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barlow IL, Rihel J (2017) Zebrafish sleep: from geneZZZ to neuronZZZ. Curr Opin Neurobiol 44: 65–71 [DOI] [PubMed] [Google Scholar]
- 14. Oikonomou G, Prober DA (2017) Attacking sleep from a new angle: contributions from zebrafish. Curr Opin Neurobiol 44: 80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Artiushin G, Sehgal A (2017) The Drosophila circuitry of sleep‐wake regulation. Curr Opin Neurobiol 44: 243–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trojanowski NF, Raizen DM (2016) Call it worm sleep. Trends Neurosci 39: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joiner WJ (2016) Unraveling the evolutionary determinants of sleep. Curr Biol 26: R1073–R1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lesku JA, Rattenborg NC, Valcu M, Vyssotski AL, Kuhn S, Kuemmeth F, Heidrich W, Kempenaers B (2012) Adaptive sleep loss in polygynous pectoral sandpipers. Science 337: 1654–1658 [DOI] [PubMed] [Google Scholar]
- 19. Rattenborg NC, Mandt BH, Obermeyer WH, Winsauer PJ, Huber R, Wikelski M, Benca RM (2004) Migratory sleeplessness in the white‐crowned sparrow (Zonotrichia leucophrys gambelii). PLoS Biol 2: E212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duboue ER, Keene AC, Borowsky RL (2011) Evolutionary convergence on sleep loss in cavefish populations. Curr Biol 21: 671–676 [DOI] [PubMed] [Google Scholar]
- 21. Siegel JM (2005) Clues to the functions of mammalian sleep. Nature 437: 1264–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Siegel JM (2009) Sleep viewed as a state of adaptive inactivity. Nat Rev Neurosci 10: 747–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mignot E (2008) Why we sleep: the temporal organization of recovery. PLoS Biol 6: e106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scharf MT, Naidoo N, Zimmerman JE, Pack AI (2008) The energy hypothesis of sleep revisited. Prog Neurobiol 86: 264–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmidt MH (2014) The energy allocation function of sleep: a unifying theory of sleep, torpor, and continuous wakefulness. Neurosci Biobehav Rev 47: 122–153 [DOI] [PubMed] [Google Scholar]
- 26. Daan S, Barnes BM, Strijkstra AM (1991) Warming up for sleep? Ground squirrels sleep during arousals from hibernation. Neurosci Lett 128: 265–268 [DOI] [PubMed] [Google Scholar]
- 27. Trachsel L, Edgar DM, Heller HC (1991) Are ground squirrels sleep deprived during hibernation? Am J Physiol 260: R1123–R1129 [DOI] [PubMed] [Google Scholar]
- 28. Schmidt MH, Swang TW, Hamilton IM, Best JA (2017) State‐dependent metabolic partitioning and energy conservation: a theoretical framework for understanding the function of sleep. PLoS ONE 12: e0185746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mackiewicz M, Shockley KR, Romer MA, Galante RJ, Zimmerman JE, Naidoo N, Baldwin DA, Jensen ST, Churchill GA, Pack AI (2007) Macromolecule biosynthesis: a key function of sleep. Physiol Genomics 31: 441–457 [DOI] [PubMed] [Google Scholar]
- 30. Ramm P, Smith CT (1990) Rates of cerebral protein synthesis are linked to slow wave sleep in the rat. Physiol Behav 48: 749–753 [DOI] [PubMed] [Google Scholar]
- 31. Nakanishi H, Sun Y, Nakamura RK, Mori K, Ito M, Suda S, Namba H, Storch FI, Dang TP, Mendelson W et al (1997) Positive correlations between cerebral protein synthesis rates and deep sleep in Macaca mulatta . Eur J Neuorsci 9: 271–279 [DOI] [PubMed] [Google Scholar]
- 32. Krueger JM, Frank MG, Wisor JP, Roy S (2016) Sleep function: toward elucidating an enigma. Sleep Med Rev 28: 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zielinski MR, Krueger JM (2011) Sleep and innate immunity. Front Biosci 3: 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kayser MS, Biron D (2016) Sleep and development in genetically tractable model organisms. Genetics 203: 21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hill AJ, Mansfield R, Lopez JM, Raizen DM, Van Buskirk C (2014) Cellular stress induces a protective sleep‐like state in C. elegans . Curr Biol 24: 2399–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tu BP, McKnight SL (2006) Metabolic cycles as an underlying basis of biological oscillations. Nat Rev Mol Cell Biol 7: 696–701 [DOI] [PubMed] [Google Scholar]
- 37. Wang Z, Ma J, Miyoshi C, Li Y, Sato M, Ogawa Y, Lou T, Ma C, Gao X, Lee C et al (2018) Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature 558: 435–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Funato H, Miyoshi C, Fujiyama T, Kanda T, Sato M, Wang Z, Ma J, Nakane S, Tomita J, Ikkyu A et al (2016) Forward‐genetics analysis of sleep in randomly mutagenized mice. Nature 539: 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi S, Lim DS, Chung J (2015) Feeding and fasting signals converge on the LKB1‐SIK3 pathway to regulate lipid metabolism in Drosophila . PLoS Genet 11: e1005263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Uebi T, Itoh Y, Hatano O, Kumagai A, Sanosaka M, Sasaki T, Sasagawa S, Doi J, Tatsumi K, Mitamura K et al (2012) Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS ONE 7: e37803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ et al (2013) Sleep drives metabolite clearance from the adult brain. Science 342: 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cirelli C, Gutierrez CM, Tononi G (2004) Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41: 35–43 [DOI] [PubMed] [Google Scholar]
- 43. Seibt J, Dumoulin MC, Aton SJ, Coleman T, Watson A, Naidoo N, Frank MG (2012) Protein synthesis during sleep consolidates cortical plasticity in vivo . Curr Biol 22: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marshall L, Helgadottir H, Molle M, Born J (2006) Boosting slow oscillations during sleep potentiates memory. Nature 444: 610–613 [DOI] [PubMed] [Google Scholar]
- 45. de Vivo L, Bellesi M, Marshall W, Bushong EA, Ellisman MH, Tononi G, Cirelli C (2017) Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science 355: 507–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, Hagenbuchle O, O'Hara BF, Franken P, Tafti M (2007) Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci USA 104: 20090–20095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diering GH, Nirujogi RS, Roth RH, Worley PF, Pandey A, Huganir RL (2017) Homer1a drives homeostatic scaling‐down of excitatory synapses during sleep. Science 355: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Porkka‐Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW (1997) Adenosine: a mediator of the sleep‐inducing effects of prolonged wakefulness. Science 276: 1265–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dudai Y, Karni A, Born J (2015) The consolidation and transformation of memory. Neuron 88: 20–32 [DOI] [PubMed] [Google Scholar]
- 50. Wagner U, Gais S, Haider H, Verleger R, Born J (2004) Sleep inspires insight. Nature 427: 352–355 [DOI] [PubMed] [Google Scholar]
- 51. von Economo C (1930) Sleep as a problem of localization. J Nerv Ment Dis 71: 249–259 [Google Scholar]
- 52. Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010) Sleep state switching. Neuron 68: 1023–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bringmann H (2018) Sleep‐active neurons: conserved motors of sleep. Genetics 208: 1279–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saper CB, Scammell TE, Lu J (2005) Hypothalamic regulation of sleep and circadian rhythms. Nature 437: 1257–1263 [DOI] [PubMed] [Google Scholar]
- 55. Rechtschaffen A, Bergmann BM (1995) Sleep deprivation in the rat by the disk‐over‐water method. Behav Brain Res 69: 55–63 [DOI] [PubMed] [Google Scholar]
- 56. Zhao Z, Zhao X, Veasey SC (2017) Neural consequences of chronic short sleep: reversible or lasting? Front Neurol 8: 235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meerlo P, Koehl M, van der Borght K, Turek FW (2002) Sleep restriction alters the hypothalamic‐pituitary‐adrenal response to stress. J Neuroendocrinol 14: 397–402 [DOI] [PubMed] [Google Scholar]
- 58. Chrousos G, Vgontzas AN, Kritikou I (2000) HPA axis and sleep In Endotext, De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A. (eds). South Dartmouth, MA: endotext.org; [Google Scholar]
- 59. Fuller PM, Sherman D, Pedersen NP, Saper CB, Lu J (2011) Reassessment of the structural basis of the ascending arousal system. J Comp Neurol 519: 933–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Krause AJ, Simon EB, Mander BA, Greer SM, Saletin JM, Goldstein‐Piekarski AN, Walker MP (2017) The sleep‐deprived human brain. Nat Rev Neurosci 18: 404–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guo F, Yu J, Jung HJ, Abruzzi KC, Luo W, Griffith LC, Rosbash M (2016) Circadian neuron feedback controls the Drosophila sleep–activity profile. Nature 536: 292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek FW (2000) The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci 20: 8138–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Angelakos CC, Watson AJ, O'Brien WT, Krainock KS, Nickl‐Jockschat T, Abel T (2017) Hyperactivity and male‐specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res 10: 572–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Alam MA, Kumar S, McGinty D, Alam MN, Szymusiak R (2014) Neuronal activity in the preoptic hypothalamus during sleep deprivation and recovery sleep. J Neurophysiol 111: 287–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Saper CB, Chou TC, Scammell TE (2001) The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci 24: 726–731 [DOI] [PubMed] [Google Scholar]
- 66. Weber F, Dan Y (2016) Circuit‐based interrogation of sleep control. Nature 538: 51–59 [DOI] [PubMed] [Google Scholar]
- 67. Weber F, Chung S, Beier KT, Xu M, Luo L, Dan Y (2015) Control of REM sleep by ventral medulla GABAergic neurons. Nature 526: 435–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Boyce R, Glasgow SD, Williams S, Adamantidis A (2016) Causal evidence for the role of REM sleep theta rhythm in contextual memory consolidation. Science 352: 812–816 [DOI] [PubMed] [Google Scholar]
- 69. Douglas CL, Vyazovskiy V, Southard T, Chiu SY, Messing A, Tononi G, Cirelli C (2007) Sleep in Kcna2 knockout mice. BMC Biol 5: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Qiu MH, Chen MC, Fuller PM, Lu J (2016) Stimulation of the pontine parabrachial nucleus promotes wakefulness via extra‐thalamic forebrain circuit nodes. Curr Biol 26: 2301–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Pedersen NP, Ferrari L, Venner A, Wang JL, Abbott SBG, Vujovic N, Arrigoni E, Saper CB, Fuller PM (2017) Supramammillary glutamate neurons are a key node of the arousal system. Nat Commun 8: 1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu J, Greco MA, Shiromani P, Saper CB (2000) Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci 20: 3830–3842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vetrivelan R, Fuller PM, Yokota S, Lu J, Saper CB (2012) Metabolic effects of chronic sleep restriction in rats. Sleep 35: 1511–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chung S, Weber F, Zhong P, Tan CL, Nguyen TN, Beier KT, Hormann N, Chang WC, Zhang Z, Do JP et al (2017) Identification of preoptic sleep neurons using retrograde labelling and gene profiling. Nature 545: 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Venner A, Anaclet C, Broadhurst RY, Saper CB, Fuller PM (2016) A novel population of wake‐promoting GABAergic neurons in the ventral lateral hypothalamus. Curr Biol 26: 2137–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Anaclet C, Pedersen NP, Ferrari LL, Venner A, Bass CE, Arrigoni E, Fuller PM (2015) Basal forebrain control of wakefulness and cortical rhythms. Nat Commun 6: 8744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Anaclet C, Lin JS, Vetrivelan R, Krenzer M, Vong L, Fuller PM, Lu J (2012) Identification and characterization of a sleep‐active cell group in the rostral medullary brainstem. J Neurosci 32: 17970–17976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, Fuller PM (2014) The GABAergic parafacial zone is a medullary slow wave sleep‐promoting center. Nat Neurosci 17: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Anaclet C, Fuller PM (2017) Brainstem regulation of slow‐wave‐sleep. Curr Opin Neurobiol 44: 139–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Arnason BB, Thornorsteinsson H, Karlsson KAE (2015) Absence of rapid eye movements during sleep in adult zebrafish. Behav Brain Res 291: 189–194 [DOI] [PubMed] [Google Scholar]
- 81. Chen A, Chiu CN, Mosser EA, Kahn S, Spence R, Prober DA (2016) QRFP and its receptors regulate locomotor activity and sleep in zebrafish. J Neurosci 36: 1823–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen S, Reichert S, Singh C, Oikonomou G, Rihel J, Prober DA (2017) Light‐dependent regulation of sleep and wake states by prokineticin 2 in zebrafish. Neuron 95: 153–168 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhdanova IV, Wang SY, Leclair OU, Danilova NP (2001) Melatonin promotes sleep‐like state in zebrafish. Brain Res 903: 263–268 [DOI] [PubMed] [Google Scholar]
- 84. Levitas‐Djerbi T, Appelbaum L (2017) Modeling sleep and neuropsychiatric disorders in zebrafish. Curr Opin Neurobiol 44: 89–93 [DOI] [PubMed] [Google Scholar]
- 85. Yokogawa T, Marin W, Faraco J, Pezeron G, Appelbaum L, Zhang J, Rosa F, Mourrain P, Mignot E (2007) Characterization of sleep in zebrafish and insomnia in hypocretin receptor mutants. PLoS Biol 5: e277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Aho V, Vainikka M, Puttonen HAJ, Ikonen HMK, Salminen T, Panula P, Porkka‐Heiskanen T, Wigren HK (2017) Homeostatic response to sleep/rest deprivation by constant water flow in larval zebrafish in both dark and light conditions. J Sleep Res 26: 394–400 [DOI] [PubMed] [Google Scholar]
- 87. Pinheiro‐da‐Silva J, Silva PF, Nogueira MB, Luchiari AC (2017) Sleep deprivation effects on object discrimination task in zebrafish (Danio rerio). Anim Cogn 20: 159–169 [DOI] [PubMed] [Google Scholar]
- 88. Pinheiro‐da‐Silva J, Tran S, Luchiari AC (2018) Sleep deprivation impairs cognitive performance in zebrafish: a matter of fact? Behav Processes 157: 656–663 [DOI] [PubMed] [Google Scholar]
- 89. Pinheiro‐da‐Silva J, Tran S, Silva PF, Luchiari AC (2017) Good night, sleep tight: the effects of sleep deprivation on spatial associative learning in zebrafish. Pharmacol Biochem Behav 159: 36–47 [DOI] [PubMed] [Google Scholar]
- 90. Prober DA, Rihel J, Onah AA, Sung RJ, Schier AF (2006) Hypocretin/orexin overexpression induces an insomnia‐like phenotype in zebrafish. J Neurosci 26: 13400–13410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chiu CN, Rihel J, Lee DA, Singh C, Mosser EA, Chen S, Sapin V, Pham U, Engle J, Niles BJ et al (2016) A zebrafish genetic screen identifies neuromedin U as a regulator of sleep/wake states. Neuron 89: 842–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen S, Chiu CN, McArthur KL, Fetcho JR, Prober DA (2016) TRP channel mediated neuronal activation and ablation in freely behaving zebrafish. Nat Methods 13: 147–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Singh C, Oikonomou G, Prober DA (2015) Norepinephrine is required to promote wakefulness and for hypocretin‐induced arousal in zebrafish. Elife 4: e07000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yelin‐Bekerman L, Elbaz I, Diber A, Dahary D, Gibbs‐Bar L, Alon S, Lerer‐Goldshtein T, Appelbaum L (2015) Hypocretin neuron‐specific transcriptome profiling identifies the sleep modulator Kcnh4a. Elife 4: e08638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lee DA, Andreev A, Truong TV, Chen A, Hill AJ, Oikonomou G, Pham U, Hong YK, Tran S, Glass L et al (2017) Genetic and neuronal regulation of sleep by neuropeptide VF. Elife 6: e25727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gandhi AV, Mosser EA, Oikonomou G, Prober DA (2015) Melatonin is required for the circadian regulation of sleep. Neuron 85: 1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hendricks JC, Finn SM, Panckeri KA, Chavkin J, Williams JA, Sehgal A, Pack AI (2000) Rest in Drosophila is a sleep‐like state. Neuron 25: 129–138 [DOI] [PubMed] [Google Scholar]
- 98. Shaw PJ, Cirelli C, Greenspan RJ, Tononi G (2000) Correlates of sleep and waking in Drosophila melanogaster . Science 287: 1834–1837 [DOI] [PubMed] [Google Scholar]
- 99. Shi M, Yue Z, Kuryatov A, Lindstrom JM, Sehgal A (2014) Identification of Redeye, a new sleep‐regulating protein whose expression is modulated by sleep amount. Elife 3: e01473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Rogulja D, Young MW (2012) Control of sleep by cyclin A and its regulator. Science 335: 1617–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bushey D, Huber R, Tononi G, Cirelli C (2007) Drosophila Hyperkinetic mutants have reduced sleep and impaired memory. J Neurosci 27: 5384–5393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G (2005) Reduced sleep in Drosophila Shaker mutants. Nature 434: 1087–1092 [DOI] [PubMed] [Google Scholar]
- 103. Stavropoulos N, Young MW (2011) insomniac and Cullin‐3 regulate sleep and wakefulness in Drosophila . Neuron 72: 964–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kume K, Kume S, Park SK, Hirsh J, Jackson FR (2005) Dopamine is a regulator of arousal in the fruit fly. J Neurosci 25: 7377–7384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Koh K, Joiner WJ, Wu MN, Yue Z, Smith CJ, Sehgal A (2008) Identification of SLEEPLESS, a sleep‐promoting factor. Science 321: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wu MN, Joiner WJ, Dean T, Yue Z, Smith CJ, Chen D, Hoshi T, Sehgal A, Koh K (2010) SLEEPLESS, a Ly‐6/neurotoxin family member, regulates the levels, localization and activity of Shaker. Nat Neurosci 13: 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Hill VM, O'Connor RM, Sissoko GB, Irobunda IS, Leong S, Canman JC, Stavropoulos N, Shirasu‐Hiza M (2018) A bidirectional relationship between sleep and oxidative stress in Drosophila . PLoS Biol 16: e2005206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Afonso DJ, Liu D, Machado DR, Pan H, Jepson JE, Rogulja D, Koh K (2015) TARANIS functions with cyclin A and Cdk1 in a novel arousal center to control sleep in Drosophila . Curr Biol 25: 1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Foltenyi K, Greenspan RJ, Newport JW (2007) Activation of EGFR and ERK by rhomboid signaling regulates the consolidation and maintenance of sleep in Drosophila . Nat Neurosci 10: 1160–1167 [DOI] [PubMed] [Google Scholar]
- 110. Haynes PR, Christmann BL, Griffith LC (2015) A single pair of neurons links sleep to memory consolidation in Drosophila melanogaster . Elife 4: e03868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Joiner WJ, Crocker A, White BH, Sehgal A (2006) Sleep in Drosophila is regulated by adult mushroom bodies. Nature 441: 757–760 [DOI] [PubMed] [Google Scholar]
- 112. Liu S, Liu Q, Tabuchi M, Wu MN (2016) Sleep drive is encoded by neural plastic changes in a dedicated circuit. Cell 165: 1347–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Donlea JM, Pimentel D, Miesenbock G (2014) Neuronal machinery of sleep homeostasis in Drosophila . Neuron 81: 860–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Pimentel D, Donlea JM, Talbot CB, Song SM, Thurston AJ, Miesenbock G (2016) Operation of a homeostatic sleep switch. Nature 536: 333–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Liu Q, Liu S, Kodama L, Driscoll MR, Wu MN (2012) Two dopaminergic neurons signal to the dorsal fan‐shaped body to promote wakefulness in Drosophila . Curr Biol 22: 2114–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Shaw PJ, Tononi G, Greenspan RJ, Robinson DF (2002) Stress response genes protect against lethal effects of sleep deprivation in Drosophila . Nature 417: 287–291 [DOI] [PubMed] [Google Scholar]
- 117. Bushey D, Hughes KA, Tononi G, Cirelli C (2010) Sleep, aging, and lifespan in Drosophila . BMC Neurosci 11: 56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Potdar S, Daniel DK, Thomas FA, Lall S, Sheeba V (2018) Sleep deprivation negatively impacts reproductive output in Drosophila melanogaster . J Exp Biol 221: jeb174771 [DOI] [PubMed] [Google Scholar]
- 119. White JG, Southgate E, Thomson JN, Brenner S (1986) The structure of the nervous system of the nematode Caenorhabditis elegans . Philos Trans R Soc Lond B Biol Sci 314: 1–340 [DOI] [PubMed] [Google Scholar]
- 120. Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You YJ, Sundaram MV, Pack AI (2008) Lethargus is a Caenorhabditis elegans sleep‐like state. Nature 451: 569–572 [DOI] [PubMed] [Google Scholar]
- 121. Cassada RC, Russell RL (1975) The dauerlarva, a post‐embryonic developmental variant of the nematode Caenorhabditis elegans . Dev Biol 46: 326–342 [DOI] [PubMed] [Google Scholar]
- 122. Iwanir S, Tramm N, Nagy S, Wright C, Ish D, Biron D (2013) The microarchitecture of C. elegans behavior during lethargus: homeostatic bout dynamics, a typical body posture, and regulation by a central neuron. Sleep 36: 385–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Monsalve GC, Van Buskirk C, Frand AR (2011) LIN‐42/PERIOD controls cyclical and developmental progression of C. elegans molts. Curr Biol 21: 2033–2045 [DOI] [PubMed] [Google Scholar]
- 124. Wu Y, Masurat F, Preis J, Bringmann H (2018) Sleep counteracts aging phenotypes to survive starvation‐induced developmental arrest in C. elegans . Curr Biol 28: 3610–3624.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Skora S, Mende F, Zimmer M (2018) Energy scarcity promotes a brain‐wide sleep state modulated by insulin signaling in C. elegans . Cell Rep 22: 953–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. You YJ, Kim J, Raizen DM, Avery L (2008) Insulin, cGMP, and TGF‐beta signals regulate food intake and quiescence in C. elegans: a model for satiety. Cell Metab 7: 249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Schwarz J, Bringmann H (2013) Reduced sleep‐like quiescence in both hyperactive and hypoactive mutants of the Galphaq Gene egl‐30 during lethargus in Caenorhabditis elegans . PLoS ONE 8: e75853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Singh K, Ju JY, Walsh MB, DiIorio MA, Hart AC (2014) Deep conservation of genes required for both Drosophila melanogaster and Caenorhabditis elegans sleep includes a role for dopaminergic signaling. Sleep 37: 1439–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Van Buskirk C, Sternberg PW (2007) Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans . Nat Neurosci 10: 1300–1307 [DOI] [PubMed] [Google Scholar]
- 130. DeBardeleben HK, Lopes LE, Nessel MP, Raizen DM (2017) Stress‐induced sleep after exposure to ultraviolet light is promoted by p53 in Caenorhabditis elegans . Genetics 207: 571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Nelson MD, Lee KH, Churgin MA, Hill AJ, Van Buskirk C, Fang‐Yen C, Raizen DM (2014) FMRFamide‐like FLP‐13 neuropeptides promote quiescence following heat stress in Caenorhabditis elegans . Curr Biol 24: 2406–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Nath RD, Chow ES, Wang H, Schwarz EM, Sternberg PW (2016) C. elegans stress‐induced sleep emerges from the collective action of multiple neuropeptides. Curr Biol 26: 2446–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Iannacone MJ, Beets I, Lopes LE, Churgin MA, Fang‐Yen C, Nelson MD, Schoofs L, Raizen DM (2017) The RFamide receptor DMSR‐1 regulates stress‐induced sleep in C. elegans . Elife 6: e19837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Turek M, Lewandrowski I, Bringmann H (2013) An AP2 transcription factor is required for a sleep‐active neuron to induce sleep‐like quiescence in C. elegans . Curr Biol 23: 2215–2223 [DOI] [PubMed] [Google Scholar]
- 135. Turek M, Besseling J, Spies JP, Konig S, Bringmann H (2016) Sleep‐active neuron specification and sleep induction require FLP‐11 neuropeptides to systemically induce sleep. Elife 5: e12499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nichols ALA, Eichler T, Latham R, Zimmer M (2017) A global brain state underlies C. elegans sleep behavior. Science 356: eaam6851 [DOI] [PubMed] [Google Scholar]
- 137. Kucherenko MM, Ilangovan V, Herzig B, Shcherbata HR, Bringmann H (2016) TfAP‐2 is required for night sleep in Drosophila . BMC Neurosci 17: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mani A, Radhakrishnan J, Farhi A, Carew KS, Warnes CA, Nelson‐Williams C, Day RW, Pober B, State MW, Lifton RP (2005) Syndromic patent ductus arteriosus: evidence for haploinsufficient TFAP2B mutations and identification of a linked sleep disorder. Proc Natl Acad Sci USA 102: 2975–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bennett HL, Khoruzhik Y, Hayden D, Huang H, Sanders J, Walsh MB, Biron D, Hart AC (2018) Normal sleep bouts are not essential for C. elegans survival and FoxO is important for compensatory changes in sleep. BMC Neurosci 19: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Trojanowski NF, Nelson MD, Flavell SW, Fang‐Yen C, Raizen DM (2015) Distinct mechanisms underlie quiescence during two Caenorhabditis elegans sleep‐like states. J Neurosci 35: 14571–14584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Goetting DL, Soto R, Van Buskirk C (2018) Food‐dependent plasticity in Caenorhabditis elegans stress‐induced sleep is mediated by TOR‐FOXA and TGF‐beta signaling. Genetics 209: 1183–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Driver RJ, Lamb AL, Wyner AJ, Raizen DM (2013) DAF‐16/FOXO regulates homeostasis of essential sleep‐like behavior during larval transitions in C. elegans . Curr Biol 23: 501–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Sanders J, Scholz M, Merutka I, Biron D (2017) Distinct unfolded protein responses mitigate or mediate effects of nonlethal deprivation of C. elegans sleep in different tissues. BMC Biol 15: 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Porkka‐Heiskanen T (2013) Sleep homeostasis. Curr Opin Neurobiol 23: 799–805 [DOI] [PubMed] [Google Scholar]
- 145. Tononi G, Cirelli C (2003) Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull 62: 143–150 [DOI] [PubMed] [Google Scholar]
- 146. Park S, Sonn JY, Oh Y, Lim C, Choe J (2014) SIFamide and SIFamide receptor defines a novel neuropeptide signaling to promote sleep in Drosophila . Mol Cells 37: 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]