

Abstract
Background
Intramuscular fat (IMF) content and composition have a strong impact on the nutritional and organoleptic properties of porcine meat. The goal of the current work was to compare the patterns of gene expression and the genetic determinism of IMF traits in the porcine gluteus medius (GM) and longissimus dorsi (LD) muscles.
Results
A comparative analysis of the mRNA expression profiles of the pig GM and LD muscles in 16 Duroc pigs with available microarray mRNA expression measurements revealed the existence of 106 differentially expressed probes (fold-change > 1.5 and q-value < 0.05). Amongst the genes displaying the most significant differential expression, several loci belonging to the Hox transcription factor family were either upregulated (HOXA9, HOXA10, HOXB6, HOXB7 and TBX1) or downregulated (ARX) in the GM muscle. Differences in the expression of genes with key roles in carbohydrate and lipid metabolism (e.g. FABP3, ORMDL1 and SLC37A1) were also detected. By performing a GWAS for IMF content and composition traits recorded in the LD and GM muscles of 350 Duroc pigs, we identified the existence of one region on SSC14 (110–114 Mb) displaying significant associations with C18:0, C18:1(n-7), saturated and unsaturated fatty acid contents in both GM and LD muscles. Moreover, we detected several genome-wide significant associations that were not consistently found in both muscles. Further studies should be performed to confirm whether these associations are muscle-specific. Finally, the performance of an eQTL scan for 74 genes, located within GM QTL regions and with available microarray measurements of gene expression, made possible to identify 14 cis-eQTL regulating the expression of 14 loci, and six of them were confirmed by RNA-Seq.
Conclusions
We have detected significant differences in the mRNA expression patterns of the porcine LD and GM muscles, evidencing that the transcriptomic profile of the skeletal muscle tissue is affected by anatomical, metabolic and functional factors. A highly significant association with IMF composition on SSC14 was replicated in both muscles, highlighting the existence of a common genetic determinism, but we also observed the existence of a few associations whose magnitude and significance varied between LD and GM muscles.
Electronic supplementary material
The online version of this article (10.1186/s12864-019-5557-9) contains supplementary material, which is available to authorized users.
Keywords: Intramuscular fat, Fatty acid content, Genome-wide association analysis, Duroc pig, Gene expression
Background
Intramuscular fat (IMF) content and fatty acids (FA) composition have important effects on the oxidative stability, tenderness and juiciness of pig meat [1]. These phenotypes are moderately heritable and, in consequence, they can be improved through artificial selection [2]. Several genome-wide association studies (GWAS) have been carried out in pigs to identify quantitative trait loci (QTL) influencing IMF content and composition traits [3–12]. Population sizes employed in these studies have oscillated between 138 [12] and 2326 [11] individuals. Indeed, IMF content and composition traits are not routinely recorded by the pig breeding industry despite their strong impact on the manufacturing of cured products because they are difficult and expensive to measure. All these studies have investigated the genomic architecture of IMF traits in a single muscle, so we do not know yet whether the genetic determinism of IMF content and composition is shared across muscles. In a number of GWAS for IMF phenotypes, genome scans for expression QTL (eQTL) have been carried out as a strategy to pinpoint potential causal mutations. Hundreds of eQTL associated with muscle gene expression phenotypes have been identified, and several of them have been shown to co-localize with QTL for traits of economic interest [8, 13–20].
In a previous study, we determined the IMF content and composition of two muscles, longissimus dorsi (LD) and gluteus medius (GM), in a commercial population of 350 Duroc pigs [21]. Phenotypic correlations between FA traits in these two muscles displayed moderate values ranging from rP = 0.28 to 0.58 [21]. These results suggested the existence of potential differences in the genetic determinism of FA composition across muscles. The goal of the current work was to investigate whether differences exist in the mRNA expression profiles of the GM and LD muscles. Moreover, we aimed to identify QTL for IMF content and composition traits in the GM and LD muscles of Duroc pigs and to establish their positional concordance.
Results
Differential mRNA expression in the gluteus medius and longissimus dorsi muscles
As shown in Table 1, Fig. 1 and Additional file 1, the comparison of the mRNA expression profiles of the GM and LD muscles based on microarray data highlighted the existence of 106 DE probes (|fold-change(FC)| > 1.5, q-value < 0.05). Amongst the genes displaying the most significant differential expression, several loci belonging to the Hox transcription factor family were either upregulated (HOXA9, HOXA10, HOXB6, HOXB7 and TBX1) or downregulated (ARX) in the GM muscle. Differences in the expression of genes with key roles in carbohydrate and lipid metabolism (e.g. FABP3, ORMDL1 and SLC37A1) were also detected. Indeed, the pathway analysis revealed that lipid and carbohydrate metabolic processes are enriched in the set of DE genes, although only at the nominal level (Additional file 2).
Table 1.
List of the twenty genes displaying the highest differential expression between the gluteus medius and longissimus dorsi muscles (threshold of significance: |FC| > 1.5, q-value < 0.05) 1
| Microarray probe | Gene | Fold Change | -log10(P) | -log10(q) |
|---|---|---|---|---|
| Ssc.27606.1.S1_at | HOXA10 | 2.36 | 14.58 | 10.68 |
| Ssc.20706.1.S1_at | HOXB6 | 2.73 | 11.62 | 7.94 |
| Ssc.14356.1.A1_at | LAMA2 | −2.28 | 10.82 | 7.29 |
| Ssc.2743.1.S1_at | MSS51 | −2.72 | 10.06 | 6.63 |
| Ssc.22336.2.A1_at | HOXB7 | 1.67 | 9.66 | 6.28 |
| Ssc.1294.1.S1_at | NIT1 | −1.71 | 7.18 | 3.87 |
| Ssc.17238.1.A1_at | ITIH4 | −1.95 | 7.09 | 3.82 |
| Ssc.4214.1.A1_at | SLC37A1 | 1.63 | 6.87 | 3.69 |
| Ssc.26748.1.A1_at | HOXA9 | 2.13 | 6.52 | 3.37 |
| Ssc.28628.1.S1_at | TIAL1 | −1.58 | 5.30 | 2.28 |
| Ssc.14245.1.A1_at | ZFP92 | −1.56 | 5.20 | 2.24 |
| Ssc.8360.1.A1_at | INPP5F | −1.76 | 5.23 | 2.24 |
| Ssc.21802.1.S1_at | CCL19 | 1.57 | 5.13 | 2.20 |
| Ssc.3838.1.S1_at | COA3 | −1.55 | 5.05 | 2.13 |
| Ssc.22770.1.A1_at | ARX | −1.64 | 4.88 | 2.03 |
| Ssc.4076.1.S1_at | PRUNE2 | −1.65 | 4.85 | 2.02 |
| Ssc.1664.1.A1_at | NRN1 | −2.17 | 4.73 | 1.95 |
| Ssc.9291.1.A1_at | PALMD | −1.84 | 4.67 | 1.94 |
| Ssc.4360.1.A1_at | FABP3 | 1.64 | 4.60 | 1.91 |
| Ssc.11423.2.A1_at | ORMDL1 | 1.54 | 4.48 | 1.85 |
1Fold-change refers to mean expression levels in gluteus medius compared to longissimus dorsi; −log10(P): decimal logarithm of the nominal P-value, −log10(q): decimal logarithm of the q-value
Fig. 1.

Volcano plot of probes differentially expressed in the gluteus medius (GM) and longissimus dorsi (LD) muscles, along with fold changes (ratio between GM/LD mean expression values) for these probes (|FC| > 1.5, q-value < 0.05)
Genome-wide association study of intramuscular fat phenotypes
Performance of a GWAS revealed the existence of several QTL displaying genome-wide significant associations with IMF phenotypes (Table 2 and Figs. 2, 3 and 4). With regard to IMF content, we found a region on SSC6 (146.5–147.7 Mb) which showed a genome-wide significant association in the LD (−log10P-value = 6.88, q-value = 0.0044, δ = − 0.69 ± 0.13) and GM (−log10P-value = 6.47, q-value = 0.01, δ = − 0.84 ± 0.16) muscles only when backfat was removed as a covariate in the statistical analysis. When considering IMF composition, the largest number of associations were found in SSC14, where we observed the existence of one region located between 110 and 114 Mb and displaying significant associations with C18:0, C18:1 (n-7), saturated and unsaturated FA contents both in the GM and the LD muscles (Fig. 2). Effect sizes of markers were negative for C18:0 (δGM = − 0.63 ± 0.10, δLD = − 0.62 ± 0.10) and saturated FA - SFA (δGM = − 0.87 ± 0.18, δLD = − 0.95 ± 0.19) and positive for C18:1 (n-7) (δGM = 0.13 ± 0.03, δLD = 0.18 ± 0.03) and unsaturated FA - UFA (δGM = 0.87 ± 0.18, δLD = 0.94 ± 0.19). We also observed significant associations between GM C18:0 and three additional SSC14 regions on 54.6–55.3 Mb, 60.6–62.9 Mb and 81.1–87.7 Mb. This latter region was also associated with LD SFA. Regarding C18:1(n-7) FA content in the LD muscle, two regions on SSC14 at 81.2–81.3 Mb and 133.1–136.7 Mb were associated with this trait (Table 2, Fig. 2). Moreover, we identified several regions showing associations that were not consistently found in both muscles (Table 2 and Figs. 3 and 4). Two QTL for C16:0 (SSC5: 76.2–77.1 Mb) and C20:3 (n-3) (SSC18: 39.4–42.3 Mb) were significant for the GM muscle but not for the LD one (Fig. 3). Moreover, the effect size of the QTL for C16:0 on SSC5 (76.2–77.1 Mb) was considerably high (δGM = − 0.87 ± 0.20). Conversely, three QTL for C14:0 (SSC9: 10.4–13.2 Mb), C16:1 (n-9) (SSC2: 21.9–22.3 Mb) and C17:0 (SSC2: 10.6–11.7 Mb) were significant for the LD but not for the GM muscle (Fig. 4, Table 2). However, the effects sizes of these three QTL were close to zero, so it is difficult to evaluate their relevance. A GWAS signal was observed for GM C14:0 on SSC9:10–13 Mb, but it did not reach genome-wide significance (Fig. 4).
Table 2.
Genome-wide significant QTL for intramuscular fat composition traits recorded in the gluteus medius (GM) and longissimus dorsi (LD) muscles of Duroc pigs1
| Main FA (gluteus medius) | ||||||||||
| Traits | SSC | N | SNP | Region (Mb) | -log10(P) | q-value | B | δ ± SE | A1 | MAF |
| GM C16:0 | 5 | 9 | H3GA0016883 | 76.2–77.1 | 4.89 | 0.047 | 0.42 | - 0.87 ± 0.20 | A | 0.07 |
| GM C18:0 | 14 | 2 | ASGA0063465 | 54.6–55.3 | 4.30 | 0.027 | 1 | - 0.44 ± 0.11 | G | 0.23 |
| 9 | ALGA0078300 | 60.6–62.9 | 4.96 | 0.008 | 0.36 | - 0.48 ± 0.11 | C | 0.23 | ||
| 8 | ALGA0079209 | 81.1–87.7 | 4.77 | 0.011 | 0.55 | 0.43 ± 0.09 | G | 0.48 | ||
| 42 | ALGA0081091 | 110.9–114.5 | 9.20 | 0.000 | 0.00 | - 0.63 ± 0.10 | A | 0.35 | ||
| GM C18:1 (n-7) | 14 | 14 | ALGA0081091 | 111.4–112.7 | 5.15 | 0.024 | 0.23 | 0.13 ± 0.03 | A | 0.35 |
| GM SFA | 14 | 37 | ALGA0081091 | 111.4–113.8 | 5.51 | 0.007 | 0.10 | - 0.87 ± 0.18 | A | 0.35 |
| GM UFA | 14 | 37 | ALGA0081091 | 111.4–113.8 | 5.52 | 0.007 | 0.10 | 0.87 ± 0.18 | A | 0.35 |
| Main FA (longissimus dorsi) | ||||||||||
| Traits | SSC | N | SNP | Region (Mb) | -log10(P) | q-value | B | δ ± SE | A1 | MAF |
| LD C18:0 | 14 | 2 | CASI0010207 | 86.8–86.8 | 4.43 | 0.035 | 1 | 0.47 ± 0.11 | G | 0.41 |
| 36 | ALGA0081091 | 110.9–113.6 | 6.62 | 0.001 | 0.01 | - 0.62 ± 0.10 | A | 0.35 | ||
| LD C18:1 (n-7) | 14 | 4 | ALGA0079221 | 81.2–81.3 | 4.45 | 0.025 | 1 | - 0.13 ± 0.03 | G | 0.45 |
| 43 | ALGA0081091 | 110.9–114.4 | 6.92 | 0.000 | 0.00 | 0.18 ± 0.03 | A | 0.35 | ||
| 6 | ALGA0082693 | 133.1–136.7 | 5.36 | 0.005 | 0.14 | - 0.13 ± 0.03 | A | 0.46 | ||
| LD SFA | 14 | 6 | ASGA0064951 | 86.4–87.7 | 4.57 | 0.028 | 0.88 | 0.81 ± 0.17 | A | 0.41 |
| 33 | ALGA0081091 | 110.9–113.6 | 5.92 | 0.004 | 0.04 | - 0.95 ± 0.19 | A | 0.35 | ||
| LD UFA | 14 | 31 | ALGA0081091 | 110.9–113.6 | 5.81 | 0.004 | 0.05 | 0.94 ± 0.19 | A | 0.35 |
| Minor FA (gluteus medius) | ||||||||||
| Traits | SSC | N | SNP | Region (Mb) | -log10(P) | q-value | B | δ ± SE | A1 | MAF |
| GM C20:3 (n-3) | 18 | 8 | ASGA0097792 | 39.4–42.3 | 6.38 | 0.005 | 0.01 | 0.12 ± 0.02 | A | 0.07 |
| Minor FA (longissimus dorsi) | ||||||||||
| Traits | SSC | N | SNP | Region (Mb) | -log10(P) | q-value | B | δ ± SE | A1 | MAF |
| LD C14:0 | 9 | 33 | M1GA0026515 | 10.4–13.2 | 5.11 | 0.014 | 0.25 | 0.09 ± 0.02 | A | 0.41 |
| LD C16:1 (n-9) | 2 | 6 | H3GA0006292 | 21.9–22.3 | 6.11 | 0.012 | 0.03 | - 0.02 ± 0.00 | A | 0.25 |
| LD C17:0 | 2 | 10 | MARC0050503 | 10.6–11.7 | 7.30 | 0.002 | 0.00 | - 0.03 ± 0.01 | A | 0.30 |
1SSC: porcine chromosome, N: Number of SNPs significantly associated with the trait under study, SNP: SNP displaying the most significant association with the trait under study, Region (Mb): region containing SNPs significantly associated with the trait under study, −log10 (P): decimal logarithm of the nominal P-value, q-value: q-value calculated with a false discovery rate approach, B: Bonferroni corrected P-values, δ: effect size of the marker and its standard error (SE), A1: minor allele, MAF: minor allele frequency
Fig. 2.

Manhattan plots depicting the genome-wide significant associations between SNP markers and C18:0 and C18:1(n-7) fatty acid contents in the gluteus medius (GM) and longissimus dorsi (LD) muscles of Duroc pigs. Negative log10 P-values of the associations between SNPs and phenotypes are plotted against the genomic location of each marker SNP. Markers on different chromosomes are denoted by different colors. The dashed line represents the genome-wide threshold of significance (q-value ≤0.05). It can be seen a strong association between SNPs on SSC14 (110–114 Mb) and C18:0 and C18:1 (n-7) recorded in both muscles (GM and LD)
Fig. 3.

Manhattan plots depicting the genome-wide significant associations between SNP markers and C16:0 and C20:3 fatty acid contents in the gluteus medius (GM) and longissimus dorsi (LD) muscles of Duroc pigs. Negative log10 P-values of the associations between SNPs and phenotypes are plotted against the genomic location of each SNP. Markers on different chromosomes are denoted by different colors. The dashed line represents the genome-wide threshold of significance (q-value ≤0.05). It can be seen that SNPs at SSC5 and SSC18 are associated, respectively, with C16:0 and C20:3 recorded in the GM muscle but not in the LD one
Fig. 4.
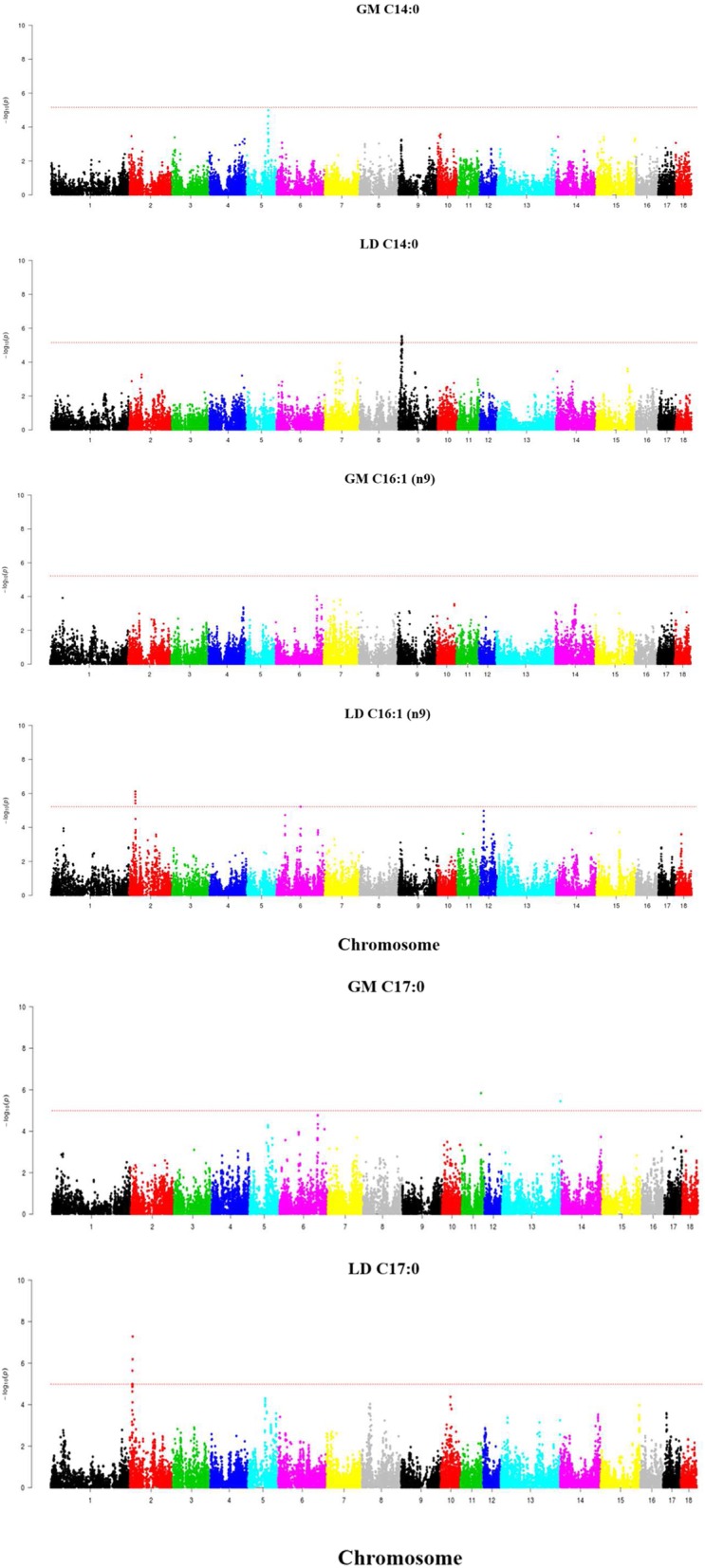
Manhattan plots depicting the genome-wide significant associations between SNPs and C14:0, C16:1(n-9) and C17:0 fatty acid contents in the gluteus medius (GM) and longissimus dorsi (LD) muscles of Duroc pigs. Negative log10 P-values of the associations between SNPs and phenotypes are plotted against the genomic location of each SNP. Markers on different chromosomes are denoted by different colors. The dashed line represents the genome-wide threshold of significance (q-value ≤0.05). It can be seen that SNPs at SSC2 (C16:1 and C17:0) and SSC9 (C14:0) display significant associations with FA composition traits recorded in the LD muscle but not in the GM one
This apparent lack of concordance between the associations found in GM and LD might be due to technical reasons. For instance, the high stringency of the correction for multiple testing could increase the rate of false negatives. Limited statistical power may also lead to the occurrence of a high type 2 error rate, so we have estimated the power of our GWAS by using the procedure reported by Purcell et al. [22]. With a sample size of 350 pigs, we should be able to detect alleles with moderate to large effects (Additional file 3), while the majority of alleles with small effects would be missed. To confirm part of the associations found, we genotyped 12 SNPs mapping to six protein-coding genes i.e. SLC38A1 (SSC5: 77.0–77.0 Mb), SLC38A4 (SSC5: 77.4–77.4 Mb), UVRAG (SSC9: 10.0–10.3 Mb), KCNIP2 (SSC14: 112.8–112.8 Mb), BLOC1S2 (SSC14: 111.3–111.3 Mb) and SCD (SSC14: 111.4–111.4 Mb) loci (Table 3). Several microRNA and lincRNA genes map to QTL regions (Additional file 4), but their functions are mostly unknown so it is difficult to make biological inferences from these co-localizations.
Table 3.
Association analyses between 12 polymorphisms in six candidate genes and intramuscular fat content and composition traits measured in the gluteus medius (GM) and longissimus dorsi (LD) muscles of Duroc pigs1
| Gene | SNP | Type | Trait | q-value | δ (SE) | A1 | MAF |
|---|---|---|---|---|---|---|---|
| SSC5 (76.2–77.1 Mb) | |||||||
| SLC38A1 | rs341329842 (5:77.02 Mb) | Splice region (T/C) | GM C16:0 | 0.000 | −0.61 (0.15) | C | 0.15 |
| LD C16:0 | 0.161 | −0.27 (0.15) | |||||
| SLC38A4 | rs333018168 (5:77.44 Mb) | Missense (G/C, Q24E) | GM C16:0 | 0.014 | −0.51 (0.19) | C | 0.13 |
| LD C16:0 | 0.266 | −0.22 (0.19) | |||||
| SSC9 (10.4–13.2 Mb) | |||||||
| UVRAG | rs321243508 (9:10.31 Mb) | Missense (A/C, K286Q) | GM C14:0 | 0.005 | 0.07 (0.02) | C | 0.43 |
| LD C14:0 | 0.001 | 0.08 (0.02) | |||||
| rs328455999 (9:10.33 Mb) | Missense (G/A, R303H) | GM C14:0 | 0.005 | 0.05 (0.02) | G | 0.26 | |
| LD C14:0 | 0.001 | 0.07 (0.02) | |||||
| SSC14 (110–114 Mb) | |||||||
| BLOC1S2 | rs335981556 (14:111.39 Mb) | Missense (G/A, L6F) | GM C18:0 | 0.001 | −0.62 (0.13) | A | 0.37 |
| LD C18:0 | 0.003 | −0.50 (0.10) | |||||
| GM SFA | 0.077 | −0.71 (0.32) | |||||
| LD SFA | 0.030 | −0.68 (0.26) | |||||
| GM UFA | 0.008 | 0.71 (0.32) | |||||
| LD UFA | 0.032 | 0.67 (0.26) | |||||
| KCNIP2 | rs320607389 (14:112.86 Mb) | Missense (G/A, A50V) | GM C18:0 | 0.001 | −0.58 (0.14) | G | 0.38 |
| LD C18:0 | 0.003 | −0.51 (0.10) | |||||
| SCD | rs698797651 (14:111.4608 Mb) | Upstream (G/GC) | GM C18:0 | 0.001 | −0.59 (0.14) | G | 0.36 |
| LD C18:0 | 0.005 | −0.49 (0.10) | |||||
| rs323081995 (14:111.4616 Mb) | 5′-UTR (T/C) | GM C18:0 | 0.001 | −0.63 (0.13) | C | 0.37 | |
| LD C18:0 | 0.003 | −0.51 (0.10) | |||||
| GM SFA | 0.049 | −0.80 (0.32) | |||||
| LD SFA | 0.029 | −0.72 (0.27) | |||||
| GM UFA | 0.049 | 0.80 (0.32) | |||||
| LD UFA | 0.031 | 0.71 (0.27) | |||||
| rs80912566 (14:111.4617 Mb) | 5′-UTR (C/T) | GM C18:0 | 0.001 | −0.61 (0.13) | T | 0.37 | |
| LD C18:0 | 0.003 | −0.51 (0.10) | |||||
| GM SFA | 0.049 | −0.83 (0.32) | |||||
| LD SFA | 0.029 | −0.70 (0.27) | |||||
| GM UFA | 0.049 | 0.83 (0.32) | |||||
| LD UFA | 0.031 | 0.69 (0.26) | |||||
| rs342182479 (14:111.4618 Mb) | 5′-UTR (G/A) | GM C18:0 | 0.001 | −0.62 (0.13) | A | 0.37 | |
| LD C18:0 | 0.003 | −0.51 (0.10) | |||||
| GM SFA | 0.049 | −0.84 (0.32) | |||||
| LD SFA | 0.029 | −0.72 (0.27) | |||||
| GM UFA | 0.049 | 0.84 (0.32) | |||||
| LD UFA | 0.031 | 0.71 (0.26) | |||||
| rs45434498 (14:111.473 Mb) | Splice region (G/A) | GM C18:0 | 0.001 | −0.61 (0.13) | A | 0.37 | |
| LD C18:0 | 0.003 | −0.52 (0.10) | |||||
| GM SFA | 0.049 | −0.84 (0.31) | |||||
| LD SFA | 0.029 | −0.75 (0.27) | |||||
| GM UFA | 0.049 | 0.84 (0.32) | |||||
| LD UFA | 0.030 | 0.74 (0.26) | |||||
| rs713641545 (14:111.474 Mb) | 3′-UTR (A/G) | GM C18:0 | 0.001 | −0.63 (0.13) | G | 0.37 | |
| LD C18:0 | 0.003 | −0.51 (0.10) | |||||
| GM SFA | 0.049 | −0.81 (0.32) | |||||
| LD SFA | 0.029 | −0.73 (0.27) | |||||
| GM UFA | 0.049 | 0.81 (0.32) | |||||
| LD UFA | 0.031 | 0.72 (0.26) | |||||
1SSC: porcine chromosome, q-value: q-value calculated with a false discovery rate approach, δ: effect size of the marker and its standard error (SE), A1: minor allele, MAF: minor allele frequency
Paralleling the results in the GWAS analysis, the two SNPs in the SLC38A1 and SLC38A4 genes showed significant associations with C16:0 in the GM muscle but not in LD (Table 3). Regarding the two SNPs in the UVRAG gene, they showed significant associations with C14:0 in both muscles (Table 3). Finally, polymorphisms in the KCNIP2, BLOC1S2 and SCD genes displayed significant associations with C18:0 (q-value < 0.005) and SFA (q-value < 0.05) in both muscles, a result that, once again, confirms part of the associations detected in the GWAS (Table 3). The polymorphism of the SCD and BLOC1S2 genes was also associated with UFA content in the GM and LD muscles (Table 3).
We have also investigated whether genes located within QTL regions are DE when comparing the mRNA expression profiles of the LD and GM muscles. If a threshold based on |FC| > 1.5 and q-value < 0.05 is considered, the only locus showing differential expression is the 5′-nucleotidase, cytosolic II (NT5C2) gene, which maps to SSC14 (114 Mb). If we consider a lowered significance threshold (|FC| > 1.2 and q-value = 0.05), then six additional genes display DE between GM and LD (Additional file 5).
Detection of expression QTL for genes located within QTL regions in the gluteus medius muscle
Performance of an eQTL scan for 74 genes, located within the GM QTL regions (as defined in Table 2) and with available microarray measurements of gene expression, made possible to identify 14 cis-eQTL regulating the expression of 14 loci (Table 4, Fig. 5). As shown in Table 4, chromosome 14 encompassed most of these cis-eQTL, which regulated genes co-localizing with two GM C18:0 QTL at 54.6–55.3 Mb (galectin 8, LGALS8 and lysosomal trafficking regulator, LYST) and 81.1–87.7 Mb (annexin A8 gene, ANXA8). The remaining cis-regulated genes, i.e. BLOC1S2, cytochrome C oxidase assembly homolog (COX15), hypoxia inducible factor 1 subunit α inhibitor (HIF1AN), F-Box and WD repeat domain containing 4 (FBXW4), KCNIP2 and armadillo-like helical domain containing 3 (ARMH3) co-localized with the SSC14 (111.4–113.8 Mb) region displaying pleiotropic effects on muscle FA composition. As shown in Table 4, we have also identified cis-regulated genes mapping to a GM C20:3 (n-3) QTL on SSC18 (39.4–42.3 Mb) i.e. FK506 binding protein 14, (FKBP14); ɣ-glutamylcyclotransferase (GGCT) and WAS/WASL interacting protein family member 3 (WIPF3).
Table 4.
List of the co-localizations between QTL for IMF traits recorded in the gluteus medius (GM) muscle and cis-eQTL regulating the mRNA levels of genes expressed in the GM muscle and mapping to QTL regions1
| SSC | N | SNPs | Region (Mb) | -log10 (P) | q-value | B | δ ± SE | A1 | MAF | Gene Symbol | Region (Mb) | Traits | Region(Mb) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14 | 7 | ASGA0063513 | 54.2–55.3 | 12.62 | 0.000 | 0.00 | 0.68 ± 0.08 | A | 0.37 | LGALS8 | 54.8 | C18:0 | 54.6–55.3 |
| 3 | H3GA0040331 | 54.9–56.2 | 3.05 | 0.039 | 0.04 | −0.25 ± 0.07 | G | 0.44 | LYST | 55.4–55.6 | |||
| 21 | MARC0041893 | 86.1–88.0 | 7.96 | 0.000 | 0.00 | 0.82 ± 0.13 | A | 0.32 | ANXA8 | 88.3 | 81.1–87.7 | ||
| 1 | DBMA0000150 | 114.0–114.0 | 2.63 | 0.023 | 0.02 | 0.32 ± 0.10 | G | 0.45 | TAF5 | 114.3 | C18:0; C18:1 (n-7); SFA; UFA |
110.9–114.5 | |
| 26 | MARC0043866 | 110.5–111.9 | 6.10 | 0.000 | 0.00 | 0.42 ± 0.07 | A | 0.49 | COX15 | 110.8 | |||
| 6 | DRGA0014486 | 111.7–112.6 | 4.23 | 0.001 | 0.00 | 0.33 ± 0.08 | G | 0.16 | BLOC1S2 | 111.3 | |||
| 42 | ALGA0081091 | 110.9–113.8 | 3.03 | 0.004 | 0.02 | −0.29 ± 0.08 | A | 0.33 | HIF1AN | 111.6 | |||
| 37 | ALGA0081091 | 111.4–113.8 | 1.98 | 0.035 | 0.24 | −0.19 ± 0.08 | A | 0.33 | FBXW4 | 112.6–112.7 | |||
| 28 | MARC0015087 | 111.4–113.8 | 3.69 | 0.005 | 0.00 | 0.39 ± 0.10 | A | 0.17 | KCNIP2 | 112.8 | |||
| 28 | ALGA0081091 | 110.9–113.8 | 3.12 | 0.002 | 0.02 | −0.27 ± 0.08 | A | 0.33 | ARMH3 | 112.8–113.0 | |||
| 18 | 9 | ASGA0079732 | 40.0–43.3 | 2.32 | 0.023 | 0.16 | 0.23 ± 0.08 | G | 0.23 | AQP1 | 42.0 | GM C20:3 (n-3) |
39.4–42.3 |
| 32 | DIAS0000749 | 41.5–43.9 | 2.25 | 0.049 | 0.18 | −0.32 ± 0.10 | A | 0.35 | GGCT | 42.4 | |||
| 21 | ALGA0116124 | 42.0–43.3 | 3.29 | 0.009 | 0.02 | 0.29 ± 0.08 | G | 0.35 | FKBP14 | 42.9 | |||
| 23 | ALGA0116124 | 42.0–43.3 | 3.27 | 0.004 | 0.03 | −0.27 ± 0.08 | G | 0.35 | WIPF3 | 42.9–43.0 |
1SSC: porcine chromosome, N: number of SNPs significantly associated with the trait under study, SNP: SNP displaying the most significant association with the trait under study, Region (Mb): region containing SNPs significantly associated with the trait under study, −log10(P): decimal logarithm of the nominal P-value, q-value: q-value calculated with a false discovery rate approach, B: Bonferroni-corrected P-value, δ: effect size of the marker and its standard error (SE), A1: minor allele, MAF: minor allele frequency.
Fig. 5.

Co-localization of cis-eQTL (right panel) for the a LGALS8 and b KCNIP2 genes and two QTL regions (left panel) for gluteus medius (GM) C18:0 traits: a SSC14, 54.6–55.3 Mb, b SSC14, 110–114 Mb. The x-axis represents the chromosomal region (Mb) containing the co-localizing QTL and eQTL, and the y-axis shows the –log10 (P-value) of the reported associations. The horizontal line indicates the threshold of significance (q-value ≤0.05). The vertical line depicts the genomic location of the LGALS8 and KCNIP2 genes
With the aim of confirming the existence of these cis-eQTL, we have used a previously published RNA-Seq data set comprising 52 of the 103 pigs analysed with microarrays [23]. As shown in Additional file 3, six cis-eQTL regulating the expression of six genes (ANXA8, BLOC1S2, COX15, GGCT, LGALS8 and FBXW4) showed significant differences across at least 2 genotypes when mRNA expression means measured with both microarrays and RNA-Seq were compared with a Student’s t-test. Moreover, both microarray and RNA-Seq methods were consistent with regard to the direction of the changes observed when comparing genotypes (Additional file 3).
The absence of an eQTL regulating SCD mRNA expression was unexpected because there are evidences that the rs80912566 (g.2228T > C) SNP is associated with the expression of this gene [24]. In Additional file 3, we have compared the GM SCD mRNA levels, measured with microarrays in TT, TC and CC pigs. Although TC individuals display a higher SCD mRNA expression than the CC and TT ones, differences are not statistically significant. A similar pattern was found when SCD mRNA expression was measured by RNA-seq (Additional file 3). We also analysed the correlation between the expression of the genes regulated by cis-eQTL and the phenotypes determined by the QTL to which they map to (Additional file 6). We found that the expression of the TAF5 gene is significantly correlated with C20:3 and saturated and unsaturated FA contents.
Discussion
Significant differences in the mRNA expression profiles of the porcine GM and LD muscles
To the best of our knowledge, no report has been published so far comparing the transcriptomic profiles of two different muscles in pigs. Our differential expression analysis revealed that several loci encoding HOX homeodomain transcription factors (HOXA9, HOXA10, HOXB6, HOXB7, ARX and TBX1) are DE between the LD and GM muscles. These transcription factors have a well-known role in morphogenesis of developing embryos and, in adult vertebrates, they are involved in a plethora of functions related to cell division and muscle contractility [25]. Similarly, the comparison of the transcriptomic profiles of gastrocnemius and quadriceps muscles in mouse revealed that several genes involved in embryogenesis (i.e. DKK3, HOXD8, HOXD9 and TBX1) are DE between these two muscles [26]. Moreover, Armstrong et al. [27] compared the expression profiles of nine lamb muscles by RNA-Seq and detected significant gene expression differences in almost all pairwise comparisons. Interestingly, the comparison of semitendinosus vs. supraspinatus mRNA levels showed that HOXD8 and HOXC10 genes are DE in these two muscles. Conceivably, HOX homeodomain transcription factors might play a critical role in the adult skeletal muscle of mice, sheep and pigs by controlling cell identity and differentiation of muscle fiber types.
The microarray analysis of gene expression also revealed that several genes related to the metabolism of glucose and lipids are DE between GM and LD muscles (Additional file 1 and Additional file 2). Consistently, Armstrong et al. [27] compared the transcriptomic profiles of the semitendinosus, semimembranosus and longissimus lumborum muscles vs. suspraspinatus and evidenced that carbohydrate metabolism pathways are enriched in the sets of DE genes. According to our results (Additional file 1), the fatty acid binding protein 3 (FABP3) mRNA, which encodes a molecule binding and transporting FA towards specific cell compartments [28], is overexpressed in the GM muscle. This finding is significant because it is known that fatty acid binding protein content in most cells is generally proportional to the rates of FA metabolism [28]. Moreover, a polymorphism of the porcine FABP3 gene has been associated with IMF content and other lipid traits (reviewed in [3, 29]). Two other relevant genes that are downregulated in the GM muscle are the Nemo-like protein kinase (NLK), whose variation has been also associated with IMF content in pigs [30], and the nuclear receptor subfamily 2 group F member 1 (NR2F1) locus, an inhibitor of lipoprotein assembly in intestinal cells [31]. The protein kinase AMP-activated non-catalytic subunit ß2 (PRKAB2) gene, which was also downregulated in GM, encodes a subunit of the AMP-activated protein kinase, an enzyme that increases the rate of ß-oxidation of FA in the skeletal muscle [32]. With regard to the glucose metabolism, the solute carrier family 37 member 1 (SLC37A1) gene, which is overexpressed in GM, encodes a glucose-6-phosphate antiporter [33]. Glucose-6-phosphate dehydrogenase uses glucose-6-phosphate as a substrate to generate NADPH that can be used in FA biosynthesis [34]. These patterns of differential expression reflect differences in the lipid metabolism of LD and GM, a feature that could explain, at least in part, the higher IMF content of the GM (Additional file 7). Noteworthy, Morales et al. [35] observed differences in the activity of two lipogenic enzymes when comparing the GM and the semimembranosus muscles in a sample of Iberian and Landrace pigs.
A genomic region on SSC6 is associated with intramuscular fat content in both GM and LD muscles
When backfat was used as a covariate to perform the GWAS for IMF content, we did not detect any significant association. In contrast, when we excluded this covariate from the statistical analysis, we found significant associations with IMF content in the GM (1 significant SNP) and LD (4 significant SNPs) muscles in a SSC6 region (146.5–147.7 Mb) which contains the leptin receptor gene (LEPR). Such association agrees well with previous studies showing that the polymorphism of the LEPR gene is associated with IMF content in Korean native × Yorkshire [36], Landrace × Iberian [37], Duroc × Iberian [38] and Duroc [12] pigs. The binding of leptin, a hormone secreted by the adipose tissue, to its receptor regulates satiety, body weight and the energy balance [39], so it could have potential effects on muscle fat deposition. Indeed, Ros-Freixedes and coworkers showed that the LEPR c.1987C > T SNP is associated with plasma leptin concentration, backfat thickness and IMF content in 853 Duroc pigs, suggesting the causal effects of this gene on fat deposition [12].
A genomic region on SSC14 displays genome-wide significant associations with FA composition traits in both GM and LD muscles
The SSC14 region comprised between 110.9–114.5 Mb encompassed most of the associations observed in our study (Table 2, Fig. 2). In both muscles, marker effect sizes were negative for C18:0 and SFA and positive for C18:1 (n-7) and UFA, suggesting the existence of one causal mutation with opposed effects on both phenotypes or of at least two causal mutations with different effects on these phenotypes. This region had been identified in previous GWAS as associated with IMF composition. Yang et al. [7] carried out a GWAS for FA composition phenotypes and detected associations between SNPs on SSC14 (121 Mb, assembly version Sscrofa10.2) and LD C18:0 content in Sutai × (White Duroc × Erhualian) F2 pigs, whilst Zhang et al. [11] reported associations of this region with the C18:1(n-9)/C18:0 ratio in a related pig population. The same region was reported by Zhang et al. [10] and Sato et al. [4] as associated with LD C18:0 in Duroc × (Landrace × Yorkshire) crossbred pigs and in Duroc pigs, respectively. Consistently, Ros-Freixedes et al. [12] described that the SSC14 (110–114 Mb, assembly version Sscrofa11.1) region is associated with GM and LD C18:1, SFA, monounsaturated FA (MUFA) and the C18:1/C18:0 ratio, whereas Van Son et al. [5] indicated that this very same region displays associations with C16:0, C16:1, C18:0, C18:1(n-9), SFA and MUFA contents in subcutaneous fat. Altogether, these results support the existence of one or several genetic determinants on SSC14 (110.9–114.5 Mb) with multiple effects on muscle FA composition. Interestingly, the annotation of the QTL regions revealed that the SSC14 (110.9–114.5 Mb) region contains, in addition to 33 protein-coding genes, three microRNAs (ssc-miR-436, ssc-miR-146b and ssc-miR-1307) and 4 lincRNA genes (Additional file 4). Studies performed in humans indicate that miR-146b inhibits the proliferation of visceral preadipocytes and stimulates their differentiation [40], while the functions of the lincRNAs are mostly unknown.
In the analysis of candidate genes (Table 3), we have observed that SNPs mapping to the KCNIP2 (rs320607389, SSC14: 112.86 Mb) and BLOC1S2 (rs335981556, SSC14: 111.39 Mb) genes show significant associations with LD and GM C18:0 contents. However, the most obvious candidate locus to explain the associations detected in the SSC14 (110.9–114.5 Mb) region is the SCD gene, which encodes an enzyme involved in the desaturation of FA. Our results show the existence of significant associations between SNPs mapping to the SCD gene and GM and LD C18:0 and SFA (Table 3). Previously, Estany et al. [24] reported that the g.2228 T > C SNP (rs80912566) located in the 5’end of the porcine SCD gene is additively associated with the desaturation C18:1/C18:0 ratio in both muscle and subcutaneous fat in Duroc pigs, a result that would be consistent with ours. In a recent study, Fernández et al. [41] analysed the association of the SCD genotype and IMF composition in three (Iberian × Landrace) × Landrace, (Iberian × Duroc) × Duroc and (Iberian × Piétrain) × Piétrain backcrosses. These authors showed that the g.2228 T > C (rs80912566) SNP in the 5’end of the SCD gene is associated with LD C18:0 (P-value = 0.001) and SFA (P-value = 5.7 × 10− 3) contents but not with C18:1(n-9) content (P-value = 0.207) in the Duroc backcross. A similar outcome was obtained in the Piétrain backcross, highlighting the consistency of such results. In the GWAS carried out in the current work, the ALGA0081091 SNP, which shows the most significant associations with GM and LD C18:0 and C18:1(n-7), is located on SSC14:111,483,985 bp while the SCD g.2228 T > C SNP (rs80912566) maps to 14: 111,461,751 bp. In other words, there are ~ 22 kb between both markers. Sato et al. [4] and Van Son et al. [5] also reported that the SSC14 (120–124 Mb, assembly version Sscrofa10.2, 110–114 Mb in the Sscrofa11.1 assembly) SNPs displaying the most significant associations with FA composition are located ~ 22 kb and 280,389 kb away from the SCD g.2228 T > C SNP, respectively, suggesting that SCD g.2228 T > C is linked to a yet to be found causal mutation with pleiotropic effects on FA traits.
The analysis of eQTL for 74 genes mapping to the SSC14 (110.9–114.5 Mb) QTL did not reveal any genetic determinant regulating SCD mRNA expression (Table 4). We also investigated differences in SCD mRNA expression amongst pigs with different g.2228 T > C SNP SCD genotypes (Additional file 3) and we did not find significant differences. Fernández et al. [41] compared the expression of the SCD mRNA in (Iberian × Duroc) × Duroc pigs with different haplotypes and found that individuals homozygous for the H1 SCD haplotype (which comprises the TT genotype) had a higher SCD mRNA expression in the LD muscle when compared with swine with other haplotypes, but differences were not statistically significant. Similar results were obtained by the same authors when analyzing SCD mRNA expression in (Iberian × Piétrain) × Piétrain pigs [41]. The g.2228 T > C SNP maps to the 5’UTR of the SCD gene, so it might have effects on the stability or the translatability of the transcript, but this needs to be confirmed with a functional assay.
By combining microarray and RNA-Seq data, we obtained evidence of eQTL regulating the expression of the BLOC1S2 gene which maps to the SSC14 (110.9–114.5 Mb) QTL (Table 4). The BLOC1S2 gene encodes a protein that forms part of the BLOC-1 complex, which is involved in the biogenesis and maturation of lysosomes and endosomes [42]. There are increasing evidences that lysosomes have a key role in the processing and sorting of exogenous and endogenous lipids [43]. Another gene of interest is COX15, which is involved in the assembly of cytochrome c oxidase, the terminal component of the mitochondrial respiratory chain [44]. The expression of these two genes did not significantly correlate with the phenotypic variation of C18:0, C18:1 (n-7), SFA or MUFA (Additional file 6). This could be due to the fact that the co-localizations detected by us do not have any functional meaning or, more likely, to the complex genomic architecture of these traits, which are determined by the action of multiple genes. On the other hand, there are many examples of genes whose expression levels do not directly correlate with protein levels and even less with protein activity levels. Therefore, although eQTL are an excellent starting point to detect sequence variability associated with gene activity, it is critical to assess also the potential consequences of regulatory polymorphisms with functional assays.
Several genomic regions on pig chromosomes 2, 5, 9 and 18 show associations with fatty acid traits that are not consistently found inº both muscles
We have also identified several genomic regions i.e. SSC2 (10.6–11.7 Mb), SSC2 (21.9–22.3 Mb), SSC5 (76.2–77.1 Mb), SSC9 (10.4–13.2 Mb) and SSC18 (39.4–42.3 Mb) displaying genome-wide significant associations with LD C17:0, LD C16:1(n-9), GM C16:0, LD C14:0 and GM C20:3(n-3), respectively (Table 2 and Figs. 3 and 4). Amongst these, only the QTL for GM C16:0 on SSC5 and the QTL for GM C20:3(n-3) on SSC18 displayed effect sizes clearly different from zero (Table 2).
The association between SSC9 (10.4–13.2 Mb) and LD C14:0 has been consistently reported in the scientific literature. Sato et al. [4] and Zhang et al. [10] also described associations between SNPs comprised within this region and LD C14:0 content recorded in Duroc swine and Duroc × (Landrace × Yorkshire) crossbred pigs, respectively. Moreover, Zhang et al. [11] showed that this SSC9 region is associated with the C16:1(n-7)/C14:0 ratio in Duroc × (Landrace × Yorkshire) swine. On the other hand, genomic regions on SSC2 (10.6–11.7 Mb) and SSC5 (76.2–77.1 Mb) have been also associated with FA traits in Erhualian and in a meta-analysis comprising five pigs populations, but such associations differ from the ones reported by us with regard to the involved phenotype [11].
Our GWAS should have enough power to detect associations produced by SNPs with moderate to large effects, while SNPs with small effects would be undetectable (Additional file 3). In a similar study based on 331 pigs and a significance threshold of 1 × 10− 6, Zhang et al. [45] showed that the power to detect a QTL that explained 5% of the phenotypic variation was ≈23.6%, but it increased to 90% when the QTL explained 10% of the variance. In another study based on half-sib families of 60 individuals and a population size of 500 individuals, Teyssèdre et al. [46] showed that, when assuming h2 = 0.3–0.6, QTL explaining 4% of the variance of FA traits could be detected with a power of 80%. In the light of these results and ours, it is reasonable to infer that QTL with modest to large effects can be detected with a population of 300–400 pigs, while the majority of QTL with small effects would be missed.
The detection of different associations for traits recorded in the GM and LD muscles might be due to technical and biological factors. The size of the Duroc population analysed in the current work is modest and the precise quantification of QTL for minority FA can be challenging. Both factors might increase the rate of false negatives (type 2 error), thus generating spurious muscle-specific associations. We aimed to take a further look at this issue by genotyping a panel of selected SNPs mapping to QTL regions and investigating their associations with FA traits in the LD and GM muscles. The genotyping of the rs341329842 (SS5: 77.0 Mb) and rs333018168 (SSC5: 77.4 Mb) SNPs mapping to the SLC38A1 and SLC38A4 genes confirmed that they are associated with C16:0 in the GM but not in the LD muscle (Table 2). These two genes are involved in the transportation of glutamine [47], an amino acid that can be used as a substrate for lipogenesis [48], so it would be worth to further explore their role on the genetic determinism of FA composition. We also typed two SNPs, rs321243508 (SSC9:10.3 Mb) and rs328455999 (SSC9:11.3 Mb), which map to the UVRAG gene. This locus encodes a key regulator of autophagy [49], a biological process with an important impact on lipid storage [50]. The polymorphism of the UVRAG gene was associated with C14:0 in both muscles (Table 3), but the significance of such association was slightly higher for LD (q-value = 0.001) than for GM (q-value = 0.005). It is difficult to evaluate the relative impact of technical and biological factors on the lack of replication of several significant associations found either in LD or in GM. Carbonetto et al. [51] measured the weight of four hindlimb muscles in mice and, by using genotypes obtained with the high-density MegaMUGA SNP panel, they found that 18 and 15 genomic regions showed associations that were shared and non-shared across the four analysed muscles, respectively. In pigs, the existence of muscle-specific genomic associations with FA traits should be further investigated because such phenomenon may have practical implications in the framework of genomic selection schemes aimed to improve IMF composition traits.
Conclusions
The comparison of the mRNA expression profiles of the LD and GM muscles has demonstrated the existence of significant differences in the mRNA expression of genes involved in the differentiation of muscle cells as well as in carbohydrate and lipid metabolism. This differential expression might be caused by differences in the body location, function and metabolism of the two porcine muscles under study. Performance of a GWAS has revealed an association between a region on SSC14 (110.9–114.5 Mb) and FA composition that happened to be highly significant in both GM and LD muscles. This region also contained cis-eQTL for genes with potential connections with lipid metabolism. In contrast, several of the associations reported in our study were not consistently found in both muscles, a finding that could be due to technical (e.g. lack of power) and/or biological factors.
Methods
Animal material and phenotype recording
Phenotypes were recorded in 350 barrows from a commercial Duroc line (Lipgen population) distributed in five half-sib families. After weaning, this pig population was transferred to the experimental test station at the Centre de Control Porcí of the Institut de Recerca i Tecnologia Agroalimentàries (IRTA). A detailed description of the experimental population and management conditions can be found in Gallardo et al. [52, 53]. Pigs were slaughtered at around 190 days of age (approximately 122 kg of live weight) following a commercial protocol in compliance with Spanish welfare regulations. Samples (200 g) of GM and LD muscles were taken immediately after slaughter to perform meat analyses at IRTA-Centre of Food Technology. A near infrared transmittance device (NIT, Infratec 1625, Tecator Hoganas, Sweden) was used to determine IMF content in the GM and LD muscles. The measurement of FA composition (C10 to C22 range) in the GM and LD muscles was achieved by gas chromatography of methyl esters [54]. A complete list of the IMF content and composition traits measured in the current experiment is shown in Additional file 7.
Microarray-based analyses of muscle gene expression
For the current study, mRNA expression in the GM and LD muscles was analysed in 16 pigs chosen at random from the Lipgen population. This sample size should be sufficient to test if both muscles have similar or different transcriptomic profiles. Gluteus medius and LD muscle samples were collected from Duroc pigs at slaughter, snap frozen in liquid nitrogen and stored at − 80 °C. Both GM and LD mRNA expression profiles were characterized with the GeneChip Porcine Genome Array (Affymetrix, Inc., Santa Clara, CA). All details about RNA isolation, microarray hybridisation and quality control of expression data are provided in Cánovas et al. [55]. Microarray data describing GM and LD mRNA expression profiles were deposited in the Gene Expression Omnibus (GEO) public repository (GEO accession numbers: GSE19275 and GSE25708). The GeneChip Porcine Genomic arrays (ThermoFisher Scientific, Barcelona, Spain) were also used to measure mRNA expression in GM samples from 103 Duroc pigs (the 16 samples mentioned before plus 87 additional samples) with the aim of performing an eQTL scan. Data pre-processing, background correction, normalization and log-transformation of expression values between samples were carried out by computing a Robust Multi-array Average (RMA) as described by Irizarry et al. [56]. Differential expression between muscles (GM vs. LD) was assessed by following the limma-trend pipeline recommendations [57, 58], where the limma’s empirical Bayes procedure was modified to incorporate a mean-variance trend, modeling the relationship between variance and gene signal intensity. Fold-change (FC) values refer to mean expression levels in GM compared to LD.
Probes showing a |FC| > 1.5 and q-value < 0.05 were considered to be differentially expressed (DE) in the GM vs. LD comparison. Probes showing a significant DE were then translated to gene equivalents by using the Affymetrix porcine annotation data (chip porcine) assembled database [59] and the Biomart database available at Ensembl repositories (https://www.ensembl.org/biomart/martview/). Pathway enrichment analysis and gene ontology annotations were performed upon the more stringent filtering of DE probes (|FC| > 1.5 and q-value < 0.05) using Panther Gene List Classification System (http://www.pantherdb.org).
Genotyping
Whole-genome genotyping of the 350 Duroc pigs was performed using the Porcine SNP60 BeadChip (Illumina, San Diego, CA) which contains probes for 62,163 single nucleotide polymorphisms (SNPs). Filtering analyses based on the quality of the genotyping results were performed with the GenomeStudio software (Illumina). By using PLINK v. 1.07 [60], we filtered SNPs with minor allele frequencies below 5%, rates of missing genotypes above 10% or showing highly significant departures from the Hardy-Weinberg expectation (threshold set at a P-value of 0.001). The SNPs that did not map to the porcine reference genome (Sscrofa11.1 assembly) and those located in sexual chromosomes were also excluded from further analyses. After these filtering steps, a subset of 32,784 SNPs were used as markers in the genome-wide association study (GWAS).
Additionally, 12 SNPs mapping to the solute carrier family 38 member 1 (SLC38A1, rs341329842) and member 4 (SLC38A4, rs333018168), UV radiation resistance associated (UVRAG, and rs328455999), biogenesis of lysosomal organelles complex 1 subunit 2 (BLOC1S2, rs335981556), potassium voltage-gated channel interacting protein 2 (KCNIP2, rs320607389) and stearoyl-CoA desaturase (SCD, rs335981556, rs698797651, rs323081995, rs80912566, rs342182479, rs45434498 and rs713641545) genes were genotyped in the Veterinary Service of Molecular Genetics (http://svgm.es/ca/Home) of the Universitat Autònoma de Barcelona by using a QuantStudio 12 K Flex real-time PCR instrument (Thermo Fisher Scientific). Primers are listed in Additional file 8. These SNPs were identified by taking into account the whole-genome sequencing of the five parental boars (our unpublished data) as well as by the RNA-Sequencing of 52 pigs selected from the population of 350 offspring individuals [23]. Polymorphisms were selected according to their co-localization with the QTL identified in the current work as well as by their potential effect on gene function (SNPs at splice sites or causing amino acid substitutions were prioritized). We also prioritized SNPs that were segregating in our resource Duroc population.
Genome-wide and gene-centric association analyses with intramuscular fat phenotypes
Statistical methods employed in the current work have been previously reported by González-Prendes et al. [61]. In this way, mixed-model association analyses were carried out with the Genome-wide Efficient Mixed-Model Association (GEMMA) software, developed by Zhou and Stephens [62]. This method corrects population structure by considering the relatedness matrix, which is built by taking into account all genome-wide SNPs as a random effect. We used the following statistical model to estimate each SNP effect on IMF content and composition traits:
| 1 |
where y is the vector of phenotypic values for all individuals; W is a matrix including a column of 1 s, the incidence of fixed effects (batch of fattening, with 4 categories) plus a covariate that depends on the trait: (1) IMF content in GM (for FA traits measured in the GM muscle), (2) IMF content in LD (for FA traits measured in the LD muscle), (3) backfat thickness (for IMF content measured in GM and LD); α is a vector of the corresponding fixed effects that includes the intercept, the batch effects and the regression coefficient on the covariate; x is a vector of marker genotypes in each individual; δ is the effect size of the marker (allele substitution effect); u is a vector of random individual genetic effects with a n-dimensional multivariate normal distribution u ∼ N (0, λ τ− 1 K), being τ− 1 the variance of the residual error, λ is the ratio between the two variance components and K a known relatedness matrix derived from SNP genotypes; and ε is the vector of errors. The statistical relevance of the systematic environmental sources of variation and the covariates were previously corroborated by Gallardo et al. [52] and Casellas et al. [63]. In this study, QTL were defined as those genomic regions containing at least 2 SNPs significantly associated with a given IMF content or composition trait. Correction for multiple testing was implemented with a false discovery rate (FDR) approach [64]. The gene-centric association analysis for the SLC38A1, SLC38A4, UVRAG, BLOC1S2, KCNIP2 and SCD loci was performed with GEMMA by using the same methods reported above. The power of the GWAS to detect associations in a population of 350 Duroc pigs was evaluated with the Genetic Power Calculator software [22]. We assumed equal allele frequencies between an unobserved causal variant and an observed genotype in our panel of SNPs under two different scenarios of linkage disequilibrium (r2 = 0.6 and r2 = 0.8). The type I error rate (α) was fixed to α = 0.00005 (equivalent to a q-value = 0.05 in our associations test). The effect size of the SNP (β) expressed in standard deviations (we assumed a normal distribution) and a minor allele frequency (MAF) equal to 0.35 were taken into account to calculate the variance explained by the causal variant in the population (q2 = β2*MAF).
Performance of genome-wide and gene-centric association analyses with gene expression phenotypes
We performed a genome scan to identify potential cis-eQTL regulating the expression of 74 genes mapping to QTL determining IMF traits recorded in the GM muscle by using a previously reported methodology [61]. Gene expression phenotypes were determined with microarrays, as reported above. Official gene names and positions of each probe included in the GeneChip Porcine Genomic array (ThermoFisher, Barcelona, Spain) were identified in the BioMart database [65]. The statistical model assumed in the GEMMA analysis was the same reported in the previous section. However, the vector of fixed effects α and its corresponding incidence matrix W included not only the batch of fattening (with 4 categories) but also a “laboratory” fixed factor (two levels) because microarray data were generated in two different laboratories. Correction for multiple testing was implemented with the FDR approach mentioned above [64]. The threshold of significance in the analysis of cis-eQTL was established in accordance with previously reported criteria [61]. We considered that two QTL and eQTL co-localize when there is at least an overlap of 1 base pair between the genomic regions containing them. The set of cis-eQTL identified with the methods reported above were independently confirmed by using RNA-Seq data from 52 of the 103 pigs employed in the current experiment. The methods involved in the RNA-Seq experiment have been reported by Cardoso et al. [23].
Additionally, we evaluated the effects of g.2228 T > C SNP SCD genotypes over the SCD mRNA expression (measured with microarrays and RNA-Seq) with a linear model considering the batch and lab effects. The significance of mean differences between TT, TC and CC genotypes were assessed with a Student’s t-test [66].
Additional files
Table S1. List of genes differentially expressed between the gluteus medius and longissimus dorsi muscles (|FC| > 1.5; q-value< 0.05). (XLSX 18 kb)
Table S2. Pathway Enrichment for genes differentially expressed between the gluteus medius and longissimus dorsi muscles (|FC| > 1.5; q-value < 0.05). (XLSX 12 kb)
Figure S1. Genetic Power Calculator plots showing the power of the GWAS analysis (x-axis) for a population of 350 Duroc pigs. The effect size (from 0 to 0.35) is plotted in the y-axis. We have assumed an allele frequency of 0.35 and two potential r2 values (0.6 and 0.8) between the unknown causal mutation and the marker SNP. Figure S2. Boxplots depicting the mRNA expression levels of 5 cis-eQTL regulated genes measured with RNA-Seq and microarrays in the gluteus medius muscle of 52 and 103 Duroc pigs, respectively. Means were compared with a Student’s t- test: P-value > 0.05 (ns); P-value ≤0.05 (*); P-value ≤0.01 (**); P-value ≤0.001 (***) and P-value ≤0.0001 (****). Figure S3. Least square means of the stearoyl-CoA desaturase (SCD) mRNA expression levels measured with microarrays (TT, N = 4; TC, N = 20 and CC, N = 18) and RNA-Seq (TT, N = 4; TC, N = 20 and CC, N = 18) in the gluteus medius muscle of Duroc pigs with known genotypes for a polymorphism located in the 5’end of the porcine SCD gene (g.2228 T > C). Means were compared with a Student’s t- test. Although in both analyses CT pigs show a higher SCD mRNA expression than the CC and TT ones, differences are non-significant (ns). (DOCX 421 kb)
Table S3. List of microRNA and long intergenic non-coding RNA genes mapping to the SSC14 QTL identified in the current work. (XLSX 10 kb)
Table S4. Genes located within QTL regions retrieved from the list of genes showing differential expression between the gluteus medius and longissimus dorsi muscles at different thresholds of fold-change (FC) and statistical significance. (XLSX 12 kb)
Table S5. Correlations between the expression of genes regulated by cis-eQTL and the variation of phenotypes determined by QTL co-localizing with the corresponding cis-eQTL (the statistical significance of the correlation is expressed as a P-value between parentheses). (DOCX 19 kb)
Table S6. Means and standard deviations (SD) of intramuscular fat and composition traits recorded in two porcine muscles (N = 350). (DOCX 14 kb)
Table S7. Primer sequences used in the amplification of SNPs mapping to candidate genes located within QTL regions associated with fatty acid composition traits. (XLSX 11 kb)
Acknowledgements
The authors are indebted to Selección Batallé S.A. for providing the animal material. We gratefully acknowledge to J. Soler (IRTA) for his collaboration in the experimental protocols.
Funding
Part of the research presented in this publication was funded by grants AGL2013–48742-C2–1-R and AGL2013–48742-C2–2-R awarded by the Spanish Ministry of Economy and Competitiveness and grant 2014 SGR 1528 from the Agency for Management of University and Research Grants of the Generalitat de Catalunya. We also acknowledge the support of the Spanish Ministry of Economy and Competitiveness for the Center of Excellence Severo Ochoa 2016–2019 (SEV-2015-0533) grant awarded to the Center for Research in Agricultural Genomics. Tainã F Cardoso was funded with a fellowship from the CAPES Foundation-Coordination of Improvement of Higher Education, Ministry of Education (MEC) of the Federal Government of Brazil. Rayner González-Prendes and Emilio Mármol-Sánchez were funded with FPU Ph.D. grants from the Spanish Ministry of Education (FPU12/00860 and FPU15/01733). Maria Ballester is financially supported by a Ramon y Cajal contract (RYC-2013-12573) from the Spanish Ministry of Economy and Competitiveness. Thanks also to the CERCA Programme of the Generalitat de Catalunya for their support. The funding bodies had no role in the design of the study, the collection, analysis, the interpretation of data or the writing of the manuscript.
Availability of data and materials
Data are available at Figshare (10.6084/m9.figshare.7473092.v2).
Abbreviations
- |FC|
Absolute value of the fold-change
- bp
Base pair
- DE
Differentially expressed
- e.g.
For example
- eQTL
Expression QTL
- FA
Fatty acids
- GEMMA
Genome-wide Efficient Mixed-Model Association
- GEO
Gene Expression Omnibus
- GM
Gluteus medius
- GWAS
Genome-wide association analysis
- i.e.
In other words
- IMF
Intramuscular fat
- Kb
Kilobase pairs
- LD
Longissimus dorsi
- lincRNA
Long intergenic noncoding RNA
- Mb
Megabase pairs
- mRNA
Messenger RNA
- QTL
Quantitative trait loci
- rP
Phenotypic correlation
- SNP
Single nucleotide polymorphism
- SSC
Porcine chromosome
- δGM
Marker effect size for traits measured in the gluteus medius muscle
- δLD
Marker effect size for traits measured in the longissimus dorsi muscle
Authors’ contributions
MA and RQ designed the experiment; RQ and JLN were responsible for the generation of animal material and phenotype recording; AMe, ACas and EMS performed the genotyping of candidate genes and EMS made the corresponding association analyses; ID measured fatty acid composition traits; ACas and AMe performed BeadChip genotyping tasks; RGP carried out the GWAS analyses for phenotypes and expression traits in collaboration with AMa, JC and TFC; RNP, ACan and RQ made RNA extractions and generated the microarray data; RNP, EMS, ACan and MB participated in the bioinformatic analyses of microarray expression data; MA, RGP and RNP wrote the paper; all authors read and approved the manuscript.
Ethics approval and consent to participate
Animal care, management procedures and blood sampling were performed following national guidelines for the Good Experimental Practices and they were approved by the Ethical Committee of the Institut de Recerca i Tecnologia Agroalimentàries (IRTA).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Rayner González-Prendes, Email: rayner.prendes@gmail.com.
Raquel Quintanilla, Email: raquel.quintanilla@irta.cat.
Emilio Mármol-Sánchez, Email: emilio.marmol@cragenomica.es.
Ramona N. Pena, Email: romi.pena@ca.udl.cat
Maria Ballester, Email: maria.ballester@irta.cat.
Tainã Figueiredo Cardoso, Email: taina.figueiredo@cragenomica.es.
Arianna Manunza, Email: arianna.manunza@gmail.com.
Joaquim Casellas, Email: joaquim.casellas@uab.cat.
Ángela Cánovas, Email: acanovas@uoguelph.ca.
Isabel Díaz, Email: isabel.diaz@irta.cat.
José Luis Noguera, Email: joseluis.noguera@irta.cat.
Anna Castelló, Email: anna.castello@uab.cat.
Anna Mercadé, Email: anna.mercader@uab.cat.
Marcel Amills, Phone: 34 93 5636600, Email: marcel.amills@uab.cat.
References
- 1.Wood JD, Enser M, Fisher AV, Nute GR, Sheard PR, Richardson RI, et al. Fat deposition, fatty acid composition and meat quality: a review. Meat Sci. 2008;78:343–358. doi: 10.1016/j.meatsci.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Sellier P, Maignel L, Bidanel JP. Genetic parameters for tissue and fatty acid composition of backfat, perirenal fat and longissimus muscle in Large White and Landrace pigs. Animal. 2010;4:497–504. [DOI] [PubMed]
- 3.Casellas J, Vidal O, Pena RN, Gallardo D, Manunza A, Quintanilla R, et al. Genetics of serum and muscle lipids in pigs. Anim Genet. 2013;44:609–619. doi: 10.1111/age.12049. [DOI] [PubMed] [Google Scholar]
- 4.Sato S, Uemoto Y, Kikuchi T, Egawa S, Kohira K, Saito T, et al. Genome-wide association studies reveal additional related loci for fatty acid composition in a Duroc pig multigenerational population. Anim Sci J. 2017;88:1482–1490. doi: 10.1111/asj.12793. [DOI] [PubMed] [Google Scholar]
- 5.Van Son M, Enger EG, Grove H, Ros-Freixedes R, Kent MP, Lien S, et al. Genome-wide association study confirm major QTL for backfat fatty acid composition on SSC14 in Duroc pigs. BMC Genomics. 2017;18:369. doi: 10.1186/s12864-017-3752-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramayo-Caldas Y, Mercadé A, Castelló A, Yang B, Rodríguez C, Alves E, et al. Genome-wide association study for intramuscular fatty acid composition in an Iberian × Landrace cross. J Anim Sci. 2012;90:2883–93. [DOI] [PubMed]
- 7.Yang B, Zhang W, Zhang Z, Fan Y, Xie X, Ai H, et al. Genome-wide association analyses for fatty acid composition in porcine muscle and abdominal fat tissues. PLoS One. 2013;8:e65554. doi: 10.1371/journal.pone.0065554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muñoz M, Rodríguez MC, Alves E, Folch JM, Ibañez-Escriche N, Silió L, et al. Genome-wide analysis of porcine backfat and intramuscular fat fatty acid composition using high-density genotyping and expression data. BMC Genomics. 2013;14:845. doi: 10.1186/1471-2164-14-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davoli R, Luise D, Mingazzini V, Zambonelli P, Braglia S, Serra A, et al. Genome-wide study on intramuscular fat in Italian Large White pig breed using the Porcine SNP60 BeadChip. J Anim Breed Genet. 2016;133:277–82. [DOI] [PubMed]
- 10.Zhang W, Zhang J, Cui L, Ma J, Chen C, Ai H, et al. Genetic architecture of fatty acid composition in the longissimus dorsi muscle revealed by genome-wide association studies on diverse pig populations. Genet Sel Evol. 2016;48:5. [DOI] [PMC free article] [PubMed]
- 11.Zhang W, Yang B, Zhang J, Cui L, Ma J, Chen C, et al. Genome-wide association studies for fatty acid metabolic traits in five divergent pig populations. Sci Rep. 2016;6:24718. doi: 10.1038/srep24718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ros-Freixedes R, Gol S, Pena RN, Tor M, Ibáñez-Escriche N, Dekkers JCM, et al. Genome-wide association study singles out SCD and LEPR as the two main loci influencing intramuscular fat content and fatty acid composition in Duroc pigs. PLoS One. 2016;11:e0152496. doi: 10.1371/journal.pone.0152496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponsuksili S, Jonas E, Murani E, Phatsara C, Srikanchai T, Walz C, et al. Trait correlated expression combined with expression QTL analysis reveals biological pathways and candidate genes affecting water holding capacity of muscle. BMC Genomics. 2008;9:367. doi: 10.1186/1471-2164-9-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wimmers K, Murani E, Ponsuksili S. Functional genomics and genetical genomics approaches towards elucidating networks of genes affecting meat performance in pigs. Brief Funct Genomics. 2010;9:251–258. doi: 10.1093/bfgp/elq003. [DOI] [PubMed] [Google Scholar]
- 15.Steibel JP, Bates RO, Rosa GJM, Tempelman RJ, Rilington VD, Ragavendran A, et al. Genome-wide linkage analysis of global gene expression in loin muscle tissue identifies candidate genes in pigs. PLoS One. 2011;6:e16766. doi: 10.1371/journal.pone.0016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cánovas A, Pena RN, Gallardo D, Ramírez O, Amills M, Quintanilla R. Segregation of regulatory polymorphisms with effects on the gluteus medius transcriptome in a purebred pig population. PLoS One. 2012;7:e35583. doi: 10.1371/journal.pone.0035583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heidt H, Cinar MU, Uddin MJ, Looft C, Jüngst H, Tesfaye D, et al. A genetical genomics approach reveals new candidates and confirms known candidate genes for drip loss in a porcine resource population. Mamm Genome. 2013;24:416–426. doi: 10.1007/s00335-013-9473-z. [DOI] [PubMed] [Google Scholar]
- 18.Ponsuksili S, Murani E, Brand B, Schwerin M, Wimmers K. Integrating expression profiling and whole-genome association for dissection of fat traits in a porcine model. J Lipid Res. 2011;52:668–678. doi: 10.1194/jlr.M013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cinar MU, Kayan A, Uddin MJ, Jonas E, Tesfaye D, Phatsara C, et al. Association and expression quantitative trait loci (eQTL) analysis of porcine AMBP, GC and PPP1R3B genes with meat quality traits. Mol Biol Rep. 2012;39:4809–4821. doi: 10.1007/s11033-011-1274-4. [DOI] [PubMed] [Google Scholar]
- 20.Puig-Oliveras A, Revilla M, Castelló A, Fernández AI, Folch JM, Ballester M. Expression-based GWAS identifies variants, gene interactions and key regulators affecting intramuscular fatty acid content and composition in porcine meat. Sci Rep. 2016;6:31803. doi: 10.1038/srep31803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quintanilla R, Pena RN, Gallardo D, Cánovas A, Ramírez O, Díaz I, et al. Porcine intramuscular fat content and composition are regulated by quantitative trait loci with muscle-specific effects. J Anim Sci. 2011;89:2963–71. [DOI] [PubMed]
- 22.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–50. [DOI] [PubMed]
- 23.Cardoso TF, Cánovas A, Canela-Xandri O, González-Prendes R, Amills M, Quintanilla R. RNA-seq based detection of differentially expressed genes in the skeletal muscle of Duroc pigs with distinct lipid profiles. Sci Rep. 2017;7:40005. [DOI] [PMC free article] [PubMed]
- 24.Estany J, Ros-Freixedes R, Tor M, Pena RN. A functional variant in the stearoyl CoA desaturase gene promoter enhances fatty acid desaturation in pork. PLoS One. 2014;9:e86177. [DOI] [PMC free article] [PubMed]
- 25.Foronda D, De Navas LF, Garaulet DL, Sánchez-Herrero E. Function and specificity of Hox genes. Int J Dev Biol. 2009;53:1409–19. [DOI] [PubMed]
- 26.de Wilde J, Hulshof MF, Boekschoten MV, de Groot P, Smit E, Mariman EC. The embryonic genes DKK3, HOXD8, HOXD9 and TBX1 identify muscle types in a diet-independent and fiber-type unrelated way. BMC Genomics. 2010;11:176. doi: 10.1186/1471-2164-11-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstrong E, Iriarte A, Nicolini P, De Los Santos J, Ithurralde J, Bielli A, et al. Comparison of transcriptomic landscapes of different lamb muscles using RNA-Seq. PLoS One. 2018;13:e0200732. doi: 10.1371/journal.pone.0200732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chmurzyńska A. The multigene family of fatty acid-binding proteins (FABPs): function, structure and polymorphism. J Appl Genet. 2006;47:39–48. doi: 10.1007/BF03194597. [DOI] [PubMed] [Google Scholar]
- 30.Supakankul P, Mekchay S. Association of NLK polymorphisms with intramuscular fat content and fatty acid composition traits in pigs. Meat Sci. 2016;118:61–5. [DOI] [PubMed]
- 31.Dai K, Hussain MM. NR2F1 disrupts synergistic activation of the MTTP gene transcription by HNF-4α and HNF-1α. J Lipid Res. 2012;53:901–8. [DOI] [PMC free article] [PubMed]
- 32.Lee WJ, Kim M, Park H-S, Kim HS, Jeon MJ, Oh KS, et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARα and PGC-1. Biochem Biophys Res Commun. 2006;340:291–5. [DOI] [PubMed]
- 33.Pan CJ, Chen SY, Jun HS, Lin SR, Mansfield BC, Chou JY. SLC37A1 and SLC37A2 are phosphate-linked, glucose-6-phosphate antiporters. PLoS One. 2011;6:e23157. [DOI] [PMC free article] [PubMed]
- 34.Stumpo DJ, Kletzien RF. Regulation of glucose-6-phosphate dehydrogenase mRNA by insulin and the glucocorticoids in primary cultures of rat hepatocytes. Eur J Biochem. 1984;144:497–502. [DOI] [PubMed]
- 35.Morales J, Baucells MD, Pérez JF, Mourot J, Gasa J. Body fat content, composition and distribution in Landrace and Iberian finishing pigs given ad libitum maize- and acorn-sorghum-maize-based diets. Anim Sci. 2003;77:215–24.
- 36.Li X, Kim SW, Choi JS, Lee YM, Lee CK, Choi BH, et al. Investigation of porcine FABP3 and LEPR gene polymorphisms and mRNA expression for variation in intramuscular fat content. Mol Biol Rep. 2010;37:3931–9. [DOI] [PubMed]
- 37.Óvilo C, Fernández A, Noguera JL, Barragán C, Letón R, Rodríguez C, et al. Fine mapping of porcine chromosome 6 QTL and LEPR effects on body composition in multiple generations of an Iberian by Landrace intercross. Genet Res. 2005;85:57–67. [DOI] [PubMed]
- 38.Muñoz G, Alcázar E, Fernández A, Barragán C, Carrasco A, de Pedro E, et al. Effects of porcine MC4R and LEPR polymorphisms, gender and Duroc sire line on economic traits in Duroc × Iberian crossbred pigs. Meat Sci. 2011;88:169–73. [DOI] [PubMed]
- 39.Barb CR, Hausman GJ, Houseknecht KL. Biology of leptin in the pig. Domest Anim Endocrinol. 2001;21:297–317. [DOI] [PubMed]
- 40.Chen L, Dai Y-M, Ji C-B, Yang L, Shi C-M, Xu G-F, et al. MiR-146b is a regulator of human visceral preadipocyte proliferation and differentiation and its expression is altered in human obesity. Mol Cell Endocrinol. 2014;393:65–74. [DOI] [PubMed]
- 41.Fernández AI, Óvilo C, Barragán C, Carmen Rodríguez M, Silió L, Folch JM, et al. Validating porcine SCD haplotype effects on fatty acid desaturation and fat deposition in different genetic backgrounds. Livest Sci. 2017;205:98–105.
- 42.John Peter AT, Lachmann J, Rana M, Bunge M, Cabrera M, Ungermann C. The BLOC-1 complex promotes endosomal maturation by recruiting the Rab5 GTPase-activating protein Msb3. J Cell Biol. 2013;201:97–111. [DOI] [PMC free article] [PubMed]
- 43.Thelen AM, Zoncu R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 2017;27:833–50. [DOI] [PMC free article] [PubMed]
- 44.Petruzzella V, Tiranti V, Fernandez P, Ianna P, Carrozzo R, Zeviani M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. [DOI] [PubMed]
- 45.Zhang J, Zhang Y, Gong H, Cui L, Huang T, Ai H, et al. Genetic mapping using 1.4M SNP array refined loci for fatty acid composition traits in Chinese Erhualian and Bamaxiang pigs. J Anim Breed Genet. 2017;134:472–83. [DOI] [PubMed]
- 46.Teyssèdre S, Elsen J-M, Ricard A. Statistical distributions of test statistics used for quantitative trait association mapping in structured populations. Genet Sel Evol. 2012;44:32. [DOI] [PMC free article] [PubMed]
- 47.Pochini L, Scalise M, Galluccio M, Indiveri C. Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Front Chem. 2014;2:61. [DOI] [PMC free article] [PubMed]
- 48.Anastasiou D, Cantley LC. Breathless cancer cells get fat on glutamine. Cell Res. 2012;22:443–6. [DOI] [PMC free article] [PubMed]
- 49.Song Z, An L, Ye Y, Wu J, Zou Y, He L, et al. Essential role for UVRAG in autophagy and maintenance of cardiac function. Cardiovasc Res. 2014;101:48–56. [DOI] [PubMed]
- 50.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–5. [DOI] [PMC free article] [PubMed]
- 51.Carbonetto P, Cheng R, Gyekis JP, Parker CC, Blizard DA, Palmer AA, et al. Discovery and refinement of muscle weight QTLs in B6 × D2 advanced intercross mice. Physiol Genomics. 2014;46:571–82. [DOI] [PMC free article] [PubMed]
- 52.Gallardo D, Pena RN, Amills M, Varona L, Ramírez O, Reixach J, et al. Mapping of quantitative trait loci for cholesterol, LDL, HDL, and triglyceride serum concentrations in pigs. Physiol Genomics. 2008;35:199–209. [DOI] [PubMed]
- 53.Gallardo D, Quintanilla R, Varona L, Díaz I, Ramírez O, Pena RN, et al. Polymorphism of the pig acetyl-coenzyme A carboxylase α gene is associated with fatty acid composition in a Duroc commercial line. Anim Genet. 2009;40:410–7. [DOI] [PubMed]
- 54.Mach N, Devant M, Díaz I, Font-Furnols M, Oliver MA, García JA, et al. Increasing the amount of n-3 fatty acid in meat from young Holstein bulls through nutrition. J Anim Sci. 2006;84:3039–48. [DOI] [PubMed]
- 55.Cánovas A, Quintanilla R, Amills M, Pena RN. Muscle transcriptomic profiles in pigs with divergent phenotypes for fatness traits. BMC Genomics. 2010;11:372. [DOI] [PMC free article] [PubMed]
- 56.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. [DOI] [PubMed]
- 57.Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, Medvedovic M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics. 2006;7:538. [DOI] [PMC free article] [PubMed]
- 58.Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed]
- 59.Carlson M. Affymetrix porcine annotation data (chip porcine). In: R package version 3.2.3. 2016.
- 60.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed]
- 61.González-Prendes R, Quintanilla R, Cánovas A, Manunza A, Figueiredo Cardoso T, Jordana J, et al. Joint QTL mapping and gene expression analysis identify positional candidate genes influencing pork quality traits. Sci Rep. 2017;7:39830. doi: 10.1038/srep39830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casellas J, Noguera JL, Reixach J, Díaz I, Amills M, Quintanilla R. Bayes factor analyses of heritability for serum and muscle lipid traits in Duroc pigs. J Anim Sci. 2010;88:2246–2254. doi: 10.2527/jas.2009-2205. [DOI] [PubMed] [Google Scholar]
- 64.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 65.Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, et al. The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Res. 2015;43:W589–W598. doi: 10.1093/nar/gkv350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lenth RV. Least-squares means: the R package lsmeans. J Stat Softw. 2016;69:1–33. doi: 10.18637/jss.v069.i01. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of genes differentially expressed between the gluteus medius and longissimus dorsi muscles (|FC| > 1.5; q-value< 0.05). (XLSX 18 kb)
Table S2. Pathway Enrichment for genes differentially expressed between the gluteus medius and longissimus dorsi muscles (|FC| > 1.5; q-value < 0.05). (XLSX 12 kb)
Figure S1. Genetic Power Calculator plots showing the power of the GWAS analysis (x-axis) for a population of 350 Duroc pigs. The effect size (from 0 to 0.35) is plotted in the y-axis. We have assumed an allele frequency of 0.35 and two potential r2 values (0.6 and 0.8) between the unknown causal mutation and the marker SNP. Figure S2. Boxplots depicting the mRNA expression levels of 5 cis-eQTL regulated genes measured with RNA-Seq and microarrays in the gluteus medius muscle of 52 and 103 Duroc pigs, respectively. Means were compared with a Student’s t- test: P-value > 0.05 (ns); P-value ≤0.05 (*); P-value ≤0.01 (**); P-value ≤0.001 (***) and P-value ≤0.0001 (****). Figure S3. Least square means of the stearoyl-CoA desaturase (SCD) mRNA expression levels measured with microarrays (TT, N = 4; TC, N = 20 and CC, N = 18) and RNA-Seq (TT, N = 4; TC, N = 20 and CC, N = 18) in the gluteus medius muscle of Duroc pigs with known genotypes for a polymorphism located in the 5’end of the porcine SCD gene (g.2228 T > C). Means were compared with a Student’s t- test. Although in both analyses CT pigs show a higher SCD mRNA expression than the CC and TT ones, differences are non-significant (ns). (DOCX 421 kb)
Table S3. List of microRNA and long intergenic non-coding RNA genes mapping to the SSC14 QTL identified in the current work. (XLSX 10 kb)
Table S4. Genes located within QTL regions retrieved from the list of genes showing differential expression between the gluteus medius and longissimus dorsi muscles at different thresholds of fold-change (FC) and statistical significance. (XLSX 12 kb)
Table S5. Correlations between the expression of genes regulated by cis-eQTL and the variation of phenotypes determined by QTL co-localizing with the corresponding cis-eQTL (the statistical significance of the correlation is expressed as a P-value between parentheses). (DOCX 19 kb)
Table S6. Means and standard deviations (SD) of intramuscular fat and composition traits recorded in two porcine muscles (N = 350). (DOCX 14 kb)
Table S7. Primer sequences used in the amplification of SNPs mapping to candidate genes located within QTL regions associated with fatty acid composition traits. (XLSX 11 kb)
Data Availability Statement
Data are available at Figshare (10.6084/m9.figshare.7473092.v2).