

Abstract
Background & Aims:
Graft dysfunction is one of the major complications after liver transplantation, but its precise mechanism remains unclear. Since steatotic liver grafts are susceptible to post-transplant dysfunction, and peroxisome proliferator-activated receptor (PPAR) α plays an important role in the maintenance of hepatic lipid homeostasis, we examined the role of PPARα in liver transplantation.
Methods:
Livers were harvested from Sv/129 wild-type (Ppara+/+) mice and PPARα-null (Ppara−/−) mice and transplanted orthotopically into syngeneic Ppara+/+ mice.
Results:
Hepatocellular damage was unexpectedly milder in transplanted Ppara−/− livers compared with Ppara+/+ ones. This was likely due to decreased lipid peroxides in the Ppara−/− livers, as revealed by the lower levels of fatty acid oxidation (FAO) enzymes, which are major sources of reactive oxygen species. Hepatic PPARα and its target genes, such as FAO enzymes and pyruvate dehydrogenase kinase 4, were strongly down-regulated after transplantation, which was associated with increases in hepatic tumor necrosis factor-α expression and nuclear factor-κB activity. Inhibiting post-transplant PPARα down-regulation by clofibrate treatment markedly augmented oxidative stress and hepatocellular injury.
Conclusions:
Down-regulation of PPARα seemed to be an adaptive response to metabolic alterations following liver transplantation. These results provide novel information to the understanding of the pathogenesis of early post-transplant events.
Keywords: Oxidative stress, Mitochondrial β-oxidation, Early graft dysfunction, Liver transplantation, PPARα
Introduction
Clinical liver transplantation (LT) has evolved greatly since its development in 1963. LT is now performed routinely in many transplant centers worldwide and is recognized as a useful strategy for the treatment of end-stage liver diseases and metabolic/genetic disorders [1,2]. However, considering the invasiveness and cost of LT and the limited supply of liver grafts available, constant improvement of post-transplant outcome is required.
Early graft dysfunction is one of the major causes that worsen the outcome of LT. The most serious type of graft dysfunction is primary non-function (PNF), occurring during the first 10 days after LT and often requiring re-transplantation. PNF is histologically characterized by massive necrosis of the liver [3]. Prevention of early graft dysfunction may contribute to the improvement of post-transplant survival, but its precise mechanism has not been fully clarified.
Several studies have demonstrated that accumulation of triglycerides (TG) in the liver is associated with a higher rate of early graft dysfunction, including PNF, and poorer LT outcome [4,5]. The mechanisms whereby steatotic liver graft is susceptible to post-transplant dysfunction are not completely understood, but several have been proposed: altered hepatic energy homeostasis, diminished portal blood flow due to distortion of sinusoidal lumen, and increased sensitivity to oxidative stress on reperfusion [5]. In another report, inhibition of fatty acid (FA) synthase improved post-transplant survival in ob/ob mice [6]. Thus, the intriguing possibility has emerged that an altered FA/TG metabolism in the liver may be associated with the pathogenesis of early graft dysfunction following LT.
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors belonging to the steroid/ thyroid hormone receptor superfamily [7]. Of three PPAR isoforms, PPARα regulates the constitutive transcription of genes encoding FA-metabolizing enzymes and mitochondrial FA oxidation (FAO) activity primarily in the liver and heart [8,9]. Administration of PPARα activators, such as the widely prescribed fibrate drugs, ameliorates hepatic steatosis through enhancing mitochondrial FAO in mice [10,11]. On the other hand, PPARα- null (Ppara−/−) mice have a lower capacity for constitutive mitochondrial FAO compared to wild-type (Ppara+/+) ones [8], leading to a higher susceptibility to hepatic steatosis under conditions in which FA is the main energy substrate, such as starvation or dietary fat overload [12]. PPARα also exhibits an anti-inflammatory effect through competitive inhibition of nuclear factor kappa B (NF-κB) [13,14]. Based on these evidences, it is hypothesized that PPARα function might be related to the development of graft dysfunction. However, the association of PPARα with the pathogenesis of early post-transplant events remains unclear.
As such, we aimed to explore the role of PPARα in the context of LT. We harvested livers from Ppara+/+ and Ppara−/− mice, transplanted them into syngeneic Ppara+/+ mice in the orthotopic position, and then investigated phenotypic changes at 1, 3, and 6 post-operative days (POD).
Materials and methods
Mice
Generation of Ppara−/− mice on a Sv/129 genetic background was described previously (Supplementary Ref. [1]). Male 12-week-old Sv/129 Ppara+/+ and Ppara−/− mice weighing 26–33 gram were used in all experiments. The mice were housed in a specific pathogen-free environment controlled for temperature and light (25 °C, 12-h light/dark cycle) and maintained with regular rodent chow (Oriental Yeast Co. Ltd., Tokyo, Japan) and tap water ad libitum. Regular rodent chow contained 5% of fats and was used as a control diet. FA composition in the control diet is shown in Supplementary Table 1. Orthotopic LT was performed according to a method by Qian et al. with minor modifications as follows (Supplementary Refs. [2,3]).
Donor operation:
After shaving and disinfecting the abdomen with alcohol, the abdominal cavity was accessed through a transverse laparotomy. Four small retractors and a retracting clamp on the xiphoid process provided exposure. The bile duct was transected. The pyloric, right adrenal, and right renal veins, as well as the hepatic artery, were ligated and divided. The liver was perfused with 2 ml of cold saline solution that was slowly injected into the portal vein. The infrahepatic vena cava was divided at the level of the splenic vein, leaving a sufficient length for cuff preparation. The freed liver was then placed into a container of cold saline solution for further preparation. The cystic duct was ligated and the gallbladder removed. Polyethylene cuffs were placed on the portal vein and the infrahepatic vena cava.
Recipient operation:
Through a midline incision, the bile duct, hepatic artery, and right adrenal vein were ligated and divided. Portal vein, infrahepatic, and suprahepatic inferior vena cava were dissected free, clamped, and transected close to the liver to preserve the maximum length for anastomosis. The native liver was removed and the graft liver was placed in orthotopic position of the recipients. The cuff technique was used for anastomosis of both the portal vein and the infrahepatic vena cava. The suprahepatic vena cava was anastomosed using a running 10–0 nylon suture. The bile duct was re-constructed with a single stent tube. Portal venous occlusion was always for less than 22 min. The hepatic artery was not re-constructed. The abdomen was closed in two layers.
All surgical procedures were carried out under sterile conditions by the same microsurgery expert (KN) with the aid of a 4–40× magnification scope. Ether was administered as a general anesthetic for all cases. Any procedures considered to have technical errors, such as unexpected massive bleeding or anastomotic failure, were excluded from the study. All animal experiments were conducted in accordance with the animal study protocols outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and approved by Shinshu University School of Medicine.
The present study consisted of three independent experiments performed in duplicate to observe recipient survival and evaluate blood and liver samples at several time points after LT. Blood was collected by cardiac puncture and centrifuged for 5 min at 8000 rpm at 4°C to isolate serum. Sera were immediately frozen and kept at −80°C until use.
In the first experiment, livers were harvested from Sv/129 Ppara+/+ and Ppara−/− mice (n = 16 for each genotype). Four livers of each genotype were not transplanted for baseline evaluation, and the remainder were immediately transplanted into syngeneic Ppara+/+ mice in the orthotopic position. Blood and transplanted livers were then taken from naïve and test mice at 1, 3, and 6 POD (n = 4 in each group) and subjected to analysis.
In the second experiment, Sv/129 Ppara+/+ and Ppara−/− mice (n = 24 for each genotype) were randomly divided into the following four groups: (1) Ppara+/+ mice fed a control diet (n = 12); (2) Ppara+/+ mice fed a control diet containing 1.0% (w/w) clofibrate (Wako Pure Chemicals Industries, Osaka, Japan) (n = 12);(3) Ppara−/− mice fed a control diet (n = 12); and (4) Ppara−/− mice fed a control diet containing 1.0% (w/w) clofibrate (n= 12). The dose of clofibrate used has been shown to sufficiently activate PPARα within 5 days of treatment without causing toxicity. Livers were harvested on day 7 after commencing such feeding. Four livers in each group were not transplanted for baseline evaluation, and the remainders were immediately transplanted into syngeneic Ppara+/+ mice in the orthotopic position. Administration of the clofibrate-containing diet was continued after LT. Blood and transplanted livers were then collected from naïve and test mice at 1 and 3 POD (n = 4 in each group) and subjected to analysis.
In the last experiment, Sv/129 Ppara+/+ mice (n = 16) were randomly divided into the following four groups and used as recipients: (1) Sham-operated mice (n = 4); (2) mice receiving livers from untreated Ppara+/+ mice (n = 4); (3) mice receiving livers from Ppara+/+ mice treated with 1.0% (w/w) clofibrate-containing diet for 7 days (n = 4); and (4) mice receiving livers from untreated Ppara+/+ mice and staring the treatment of 1.0% clofibrate-containing diet after LT(n = 4). Blood and livers were collected at 6 POD and subjected to analysis.
Immunoblot analysis
Preparation of nuclear fractions and measurement of protein concentrations were described previously (Supplementary Refs. [4,5]). For analysis of PPARs, retinoid X receptor α (RXRα), and NF-κB, nuclear fractions (100 μg of protein) were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS- PAGE). For analysis of other proteins, whole liver lysates (50–100 μg of protein) were subjected to 10% SDS-PAGE (Supplementary Refs. [6–8]). One sample was loaded for each electrophoresis and all samples (n = 4 in each group) were examined. After SDS-PAGE, proteins were transferred to nitrocellulose membranes and incubated with a primary antibody followed by alkaline phosphatase-conjugated goat anti-rabbit IgG. Antibodies raised against acyl-coenzyme A (CoA) oxidase (ACO), cytochrome P450 4A10(CYP4A10), catalase, long-chain acyl-CoA synthase (LACS), carnitine palmitoyl-CoA transferase (CPT)-I, and medium-chain acyl-CoA dehydrogenase (MCAD) have been described previously (Supplementary Ref.[7]). Antibodies against other proteins were purchased commercially (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Actin or histone H1 bands were used as loading controls. Band intensity was measured densitometrically, normalized to that of actin or histone H1, and expressed as fold change relative to that of naïve Ppara+/+ mice fed a control diet.
Quantitative polymerase chain reaction (PCR)
Total liver RNA was extracted from all mice (n = 4in each group) using an RNeasy Mini Kit (Qiagen, Tokyo,Japan), and cDNAwas generated by SuperScript II reverse transcriptase (Gibco BRL, Paisley, Scotland). Quantitative PCR was performed using a SYBR green PCR kit and ABI Prism 7700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The primer pairs used are shown in Supplementary Table 2. Measured mRNA levels were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA and expressed as fold change relative to those of naïve Ppara+/+ mice fed a control diet.
Histological analysis
Small blocks of liver tissue from all mice (n = 4 in each group) were immediately fixed in 4% paraformaldehyde in phosphate-buffered saline and embedded in paraffin. Sections (4 μm thick) were stained by the hematoxylin and eosin method. At least three discontinuous liver sections were evaluated for each mouse. The area of hepatocellular necrosis was quantified in each section as described elsewhere (Supplementary Ref [4]) and expressed as a percentage.
Other methods
Mitochondrial FAO activity, pyruvate dehydrogenase (PDH) activity, and caspase 3 activity were determined as described elsewhere (Supplementary Refs. [4,8]). Serum concentrations of aspartate and alanine aminotransferase (AST and ALT) were measured using a GOT and GPT-test kit (Wako), respectively. Total hepatic lipids were extracted according to a method by Folch et al. (Supplementary Ref. [9]), and levels oflipid peroxides [4-hydroxynonenal (4-HNE) and malondial- dehyde (MDA)] were assessed with a LPO-586 kit(OXIS International, Portland, OR, USA).
Statistical analysis
Results are expressed as mean ± standard deviation (SD). Statistical analyses were performed using the one-way ANOVA with Dunnett’s test (comparison between time points in the same mouse group) and Tukey’s test (comparison between mouse groups at the same time point), respectively. A p value of less than 0.05 was considered to be statistically significant.
Results
Post-transplant hepatocellular damage is milder in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers
All mice receiving Ppara+/+ or Ppara−/− livers survived for more than 7 days without any gross abnormalities. There were no significant differences in body weight, transplanted liver weight, or liver ischemic time between mice receiving Ppara+/+ livers and those receiving Ppara−/− livers (Supplementary Table 3). Serum levels of AST and ALT were markedly increased at 1 POD and gradually decreased afterwards in both groups (Supplementary Fig. 1A). However, these levels were lower in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers at 1 and 3 POD (Supplementary Fig. 1). Histologically, small dispersed foci of hepatocellular necrosis were observed in both groups at 1 POD (arrows in Supplementary Fig. 1B), but were undetectable at 6 POD. Marked infiltration of leukocytes, bile duct damage, cholestasis, endotheliitis, or thrombus were not found. Although hepatic caspase 3 activity did not differ between groups, the total area of hepatocellular necrosis was smaller in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers at 1 and 3 POD (Supplementary Fig. 1C). These results suggest that posttransplant hepatocellular damage within 1 week was unexpectedly milder in mice receiving Ppara−/− livers.
Post-transplant hepatic oxidative stress is milder in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers
To determine the mechanism of lower hepatic damage in the Ppara−/− group, the amount of hepatic lipid peroxides was assessed. Hepatic levels of HNE and MDA + 4-HNE were increased in both groups at 1 POD but decreased afterwards (Fig. 1A). As well as serum AST and ALT, hepatic lipid peroxide levels were lower in mice receiving Ppara−/− livers than in those receiving Ppara+/+ at 1 and 3 POD (Fig. 1A).
Fig. 1. Post-transplant hepatic oxidative stress was milder in mice receiving Ppara-null (−/−) livers than in those receiving wild-type (+/+) livers.
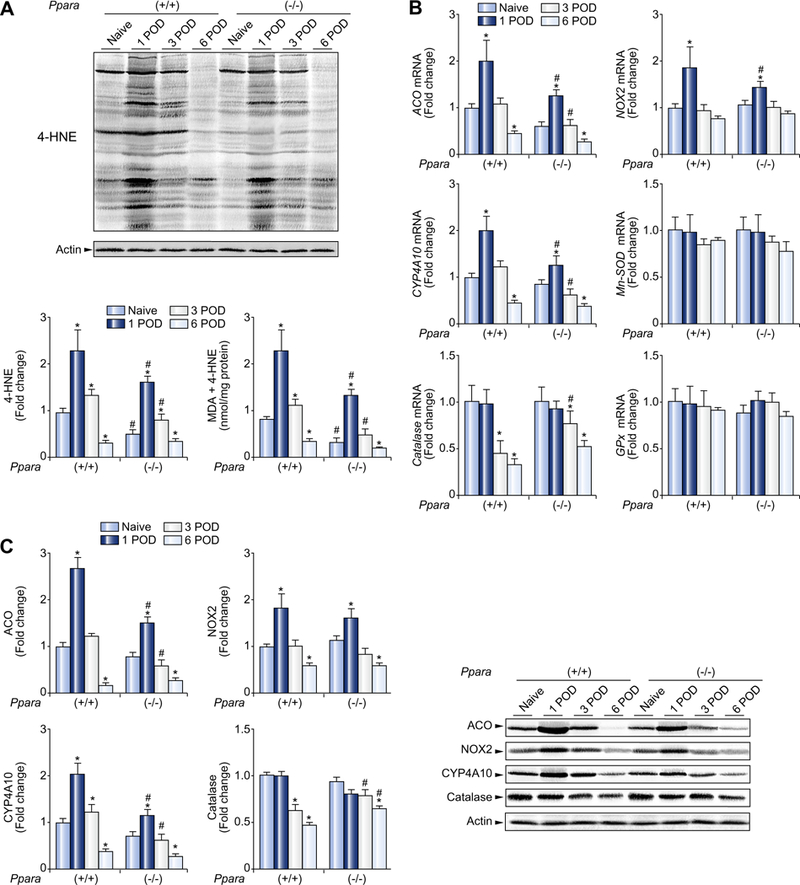
(A) Immunoblot analysis of 4-HNE and quantification of MDA + 4-HNE levels in the liver. Whole liver lysates (100 μg of protein) obtained from one mouse in each group were loaded onto the gels. Actin was used as the loading control. Band intensity was quantified densitometrically, normalized to that of actin, and subsequently normalized to that of naïve Ppara+/+ mice. Measurement of hepatic MDA + 4-HNE is described in Materials and methods. Results are expressed as mean ±SD(n = 4in each group). POD, post-operative day; *p <0.05 vs. naïve mice of the same genotype; #p <0.05 vs. Ppara+/+ mice at the same time point. (B) Hepatic mRNA levels of ROS-generating and ROS-eliminating enzymes. The mRNA levels were normalized to those of GAPDH mRNA, and subsequently normalized to those of naïve Ppara+/+ mice. Results are expressed as mean ± SD (n = 4 in each group). Abbreviations are the same as panel (A). (C) Immunoblot analysis of selected ROS-related enzymes. The same samples used in panel (A) (whole liver lysate, 50 μg of protein) were loaded onto the gels. Band intensity was quantified densitometrically, normalized to that of actin, and subsequently normalized to that of naïve Ppara+/+ mice. Results are expressed as mean ±SD (n = 4 in each group). *p <0.05 vs. naïve mice of the same genotype; #p <0.05 vs. Ppara+/+ mice at the same time point.
Hepatic mRNA levels of typical reactive oxygen species (ROS)- generating enzymes, such as ACO, NADPH oxidase (NOX) 2, and CYP4A10, were all increased at 1 POD (Fig. 1B). Of these, the mRNA levels of ACO and CYP4A10, which are also key enzymes of peroxisomal and microsomal FAO, respectively, were lower in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers at 1 and 3 POD (Fig. 1B). When hepatic expression of ROS-eliminating enzymes was examined, the mRNA level of catalase, but not manganese superoxide dismutase (Mn-SOD) or glutathione peroxidase (GPx), was higher in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers at 3 POD (Fig. 1B). These differences were confirmed at the protein level by immunoblot analysis (Fig. 1C).
Mitochondrial FAO is another important source of ROS in the liver [15]. As expected, hepatic mRNAs encoding LCAS, CPT-I, and MCAD were lower in mice receiving Ppara−/− livers than in those receiving Ppara+/+ livers at 1 and 3 POD (Fig. 2A). Similar results were obtained by immunoblot analysis (Fig. 2B). Actual FAO activity was also lower in mice receiving Ppara−/− livers (Fig. 2C).
Fig. 2. Post-transplant hepatic mitochondrial FAO enzymes and PDK4 were down-regulated in mice receiving wild-type (+/+) or Ppara-null (−/−) livers.
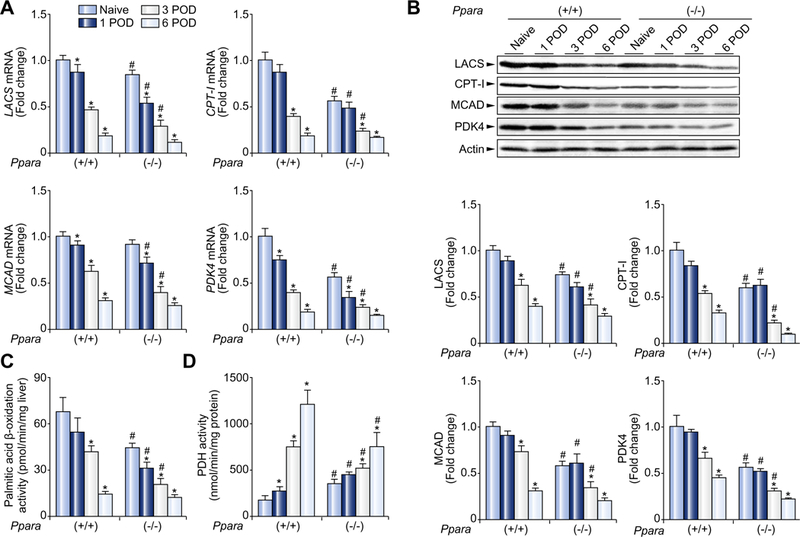
(A) Hepatic mRNA levels of mitochondrial FAO enzymes and PDK4. The mRNA levels were normalized to those of GAPDH mRNA, and subsequently normalized to those of naïve Ppara+/+ mice. Results are expressed as mean ±SD (n = 4 in each group). POD, post-operative day; *p <0.05 vs. naïve mice of the same genotype; #p <0.05 vs. Ppara+/+ mice at the same time point. (B) Immunoblot analysis of mitochondrial FAO enzymes and PDK4. The same samples used in Fig. 1A (whole liver lysate, 50 μg of protein) were loaded onto the gels. Band intensity was quantified densitometrically, normalized to that of actin, and subsequently normalized to that of naïve Ppara+/+ mice. Results are expressed as mean ±SD(n = 4in each group). Abbreviations are the same as panel (A). (C and D) Activities of mitochondrial β-oxidation and PDH. Results are expressed as mean ±SD (n = 4 in each group). Abbreviations are the same as panel (A).
Based on the above results milder post-transplant hepatocellular damage in Ppara−/− livers was likely due to lowered oxidative stress that was associated with decreased expression of ACO and CYP4A10 and lower mitochondrial FAO activity.
Mitochondrial FAO are down-regulated after LT
As shown in Fig. 2, mitochondrial FAO activity was significantly decreased after LT in mice receiving Ppara+/+ and Ppara−/− livers. In addition to mitochondrial FAO, PDH plays an important role in providing acetyl-CoA to the citric acid cycle. The expression of PDH kinase (PDK) 4, a major down-regulator of PDH activity in the liver, was decreased after LT in both groups (Fig. 2A and B). PDH activity had a tendency to increase after LT in both groups (Fig. 2D). These alterations suggest that the major source of acetyl-CoA supply was shifted from FAO to glycolysis following LT.
PPARα is down-regulated after LT
The expression of several key genes down-regulated after LT, such as ACO, CYP4A10, LACS, CPT-I, MCAD, and PDK4, and mitochondrial FAO activity is known to be tightly regulated by PPARα [7,8,11,16]. Hepatic levels of PPARα mRNA and protein were both significantly diminished at 3 and 6 POD (Fig. 3A and B). The same was true for RXRα, a heterodimeric partner of PPARα (Fig. 3A and B), but hepatic levels of PPAR β and PPARαγ did not change following LT (Supplementary Fig. 2). The mRNA levels of PPARα and RXRα are known to be negatively regulated by NF-κB and tumor necrosis factor (TNF) α [17–20]. Accordingly, nuclear levels of the NF-κB p50 subunit, as well as the mRNA levels of TNFα and its receptor TNR1, were markedly increased after LT (Fig. 3C and D).
Fig. 3. Post-transplant hepatic PPARα levels were down-regulated.
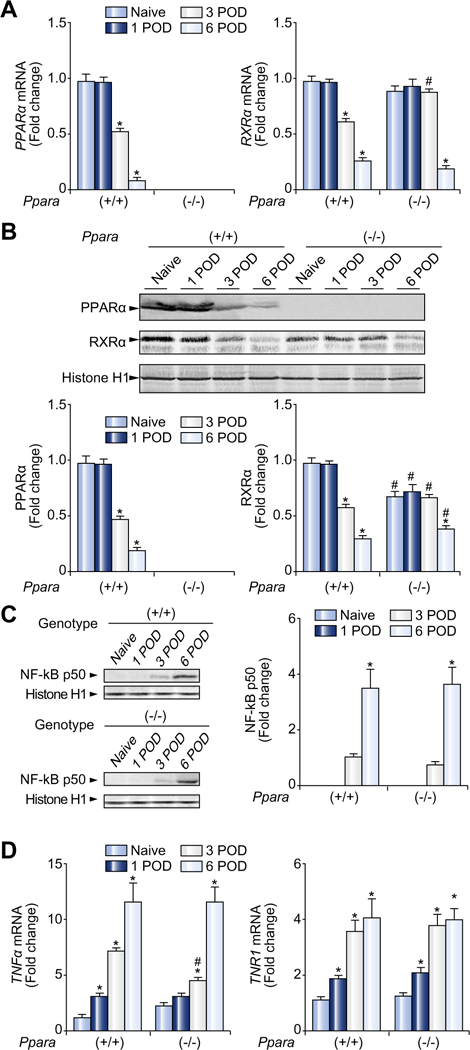
(A) Hepatic mRNA levels of PPARα and RXRα. The mRNA levels were normalized to those of GAPDH mRNA, and subsequently normalized to those of naïve Ppara+/+ mice. Results are expressed as mean ± SD (n = 4 in each group). POD, post-operative day; *p <0.05 vs. naïve mice of the same genotype; #p <0.05 vs. Ppara+/+ mice at the same time point. (B) Immunoblot analysis of PPARα and RXRα. Nuclear fractions (100 μg of protein) obtained from one mouse in each group were loaded. Histone H1 was used as the loading control. Band intensity was quantified densitometrically, normalized to that of histone H1, and subsequently normalized to that of naïve Ppara+/+ mice. Results are expressed as mean ± SD (n = 4 in each group). Abbreviations are the same as panel (A). (C) Immunoblot analysis of the NF-κB p50 subunit. The same samples used in panel (B) (nuclear fraction, 100 μg of protein) were loaded. Band intensity was quantified densitometrically, normalized to that of histone H1, and subsequently normalized to that of Ppara+/+ mice at 3 POD because the band was undetectable for naïve Ppara+/+ and Ppara−/− mice at 1 POD. Results are expressed as mean ± SD (n = 4 in each group). *p <0.05 vs. 3 POD of the same genotype; #p <0.05 vs. Ppara+/+ mice at the same time point. (D) Hepatic mRNA levels of TNFα and its receptor (TNR1). The same samples used in panel (A) were adopted. Results are expressed as mean ± SD (n = 4 in each group). Abbreviations are the same as panel (A).
inhibition of spontaneous down-regulation of PPARα after LT increases oxidative stress and causes severe hepatocellular damage
In the light of these observations, we hypothesized that down-regulation of PPARα is an adaptive response to various metabolic changes following LT, such as increased oxidative stress. To verify our hypothesis, we harvested livers from Ppara+/+ and Ppara−/− mice treated with cloflbrate, a typical PPARα activator, for 7 days, transplanted them into syngeneic Ppara+/+ mice, and continued clofibrate treatment to interrupt post-transplant decreases in PPARα. Although livers from clofibrate-treated Ppara+/+ mice tended to be larger than those from other groups of mice, ischemic time did not differ among groups (Supplementary Table 4). Surprisingly, all of the clofibrate-treated mice receiving Ppara+/+ livers died within 5 days after LT. All mice in the other groups survived for more than 7 days without any gross abnormalities (Fig. 4A). Clofibrate treatment notably increased serum AST and ALT levels, total area of hepatocellular necrosis, and hepatic caspase 3 activity at 1 and 3 POD in a PPARα-dependent fashion (Fig. 4B and D). Microscopically, severe hepatocellular degeneration and necrosis were evident in clofibrate-treated mice receiving Ppara+/+ livers only (Fig. 4C).
Fig. 4. Cloflbrate treatment caused severe post-transplant hepatocellular damage in mice receiving wild-type (+/+) livers.
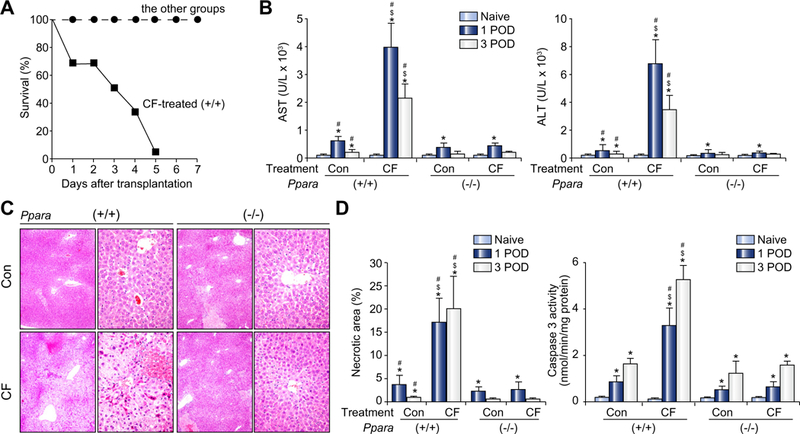
(A) Survival of recipients. Prior to liver donation, a control diet with or without 1.0% (w/w) cloflbrate (CF) was administered to 12-week-old male Sv/129 Ppara+/+ or Ppara−/− mice for 7 days (n = 12 in each group). Livers were harvested and orthotopically transplanted into syngeneic Ppara+/+ mice. Administration of CF was continued after transplantation. POD, post-operative day (POD). (B) Serum AST and ALT levels. Results are expressed as mean ± SD (n = 4 in each group). Con, control diet; CF, 1.0% CF-containing diet; (+/+), Ppara+/+; (−/−), Ppara−/−; *p <0.05 vs. naïve mice of the same genotype and treatment; #p <0.05 vs. Ppara−/− mice at the same time point and treatment; $p <0.05 vs. control diet treatment at the same time point and genotype. (C) Representative light micrographs of the liver at 1 POD stained by the hematoxylin and eosin method. Massive hepatocellular degeneration and necrosis were observed in CF-treated mice receiving Ppara+/+ livers only. Upper and lower rows in each group show a magnification of 100 × and 400 ×, respectively. Abbreviations are the same as panel (B). (D) Quantification of the total area of hepatocellular necrosis and measurement of hepatic caspase 3 activity. Results are expressed as mean ± SD (n = 4 in each group). Abbreviations are the same as panel (B).
Clofibrate treatment markedly augmented hepatic levels of lipid peroxides (Supplementary Fig. 3A) through increases in the mRNA levels of PPARα, ROS-generating enzymes (ACO and CYP4A10), and mitochondrial FAO enzymes (LACS, CPT-I, and MCAD), as well as by the enhancement of mitochondrial FAO activity in a PPARα-dependent manner (Supplementary Fig. 3B- D). Furthermore, post-transplant PPARα activation inhibited a rise in PDH activity by maintaining a high expression level of PDK4 (Supplementary Fig. 3C and D).
Cyclophilin D and Bax play a critical role in oxidative stressinduced hepatocyte necrosis and apoptosis, respectively [21,22]. Clofibrate treatment significantly increased these proteins in mice receiving Ppara+/+ livers (Supplementary Fig. 4). Expression of the anti-apoptotic proteins Bcl-2 and Bcl-xL was not affected by the treatment (Supplementary Fig. 4).
To further examine the significance of spontaneous down-regulation of PPARα after LT, we compared phenotypic changes between Ppara+/+ mice receiving livers from untreated Ppara+/+ mice and started clofibrate treatment only after LT, and those receiving livers from clofibrate-treated Ppara+/+ mice and continued the control diet after LT. In 6 POD, there were no significant differences in serum AST and ALT levels between mice receiving clofibrate-treated mouse livers and mice receiving untreated mouse livers. However, clofibrate treatment after LT inhibited PPARα down-regulation, increased hepatic mRNA levels of ACO, CYP4A10, MCAD, and PDK4 and lipid peroxide contents, and increased serum AST and ALT concentrations (Supplementary Fig. 5).
Collectively, we witnessed that inhibiting spontaneous down-regulation of PPARα stimulated ROS-generating pathways, augmented hepatic oxidative stress, and consequently resulted in hepatocellular necrosis and apoptosis after LT.
Discussion
The present study was designed to evaluate the role of PPARα in the context of LT. We obtained an unexpected and interesting result that PPARα was markedly and spontaneously down-regulated after LT. Inhibiting post-transplant PPARα down-regulation aggravated oxidative stress and caused hepatocellular damage.
FAO may contribute to ROS generation through several routes: (1) mitochondrial β-oxidation results in large amounts of acetyl-CoA that are consumed in the respiratory chain and lead to increased ROS generation; (2) the initial step of peroxisomal β- oxidation catalyzed by ACO is accompanied with the production of hydrogen peroxide due to a direct transfer of electrons to molecular oxygen; and (3) FA ω-oxidation in the endoplasmic reticulum catalyzed mainly by CYP4A generates ROS through an inefficient transfer of electrons from a flavoprotein [15]. In this study, the alterations in enzymes involved in the above pathways were confirmed, as well as those that result from lipid peroxidation and hepatic damage. Based on these observations, we speculated that spontaneous down-regulation of PPARα and the related metabolic alterations in the early post-transplant period are an adaptive response to minimize FA-derived ROS generation. In support of this idea, clofibrate administration to inhibit posttransplant PPARα down-regulation augmented ROS generation by enhancing FAO and subsequently resulted in hepatocellular injury. In fact, it has been reported that forced overexpression of PPARα in mouse hearts increased FA utilization and significantly delayed recovery of cardiac function after ischemia/reperfusion [23]. Therefore, we suppose that excessive PPARα activation may be harmful after LT.
PDH activity was increased following LT in correlation with decreased mitochondrial FAO activity, suggesting a switching of energy source preference in the transplanted livers. However, such alterations were also seen in Ppara−/− livers, indicating the presence of important transcription factors other than PPARα in this process. Hypoxia-inducible transcription factor 1 is a promising candidate, since its activation is reported to suppress the transcriptional activity of FAO enzymes [24].
In this study, activation of NF-κB was considered to be one of the possible factors contributing to post-transplant down-regulation of PPARα. Since oxidative stress can activate NF-κB [14,25], we can speculate that a transient increase in hepatic oxidative stress just after LT triggered activation of NF-κB. This notion is corroborated by a previous study showing that although down-regulation of PPARα was observed in hearts after repetitive ischemia/reperfusion, it did not occur in mice overexpressing extracellular SOD [26]. Further studies are needed to clarify the precise molecular mechanism of spontaneous PPARα down-regulation after LT.
There are some studies showing the detrimental effects of PPARα deficiency or the protective effects of PPARα activation in hepatic ischemia/reperfusion injury models using both lean and steatotic livers [27–30]. Notably, these models in general produced a more severe injury than our transplantation model and the protection by PPARα likely related to down-regulation of the pro-inflammatory response. Thus, it is plausible to consider that, in more severe liver injury models, the protective effects of PPARα activation may overrule the benefit that could come from the down-regulation of FA-oxidizing enzymes. However, in this study, we used the mouse transplantation model established by Qian et al. [31]. This model was first reported in 1991 and generally recognized as a valid liver transplantation model [32–40]. Unlike the conventional ischemia/reperfusion models, liver grafts removed from recipients were perfused and kept in cold Ringer’s solution or physiological saline until finishing removal of donors’ livers and the suture of vasculatures was completed as soon as possible to minimize total ischemic time. Therefore, this transplantation model demonstrates milder liver injury than the traditional ischemia/reperfusion model. We thus consider that this model resembles the situations of clinical LT more closely than the conventional ischemia/reperfusion model and that similar metabolic changes might occur in liver grafts in humans.
The validity of using steatotic liver grafts in humans is still controversial [41–44]. Since FA accumulated in hepatocytes may serve as a ligand for PPARα [7,45–47], we postulate that persistent PPARα activation might be one of the possible mechanisms of early dysfunction in human steatotic liver grafts. In agreement with this hypothesis, it has been documented that, although severe coagulation necrosis appeared in steatotic livers from ob/ob mice after LT, pre-treatment with cerulenin to suppress FA synthesis down-regulated hepatic PPARα and improved post-transplant survival [6]. Therefore, further experimentation to examine whether PPARα inhibitors can attenuate early posttransplant injury in steatotic liver grafts might corroborate the importance of PPARα down-regulation following LT. Additionally, future studies using human samples will provide a clue to uncover the exact relationship among lipid metabolism, PPARα, and the development of early post-transplantation complications.
Supplementary Material
Acknowledgement
We thank Mr. Trevor Ralph for his editorial assistance.
Abbreviations:
- ACO
acyl-coenzyme A oxidase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CoA
coenzyme A
- CPT
carnitine palmitoyl-CoA transferase
- CYP
cytochrome P450
- FA
fatty acid
- FAO
fatty acid β-oxidation
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GPx
glutathione peroxidase
- HNE
hydroxynonenal
- LACS
long-chain acyl-CoA synthase
- LT
liver transplantation
- MCAD
medium-chain acyl-CoA dehydrogenase
- MDA
malondialdehyde
- Mn-SOD
manganese-superoxide dismutase
- NF-κB
nuclear factor kappa B
- NOX
NADPH oxidase
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- PCR
polymerase chain reaction
- PDH
pyruvate dehydrogenase
- PDK
PDH kinase
- PNF
primary non-function
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- RXR
retinoid Xreceptor
- SD
standard deviation
- TG
triglycerides
- TNF
tumor necrosis factor
- TNR
TNF receptor
Footnotes
Conflict of interest
The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi: 10.1016/j.jhep.2011.08.021.
References
- [1].Berg CL, Steffick DE, Edwards EB, Heimbach JK, Magee JC, Washburn WK, et al. Liver and intestine transplantation in the United States 1998–2007. Am J Transplant 2009;9:907–931. [DOI] [PubMed] [Google Scholar]
- [2].Mehrabi A, Fonouni H, Müller SA, Schmidt J. Current concepts in transplant surgery: liver transplantation today. Langenbecks Arch Surg 2008;393:245–260. [DOI] [PubMed] [Google Scholar]
- [3].Nissen NN, Colquhoun SD. Graft failure: etiology, recognition, and treatmentIn: Busuttil RW, Klintmalm GB, editors. Transplantation of the liver. Philadelphia: Elsevier Saunders; 2005. p. 915–928. [Google Scholar]
- [4].Marsman WA, Wiesner RH, Rodriguez L, Batts KP, Porayko MK, Hay JE, et al. Use of fatty donor liver is associated with diminished early patient and graft survival. Transplantation 1996;62:1246–1251. [DOI] [PubMed] [Google Scholar]
- [5].Burke A, Lucey MR. Non-alcoholic fatty liver disease, non-alcoholic steato- hepatitis and orthotopic liver transplantation. Am J Transplant 2004;4:686–693. [DOI] [PubMed] [Google Scholar]
- [6].Chavin KD, Fiorini RN, Shafizadeh S, Cheng G, Wan C, Evans Z, et al. Fatty acid synthase blockade protects steatotic livers from warm ischemia reperfusion injury and transplantation. Am J Transplant 2004;4:1440–1447. [DOI] [PubMed] [Google Scholar]
- [7].Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 1999;20:649–688. [DOI] [PubMed] [Google Scholar]
- [8].Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα). J Biol Chem 1998;273:5678–5684. [DOI] [PubMed] [Google Scholar]
- [9].Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor α-associated with age-dependent cardiac toxicity. J Biol Chem 2000;275:22293–22299. [DOI] [PubMed] [Google Scholar]
- [10].Harano Y, Yasui K, Toyama T, Nakajima T, Mitsuyoshi H, Mimani M, et al. Fenofibrate, a peroxisome proliferator-activated receptor α agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int 2006;26:613–620. [DOI] [PubMed] [Google Scholar]
- [11].Nakajima T, Tanaka N, Kanbe H, Hara A, Kamijo Y, Zhang X, et al. Bezafibrate at clinically relevant doses decreases serum/liver triglycerides via down-regulation of sterol regulatory element-binding protein-1c in mice. a novel peroxisome proliferator-activated receptor α-independent mechanism. Mol Pharmacol 2009;75:782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J Clin Invest 1999;103:1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors (PPARs): nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm Res 2000;49:497–505. [DOI] [PubMed] [Google Scholar]
- [14].Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K, et al. Peroxisome proliferator-activated receptor α protects against alcohol- induced liver damage. Hepatology 2004;40:972–980. [DOI] [PubMed] [Google Scholar]
- [15].Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor α target genes. Cell Mol Life Sci 2004;61:393–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nanji AA, Dannenberg AJ, Jokelainen K, Bass NM. Alcoholic liver injury in the rat is associated with reduced expression of peroxisome proliferator-α (PPARα)-regulated genes and is ameliorated by PPARα activation. J Pharmacol Exp Ther 2004;310:417–424. [DOI] [PubMed] [Google Scholar]
- [18].Glosli H, Gudbrandsen OA, Mullen AJ, Halvorsen B, Røst TH, Wergedahl H, et al. Down-regulated expression of PPARα target genes;reduced fatty acid oxidation and altered fatty acid composition in the liver of mice transgenic for hTNFα. Biochim Biophys Acta 2005;1734:235–246. [DOI] [PubMed] [Google Scholar]
- [19].Razeghi P, Wang ME, Youker KA, Golfman L, Stepkowski S, Taegtmeyer H. Lack of NF-κB1 (p105/p50) attenuates unloading-induced downregulation of PPARα and PPARα-regulated gene expression in rodent heart. Cardiovasc Res 2007;74:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim MS, Sweeney TR, Shigenaga JK, Chui LG, Moser A, Grunfeld C, et al. Tumor necrosis factor and interleukin 1 decrease RXRα, PPARα, PPARγ, LXRα, and the coactivators SRC-1, PGC-1α, and PGC-1 β in liver cells. Metabolism 2007;56:267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434:658–662. [DOI] [PubMed] [Google Scholar]
- [22].Tsujimoto Y, Nakagawa T, Shimizu S. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta 2006;1757:1297–1300. [DOI] [PubMed] [Google Scholar]
- [23].Sambandam N, Morabito D, Wagg C, Finck BN, Kelly DP, Lopaschuk GD. Chronic activation of PPARα is detrimental to cardiac recovery after ischemia. Am J Physiol Heart Circ Physiol 2006;290:H87–95. [DOI] [PubMed] [Google Scholar]
- [24].Belanger AJ, Luo Z, Vincent KA, Akita GY, Cheng SH, Gregory RJ, et al. Hypoxia-inducible factor 1 mediates hypoxia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated receptor α/retinoid X receptor. Biochem Biophys Res Commun 2007;364:567–572. [DOI] [PubMed] [Google Scholar]
- [25].Okiyama W, Tanaka N, Nakajima T, Tanaka E, Kiyosawa K, Gonzalez FJ, et al. Polyenephosphatidylcholine prevents alcoholic liver disease in PPARα-null mice through attenuation of increases in oxidative stress. J Hepatol 2009;50:1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, et al. Downregulation of peroxisome proliferator-activated receptor-α gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation 2005;112:407–415. [DOI] [PubMed] [Google Scholar]
- [27].Okaya T, Lentsch AB. Peroxisome proliferator-activated receptor-α regulates postischemic liver injury. Am J Physiol Gastrointest Liver Physiol 2004;286:G606–G612. [DOI] [PubMed] [Google Scholar]
- [28].Teoh NC, Williams J, Hartley J, Yu J, McCuskey RS, Farrell GC. Short-term therapy with peroxisome proliferation-activator receptor-alpha agonist Wy-14, 643 protects murine fatty liver against ischemia-reperfusion injury. Hepatology 2010;51:996–1006. [DOI] [PubMed] [Google Scholar]
- [29].Ramsey HE, Da Silva CG, Longo CR, Csizmadia E, Studer P, Patel VI, et al. A20 protects mice from lethal liver ischemia/reperfusion injury by increasing peroxisome proliferator-activated receptor-alpha expression. Liver Transpl 2009;15:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Massip-Salcedo M, Zaouali MA, Padrissa-Altés S, Casillas-Ramirez A, Rodés J, Roselló-Catafau J, et al. Activation of peroxisome proliferator-activated receptor-alpha inhibits the injurious effects of adiponectin in rat steatotic liver undergoing ischemia-reperfusion. Hepatology 2008;47:461–472. [DOI] [PubMed] [Google Scholar]
- [31].Qian SG, Fung JJ, Demetris AV, Ildstad ST, Starzl TE. Orthotopic liver transplantation in the mouse. Transplantation 1991;52:562–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Birsner JH, Wan C, Cheng G, Evans ZP, Polito CC, Fiorini RN, et al. Steatotic liver transplantation in the mouse: a model of primary nonfunction. J Surg Res 2004;120:97–101. [DOI] [PubMed] [Google Scholar]
- [33].Li W, Lu L, Wang Z, Wang L, Fung JJ, Thomson AW, et al. Il-12 antagonism enhances apoptotic death of T cells within hepatic allografts from Flt3 ligand-treated donors and promotes graft acceptance. J Immunol 2001;166:5619–5628. [DOI] [PubMed] [Google Scholar]
- [34].Fu F, Thai NL, Li Y, Lu L, Thomson AW, Fung JJ, et al. Second-set rejection of mouse liver allografts is dependent on radiation-sensitive nonparenchymal cells of graft bone marrow origin. Transplantation 1996;61:1228–1233. [DOI] [PubMed] [Google Scholar]
- [35].Sugioka A, Morita M, Kato T, Hoshimoto S, Fujita J, Morise Z, et al. FK 506 inhibits tolerance induction in mice liver transplantation. Transplant Proc 2005;37:146–147. [DOI] [PubMed] [Google Scholar]
- [36].Qian S, Wang Z, Lee Y, Chiang Y, Bonham C, Fung J, et al. Transplant Proc 2001;33:226. [DOI] [PubMed] [Google Scholar]
- [37].Sugioka A, Morita M, Fujita J, Hasumi A, Shiroishi T. Graft acceptance and tolerance induction in mouse liver transplantation using wild mice. Transplant Proc 2001;33:137–139. [DOI] [PubMed] [Google Scholar]
- [38].Conzelmann L, Schemmer P, Zhong Z, Smutney O, Bunzendahl H, Thurman R. Orthotopic liver transplantation in knockout mice. is TNFalpha involved in early graft injury and regeneration? Transplant Proc 2002;34:2299–2300. [DOI] [PubMed] [Google Scholar]
- [39].Jiang X, Morita M, Sugioka A, Harada M, Kojo S, Wakao H, et al. The importance of CD25+ CD4+ regulatory T cells in mouse hepatic allograft tolerance. Liver Transpl 2006;12:1112–1118. [DOI] [PubMed] [Google Scholar]
- [40].Morita M, Fujino M, Jiang G, Kitazawa Y, Xie L, Azuma M, et al. PD-1/B7-H1 interaction contribute to the spontaneous acceptance of mouse liver allograft. Am J Transplant 2010;10:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].de Meijer VE, Kalish BT, Puder M, Ijzermans JN. Systematic review and metaanalysis of steatosis as a risk factor in major hepatic resection. Br J Surg 2010;97:1331–1339. [DOI] [PubMed] [Google Scholar]
- [42].McCormack L, Dutkowski P, El-Badry AM, Clavien PA. Liver transplantation using fatty livers: always feasible? J Hepatol 2011;54:1055–1062. [DOI] [PubMed] [Google Scholar]
- [43].Clavien PA, Oberkofler CE, Raptis DA, Lehmann K, Rickenbacher A, El-Badry AM. What is critical for liver surgery and partial liver transplantation: size or quality? Hepatology 2010;52:715–729. [DOI] [PubMed] [Google Scholar]
- [44].Noujaim HM, de Goyet J, Montero EF, Ribeiro CM, Capellozzi VL, Crescentini F, et al. Expanding postmortem donor pool using steatotic liver grafts: a new look. Transplantation 2009;87:919–925. [DOI] [PubMed] [Google Scholar]
- [45].Tanaka N, Sano K, Horiuchi A, Tanaka E, Kiyosawa K, Aoyama T. Highly purified eicosapentaenoic acid treatment improves nonalcoholic steatohepatitis. J Clin Gastroenterol 2008;42:413–418. [DOI] [PubMed] [Google Scholar]
- [46].Tanaka N, Zhang X, Sugiyama E, Kono H, Horiuchi A, Nakajima T, et al. Eicosapentaenoic acid improves hepatic steatosis independent of PPARα activation through inhibition of SREBP-1 maturation in mice. Biochem Pharmacol 2010;80:1601–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tanaka N, Moriya K, Kiyosawa K, Koike K, Gonzalez FJ, Aoyama T. PPARα activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J Clin Invest 2008;118:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.