

Abstract
Myasthenia gravis (MG) is an autoantibody-mediated disease that compromises the acetylcholine receptors or associated structures of the postsynaptic membrane of the neuromuscular junction. This leads to impaired neuromuscular transmission and subsequent fluctuating fatigability and weakness of ocular, bulbar, and limb skeletal muscles. Over the past few decades, there have been significant advances in our understanding of the disease pathophysiology and improvements in prognosis due to intensive care medicine and immunomodulation. Despite this, an estimated 10–20% of patients with MG do not achieve an adequate response, are intolerant to conventional treatment, or require chronic treatment with intravenous immunoglobulins or plasma separation procedures. Such patients are regarded as having MG that is ‘refractory’ to treatment and may represent a distinct clinical subgroup. Because the majority of patients with MG have well-controlled disease, the burden of illness in the minority with refractory disease is poorly understood and may be underestimated. However, clinically these patients are liable to experience extreme fatigue, considerable disability owing to uncontrolled symptoms, and frequent myasthenic crises and hospitalizations. Both acute adverse effects and an increased risk of comorbidity from treatment regimens may contribute to reduced quality of life. As yet, little is known concerning the impact of refractory MG on mental health and health-related quality of life. This review aims to highlight the burden of disease and unmet needs in patients with refractory MG.
Keywords: burden, definition, disability, quality of life, refractory myasthenia gravis, side effects, tolerability, treatment, unmet need
Introduction
Myasthenia gravis (MG) is a rare, antibody-mediated autoimmune disease of the neuromuscular junction, resulting in fluctuating fatigability and weakness of ocular, bulbar, and limb skeletal muscles. Autoantibodies against components of the postsynaptic neuromuscular endplate [acetylcholine receptor (AChR; most common); muscle-specific kinase (MuSK), and lipoprotein-related protein 4] are involved in the underlying pathogenesis1 and are well accepted as diagnostic markers.2,3 Increasing evidence also suggests a role for anti-agrin autoantibodies, although this remains to be confirmed in humans.4–6
In the majority of patients with MG the disease can be managed via treatment with acetylcholinesterase (AChE) inhibitors, glucocorticosteroids, and/or conventional immunosuppressants, along with thymectomy in some cases. However, a subgroup of patients experience MG that is extremely difficult to control; this is often termed refractory MG and may arise from either a suboptimal response or intolerance to therapy. At present, there is no single accepted definition of refractory MG and a variety of definitions can be found in the published literature (reviewed by Mantegazza and Antozzi;7 summarized in Table 1). Depending on the definition used, the prevalence of refractory MG ranges from approximately 10% to 20%.3,8–10 Patients with refractory MG have been shown typically to be female, to be younger at disease onset, to have a history of thymoma, or to be MuSK antibody-positive.7,9,11
Table 1.
Commonly used definitions for refractory MG (adapted from Mantegazza and Antozzi7).
| Number | Definition | Attributes |
|---|---|---|
| 1 | Failure to respond adequately to conventional treatment | Insufficient response (e.g. persistent moderate to severe weakness) to maximal safe doses of steroids and at least one immunosuppressive drug at adequate dose and duration, with sufficient symptomatic treatment |
| 2 | Inability to reduce immuno-suppressive therapy without clinical relapse or need for ongoing rescue therapy (e.g. IVIg or PE/IA) | Possible sufficient initial response to immuno-suppressive therapy; however, the duration of such therapies has to be restricted owing to the potential for profound side effects associated with their use (particularly in the case of corticosteroids) |
| 3 | Severe or intolerable adverse effects from immunosuppressive or symptomatic therapy | More accurately described as ‘treatment intolerant’; however, the inability to effectively treat MG using conventional immunosuppressive agents has the same result as being treatment refractory |
| 4 | Comorbid conditions restricting use of conventional therapies | Again, ‘treatment intolerant’ |
| 5 | Frequent myasthenic crises even while on immunosuppressive and symptomatic therapy | Life-threatening events, characterized by respiratory or bulbar weakness, or paralysis requiring urgent hospitalization |
IA, immunoadsorption; IVIg, intravenous immunoglobulin G; MG, myasthenia gravis; PE, plasma exchange.
The purpose of this narrative review is to highlight the burden caused by refractory MG, with the aim of understanding unmet needs in this patient population.
Burden of refractory myasthenia gravis
To understand the burden that refractory MG places on patients, it is necessary to consider the clinical symptoms of the disease, the side effects of medications and surgery, the psychiatric burden, and the impact on quality of life (QoL). However, published information is limited, possibly owing to the rarity of this disease. The absence of an accepted standard definition of refractory MG is a further complicating factor. Details of the clinical characteristics used to define refractory MG in each publication cited in this article are provided in Table S1, which illustrates the variety of definitions used.
Clinical symptoms
The detailed case histories available in the literature highlight the multiple symptoms that can affect patients with refractory MG in their daily life. Individual examples include severe bulbar weakness, resulting in difficulties with swallowing and speaking12 or the need for a feeding tube,13 fatigable muscle weakness, dysphagia, dysarthria, and dyspnea,14 as well as diplopia and ptosis.15 In very severe cases, patients with refractory MG are so severely disabled that they are bedridden or mechanically ventilated.16
The symptoms of refractory MG may have consequences beyond the initial medical aspects, as illustrated in case studies; for example, patients may experience malnutrition due to escalating dysphagia.17 The prevalence of sleep-disordered breathing and obstructive sleep apnea is higher in patients with MG than in the general population,18 and sleep disturbance in patients with MG has been shown to correlate with lack of remission and the need for immunosuppressive agents, suggesting a higher prevalence in patients with refractory MG.19 Ongoing disease can also interfere with the ability to work because of functionally limiting fatigable weakness after short periods20 or diplopia,15,21 which can restrict the patient’s independence and flexibility because of an inability to drive. A study of 917 patients in Japan showed a significant positive correlation between insufficient control of MG symptoms (a definition of refractory MG) and unemployment or unwilling job transfer.22 Inability to work obviously has a subsequent financial impact on the patient and their family.23
Several studies have shown that myasthenic crises and exacerbations are more common in patients with refractory MG than in those whose disease is controlled.24,25 In a retrospective multicenter study, 13 patients with refractory MG (defined as failure to respond to thymectomy and ⩾2 immuno-suppressive drugs including steroids) experienced a total of 10 intensive care unit visits in the course of 1 year, while seven patients with nonrefractory MG had just two visits; rates of hospitalization because of disease aggravation were also higher in patients with refractory MG [Figure 1(a)].24 In another retrospective analysis, 13 patients with refractory MG (defined as an inadequate clinical response to or intolerance of immunosuppressive treatments) reported 44 acute severe exacerbations of MG (defined as diffuse extremity paresis, dysarthria, dysphagia, or shortness of breath, any of which significantly affected activities of daily living) over a total of 35 patient-years [Figure 1(a)].26 In addition, a prospective observational study in patients with severe refractory MG [Myasthenia Gravis Foundation of America (MGFA) classification27 IIIb–IVb; n = 6] reported two to five myasthenic crises requiring artificial ventilation in each of two patients, and more than five such crises in each of two patients; the remaining two patients had one or two crises.28 In the phase III study of eculizumab in patients with refractory generalized MG, 78% of patients had a history of MG exacerbations and 18% had experienced a myasthenic crisis in the 2 years before study initiation. Furthermore, almost a quarter of patients with refractory generalized MG had previously required ventilator support during the course of their MG.29
Figure 1.
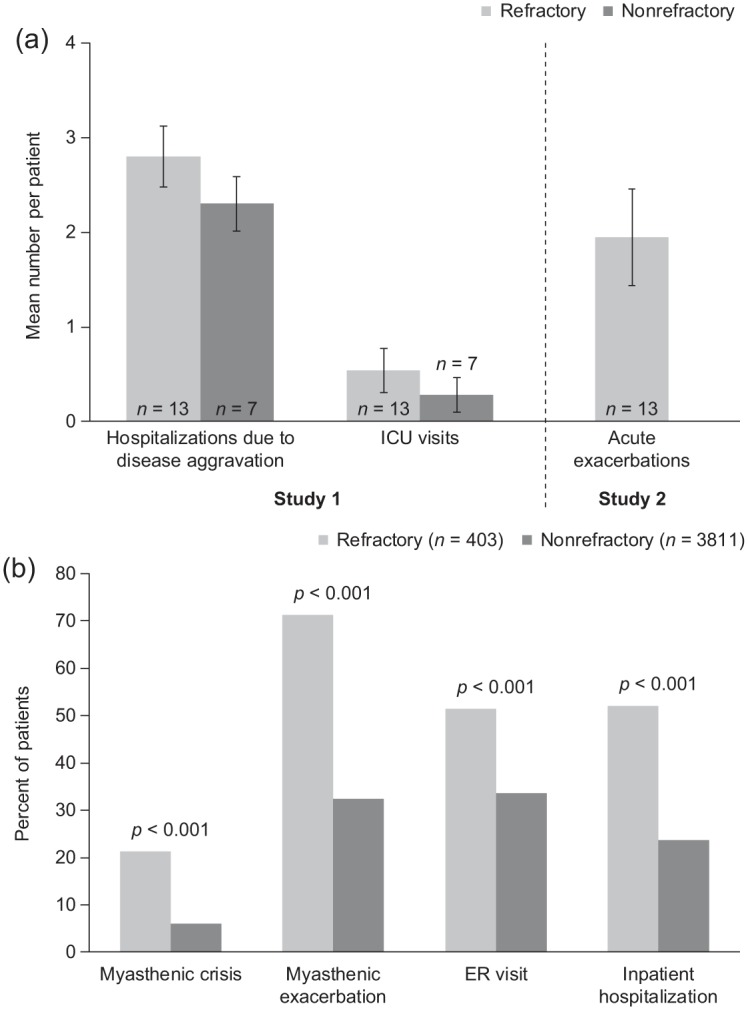
Clinical event rates in patients with MG with and without refractory illness.
(a) Annual mean (± standard error) per patient number of hospitalizations and ICU visits in patients with refractory or nonrefractory MG (Study 1),24 and acute exacerbations in patients with refractory MG (Study 2).26 (b) Unadjusted percentages of patients who experienced myasthenic crises, myasthenic exacerbations, ER visits, and inpatient hospitalizations over a 1-year period.25
ER, emergency room; ICU, intensive care unit; MG, myasthenia gravis.
The experiences reported in these studies are supported by an analysis of health plan databases conducted in the United States of America (USA; refractory MG, n = 403; nonrefractory MG, n = 3811; non-MG control patients, n = 403).25 Over 1 year, compared with patients with non-refractory MG, significantly more patients with refractory MG had at least one myasthenic crisis [adjusted odds ratio (OR) 4.0, 95% confidence interval (CI) 3.0–5.3; p < 0.001] and at least one exacerbation [adjusted OR 4.7, 95% CI 3.7–6.0; p < 0.001; Figure 1(b)]. In addition, patients with refractory MG were almost twice as likely to visit an emergency room and 3.5-times more likely to require inpatient hospitalization than patients with nonrefractory disease (p < 0.001 for both).25 Other studies have noted that patients with refractory MG frequently require multiple intubations during periods of worsening symptoms.15 Because of wide country-specific variations in treatment availability, costs of therapy will not be considered in this review; however, the potential economic impact of refractory MG due to such events is clear from the above reports.
Assessment of disease severity in refractory MG
Patients with refractory MG have a marked disease burden in terms of disability, as supported by the case histories, small studies, and phase II/III clinical trials summarized in this section. Several scales and instruments have been used to assess severity of illness in patients with refractory MG, and these are summarized below.
MGFA clinical classification
The MGFA clinical classification27 reflects the worst pretreatment clinical condition experienced by a patient, rather than the current clinical situation. It categorizes MG into five classes according to the degree of muscle weakness, from class I (any ocular weakness) to class V (requirement for intubation). Classes II–IV rate the mild to severe weakness of muscles other than ocular muscles, subdivided according to body location into ‘a’ (predominantly limb or axial) and ‘b’ (predominantly oropharyngeal or respiratory). Patients with refractory MG have been shown to have a higher MGFA classification (classes III–V), reflecting their greater disability compared with patients with nonrefractory MG (classes II–IV).24
Quantitative MG score
The quantitative MG (QMG) score is used in clinical studies as a measure of disease severity. The validated scale is clinician administered and comprises 13 items that quantitatively assess the endurance or fatigability of different muscle groups.27 The total score ranges from 0 to 39, with higher scores indicating greater disease severity. A ⩾3-point difference in the QMG score is considered clinically meaningful and scores of 10–16 and >16 indicate mild and moderate disease, respectively.30 In clinical studies of patients with nonrefractory generalized MG, baseline mean QMG scores have ranged from 10.4 to 13.3 (i.e. mild MG).31–33 In contrast, patients with refractory MG have high levels of disability as a consequence of their persistent clinical symptoms. Scores ranging from 7 to 28 (considered to be mild to severe MG, respectively) have been reported in a few small case series of patients with refractory MG.34–36 A median (range) score of 21 (12–28) corresponding to mainly moderate to severe MG was reported in 13 patients with residual ciclosporin- and prednisolone-resistant MG;35 and a mean ± standard deviation (SD) score of 20.7 ± 4.9 (indicating mainly moderate to severe MG; median not provided) was reported in a larger follow-up study (n = 79) by the same investigators.37 A mean ± SD baseline QMG score of 17.1 ± 5.3 (range 6–34; that is, mild to severe MG) was reported in the phase III clinical study of eculizumab in patients (n = 125) with refractory generalized MG who were categorized as having MGFA class II–IV disease and who had an MG activities of daily living (MG-ADL) score of ⩾6, suggesting moderate to severe impairment of activities of daily living.29,38 Thus, in general, QMG scores reported in these studies suggest higher disability in patients with refractory MG compared with those with nonrefractory MG.
MG activities of daily living
The MG-ADL profile is an eight-item patient-reported scale that assesses MG symptom severity (ocular, oropharyngeal, respiratory, and extremity function) and effects on daily activities.39 Each item is scored from 0 to 3, with higher scores indicating greater limitations. A 2-point difference is considered clinically relevant.40 In a study of eight patients with refractory MG, the mean MG-ADL score was approximately 8;34 in a second study, three patients had MG-ADL scores of 8–15.41 Both studies indicate disability interfering with functioning during normal daily activities. Studies assessing the effect of immunosuppressants [mycophenolate mofetil (MMF) and methotrexate] in patients with nonrefractory MG31,33 have reported notably lower baseline MG-ADL scores (indicating less impact on daily living) than in patients with refractory MG.34,41,42
Manual muscle test and myasthenia muscle score
The MG-specific manual muscle test (MG-MMT)43 has been used in a few studies of refractory MG to evaluate muscle strength. Without the use of instruments, the MMT determines the strength of 12 bilateral muscle groups and six ocular or axial muscles that are usually affected by MG, with scores from 0 (normal) to 4 (paralysis). The total score ranges from 0 to 120, with higher scores indicating more severe disease. In one small rituximab study, patients with refractory MG had baseline total MMT scores ranging from 4 to 27.44 The myasthenia muscle score45 comprises nine items based on clinical examination of muscle function, with individual scores ranging from 10 to 15 and the total score from 0 to 100, with lower scores indicating greater disease severity. Two small studies of the efficacy of rituximab in treating refractory MG reported baseline myasthenia muscle scores of 27–90.46,47
The wide ranges in scores for these two measures observed in patients with refractory MG may reflect the variety of reasons underlying the classification of refractory disease in individual patients; for example, some patients may be deemed to have refractory disease because, even though a drug was effective, its side effects rendered it intolerable. Scores in these measures do not necessarily reflect whether or not the disease is refractory.
MG composite
The myasthenia gravis composite (MGC) is a validated, 10-item instrument comprising components of the QMG score, the MG-ADL scale, and the MMT, which cover the functional domains most frequently affected by MG and have been found to be the most responsive in MG trials.48 Unlike the QMG and MGFA, the MGC is a weighted scale that takes into account both physician-assessed parameters and patient history. Assessment using the MGC provides a score of 0 (not affected) to 50 (severely affected), with a ⩾3-point change accepted as being clinically meaningful.33 A mean ± SD baseline MGC score of 19.6 ± 6.1 was reported in the phase III eculizumab study in patients with refractory generalized MG and MG-ADL scores of ⩾6.29 Estimated scores of 12–34 were noted in a small retrospective study of eight patients with refractory MG selected for treatment with cyclophosphamide,49 and median baseline scores of 11.5–12.0 (range 2.0–28.0) were reported in patients with refractory MG enrolled in a phase II study of adjunctive belimumab.50
Side effects of treatments for refractory MG
Overall, two approaches are used for pharmacotherapy in MG: (1) symptomatic therapy promoting neuromuscular transmission (e.g. AChE inhibitors); and (2) immunosuppression of the pathological immune processes underlying the disease.2 Immunosuppressants are divided into basic immunotherapy (glucocorticoids, azathioprine, ciclosporin A, methotrexate, MMF, and tacrolimus) and escalation therapies (monoclonal antibodies such as rituximab and eculizumab, and cyclophosphamide). Intervention therapies [IVIg, plasmapheresis/plasma exchange (PE), or immunoadsorption (IA)] are used in situations of imminent or apparent myasthenic crisis.2 Patients with MG and mild to moderate symptoms usually achieve full remission or a substantial improvement following treatment with AChE inhibitors, glucocorticosteroids, or conventional immunosuppressants (azathioprine, ciclosporin A, methotrexate, MMF, and tacrolimus).3 For example, glucocorticosteroids have been shown to improve clinical symptoms within 4–8 weeks in 70–80% of patients.51,52 However, for patients not achieving an adequate response to the above-mentioned drugs, or who experience intolerable side effects, options include high-dose steroids, escalation therapy, or intervention therapy.
Medical treatment
The acute and long-term side effects and contraindications associated with medications used to treat MG are summarized in Table S2, with examples of adverse events (AEs) reported in case studies and clinical trials in patients with MG. Crucially, the substantial side-effect burden can reduce patients’ persistence with treatment or result in patients stopping therapy completely. Furthermore, immunomodulatory treatments for refractory MG can be associated with severe bacterial and viral infections, the most serious of which is progressive multifocal leukoencephalopathy (PML), and mycosis.
Glucocorticosteroids
Patients with refractory MG often receive high-dose steroids for extended periods. Steroids are associated with several well-documented serious long-term AEs (Table S2), including osteoporosis and diabetes.49,53 Treatment strategies in patients with MG aim to reduce the steroid dose when possible through the use of steroid-sparing immunosuppressants (azathioprine, MMF, ciclosporin, tacrolimus, and methotrexate). However, these are themselves associated with significant AEs.
Steroid-sparing immunosuppressants
Azathioprine is frequently administered in patients with MG; however, it is associated with a range of common AEs (Table S2), as well as increased cutaneous photosensitivity that may result in a higher risk of skin cancer.54,55 Although MMF is an alternative treatment option to azathioprine, the risk of PML must be considered; a number of cases have been reported in other therapeutic areas, predominantly in patients with systemic lupus erythematosus or following renal transplantation.56,57 Other, less frequently used steroid-sparing immunosuppressants are ciclosporin, tacrolimus, and methotrexate, which are all associated with a wide range of AEs (Table S2).
Escalation therapies
Eculizumab is approved for use in generalized MG (USA) and refractory generalized MG (European Union) in patients who are anti-AChR antibody-positive. Commonly occurring AEs have been reported with similar frequency with eculizumab and placebo treatment29 (Table S2). Eculizumab has been found to increase patient susceptibility to meningococcal infections owing to its mechanism of action; life-threatening and fatal meningococcal infections have been reported in patients receiving eculizumab58 (see Table S2 for further information). To date, no information is available from randomized controlled trials regarding the safety of rituximab in patients with MG. Rituximab can be associated with prolonged B-cell depletion, especially when administered in combination with other immunosuppressants, leading to an increased risk of infection. Cases of PML have been reported in patients with other autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus, who have been treated with rituximab.59,60 With regard to MG, two cases of PML have been reported46,61 (see Table S2 for further details). There is a paucity of published safety data on the use of cyclophosphamide in MG; however, treatment with cyclophosphamide is associated with nephrotoxicity.20,62
Intervention therapies
IVIg and PE/IA are recommended as short-term treatments in patients with MG who experience myasthenic exacerbations and crises. They may also be administered as maintenance therapy in patients with refractory MG. The benefits and risks of long-term use have not been extensively studied, although IVIg treatment has been reported to be associated with a range of delayed AEs (Table S2). Complications associated with PE/IA primarily arise because of the need for catheter placement (see Table S2 for examples). A comparison of PE and IA in patients with MG noted that allergic reactions or hypocoagulability occurred more frequently in those receiving PE (37% in PE versus 4% in IA).63
Other effects of medical treatment
In addition to the wide variety of acute drug-related AEs, as described above, some drugs used to treat MG are associated with the risk of developing comorbid conditions such as dyslipidemia, obesity, osteoporosis, and cardiac arrhythmias (Table S2).11,25 Of particular concern is steroid-induced diabetes or worsening of existing diabetes.64 Comorbidities are common in patients with MG, with comorbidity rates of 65–73% reported in some studies.65,66 However, the increased requirement for medication in patients with refractory MG may further exacerbate this risk. For example, in a study of 76 patients with refractory MG, 29% were reported to have diabetes;11 in the wider MG population, the prevalence of diabetes has been reported to be 3–20%.65–67 A further factor to consider is potential drug–drug interactions, which should be appraised on a patient-by-patient basis.
Thymectomy
Thymectomy has long been recognized as a treatment option for nonthymomatous MG and refractory nonthymomatous MG. Three surgical approaches are used: video-assisted thoracoscopic surgery (VATS), robotic-assisted thoracoscopic surgery (RATS), and extended transsternal thymectomy. Interestingly, the positive effect of thymectomy on MG was only recently confirmed in a large randomized study that included 126 patients receiving extended transsternal thymectomy.32
Despite the benefits of thymectomy, the surgical procedure of transsternal thymectomy is not without risk for patients because of the need for open thoracotomy. For transsternal thymectomy procedures conducted between 1987 and 1998, major complications associated with surgery were reported to occur in approximately 10–20% of patients.68,69 In a retrospective comparison of surgery performed between 1980 and 2005 via either transsternal (n = 84) or thoracoscopic (n = 22) approaches, the perioperative morbid-ity rate was approximately 20% for both approaches.70 Surgical complications (12%), pneumonia (6%), thrombosis (1%), lesion of the phrenic nerve (7%), pleural effusion (2%), and wound infection (1%) were reported across the two groups; postoperative bleeding necessitating urgent revision after transsternal thymectomy also occurred in one patient.70 In a retrospective analysis of transsternal thymectomy procedures conducted between 1986 and 1989 (n = 26), early complications of sternal osteomyelitis requiring intravenous antibiotics and surgical debridement were reported in one patient; no cases of pneumonia or phrenic nerve palsy were observed.71 Late postoperative complications were also reported, including re-operations, sternal instabilities (9%), and residual thoracic pain (26%).71 A total of seven patients (30%) had hypertrophic scars, one of whom (a young woman) was cosmetically disturbed; however, no patient considered correction of the scar.71 Injury to the innominate vein has also been reported.72,73
The optimal surgical technique for thymectomy in patients with MG is subject to ongoing debate. A retrospective review of a prospectively maintained database of 1002 thymectomy patients in a single institution from 1941 to 2013 reported that although the VATS technique had the best odds of achieving complete stable remission, this was not statistically significant and was probably related to the small number of patients in that cohort.74 The authors concluded that the optimal approach and degree of resection required remain controversial. In contrast, a meta-analysis comparing VATS with open transsternal thymectomy, which included 12 studies with a total of 1173 patients, reported advantages for VATS compared with open thymectomy.75 The VATS approach was associated with less intraoperative blood loss and a shorter hospital length of stay, and lower rates of total complications (OR 0.59, 95% CI 0.37–0.94; p = 0.03) and myasthenic crisis (OR 0.51, 95% CI 0.28–0.92; p = 0.03). However, there were no significant differences in operation time, intensive care unit time, or rates of pneumonia.75 In addition, a literature analysis of VATS and RATS concluded that both techniques offer good and safe operative and perioperative outcomes, with little difference between the surgical approaches.76 Considering the evidence presented above, in our opinion, VATS and RATS are superior to open transsternal thymectomy; however, long-term data are currently lacking.
Repeat thymectomy may be considered in patients with refractory MG. In a review of refractory MG, Silvestri and Wolfe concluded that even patients with negative imaging findings for retained thymic tissue may experience potential benefits from repeat thymectomy following careful selection of appropriate patients.8 In our opinion, however, there should be imaging evidence for remaining thymic tissue (detected by computed tomography, positron-emission computed tomography, or magnetic resonance imaging) before performing repeat thymectomy. In addition, it is important to note that any thymectomy procedure should be performed when a patient is in a stable clinical condition.2,77
Although thymectomy has potential benefits in terms of clinical improvement,8,78 each thymectomy procedure is associated with considerable morbidity. The burden of surgery is particularly relevant given that these patients are experiencing a second surgical procedure (possibly major surgery) against a background of the severe disabling symptoms of refractory MG with the complicating factor of a potentially higher risk of peri- and postoperative events.79,80 Chang and colleagues reported that postoperative AEs were significantly increased in patients with MG who had a history of hospitalization for MG, thymectomy, emergency care for MG, or short-term immunotherapy (IVIg and plasmapheresis);80 these are all factors associated with refractory MG, as detailed above. Furthermore, postoperative complications can arise with any type of surgery in patients with refractory MG.80
Postoperative myasthenic crisis (POMC) is a serious complication associated with thymectomy. In a case series of thoracoscopic complete thymectomy in patients with refractory nonthymomatous MG, two cases of POMC were reported and the authors speculated that, ‘the patients’ unstable clinical condition may have contributed to an increased risk of postoperative exacerbation of myasthenic symptoms.’81 Subsequently, Osserman stage >IIB,82 bulbar symptoms, preoperative plasmapheresis, and a symptom duration of >2 years have been reported to be associated with the occurrence of POMC.83 Presurgically, unstable MG has also been found to be an independent predictor for POMC,84 indicating an increased risk of POMC in patients with refractory MG.
Psychiatric burden
Published data on the psychiatric burden of refractory MG are scarce and, to date, specific rating scales, such as the Hamilton depression rating scale, have not been used in studies of refractory MG. However, it is possible to extrapolate data from studies in the wider MG population to obtain an indication of the effects of refractory MG on mental health.
A cross-sectional study in Turkey (n = 42) reported that greater MG disease severity was associated with worse Hamilton depression rating scale and Hamilton anxiety rating scale scores.85 However, in this study, the most severely affected patients were those with Osserman stage IIB MG (i.e. moderately generalized, inadequate treatment response, bulbar involvement, no crisis).
It is well accepted that the short-term use of corticosteroids can induce mania; however, in the long term, systemic steroid use has also been associated with depressive symptoms.86,87 A large Japanese cross-sectional study of 287 patients with MG (MGFA class I/II/III/IV/V: 68/125/60/12/22, respectively; mean ± SD baseline QMG: 7.0 ± 5.1) reported that the dose of oral steroids for MG treatment was a major factor independently associated with depressive state in MG (assessed using the Beck depression inventory, second edition).88 Furthermore, an ‘unchanged’ or ‘worsened’ disease state after treatment and MGC scores were independently associated with depression.88 As patients with refractory MG are often unable to reduce immunosuppressive therapy (including corticosteroids) without clinical relapse, have an inadequate response to standard therapies, and have worse disease assessment scores, this subpopulation may be more at risk of developing mental illness.
There is a need for further evaluation of the psychiatric burden of refractory MG to inform clinical practice on the optimum psychiatric management of affected patients.
Assessment of QoL
Various outcome measures have been used to assess health-related (HR)-QoL in patients with MG. Although widely used in a variety of therapeutic areas, the generic Medical Outcomes Study short-form (36-item) health survey (SF-36) is not disease specific and is less commonly used now that disease-specific measures are available. The 60-item MG-specific QoL instrument (MG-QoL) has been reported to perform better than the SF-36 in terms of demonstrating disease change over time in patients with MG.89 However, this 60-item instrument was subsequently shortened to 15 items (MG-QoL15).90 The MG-QoL15 can be quickly and easily administered and interpreted, thus increasing its utility in daily practice and clinical trials.90 Each item is scored from 0 to 4; a higher score indicates lower QoL and a 7- to 8-point reduction has been established as a clinically meaningful improvement.90
Despite the availability of disease-specific rating scales, there are limited publications on HR-QoL in patients with refractory MG. However, a few reports are available of small studies describing MG-QoL scores in these patients.34,41,42 In one study, eight patients with refractory MG had a mean MG-QoL15 score of approximately 37.34 In a second study, three patients had MG-QoL15 scores of 30–36.41 These studies indicate considerable detrimental effects on QoL. In a study of 62 patients with moderate to severe MG and worsening symptoms who required IVIg or plasmapheresis, mean ± SD MG-QoL15 scores were 26 ± 13 to 28 ± 14.42 An important finding in this study was that the mean ± SD MG-QoL15 score was significantly higher, indicating worse QoL, in patients with a QMG score of ⩾17 versus <17 (MG-QoL15 score 42 ± 7 versus 28 ± 12, respectively; p = 0.001; Figure 2).42 In agreement with these smaller studies, the phase III eculizumab REGAIN study reported a mean ± SD MG-QoL15 score of 32.1 ± 12.5 at baseline,29 further confirming the negative effects of refractory MG on HR-QoL.
Figure 2.
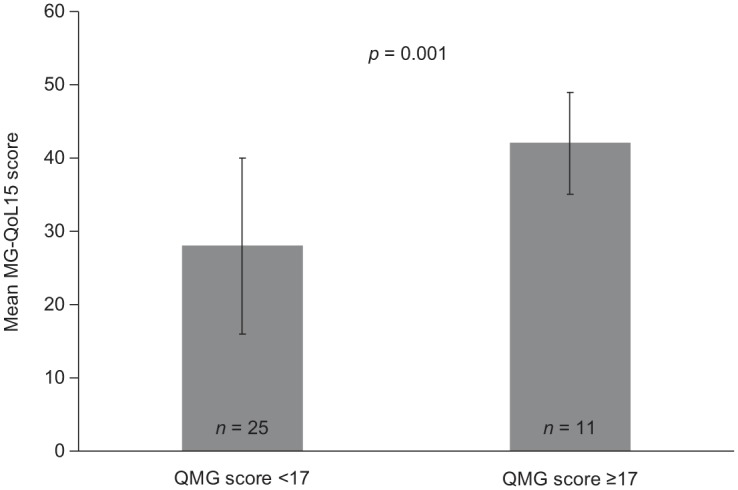
Mean (± standard deviation) MG-QoL15 scores in patients with different degrees of severity of myasthenia gravis.42
Higher scores on the QMG instrument indicate greater disease severity; higher scores on the MG-QoL15 indicate worse health-related quality of life.
MG-QoL15, 15-item myasthenia gravis quality of life questionnaire; QMG, quantitative myasthenia gravis.
Studies assessing the relationship between disease severity, disability, and QoL provide further support for the burden of refractory MG on HR-QoL. In a study of 102 patients with MG (MGFA class I–IV), Raggi and colleagues observed concordance between MG disease severity, disability, and HR-QoL, including both physical and mental health aspects, as assessed using the SF-36.91 In addition, the MG-QoL15 score has been reported to be positively correlated with skeletal muscle weakness, hospital anxiety and depression scale (HADS) total score, HADS anxiety subscale score, and HADS depression subscale score.92 These increased levels of muscle weakness, anxiety, and depression (i.e. higher scores) were associated with lower HR-QoL. In contrast, remission and absence of generalized symptoms are favor-able factors for HR-QoL.93
These studies, together with the small number of reports in patients with refractory MG summarized above, suggest worse HR-QoL for such patients compared with those with nonrefractory MG.
Perspectives and conclusions
Refractory disease occurs in 10–20% of patients with MG;3,8–10 however, this review highlights the breadth and degree of the burden of refractory MG on patients, which is likely to include an increased risk of disability, serious drug- or surgery-associated AEs, myasthenic crises, MG-related hospitalization, and comorbidities. This profile is likely to be accompanied by increased unemployment and worse mental health and HR-QoL. Combinations of symptoms and their consequences amplify the burden on the patient.
This subset of patients has previously been described as being distinct from those with non-refractory disease, with the possibility of underlying biological differences between the two groups.9,25 At present, although certain patient and clinical characteristics have been associated with an increased risk of being refractory to treatment, there are no reliable biomarkers to determine, early on, which patients will respond to therapy. Patients with anti-MuSK antibody-positive MG are unlikely to respond to AChE inhibitors94 and there is some evidence that genetic associations might explain the variability of the response to drugs, including glucocorticoids and azathioprine, among patients with MG.95–97 Further work in this area may help to predict which patients will have disease that is refractory to treatment or experience intolerable AEs to certain drugs in clinical practice.
A number of other unmet needs can be identified in refractory MG. For the clinical management of patients, a consistent definition of ‘refractory MG’ needs to be reached. Definitions vary widely in the published literature and clinical trials, making it difficult to compare study results and derive implications for clinical management in individual situations. Previously used definitions comprise a heterogeneous spectrum, based on disease severity (disease-defined criteria) such as: MGFA class; fluctuations; lack of therapeutic effect of two or three immunosuppressants, PE/IA or IVIg; repeat hospitalization or regular planned hospitalizations; and deterioration lasting <24 months; or drug-defined criteria such as resistance, toxicity, and contraindications to therapy; or a combination of both disease- and drug-defined criteria. Older definitions also fail to take into account the improved outcomes achievable with IVIg, plasmapheresis, and newer drugs.98 One possibility is to create a composite of disease-course and drug-related criteria and to specify a frequency of hospitalization (per year) and distinct period of deterioration that defines ‘refractory’ disease. A multiaxial classification system could include the number and type of immunosuppressive drugs; duration and dose of steroid therapy; exacerbations or need for escalation strategies; and intolerable side effects, taking into account that full remission of MG symptoms should be the therapeutic goal. It should be borne in mind that the definition needs to be applicable to all healthcare systems; therefore, ‘refractory’ disease should not be based on the use of specific drugs, as not all are universally available.
Thus, we would like to propose the following set of disease- and drug-related criteria for refractory MG:
Persistent impairment in activities of daily living; or
at least one imminent or apparent myasthenic crisis per year (not related to inconsequent medication, infection, or use of drugs that induce deterioration of MG) with a need for PE, IA, or IVIg; or
the need for PE, IA, or IVIg at regular intervals; or
persistent intolerable side effects;
despite adequate standard treatment (thymectomy if indicated; AChE inhibitors at maximum tolerable doses; steroids at a dose reduced below the Cushing level within 1 year; and at least one steroid-sparing immunosuppressant) for ⩾1 year.
Refractory MG is uncommon, even in tertiary clinical centers. An online focus group comprising patients with refractory MG convened by the MGFA has identified a need for both patient and physician education on the condition.23 There is a need for greater physician awareness of this subset of patients and, in particular, each individual’s unmet needs, as the disease course and response to treatment can vary widely, necessitating perseverance in identifying the optimal treatment regimen. The need for management by a multidisciplinary team is highlighted not only by the physical and psychological needs of patients with refractory MG, but also by the potential for multiple comorbidities.
At present, there is a lack of evidence-based guidelines for the treatment of refractory MG. Treatment options for patients who fail to achieve remission with standard therapies are limited, and there is a need for effective, steroid-sparing, well-tolerated therapies. A recently published review of available and potential therapeutic options highlighted the promise of biologics as targeted immunotherapy for MG.98 However, few clinical trials appear to be ongoing or planned specifically in patients with refractory MG. In a recently reported study in patients with active generalized MG despite standard therapy, adjunctive belimumab failed to demonstrate a significant improvement in QMG score.50 Rituximab has shown promise in small studies and individual cases of refractory MG. Although recently reported results from a phase II study in patients with anti-AChR antibody-positive MG (not specifically refractory disease) treated with a stable standard immunosuppressive regimen showed that adjunctive rituximab was not effective in reducing the dose of steroid required to control the disease,99 disease severity at baseline was relatively low in this study (60% of patients were assessed as being MGFA class II), which may have limited the ability to detect a treatment effect. However, a meta-analysis of small observational studies suggested that rituximab may be more effective in patients with anti-MuSK antibody-positive MG than in those with anti-AChR antibody-positive MG.100 A subsequent study of patients with anti-MuSK antibody-positive MG reported that compared with the control group, patients receiving rituximab were more likely to achieve the study’s primary clinical endpoint (a composite of MGFA postintervention status score and number and dosages of other immunosuppressant therapies administered).101 Ongoing studies are also examining the efficacy of: rozanolixizumab (an Fc receptor antagonist) in patients with moderate to severe MG who would be considered for IVIg or PE treatment (ClinicalTrials.gov identifier: NCT03052751); abatacept (a naïve T-cell activation inhibitor) in patients who have an inadequate response to conventional immunotherapy (ClinicalTrials.gov identifier: NCT03059888); and bortezomib (a proteasome inhibitor) in patients with therapy-refractory MG (ClinicalTrials.gov identifier: NCT02102594). Other targeted agents being investigated in generalized MG include RA101495 (a subcutaneously administered peptide inhibitor of complement component 5);102 CFZ533 (an anti-CD40 monoclonal antibody);103 and ARGX-113/efgartigimod (an Fc receptor antagonist). Results from controlled studies of these agents in patients with MG are keenly awaited.
In addition, knowledge regarding the psychological impact and patient-reported outcomes in refractory MG is sparse. The past decade has seen a growing awareness across therapeutic areas of the need to incorporate assessments of patient-reported outcomes, and particularly HR-QoL, in clinical trials. The MG-QoL15, a simplified disease-specific instrument to assess HR-QoL in MG,90 has been used in several clinical studies to assess patients’ perceptions of a range of everyday activities and needs that might be affected by MG. A modified version (MG-QoL15r) has recently been developed, which the authors state performs slightly better than the original instrument.104 Wider use of this instrument in clinical trials should improve our understanding of HR-QoL in patients with refractory MG, and its use in clinical practice, along with routine assessment of psychiatric burden, may help to inform disease-management decisions. A large ongoing study in Germany (Myasthenia Gravis and Psyche; ClinicalTrials.gov identifier: NCT03205306; estimated enrollment of 4300 patients) has been specifically designed to characterize psychiatric comorbidities in patients with MG, with outcomes including anxiety disorders, depression, and HR-QoL. In addition, the researchers are examining how particular aspects of the disease influence HR-QoL, and whether different concepts of coping with the disease have different effects on HR-QoL. It would be of interest to analyze patients with refractory MG separately, if possible, in this study.
As with most chronic refractory diseases, living with refractory MG is frustrating for the patient and, as observed in the recent MGFA report from the focus group, ‘… it is particularly frustrating [for people with refractory disease] to read that most people with MG are well managed on treatment, or have ‘normal’ lives, when their experiences are quite the opposite’.23 There is a clear need for greater awareness of the burden of refractory MG and focused research into its impact and treatment.
Supplemental Material
Supplemental material, Schneider-Gold_Refractory_MG__review_suppl_file_updated for Understanding the burden of refractory myasthenia gravis by Christiane Schneider-Gold, Tim Hagenacker, Nico Melzer and Tobias Ruck in Therapeutic Advances in Neurological Disorders
Acknowledgments
Editorial support in the development of the article was provided by Katherine Oldfield of Anthemis Consulting Ltd., funded by Alexion Pharmaceuticals, Inc. Alexion Pharmaceuticals provided a courtesy medical review of the final draft.
Footnotes
Funding: Alexion Pharmaceuticals funded editorial support for this article.
Conflict of interest statement: Dr Schneider-Gold, Dr Hagenacker, and Dr Melzer have received speaker’s honoraria and honoraria for attendance at advisory boards from Alexion Pharmaceuticals. Dr Ruck has received speaker’s honoraria from Alexion Pharmaceuticals.
Supplemental material: Supplemental material for this article is available online.
Contributor Information
Christiane Schneider-Gold, Department of Neurology, St. Josef-Hospital, Ruhr-University Bochum, Gudrunstrasse 56, Bochum, D-44791, Germany.
Tim Hagenacker, University Hospital Essen, Essen, Germany.
Nico Melzer, University of Münster, Münster, Germany.
Tobias Ruck, University of Münster, Münster, Germany.
References
- 1. Verschuuren JJ, Huijbers MG, Plomp JJ, et al. Pathophysiology of myasthenia gravis with antibodies to the acetylcholine receptor, muscle-specific kinase and low-density lipoprotein receptor-related protein 4. Autoimmun Rev 2013; 12: 918–923. [DOI] [PubMed] [Google Scholar]
- 2. Melzer N, Ruck T, Fuhr P, et al. Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society. J Neurol 2016; 263: 1473–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol 2015; 14: 1023–1036. [DOI] [PubMed] [Google Scholar]
- 4. Drachman DB, Kaminski HJ. Neuromuscular junction as Achilles’ heel: yet another autoantibody? Neurology 2014; 82: 1942–1943. [DOI] [PubMed] [Google Scholar]
- 5. Gasperi C, Melms A, Schoser B, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology 2014; 82: 1976–1983. [DOI] [PubMed] [Google Scholar]
- 6. Yan M, Liu Z, Fei E, et al. Induction of anti-agrin antibodies causes myasthenia gravis in mice. Neuroscience 2018; 373: 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord 2018; 11: 1756285617749134. Erratum: Ther Adv Neurol Disord 2018; 11: 1756286418765591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Silvestri NJ, Wolfe GI. Treatment-refractory myasthenia gravis. J Clin Neuromuscul Dis 2014; 15: 167–178. [DOI] [PubMed] [Google Scholar]
- 9. Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med 2013; 86: 255–260. [PMC free article] [PubMed] [Google Scholar]
- 10. Zebardast N, Patwa HS, Novella SP, et al. Rituximab in the management of refractory myasthenia gravis. Muscle Nerve 2010; 41: 375–378. [DOI] [PubMed] [Google Scholar]
- 11. Sudulagunta SR, Sepehrar M, Sodalagunta MB, et al. Refractory myasthenia gravis – clinical profile, comorbidities and response to rituximab. Ger Med Sci 2016; 14: Doc12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rahul LY, Dakua T, Mukherjee A. Refractory myasthenia gravis treated successfully with rituximab: a case report. Ann Indian Acad Neurol 2017; 20: 436–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evoli A, Tonali PA, Padua L, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain 2003; 126: 2304–2311. [DOI] [PubMed] [Google Scholar]
- 14. Kanth KM, Solorzano GE, Goldman MD. PML in a patient with myasthenia gravis treated with multiple immunosuppressing agents. Neurol Clin Pract 2016; 6: e17–e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gladstone DE, Brannagan TH, III, Schwartzman RJ, et al. High dose cyclophosphamide for severe refractory myasthenia gravis. J Neurol Neurosurg Psychiatry 2004; 75: 789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lebrun C, Bourg V, Tieulie N, et al. Successful treatment of refractory generalized myasthenia gravis with rituximab. Eur J Neurol 2009; 16: 246–250. [DOI] [PubMed] [Google Scholar]
- 17. Cereda E, Beltramolli D, Pedrolli C, et al. Refractory myasthenia gravis, dysphagia and malnutrition: a case report to suggest disease-specific nutritional issues. Nutrition 2009; 25: 1067–1072. [DOI] [PubMed] [Google Scholar]
- 18. Nicolle MW, Rask S, Koopman WJ, et al. Sleep apnea in patients with myasthenia gravis. Neurology 2006; 67: 140–142. [DOI] [PubMed] [Google Scholar]
- 19. Martínez-Lapiscina EH, Erro ME, Ayuso T, et al. Myasthenia gravis: sleep quality, quality of life, and disease severity. Muscle Nerve 2012; 46: 174–180. [DOI] [PubMed] [Google Scholar]
- 20. Meriggioli MN, Rowin J. Treatment of myasthenia gravis with mycophenolate mofetil: a case report. Muscle Nerve 2000; 23: 1287–1289. [DOI] [PubMed] [Google Scholar]
- 21. Robeson KR, Kumar A, Keung B, et al. Durability of the rituximab response in acetylcholine receptor autoantibody-positive myasthenia gravis. JAMA Neurol 2017; 74: 60–66. [DOI] [PubMed] [Google Scholar]
- 22. Nagane Y, Murai H, Imai T, et al. Social disadvantages associated with myasthenia gravis and its treatment: a multicentre cross-sectional study. BMJ Open 2017; 7: e013278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Myasthenia Gravis Foundation of America. Living with refractory MG, http://www.myasthenia.org/LinkClick.aspx?fileticket=fKp4Wob0OJY%3D&tabid=75 (2017, accessed 16 May 2018).
- 24. Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve 2012; 46: 687–691. [DOI] [PubMed] [Google Scholar]
- 25. Engel-Nitz NM, Boscoe A, Wolbeck R, et al. Burden of illness in patients with treatment refractory myasthenia gravis. Muscle Nerve 2018; 58: 99–105. [DOI] [PubMed] [Google Scholar]
- 26. Sorgun MH, Sener HO, Yucesan C, et al. Intravenous immunoglobulin for prophylaxis of acute exacerbation in myasthenia gravis. Neurol Sci 2014; 35: 891–896. [DOI] [PubMed] [Google Scholar]
- 27. Jaretzki A, III, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis. Recommendations for clinical research standards. Ann Thorac Surg 2000; 70: 327–334. [DOI] [PubMed] [Google Scholar]
- 28. Blaha M, Pit’ha J, Blaha V, et al. Extracorporeal immunoglobulin elimination for the treatment of severe myasthenia gravis. J Biomed Biotechnol 2010; 2010: 419520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Howard JF, Jr, Utsugisawa K, Benatar M, et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a phase 3, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol 2017; 16: 976–986. [DOI] [PubMed] [Google Scholar]
- 30. Katzberg HD, Barnett C, Merkies IS, et al. Minimal clinically important difference in myasthenia gravis: outcomes from a randomized trial. Muscle Nerve 2014; 49: 661–665. [DOI] [PubMed] [Google Scholar]
- 31. Pasnoor M, He J, Herbelin L, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology 2016; 87: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med 2016; 375: 511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008; 71: 394–399. [DOI] [PubMed] [Google Scholar]
- 34. Jing S, Song Y, Song J, et al. Responsiveness to low-dose rituximab in refractory generalized myasthenia gravis. J Neuroimmunol 2017; 311: 14–21. [DOI] [PubMed] [Google Scholar]
- 35. Ponseti JM, Azem J, Fort JM, et al. Benefits of FK506 (tacrolimus) for residual, cyclosporin- and prednisone-resistant myasthenia gravis: one-year follow-up of an open-label study. Clin Neurol Neurosurg 2005; 107: 187–190. [DOI] [PubMed] [Google Scholar]
- 36. Stieglbauer K, Pichler R, Topakian R. 10-year-outcomes after rituximab for myasthenia gravis: efficacy, safety, costs of inhospital care, and impact on childbearing potential. J Neurol Sci 2017; 375: 241–244. [DOI] [PubMed] [Google Scholar]
- 37. Ponseti JM, Azem J, Fort JM, et al. Long-term results of tacrolimus in cyclosporin- and prednisolone-dependent myasthenia gravis. Neurology 2005; 64: 1641–1643. [DOI] [PubMed] [Google Scholar]
- 38. Vissing J, O’Brien F, Wang JJ, et al. Correlation between myasthenia gravis-activities of daily living (MG-ADL) and quantitative myasthenia gravis (QMG) assessments of anti-acetylcholine receptor antibody-positive refractory generalized myasthenia gravis in the phase 3 REGAIN study. Muscle Nerve 2018; 58: E21–E22. [DOI] [PubMed] [Google Scholar]
- 39. Wolfe GI, Herbelin L, Nations SP, et al. Myasthenia gravis activities of daily living profile. Neurology 1999; 52: 1487–1489. [DOI] [PubMed] [Google Scholar]
- 40. Muppidi S. The myasthenia gravis-specific activities of daily living profile. Ann N Y Acad Sci 2012; 1274: 114–119. [DOI] [PubMed] [Google Scholar]
- 41. Bourque PR, Pringle CE, Cameron W, et al. Subcutaneous immunoglobulin therapy in the chronic management of myasthenia gravis: a retrospective cohort study. PLoS One 2016; 11: e0159993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barnett C, Wilson G, Barth D, et al. Changes in quality of life scores with intravenous immunoglobulin or plasmapheresis in patients with myasthenia gravis. J Neurol Neurosurg Psychiatry 2013; 84: 94–97. [DOI] [PubMed] [Google Scholar]
- 43. Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Ann N Y Acad Sci 2003; 998: 440–444. [DOI] [PubMed] [Google Scholar]
- 44. Beecher G, Anderson D, Siddiqi ZA. Rituximab in refractory myasthenia gravis: extended prospective study results. Muscle Nerve 2018; 58: 452–455. [DOI] [PubMed] [Google Scholar]
- 45. Gajdos P, Simon N, de Rohan-Chabot P, et al. Long-term effects of plasma exchange in myasthenia. Results of a randomized study. Presse Med 1983; 12: 939–942. [PubMed] [Google Scholar]
- 46. Afanasiev V, Demeret S, Bolgert F, et al. Resistant myasthenia gravis and rituximab: a monocentric retrospective study of 28 patients. Neuromuscul Disord 2017; 27: 251–258. [DOI] [PubMed] [Google Scholar]
- 47. Landon-Cardinal O, Friedman D, Guiguet M, et al. Efficacy of rituximab in refractory generalized anti-AChR myasthenia gravis. J Neuromuscul Dis 2018; 5: 241–249. [DOI] [PubMed] [Google Scholar]
- 48. Burns TM, Conaway M, Sanders DB; MG Composite and MG-QOL15 Study Group. The MG Composite: a valid and reliable outcome measure for myasthenia gravis. Neurology 2010; 74: 1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Buzzard KA, Meyer NJ, Hardy TA, et al. Induction intravenous cyclophosphamide followed by maintenance oral immunosuppression in refractory myasthenia gravis. Muscle Nerve 2015; 52: 204–210. [DOI] [PubMed] [Google Scholar]
- 50. Hewett K, Sanders DB, Grove RA, et al. ; BEL115123 Study Group. Randomized study of adjunctive belimumab in participants with generalized myasthenia gravis. Neurology 2018; 90: e1425–e1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol 1984; 15: 291–298. [DOI] [PubMed] [Google Scholar]
- 52. Schneider-Gold C, Gajdos P, Toyka KV, et al. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev 2005; 2: CD002828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kovács E, Dankó K, Nagy-Vince M, et al. Long-term treatment of refractory myasthenia gravis with subcutaneous immunoglobulin. Ther Adv Neurol Disord 2017; 10: 363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. O’Donovan P, Perrett CM, Zhang X, et al. Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science 2005; 309: 1871–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol 2008; 159: 198–204. [DOI] [PubMed] [Google Scholar]
- 56. Molloy ES. PML and rheumatology: the contribution of disease and drugs. Cleve Clin J Med 2011; 78(Suppl. 2): S28–S32. [DOI] [PubMed] [Google Scholar]
- 57. Schmedt N, Andersohn F, Garbe E. Signals of progressive multifocal leukoencephalopathy for immunosuppressants: a disproportionality analysis of spontaneous reports within the US Adverse Event Reporting System (AERS). Pharmacoepidemiol Drug Saf 2012; 21: 1216–1220. [DOI] [PubMed] [Google Scholar]
- 58. Alexion Pharmaceuticals Inc. SOLIRIS® (eculizumab) injection, for intravenous use (highlights of prescribing information). New Haven, USA: Alexion Pharmaceuticals Inc, 2017. [Google Scholar]
- 59. Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports Project. Blood 2009; 113: 4834–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berger JR, Malik V, Lacey S, et al. Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event. J Neurovirol 2018; 24: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tandan R, Hehir MK, II, Waheed W, et al. Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve 2017; 56: 185–196. [DOI] [PubMed] [Google Scholar]
- 62. Stieglbauer K, Topakian R, Schäffer V, et al. Rituximab for myasthenia gravis: three case reports and review of the literature. J Neurol Sci 2009; 280: 120–122. [DOI] [PubMed] [Google Scholar]
- 63. Schneider-Gold C, Krenzer M, Klinker E, et al. Immunoadsorption versus plasma exchange versus combination for treatment of myasthenic deterioration. Ther Adv Neurol Disord 2016; 9: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Suh S, Park MK. Glucocorticoid-induced diabetes mellitus: an important but overlooked problem. Endocrinol Metab (Seoul) 2017; 32: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cacho Diaz B, Flores-Gavilán P, García-Ramos G, et al. Myasthenia gravis and its comorbidities. J Neurol Neurophysiol 2015; 6: 1000317. [Google Scholar]
- 66. Tanovska N, Novotni G, Sazdova-Burneska S, et al. Myasthenia gravis and associated diseases. Open Access Maced J Med Sci 2018; 6: 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wakata N, Nemoto H, Konno S, et al. Myasthenia gravis and diabetes mellitus: a 35-year retrospective study. Intern Med 2007; 46: 557–559. [DOI] [PubMed] [Google Scholar]
- 68. Kas J, Kiss D, Simon V, et al. Decade-long experience with surgical therapy of myasthenia gravis: early complications of 324 transsternal thymectomies. Ann Thorac Surg 2001; 72: 1691–1697. [DOI] [PubMed] [Google Scholar]
- 69. Kattach H, Anastasiadis K, Cleuziou J, et al. Transsternal thymectomy for myasthenia gravis: surgical outcome. Ann Thorac Surg 2006; 81: 305–308. [DOI] [PubMed] [Google Scholar]
- 70. Bachmann K, Burkhardt D, Schreiter I, et al. Long-term outcome and quality of life after open and thoracoscopic thymectomy for myasthenia gravis: analysis of 131 patients. Surg Endosc 2008; 22: 2470–2477. [DOI] [PubMed] [Google Scholar]
- 71. Roth T, Ackermann R, Stein R, et al. Thirteen years follow-up after radical transsternal thymectomy for myasthenia gravis. Do short-term results predict long-term outcome? Eur J Cardiothorac Surg 2002; 21: 664–670. [DOI] [PubMed] [Google Scholar]
- 72. Zieliński M, Kuzdzał J, Staniec B, et al. Extended rethymectomy in the treatment of refractory myasthenia gravis: original video-assisted technique of resternotomy and results of the treatment in 21 patients. Interact Cardiovasc Thorac Surg 2004; 3: 376–380. [DOI] [PubMed] [Google Scholar]
- 73. Özkan B, Toker A. Catastrophes during video-assisted thoracoscopic thymus surgery for myasthenia gravis. Interact Cardiovasc Thorac Surg 2016; 23: 450–453. [DOI] [PubMed] [Google Scholar]
- 74. Kaufman AJ, Palatt J, Sivak M, et al. Thymectomy for myasthenia gravis: complete stable remission and associated prognostic factors in over 1000 cases. Semin Thorac Cardiovasc Surg 2016; 28: 561–568. [DOI] [PubMed] [Google Scholar]
- 75. Qi K, Wang B, Wang B, et al. Video-assisted thoracoscopic surgery thymectomy versus open thymectomy in patients with myasthenia gravis: a meta-analysis. Acta Chir Belg 2016; 116: 282–288. [DOI] [PubMed] [Google Scholar]
- 76. Fok M, Bashir M, Harky A, et al. Video-assisted thoracoscopic versus robotic-assisted thoracoscopic thymectomy: systematic review and meta-analysis. Innovations (Phila) 2017; 12: 259–264. [DOI] [PubMed] [Google Scholar]
- 77. Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology 2016; 87: 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ng JK, Ng CS, Underwood MJ, et al. Does repeat thymectomy improve symptoms in patients with refractory myasthenia gravis? Interact Cardiovasc Thorac Surg 2014; 18: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Alshaikh JT, Amdur R, Sidawy A, et al. Thymectomy is safe for myasthenia gravis patients: analysis of the NSQIP database. Muscle Nerve 2016; 53: 370–374. [DOI] [PubMed] [Google Scholar]
- 80. Chang YW, Chou YC, Yeh CC, et al. Outcomes after major surgery in patients with myasthenia gravis: a nationwide matched cohort study. PLoS One 2017; 12: e0180433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pompeo E, Nofroni I, Iavicoli N, et al. Thoracoscopic completion thymectomy in refractory nonthymomatous myasthenia. Ann Thorac Surg 2000; 70: 918–923. [DOI] [PubMed] [Google Scholar]
- 82. Osserman KE, Genkins G. Studies in myasthenia gravis: review of a twenty-year experience in over 1200 patients. Mt Sinai J Med 1971; 38: 497–537. [PubMed] [Google Scholar]
- 83. Leuzzi G, Meacci E, Cusumano G, et al. Thymectomy in myasthenia gravis: proposal for a predictive score of postoperative myasthenic crisis. Eur J Cardiothorac Surg 2014; 45: e76–e78. [DOI] [PubMed] [Google Scholar]
- 84. Ando T, Omasa M, Kondo T, et al. Predictive factors of myasthenic crisis after extended thymectomy for patients with myasthenia gravis. Eur J Cardiothorac Surg 2015; 48: 705–709. [DOI] [PubMed] [Google Scholar]
- 85. Aysal F, Karamustafalioğlu O, Özçelik B, et al. The relationship of symptoms of anxiety and depression with disease severity and treatment modality in myasthenia gravis: a cross-sectional study. Noro Psikiyatr Ars 2013; 50: 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Brown ES, Suppes T. Mood symptoms during corticosteroid therapy: a review. Harv Rev Psychiatry 1998; 5: 239–246. [DOI] [PubMed] [Google Scholar]
- 87. Bolanos SH, Khan DA, Hanczyc M, et al. Assessment of mood states in patients receiving long-term corticosteroid therapy and in controls with patient-rated and clinician-rated scales. Ann Allergy Asthma Immunol 2004; 92: 500–505. [DOI] [PubMed] [Google Scholar]
- 88. Suzuki Y, Utsugisawa K, Suzuki S, et al. Factors associated with depressive state in patients with myasthenia gravis: a multicentre cross-sectional study. BMJ Open 2011; 1: e000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mullins LL, Carpentier MY, Paul RH, et al. Muscle Study Group. Disease-specific measure of quality of life for myasthenia gravis. Muscle Nerve 2008; 38: 947–956. [DOI] [PubMed] [Google Scholar]
- 90. Burns TM, Conaway MR, Cutter GR, et al. Muscle Study Group. Less is more, or almost as much: a 15-item quality-of-life instrument for myasthenia gravis. Muscle Nerve 2008; 38: 957–963. [DOI] [PubMed] [Google Scholar]
- 91. Raggi A, Leonardi M, Antozzi C, et al. Concordance between severity of disease, disability and health-related quality of life in myasthenia gravis. Neurol Sci 2010; 31: 41–45. [DOI] [PubMed] [Google Scholar]
- 92. Braz NFT, Rocha NP, Vieira ÉLM, et al. Muscle strength and psychiatric symptoms influence health-related quality of life in patients with myasthenia gravis. J Clin Neurosci 2018; 50: 41–44. [DOI] [PubMed] [Google Scholar]
- 93. Boldingh MI, Dekker L, Maniaol AH, et al. An up-date on health-related quality of life in myasthenia gravis – results from population-based cohorts. Health Qual Life Outcomes 2015; 13: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hatanaka Y, Hemmi S, Morgan MB, et al. Nonresponsiveness to anticholinesterase agents in patients with MuSK-antibody-positive MG. Neurology 2005; 65: 1508–1509. [DOI] [PubMed] [Google Scholar]
- 95. Colleoni L, Galbardi B, Barzago C, et al. A novel ABCC6 haplotype is associated with azathioprine drug response in myasthenia gravis. Pharmacogenet Genomics 2017; 27: 51–55. [DOI] [PubMed] [Google Scholar]
- 96. Lantsova VB, Gerasimov AS, Sepp EK. The role of ADRB2 in myasthenia: genetic and immunological factors. Bull Exp Biol Med 2013; 154: 351–353. [DOI] [PubMed] [Google Scholar]
- 97. Xie Y, Li HF, Sun L, et al. The role of osteopontin and its gene on glucocorticoid response in myasthenia gravis. Front Neurol 2017; 8: 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Dalakas MC. Immunotherapy in myasthenia gravis in the era of biologics. Nat Rev Neurol 2019; 15: 113–124. [DOI] [PubMed] [Google Scholar]
- 99. Nowak R. B cell targeted treatment in myasthenia gravis (BeatMG) – a phase 2 trial of rituximab in myasthenia gravis: topline results. In: The American Academy of Neurology Annual Meeting, Philadelphia, Pennsylvania, 4–10 May 2018, abstract P4.478. [Google Scholar]
- 100. Iorio R, Damato V, Alboini PE, et al. Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol 2015; 262: 1115–1119. [DOI] [PubMed] [Google Scholar]
- 101. Hehir MK, Hobson-Webb LD, Benatar M. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology 2017; 89: 1069–1077. [DOI] [PubMed] [Google Scholar]
- 102. Howard JF, Kaminski HJ, Nowak RJ, et al. RA101495, a subcutaneously administered peptide inhibitor of complement component 5 (C5) for the treatment of generalized myasthenia gravis (gMG): phase 1 results and phase 2 design. Neurology 2018; 90(Suppl. 15): S31.006. [Google Scholar]
- 103. Ristov J, Espie P, Ulrich P, et al. Characterization of the in vitro and in vivo properties of CFZ533, a blocking and non-depleting anti-CD40 monoclonal antibody. Am J Transplant. 2018; 18: 2895–2904. [DOI] [PubMed] [Google Scholar]
- 104. Burns TM, Sadjadi R, Utsugisawa K, et al. International clinimetric evaluation of the MG-QOL15, resulting in slight revision and subsequent validation of the MG-QOL15r. Muscle Nerve 2016; 54: 1015–1022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Schneider-Gold_Refractory_MG__review_suppl_file_updated for Understanding the burden of refractory myasthenia gravis by Christiane Schneider-Gold, Tim Hagenacker, Nico Melzer and Tobias Ruck in Therapeutic Advances in Neurological Disorders