

Abstract
Background
Prostate cancer (PCa) is a malignancy cause of cancer deaths and frequently diagnosed in male. This study aimed to identify tumor suppressor genes, hub genes and their pathways by combined bioinformatics analysis.
Methods
A combined analysis method was used for two types of microarray datasets (DNA methylation and gene expression profiles) from the Gene Expression Omnibus (GEO). Differentially methylated genes (DMGs) were identified by the R package minfi and differentially expressed genes (DEGs) were screened out via the R package limma. A total of 4451 DMGs and 1509 DEGs, identified with nine overlaps between DMGs, DEGs and tumor suppressor genes, were screened for candidate tumor suppressor genes. All these nine candidate tumor suppressor genes were validated by TCGA (The Cancer Genome Atlas) database and Oncomine database. And then, the gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway (KEGG) enrichment analyses were performed by DAVID (Database for Annotation, Visualization and Integrated Discovery) database. Protein–protein interaction (PPI) network was constructed by STRING and visualized in Cytoscape. At last, Kaplan–Meier analysis was performed to validate these genes.
Results
The candidate tumor suppressor genes were IKZF1, PPM1A, FBP1, SMCHD1, ALPL, CASP5, PYHIN1, DAPK1 and CASP8. By validation in TCGA database, PPM1A, DAPK1, FBP1, PYHIN1, ALPL and SMCHD1 were significant. The hub genes were FGFR1, FGF13 and CCND1. These hub genes were identified from the PPI network, and sub-networks revealed by these genes were involved in significant pathways.
Conclusion
In summary, the study indicated that the combined analysis for identifying target genes with PCa by bioinformatics tools promote our understanding of the molecular mechanisms and underlying the development of PCa. And the hub genes might serve as molecular targets and diagnostic biomarkers for precise diagnosis and treatment of PCa.
Electronic supplementary material
The online version of this article (10.1186/s12935-019-0753-x) contains supplementary material, which is available to authorized users.
Keywords: Bioinformatics, Prostate cancer, Differentially expressed gene, DNA methylation
Background
DNA methylation, one of the most important epigenetic factors, has been studied extensively over several decades, and its influence in a variety of human diseases, most malignancy tumor, diagnostic biomarkers and therapeutic targets, has been firmly measured and evaluated [1–6]. Recently epigenetic study has identified that DNA methylation is an important biological mechanism for tumor occur and development [5]. In CpG islands, aberrant methylation could influence the functions of tumor suppressor genes by altering their expression levels. CpG islands are located in or near promoter regions of the genome, aberrant methylation genes in CpG islands are often hypermethylated and may cause silencing of tumor suppressor genes [7]. Variations of DNA methylation exist at CpG islands, including gene hypermethylation-low regulation and hypomethylation-high regulation.
Several studies that have done to investigate DNA methylation in gene body has positively correlated with gene expression by increasing transcription activity [8]. This may be caused by blocking the intragenic promoter activity or affecting the methylation status of repetitive sequence within the transcription unit [9]. So DNA methylation in gene body may be an interesting additional therapy target for cancer diagnosing and treatment.
Prostate cancer (PCa) is the second most frequently diagnosed male-specific malignancy tumor in western countries. According to the World Health Organization’s International Agency for Research on Cancer, 1.1 million men were diagnosed with prostate cancer worldwide in 2012, accounting for 15% of all cancer diagnosed in men [6]. PCa is considered as a heterogeneous disease [10]. Accumulating evidence has also demonstrated that multiple genes and cellular pathways participate together in the occurrence and development of PCa. Tumor led by epigenetic mutation through cells may grow and reproduce uncontrollably [11]. Hypomethylation of CpG can cause chromosome instability [12]. Illumina Infinium 450 k microarray and DNA microarray have utilized to investigate DNA methylation and gene expression in molecular mechanism, biological process, molecular diagnosis, tumor molecular, biomarker and drug targets discovery [13–15].
Many gene expression profiling analysis and aberrant methylation analysis were introduced for differentially expressed genes (DEGs) and differentially methylated genes (DMGs) [16]. However, separated analysis of DEGs and DMGs are limited [17, 18]. So it is necessary to make jointly analyze for both gene expression profiling microarray and gene methylation profiling microarray in PCa. In this study, gene methylation profiling datasets and gene expression profiling datasets were analyzed by bioinformatics tools for screening the DMGs and DEGs. Later, the overlapping of hypermethylation genes, down-regulated genes and tumor suppressor genes were used to identify the candidate tumor suppressor genes. For validating these candidate tumor suppressor genes, TCGA database was used to identify the CpG islands and Oncomine database was used to validate the analyzing result. At last, the biological functions and pathways analyzing were discussed for the molecular mechanism.
Methods
Datasets
In this study, the gene methylation profiling datasets and gene expression profiling datasets were downloaded from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo) database. All these gene methylation profiling datasets were based on GPL13534 platform (Illumina HumanMethylation450 BeadChip). For the gene expression profiling datasets were based on GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array). For each dataset, only the samples associated with PCa were selected (Table 1).
Table 1.
Datasets for gene methylation profiling and gene expression profiling associated with PCa
| Dataset | Platform | Tumor tissue samples | Normal tissue samples |
|---|---|---|---|
| Gene methylation profiles | |||
| GSE52955 | GPL13534 | 25 | 5 |
| GSE73549 | GPL13534 | 57 | 15 |
| GSE76938 | GPL13534 | 73 | 63 |
| GSE84749 | GPL13534 | 20 | 4 |
| Gene expression profiles | |||
| GSE26910 | GPL570 | 6 | 6 |
| GSE30174 | GPL570 | 70 | 10 |
| GSE46602 | GPL570 | 36 | 14 |
| GSE55945 | GPL570 | 13 | 8 |
| GSE69223 | GPL570 | 15 | 15 |
Data preprocessing and analyzing
R package affy was used to explore oligonucleotide array analysis by the robust multiarray average (RMA) algorithm [19]. While R package methylumiIlluminaHumanMethylation450kmanifest, limma, minfi, watermelon and IlluminaHumanMethylation450kanno.ilmn12.hg19 were used to analyze the gene methylation profiling data and gene expression profiling data in order to identify DMGs and DEGs. And the R package limma in RStudio 1.1.453 was installed to identify genes that were differentially expressed between normal and tumor samples. The Benjamini and Hochberg (BH) procedure were obtained to control the False Discovery Rate (FDR) [20]. Then the log2-fold change (log2FC) was calculated. The adjusted P value < 0.05 and |log2FC| > 2.0 were considered as the cutoff value for DMGs and DEGs screening.
Functional and pathway enrichment analysis of DMGs and DEGs
In order to analyze the DMGs and DEGs for the functional enrichment, GO enrichment and KEGG pathway analysis were performed using DAVID (https://david.ncifcrf.gov). In this paper, DMGs and DEGs were uploaded to online analysis tool to systematically investigate biological meanings behind these genes [21]. Gene ontology analysis (GO) is a common useful method for annotating genes for identifying biological process (BP), cellular component (CC) and molecular function (MF) [22, 23]. At the same time, Kyoto Encyclopedia of Genomes (http://www.genome.jp) pathway enrichment analysis was conducted for candidate genes and prostate specific antigen (PSA) [24].
Protein–protein interaction (PPI) network construction and module analysis
STRING database (https://string-db.org) was used for protein–protein interaction (PPI) analysis in order to investigate the molecular mechanisms. STRING database (version 10.5) covers 9,643,763 proteins from 2031 organisms. While network analysis is a useful method for uncovering all kinds of protein–protein interactions networks. It can measure networks by nodes, edges, degrees and network structures, so it can help us to identify hub genes and key protein community. Then, PPI networks were constructed by Cytoscape software (http://www.cytoscape.org). Cytoscape is an open source software platform for visualizing molecular interaction networks, biological pathways and integrating these networks with annotations, gene expression profiles and other state data. A plugin named cytoHubba was introduced to screen hub genes of PPI network in Cytoscape. CytoHubba can provide 12 topological analysis methods including Betweenness, BottleNeck, Closeness, Clustering Coefficient, Degree, DMNC, EcCentricity, EPC, MCC, MNC, Radiality and Stress based on shortest paths [25].
Results
Normalization of gene methylation profiling and gene expression profiling
In this study, the five gene expression profiling dataset (Fig. 1) and four gene methylation profiling dataset (Fig. 2) were separately analyzed by R package affy and online GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) for screening DEGs and DMGs. All the gene expression profiling microarray chips were based on affymetrix GPL 570 platform. On affymetrix arrays, genes are represented by one or more probe sets, which are short oligonucleotides covering distinct sections of the gene synthesised in place through photolithography [26].
Fig. 1.

Data distribution of gene expression profiling data (a GSE26910; b GSE69223; c GSE55945; d GSE46602; e GSE30174)
Fig. 2.

Data distribution of methylation profiling data (a GSE52955; b GSE73549; c GSE84749; d GSE76938)
While on Illumina BeadChips arrays, 50 base pairs Infinium methylation probes synthesised by bisulfite conversion of unmethylated cytosines, which are randomly dispersed over the array [27]. This random allocation means that each probes was represented a random number of times on each array.
Microarrays and bead chips technologies depend on a complicated set of reagents and hardware, along with highly trained personnel, to produce accurate measurements. Both biological and non-biological factors will affect the results during the experiment when a series of complicated set of reagents and hardware varied [28]. So batch effects in different microarrays and bead chips should be focused after high-throughput experiments. Batch effects may occur at different laboratories, seasons and days. In order to eliminate batch effect problem, the surrogate variable analysis was conducted to reduce the batch effects depend on R package sva (Fig. 3). The sva package contains functions for removing batch effects and other unwanted variation in high-throughput experiment. The value distribution of methylation profiling data and the normalization of gene methylation profiling data were shown in Fig. 4.
Fig. 3.

Reject batch effects for gene expression profiling data
Fig. 4.

Normalization of gene methylation profiling (a1 GSE76938 gene methylation profiling data value distribution; a2 normalization of GSE76938 gene methylation profiling data; b1 GSE73549 gene methylation profiling data value distribution; b2 normalization of GSE73549 gene methylation profiling data; c1 GSE84749 gene methylation profiling data value distribution; c2 normalization of GSE84749 gene methylation profiling data; d1 GSE52955 gene methylation profiling data value distribution; d2 normalization of GSE52955 gene methylation profiling data)
Identification of DEGs and DMGs in PCa
After data normalizing and removing batch effects, total of 1331 DEGs were identified, using P < 0.05 and log2-fold change |log2FC| > 2.0 criteria, of which, 938 up-regulated genes and 393 down-regulated genes (Fig. 5). For DMGs, total of 3261 DMGs were identified according P < 0.05, log2-fold change |log2FC| > 2.0 and fwer < 0.5 criteria, of which, 2699 hypermethylation genes and 562 hypomethylation genes. The result has been shown as the volcano plot in Fig. 6.
Fig. 5.

Volcano plot of DEGs in gene expression datasets
Fig. 6.

Volcano plot of DMGs in gene methylation datasets (a GSE52955; b GSE73549; c GSE76938; d GSE84749)
Aberrantly methylated-differentially expressed genes in PCa
Then, totally 62 hypermethylation-low genes were obtained by overlapping 2699 hypermethylation genes and 393 down-regulated genes in Fig. 7. And 41 hypomethylation-high genes were obtained by overlapping 362 hypomethylation genes and 938 up-regulated genes in Fig. 7.
Fig. 7.

Venn diagram [29] for aberrantly methylated-differentially expressed genes by overlapping gene expression datasets and gene methylation datasets (a hypermethylation and down-regulated genes; b hypomethylation and up-regulated genes)
Integrating dataset for screening candidate tumor suppressor genes
Then, totally 9 candidate tumor suppressor genes were obtained by overlapping 2699 hypermethylation genes, 393 down-regulated genes and 1217 human tumor suppressor genes [30] in Fig. 8.
Fig. 8.
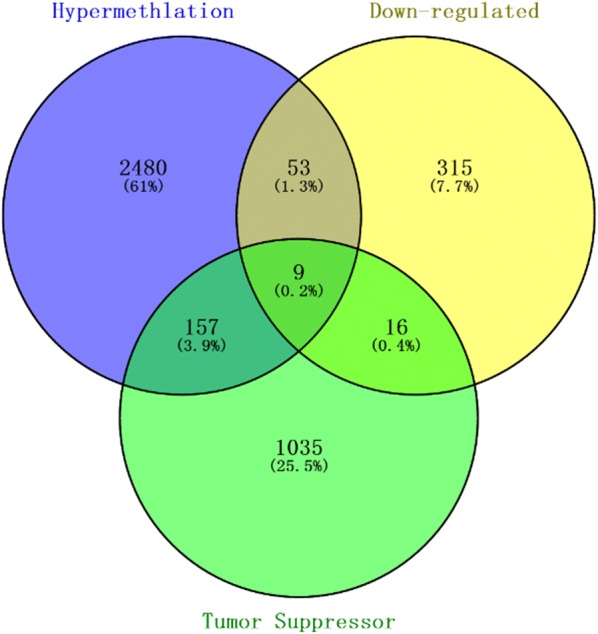
Venn diagram [29] for candidate tumor suppressor genes by overlapping hypermethylation genes, down-regulated genes and tumor suppressor genes
The heat map of nine candidate tumor suppressor genes (IKZF1, PPM1A, FBP1, SMCHD1, ALPL, CASP5, PYHIN1, DAPK1 and CASP8) was shown as Fig. 9. Although these tumor suppressor genes were hypermethylation-low genes, these genes were not significant in some samples for TCGA database.
Fig. 9.

Nine candidate tumor suppressor genes
Validation of candidate tumor suppressor genes in TCGA database
To further investigate the candidate tumor suppressor genes, TCGA database was used to validate these results. The outcome was shown as Fig. 10. The candidate tumor suppressor genes were separately significant in tumor group and normal group except IKZF1, CASP5 and CASP8, which significantly indicate that PPM1A, DAPK1 and FBP1 were hypermethylation in tumor samples, at the same time, PYH1N1, ALPL and SMCHD1 were hypermethylation in normal samples. And then, in order to further confirm these candidate tumor suppressor genes the MethPrimer [31] and cpgplot software were used to predict the CpG islands in Figs. 11 and 12. At the same time, pan-cancer analysis also shows that PPM1A, DAPK1, FBP1, PYHIN1, ALPL and SMCHD1 have significant amplification in PCa.
Fig. 10.

PCa-related candidate tumor suppressor genes expression in TCGA database
Fig. 11.

Pan-cancer analysis of PCa-related candidate tumor suppressor genes in normal samples
Fig. 12.

Pan-cancer analysis of PCa-related candidate tumor suppressor genes in tumor samples
Integrated Oncomine database and survival analysis for candidate tumor suppressor genes
To confirm the candidate tumor suppressor genes expression between tumor and normal tissues in multiple cancers, the Oncomine database was performed to analyze the different expression. Using P < 0.01 and |log2FC| > 1.5 criteria, a total of 455, 455, 444, 398, 341, 407, 342, 453 and 398 unique analyses for IKZF1, PPM1A, FBP1, SMCHD1, ALPL, CASP5, PYHIN1, DAPK1 and CASP8 were shown in Fig. 13. In 80 studies, FBP1 was ranked by the top 10% of gene rank indicating significant statistical differences, 6 of which revealed higher expression level in tumor than normal tissues. For higher expression of CASP5, there was only one dataset listed. Up-regulated KAPK1 was founded in cancers based on sixty-one significant analyses.
Fig. 13.

The landscape of the candidate tumor suppressor genes in human cancers (the more intense red indicates over-expression; the more intense blue indicates under-expressed gene.)
To further confirm the candidate tumor suppressor genes, Kaplan–Meier analysis was performed in Fig. 14. All the low expression values of tumor suppressor genes are all significantly associated with poor prognosis while a high expression of tumor suppressor genes are associated with good prognosis.
Fig. 14.

Kaplan-Meier survival analysis of candidate tumor suppressor genes associated with PCa [32]
The Gleason Score (GS) system for grading PCa is a standard evaluation method that has different stratification: GS ≤ 6, 3 + 4, 4 + 3, 8, 4 + 5, 5 + 4, 10, respectively to Gleason Grading Group 1, 2, 3, 4, and 5 [33]. Expression of candidate tumor suppressor genes according Gleason Score system is shown in Fig. 15. A low Gleason score (≤ 6) indicates good prognosis without risk of lymphatic metastasis whereas a high Gleason score (> 8) is associated with distal metastasis.
Fig. 15.

Expression of candidate tumor suppressor genes according Gleason Score system
The expression of IKZF1, PPM1A, FBP1, SMCHD1, ALPL, CASP5, PYHIN1, DAPK1 and CASP8 candidate tumor suppressor genes was significantly deregulated in PCa by Gleason Score (Table 2). The significant genes observed were PYHIN1, IKZF1, CASP8, DAPK1 and SMCHD1 expression in tumor samples. Furthermore, FBP1 was deregulated at Gleason Score 6–9.
Table 2.
Expression of candidate tumor suppressor genes in different PCa stage
| Candidate tumor suppressor gene | Gleason Score (GS) | ||||
|---|---|---|---|---|---|
| GS = 6 | GS = 7 | GS = 8 | GS = 9 | GS = 10 | |
| IKZF1 | No sig | No sig | No sig | No sig | Up |
| PPM1A | No sig | No sig | No sig | Down | Down |
| FBP1 | Up | Up | Up | Up | Down |
| SMCHD1 | No sig | Down | No sig | No sig | Up |
| ALPL | Down | Down | Down | Down | Down |
| CASP5 | Down | Down | No sig | Down | No sig |
| PYHIN1 | No sig | No sig | No sig | No sig | Up |
| DAPK1 | Up | Up | Up | Up | Up |
| CASP8 | No sig | Up | Up | Up | Up |
Up: up-regulated; down: down-regulated; no sig: no significant
Gene ontology enrichment analysis and KEGG pathway analysis
In order to uncover these genes affected by altered DNA methylation status, all hypermethylation-low expression genes and hypomethylation-high expression genes were uploaded to DAVID database to identify overrepresented BP categories and KEGG pathways. The top 10 significant GO enrichments of biological processes were illustrated in Table 3.
Table 3.
GO terms analysis of aberrantly methylated-differentially expressed genes in PCa
| Category | Term | P-value |
|---|---|---|
| Hypermethylation-low expression | Cytokine-mediated signaling pathway | 1.38E−04 |
| Benzene-containing compound metabolic process Kynurenine metabolic process |
3.36E−04 | |
| Positive regulation of cysteine-type endopeptidase activity involved in apoptotic process | 3.36E−04 | |
| Indolalkylamine catabolic process | 3.78E−04 | |
| Response to vitamin | 4.19E−04 | |
| Cellular response to mechanical stimulus | 4.19E−04 | |
| Tryptophan catabolic process | 4.72E−04 | |
| Tryptophan metabolic process | 5.11E−04 | |
| Positive regulation of transcription, DNA-templated | 5.11E−04 | |
| Hypomethylation-high expression | Regulation of transforming growth factor beta2 production | 1.43E−02 |
| Regulation of receptor recycling | 6.14E−04 | |
| Membrane raft assembly | 1.43E−02 | |
| Telencephalon cell migration | 1.43E−02 | |
| Regulation of endothelial cell chemotaxis to fibroblast growth factor | 1.63E−02 | |
| Positive regulation of vesicle fusion | 1.43E−02 | |
| Negative regulation of protein depolymerization | 8.40E−04 | |
| Positive regulation of receptor binding | 1.43E−02 | |
| Nodal signaling pathway | 1.83E−02 | |
| Positive regulation of vascular endothelial cell proliferation | 2.03E−02 |
KEGG pathways analysis were also significantly enriched in PCa genes in Table 4. And the relationships between these pathways were shown in Fig. 16. Furthermore, GSEA (Gene Set Enrichment Analysis) was conducted to verify KEGG enrichment analysis in Fig. 16. All of these pathways were significantly enriched in Pathway in cancer, Wnt signaling pathway, Pancreatic cancer and Melanoma (Fig. 17). And most of these genes were up-regulated, which show a credible knowledge of PCa.
Table 4.
KEGG pathway analysis of aberrantly methylated-differentially expressed genes in PCa
| Category | Pathway | P-value |
|---|---|---|
| Hypermethylation-low expression | NOD-like receptor signaling pathway | 7.37E−04 |
| Pentose phosphate pathway | 3.64E−03 | |
| Viral carcinogenesis | 3.76E−03 | |
| Tryptophan metabolism | 6.84E−03 | |
| Measles | 8.68E−03 | |
| Hepatitis B | 1.05E−02 | |
| Legionellosis | 1.26E−02 | |
| Cytosolic DNA-sensing pathway | 1.69E−02 | |
| Influenza A | 1.71E−02 | |
| Inflammatory bowel disease (IBD) | 1.74E−02 | |
| Hypomethylation-high expression | Pathways in cancer | 1.47E−04 |
| MAPK signaling pathway | 1.67E−04 | |
| Wnt signaling pathway | 2.01E−04 | |
| Colorectal cancer | 2.78E−04 | |
| Pancreatic cancer | 3.35E−04 | |
| Melanoma | 4.15E−04 | |
| AGE-RAGE signaling pathway in diabetic complications | 1.16E−03 | |
| Insulin resistance | 1.44E−03 | |
| FoxO signaling pathway | 2.55E−03 | |
| Hepatitis B | 3.32E−03 |
Fig. 16.

KEGG pathway analysis of aberrantly methylated-differentially expressed genes in PCa
Fig. 17.

Pathway enrichment identified by GSEA of aberrantly methylated-differentially expressed genes in PCa
PPI network construction and module analysis
All hypermethylation-low expressed genes and hypomethylation-high expressed genes were uploaded to the STRING database for constructing PPI network. Moreover, all these nodes and edges were analyzed using Cytoscape plug-ins called cytoHubba. For hypermethylation-low expressed genes, PPI network was shown in Fig. 18a and top modules were displayed in Fig. 18b. Hub genes were CASP1, STAT4 and IRF7.
Fig. 18.

PPI network and hypermethylation-low expression genes (a PPI network; b top module; c hub genes)
For hypomethylation-high expressed genes, PPI network was shown in Fig. 19a and top modules were displayed in Fig. 19b. Hub genes were FGFR1, FGF13 and CCND1. Biological process of significant hub genes were demonstrated in Table 5.
Fig. 19.

PPI network and hypomethylation-high expression genes (a PPI network; b top module; c hub genes)
Table 5.
GO terms analysis of hub genes in PCa
| Category | Term | P-value |
|---|---|---|
| Hypermethylation-low expression | Negative regulation of myeloid cell apoptotic process | 1.05E−03 |
| Positive regulation of tumor necrosis factor-mediated signaling pathway | 1.20E−03 | |
| Regulation of macrophage apoptotic process | 1.05E−03 | |
| Regulation of myd88-independent toll-like receptor signaling pathway | 1.35E−03 | |
| Protein autoprocessing | 1.50E−03 | |
| Establishment of viral latency | 1.50E−03 | |
| Interleukin-23-mediated signaling pathway | 1.50E−03 | |
| Cellular response to interferon-gamma | 1.01E−04 | |
| Interleukin-21-mediated signaling pathway | 1.35E−03 | |
| Regulation of myeloid leukocyte differentiation | 1.65E−03 | |
| Hypomethylation-high expression | Telencephalon cell migration | 1.05E−03 |
| Regulation of endothelial cell chemotaxis to fibroblast growth factor | 1.20E−03 | |
| Positive regulation of vascular endothelial cell proliferation | 1.50E−03 | |
| Response to UV-A | 1.05E−03 | |
| Cerebral cortex cell migration | 1.05E−03 | |
| Regulation of collateral sprouting | 1.20E−03 | |
| Neuron migration | 1.23E−05 | |
| Positive regulation of protein serine/threonine kinase activity | 1.05E−04 | |
| Mitotic G1 DNA damage checkpoint | 1.65E−03 | |
| Regulation of cardiac muscle cell action potential involved in regulation of contraction | 1.35E−03 |
Interlink between candidate genes with prostate specific antigen (PSA)
Prostate specific antigen (PAS) as a biomarker was discovered for diagnosing being and malignant prostate disease in 1960 [34]. Later, cumulative research has uncovered the antigens in the prostate and semen [35–43]. Prostate specific antigen is a protein expressed by multiple non-prostatic tissues in men and women. To obtain the relationship between candidate genes with PSA, a combined bioinformatics tools were utilized. Firstly, the Universal Protein Resource (UniProt) was used to get the PAS amino acid sequence (Additional file 1). Secondly, the PAS amino acid sequence and candidate genes were uploaded to BlastKOALA (https://www.kegg.jp/blastkoala/) for genome annotation. Finally, all the annotated genes were uploaded to KEGG PATHWAY Database (https://www.kegg.jp/kegg/pathway.html) for mapping pathway in Fig. 20. CASP8, CCND1, DAPK1 and PSA are involved in pathways in cancer. CASP8 (Caspase-8) paly essential role in apoptosis [44]. Apoptosis is regulated by CASP8 [45]. Some clinical study has confirmed that overexpression of CCND1 (Cyclin D1) is a common biomarker for treatment [46] and being ignored to cisplatin resistance in prostate cancer [47, 48]. Death associated protein kinase 1 (DAPK1) plays a critical role in apoptosis. The methylation of DAPK1 has interlink with cancer.
Fig. 20.

Pathways for candidate genes and PSA (red: candidate genes; yellow: PSA)
Discussion
Since microarray and high-throughput sequencing can provide expression levels of thousands of genes in human genome simultaneously, it has been widely used to predict the potential therapeutic targets for PCa. In this study, we conducted a combined analysis of two types of microarray chips (DNA methylation and gene expression profile datasets) of PCa for uncovering the epigenetic and genetic mechanisms in PCa using bioinformatics analysis tools. By overlapping DEGs, DMGs and TSGs (tumor suppressor genes), we identified some candidate tumor suppressor genes that can provide new ideas for diagnosis, therapy and biomarker studies in PCa. In order to better understand the molecular mechanism of candidate tumor suppressor genes and hub genes, GO, KEGG pathway and PPI analysis were further performed.
The results demonstrated that these hypermethylation-low expressed genes were enriched in cytokine-mediated signaling pathway, benzene-containing compound metabolic process, kynurenine metabolic process, positive regulation of cysteine-type endopeptidase activity involved in apoptotic process, indolalkylamine catabolic process, response to vitamin, cellular response to mechanical stimulus, tryptophan catabolic process, tryptophan metabolic process and positive regulation of transcription, DNA-templated.
This indicated that cytokines is associated with prostate cancer. Some evidence shows the role of cytokines in prostate carcinogenesis. Cytokines are widely recognized as crucial factors in cancer development. In the progress of cancer development, cytokines were released by other immune cells. And cytokines enhance therapeutic resistances through EMT activation in tumor microenvironment [49]. Kynurenine has relationship with immune escape of tumor cells [50]. Srekumar et al. found that Kynurenine was associated with prostate cancer (PCa) progression [51]. More and more experimental researches have indicated that vitamin D has the effects of anti-prostate tumor [52, 53]. Like vitamin D, vitamin C was also natural product that has the property of antioxidant [54]. So vitamin C also plays an important role for antitumor due to characterize of antioxidant [55].
Furthermore, the enriched KEGG pathways of hypermethylation-low expressed genes have significant enrichment in NOD-like receptor signaling pathway, Pentose phosphate pathway, Viral carcinogenesis, Tryptophan metabolism, Measles_Homo sapiens, Hepatitis B, Legionellosis, Cytosolic DNA-sensing pathway and Influenza A and Inflammatory bowel disease (IBD). Cumulative evidence revealed that the pentose phosphate pathway (PPP) is a metabolic pathway, parallel to glycolysis that generates NADPH, nucleotides and nucleic acids [56]. Heritable factors and environmental factors may cause prostate cancer together. Recent research focus on the role of viral infections in prostate cancer [57]. Cytosolic DNA sensing is associated to the secretion of cytokines [58]. Cytosolic DNA sensing mediates robust antimicrobial. Cancer cells often acquire genetic or epigenetic alterations [59, 60]. Cytosolic DNA sensing is important for tumor control. In cytosolic DNA sensing, transcriptional and post-translational signaling modules enable the release of immunomodulatory cytokines [58].
After constructing PPI network for hypomethylation-high expression genes, a novel Cytoscape plugin called cytoHubba was introduced for ranking nodes in a network by the network. CytoHubba provides 12 topological analysis methods including Betweenness, BottleNeck Closeness, Clustering Coefficient, Degree, DMNC, EcCentricity, EPC, MCC, MNC, Radiality and Stress [23]. The hub genes appeared to be FGFR1, FGF13 AND CCND1.
Type 1 fibroblast growth factor receptor binding (FGFR1) was an important factor in tumor initiation and progression in prostate cancer. The activation of FGFR1 regulate the EMT in cancer progression [61]. Amplification of FGFR1 has been well studied [62, 63]. And epithelial mesenchymal-transition (EMT) may play a crucial role during in tumor metastasis and progression [64]. Recent studies have indicated that inducing EMT can affect tumor microenvironment [65].
Fibroblast growth factor 13 (FGF13) is overexpressed in several types of cancer [66, 67]. FGF13 inhibit ribosomal RNA synthesis, and may be an interplay with p53 involving a nucleolus-dependent mechanism [68]. It is possible that FGF13 may similarly signal to p53 by the same underlying molecular mechanism. FGF13 expression would activate p53, whereas the up-regulation of miR-504 would antagonize such an effect. It will be important to explore this issue directly in future studies [69]. Although the augmented FGF13 expression in tumors is unlikely to be a cancer driver, it is not merely a passenger, because it allows the cancer cells to cope with undesirable side effects of oncogene activation [68]. So FGF 13 may be reviewed as a cancer switch [70]. Cycline D1 (CCND1) plays a significant role in cell cycle. The over-expression of CCND1 in human tumors has been indicated as proto-oncogenes [71–77].
For tumor suppressor genes, IKAROS family zinc finger 1 (IKZF1) is a key regulator factor that enhanced immune infiltrate recruitment and tumor sensitivity in several tumors. Overexpression of IKZF1 can activate autoimmune susceptibility via infiltrating NKG2D+, CD8+ T cells [78]. Protein phosphatase, Mg2+/Mn2+ dependent 1A (PPM1A) is a phosphatase that has been the function of dephosphorylating TGF-β–activated P-Smad2/3, p38 and regulating several tumor-related signaling pathways [79, 80]. As a phosphatase PPM1A plays a significant role in cell cycle progression, cell proliferation, and apoptosis [81–83]. Previous study indicated that metastatic prostate cancer had lower PPM1A expression compared with primary tumor [84]. Overexpression of PPM1A has been reported to activate the expression of tumor suppressor gene TP53 [82] and increased PPM1A expression inhibited the activity of NF-kB in promoting prostate cancer invasion and metastasis [85]. Fructose-bisphosphatase 1 (FBP1) palys negative regulation roles in glycolysis and affects some process of survival, proliferation and metastasis in tumor cells [86, 87]. Some studies also showed that overexpressed FBP1 in prostate cancers can be as a tumor biomarker [88, 89]. Structural maintenance of chromosomes flexible hinge domain containing 1 (SMCHD1) is a chromatin protein associated with epigenetic modifier [90]. Alkaline phosphatase (ALPL) is a non-specific hydrolase and plays crucial role in regulating phosphate metabolism involved in cell cycle, growth, apoptosis and signal transduction pathways [91]. The abnormal expression of ALP can be a biomarker for prostate cancer [92]. The expression of caspase-5 (CACP5) is very low in many normal tissues, while its expression plays an important role in cell apoptosis [93]. Pyrin and HIN domain family member 1 (PYHIN1) has the function of tumor suppression [94]. Death associated protein kinase 1 (DAPK1) is a kinase regulated neuronal apoptosis by calcium-calmodulin. So far, a large number of studies have demonstrated that DAPK1 acts as a positive mediator interact with several proteins, such as DAPK1-p53 signaling, DAPK1-tau signaling, and DAPK1-DANGER signaling [95–98]. Caspase-8 (CASP8) acts as an environment sensor in the control of cell death. CASP8 induced a wide range of biological process, such as the control of apoptosis and necroptosis [99].
All the genes and pathways in this investigation are all based on bioinformatics methods. So no clinical samples were used to validate the data. But a shot literature review was introduced to confirm out findings.
Conclusions
In this study, a joint bioinformatics analysis method was used for indicating candidate tumor suppressor genes and pathways in PCa by combined gene methylation microarrays and gene expression microarray, this may provide a set of useful targets for future investigation into the molecular mechanisms and biomarkers. Candidate tumor suppressor genes were IKZF1, PPM1A, FBP1, SMCHD1, ALPL, CASP5, PYHIN1, DAPK1 and CASP8. Hub genes were FGFR1, FGF13 and CCND1 that may contribute to the finding of molecular mechanisms underlying the initiation and development of PCa. Meanwhile, there were some limitations should be declared in this work. The clinical gene methylation profiling and gene expression profiling datasets were not analyzed in this study. Besides, the hub genes on gene expression were only validated in TCGA database.
Additional file
Additional file 1. Supplementary S1-PAS protein sequence.
Authors’ contributions
YT and SD conceived the study; YS collected the data; YT wrote the R code; YT wrote the paper. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The authors declare that the data supporting the findings of this study are available within the article.
Consents for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This work is supported by Basic science and frontier project of Chongqing Municipal Science and Technology Commission (cstc2016jcyjA0582).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- PCa
prostate cancer
- GEO
Gene Expression Omnibus
- DEG
differentially expressed gene
- DMG
differentially methylated gene
- TSG
tumor suppressor gene
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- GO
gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- PPI
protein–protein interaction
- STRING
Search Tool for the Retrieval of Interacting Genes
- TCGA
The Cancer Genome Atlas
- BP
biological process
- BH
Benjamini and Hochberg
- log2FC
log2-fold change
Contributor Information
Yanqiu Tong, Email: yqtong@126.com.
Yang Song, Email: songyangcqmu@126.com.
Shixiong Deng, Email: dengshixiong1963@163.com.
References
- 1.Jones PA. DNA methylation and cancer. Cancer Res. 1986;46:461–466. [PubMed] [Google Scholar]
- 2.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 3.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 4.Rakyan VK, Down TA, Balding DJ, et al. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12(8):529. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lo KW, Huang DP. Genetic and epigenetic changes in nasopharyngeal carcinoma. Semin Cancer Biol. 2002;12(6):451–462. doi: 10.1016/s1044579x02000883. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. GLOBOCAN 2012: estimated cancer incidence, mortality, and prevalence worldwide in 2012; 2012. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed 1 Apr 2018.
- 7.Wang Qian, Wang Gang, Liu Chaoxu, He Xianli. Prognostic value of CpG island methylator phenotype among hepatocellular carcinoma patients: a systematic review and meta-analysis. Int J Surg. 2018;54:92–99. doi: 10.1016/j.ijsu.2018.04.033. [DOI] [PubMed] [Google Scholar]
- 8.Yang X, Han H, De Carvalho D, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):585. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aran V, Victorino AP, Thuler LC, Ferreira CG. Colorectal cancer: epidemiology, disease mechanisms and interventions to reduce onset and mortality. Clin Colorectal Cancer. 2016;15(3):195–203. doi: 10.1016/j.clcc.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 11.Decarvalho D, Sharma S, You JS, et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell. 2012;21(5):655. doi: 10.1016/j.ccr.2012.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4(2):143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 13.Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol. 2008;5:588–599. doi: 10.1038/ncponc1187. [DOI] [PubMed] [Google Scholar]
- 14.Nannini M, Pantaleo MA, Maleddu A, et al. Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives. Cancer Treat Rev. 2009;35:201–209. doi: 10.1016/j.ctrv.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 15.Bustin SA, Dorudi S. Gene expression profiling for molecular staging and prognosis prediction in colorectal cancer. Exp Rev Mol Diagn. 2004;4:599–607. doi: 10.1586/14737159.4.5.599. [DOI] [PubMed] [Google Scholar]
- 16.Carmona FJ, Azuara D, Berenguer-Llergo A, Fernandez AF, Biondo S, de Oca J, Rodriguez-Moranta F, Salazar R, Villanueva A, Fraga MF, et al. DNA methylation biomarkers for noninvasive diagnosis of colorectal cancer. Cancer Prev Res. 2013;6(7):656–665. doi: 10.1158/1940-6207.CAPR-12-0501. [DOI] [PubMed] [Google Scholar]
- 17.Lascorz J, Hemminki K, Forsti A. Systematic enrichment analysis of gene expression profiling studies identifies consensus pathways implicated in colorectal cancer development. J Carcinog. 2011;10:7. doi: 10.4103/1477-3163.78268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwaya T, Yokobori T, Nishida N, et al. Downregulation of miR-144 is associated with colorectal cancer progression via activation of mTOR signaling pathway. Carcinogenesis. 2012;33(12):2391–2397. doi: 10.1093/carcin/bgs288. [DOI] [PubMed] [Google Scholar]
- 19.Irizarry RA. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 20.Benjamini Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
- 21.Glynn Dennis J, Sherman BT, Hosack DA, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):3. [PubMed] [Google Scholar]
- 22.Gene Ontology C. The gene ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:322–326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chin CH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilder SP, Kaisaki PJ, Argoud K, et al. Comparative analysis of methods for gene transcription profiling data derived from different microarray technologies in rat and mouse models of diabetes. BMC Genomics. 2009;10(1):63. doi: 10.1186/1471-2164-10-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michael KL, Taylor LC, Schultz SL, Walt DR. Randomly ordered addressable high-density optical sensor arrays. Anal Chem. 1998;70(7):1242–1248. doi: 10.1021/ac971343r. [DOI] [PubMed] [Google Scholar]
- 28.Leek JT, Scharpf RB, Bravo HC, et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010;11(10):733–739. doi: 10.1038/nrg2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bardou P, Mariette J, Escudié F, et al. jvenn: an interactive Venn diagram viewer. BMC Bioinform. 2014;15(1):1–7. doi: 10.1186/1471-2105-15-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Min Z, Kim P, Mitra R, et al. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016;44(Database issue):D1023–D1031. doi: 10.1093/nar/gkv1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 32.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Rodriguez IP, Chakravarthi BVSK, Varambally S. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Epstein JI, Zelefsky MJ, Sjoberg DD, Nelson JB, Egevad L, Magi-Galluzzi C. A contemporary prostate cancer grading system: a validated alternative to the Gleason score. Eur Urol. 2015;69:428–435. doi: 10.1016/j.eururo.2015.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flocks RH, Urich VC, Patel CA, et al. Studies on the antigenic properties of prostatic tissue. J Urol. 1960;84(1):134. doi: 10.1016/S0022-5347(17)65503-4. [DOI] [PubMed] [Google Scholar]
- 35.Pérez-Ibave DC, Burciaga-Flores CH, Elizondo-Riojas MÁ. Prostate-specific antigen (PSA) as a possible biomarker in non-prostatic cancer: a review. Cancer Epidemiol. 2018;54:48–55. doi: 10.1016/j.canep.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Flocks RH, et al. Studies on spermagglutinating antibodies in antihuman prostate sera. J Urol. 1962;87:475–478. doi: 10.1016/S0022-5347(17)64982-6. [DOI] [PubMed] [Google Scholar]
- 37.Catalona WJ. History of the discovery and clinical translation of prostate-specific antigen. Asian J Urol. 2014;1(1):12–14. doi: 10.1016/j.ajur.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tokudome S, Ando R, Koda Y. Discoveries and application of prostate-specific antigen, and some proposals to optimize prostate cancer screening. Cancer Manag Res. 2018;8:45–47. doi: 10.2147/CMAR.S98326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ablin RJ, et al. Precipitating antigens of the normal human prostate. J Reprod Fertil. 1970;22(3):573–574. doi: 10.1530/jrf.0.0220573. [DOI] [PubMed] [Google Scholar]
- 40.Li TS, Beling CG. Isolation and characterization of two specific antigens of human seminal plasma. Fertil Steril. 1973;24(2):134–144. doi: 10.1016/s0015-0282(16)39496-1. [DOI] [PubMed] [Google Scholar]
- 41.Sokoll L J, Chan D W. Prostate-specific antigen. Its discovery and biochemical characteristics. Urologic Clinics of North America. 1997; 24(2):253-259. [DOI] [PubMed]
- 42.Sensabaugh GF. Isolation and characterization of a semen-specific protein from human seminal plasma: a potential new marker for semen identification. J Forensic Sci. 1978;23(1):106–115. [PubMed] [Google Scholar]
- 43.Sensabaugh GF, Blanke ET. Seminal plasma protein p30: simplified purification and evidence for identity with prostate specific antigen. J Urol. 1990;144(6):1523–1526. doi: 10.1016/s0022-5347(17)39790-2. [DOI] [PubMed] [Google Scholar]
- 44.Fu G, Tang J, Wang M, et al. CASP8 promoter polymorphism, mRNA expression and risk of prostate cancer among Chinese men. J Biomed Res. 2011;25(2):128–134. doi: 10.1016/S1674-8301(11)60016-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 46.Noel EE, Yeste-Velasco M, Mao X, et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am J Pathol. 2010;176(6):69. doi: 10.2353/ajpath.2010.090780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin? Cell Cycle. 2008;7:1353–1359. doi: 10.4161/cc.7.10.5930. [DOI] [PubMed] [Google Scholar]
- 48.Houldsworth J, Korkola JE, Bosl GJ, et al. Biology and genetics of adult male germ cell tumors. J Clin Oncol. 2006;24(35):5512–5518. doi: 10.1200/JCO.2006.08.4285. [DOI] [PubMed] [Google Scholar]
- 49.Tan C, Hu W, He Y, et al. Cytokine-mediated therapeutic resistance in breast cancer. Cytokine. 2018;108:151–159. doi: 10.1016/j.cyto.2018.03.020. [DOI] [PubMed] [Google Scholar]
- 50.Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20(10):469–473. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 51.Sreekumar A, Poisson LM, Rajendiran TM, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457(7231):910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 52.Sherman MH, Yu RT, Engle DD, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159(1):80. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bristol ML, Di X, Beckman MJ, et al. Dual functions of autophagy in the response of breast tumor cells to radiation: cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D3. Autophagy. 2012;8(5):739. doi: 10.4161/auto.19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai-Ning Z, Ya L, Xiao M, et al. Insight into the roles of vitamins C and D against cancer: myth or truth? Cancer Lett. 2018;431:161–170. doi: 10.1016/j.canlet.2018.05.039. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Zhang JJ, Xu DP, et al. Bioactivities and health benefits of wild fruits. Int J Mol Sci. 2016;17(8):1258. doi: 10.3390/ijms17081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Whitburn J, Rao S, Tabata S, et al. The prostate cancer-bone environment causes upregulation of the pentose phosphate pathway. Eur Urol Suppl. 2017;16(3):e152–e153. [Google Scholar]
- 57.Abidi SH, Bilwani F, Ghias K, et al. Viral etiology of prostate cancer: genetic alterations and immune response. A literature review. Int J Surg. 2018;52:136. doi: 10.1016/j.ijsu.2018.02.050. [DOI] [PubMed] [Google Scholar]
- 58.Claire VB, Sandra D, Formenti SC, et al. Cytosolic dna sensing in organismal tumor control. Cancer Cell. 2018;34(3):361–378. doi: 10.1016/j.ccell.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 59.Ying KC, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol. 2016;14(6):360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marinho FV, Benmerzoug S, Oliveira SC, et al. The emerging roles of STING in bacterial infections. Trends Microbiol. 2017;25:906–918. doi: 10.1016/j.tim.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li Q, Ingram L, Kim S, et al. Paracrine fibroblast growth factor initiates oncogenic synergy with epithelial FGFR/Src transformation in prostate tumor progression 12. Neoplasia. 2018;20(3):233–243. doi: 10.1016/j.neo.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bova GS, Kallio HML, Annala M, et al. Integrated clinical, whole-genome, and transcriptome analysis of multisampled lethal metastatic prostate cancer. Cold Spring Harb Mol Case Stud. 2016;2(3):a000752. doi: 10.1101/mcs.a000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.François B, Catherine N, Gwenaëlle G, et al. FGFR1 and WT1 are markers of human prostate cancer progression. BMC Cancer. 2006;6(1):272. doi: 10.1186/1471-2407-6-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu M, Polyak K. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 66.Hoek K, Rimm DL, Williams KR, et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004;64(15):5270–5282. doi: 10.1158/0008-5472.CAN-04-0731. [DOI] [PubMed] [Google Scholar]
- 67.Missiaglia E, Dalai I, Barbi S, et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28(2):245–255. doi: 10.1200/JCO.2008.21.5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bublik DR, Bursać S, Sheffer M, et al. Regulatory module involving FGF13, miR-504, and p53 regulates ribosomal biogenesis and supports cancer cell survival. Proc Natl Acad Sci USA. 2016;114(4):201614876. doi: 10.1073/pnas.1614876114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Manfredi JJ. Tumor suppression by p53 involves inhibiting an enabler, FGF13. Proc Natl Acad Sci USA. 2017;114(4):632. doi: 10.1073/pnas.1619815114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuuring E. The involvement of the chromosome 11q13 region in human malignancies: cyclin D1, and EMS1, are two new candidate oncogenes-a review. Gene. 1995;159(1):83–96. doi: 10.1016/0378-1119(94)00562-7. [DOI] [PubMed] [Google Scholar]
- 72.Ormandy CJ, Musgrove EA, Hui R, et al. Cyclin D1, EMS1 and 11q13 amplification in breast cancer. Breast Cancer Res Treat. 2003;78(3):323–335. doi: 10.1023/a:1023033708204. [DOI] [PubMed] [Google Scholar]
- 73.Bertoni F, Rinaldi A, Zucca E, et al. Update on the molecular biology of mantle cell lymphoma. Hematol Oncol. 2006;24(1):22–27. doi: 10.1002/hon.767. [DOI] [PubMed] [Google Scholar]
- 74.Jin M, Inoue S, Umemura T, et al. Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II. Lung Cancer. 2001;34(2):207–218. doi: 10.1016/s0169-5002(01)00225-2. [DOI] [PubMed] [Google Scholar]
- 75.Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009;220(2):292–296. doi: 10.1002/jcp.21791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Musgrove EA. Cyclins: roles in mitogenic signaling and oncogenic transformation. Growth Fact. 2006;24(1):13. doi: 10.1080/08977190500361812. [DOI] [PubMed] [Google Scholar]
- 77.Shamma A, Doki Y, Shiozaki H, et al. Cyclin D1 overexpression in esophageal dysplasia: a possible biomarker for carcinogenesis of esophageal squamous cell carcinoma. Int J Oncol. 2000;16(2):261. doi: 10.3892/ijo.16.2.261. [DOI] [PubMed] [Google Scholar]
- 78.Chen James C, Perez-Lorenzo Rolando, Saenger Yvonne M, Drake Charles G, Christiano Angela M. IKZF1 enhances immune infiltrate recruitment in solid tumors and susceptibility to immunotherapy. Cell Systems. 2018;7(1):92–103. doi: 10.1016/j.cels.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 79.Fan Jie, Yang Michelle X, Ouyang Qi, Deliang Fu, Zude Xu, Liu Xiuping, Mino-Kenudson Mari, Geng Jiang, Tang Feng. Phosphatase PPM1A is a novel prognostic marker in pancreatic ductal adenocarcinoma. Hum Pathol. 2016;55:151–158. doi: 10.1016/j.humpath.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 80.Lin X, Duan X, Liang YY, Su Y, Wrighton KH, Long J, Hu M, Davis CM, Wang J, Brunicardi FC, Shi Y, Chen YG, Meng A, Feng XH. PPM1A functions as a Smad phosphatase to terminate TGFbeta signaling. Cell. 2006;125(5):915–928. doi: 10.1016/j.cell.2006.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng A, Kaldis P, Solomon MJ. Dephosphorylation of human cyclin-dependent kinases by protein phosphatase type 2C alpha and beta 2 isoforms. J Biol Chem. 2000;275:34744–34749. doi: 10.1074/jbc.M006210200. [DOI] [PubMed] [Google Scholar]
- 82.Ofek P, Ben-Meir D, Kariv-Inbal Z, et al. Cell cycle regulation and p53 activation by protein phosphatase 2C alpha. J Biol Chem. 2003;278:14299–14305. doi: 10.1074/jbc.M211699200. [DOI] [PubMed] [Google Scholar]
- 83.Shohat M, Ben-Meir D, Lavi S. Protein phosphatase magnesium dependent 1 A (PPM1A) plays a role in the differentiation and survival processes of nerve cells. PLoS ONE. 2012;7:e32438. doi: 10.1371/journal.pone.0032438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lu X, An H, Jin R, et al. PPM1A is a RelA phosphatase with tumor suppressor-like activity. Oncogene. 2014;33(22):2918. doi: 10.1038/onc.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu X, An H, Jin R, et al. PPM1A is a RelA phosphatase with tumor suppressor-like activity. Oncogene. 2014;33:2918–2927. doi: 10.1038/onc.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dong C, Yuan T, Wu Y, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer Canc. Cell. 2013;23:316–331. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 88.Lapointe J, Li C, Higgins JP, Van De Rijn M, Bair E, Montgomery K, Ferrari M, Egevad L, Rayford W, Bergerheim U, Ekman P, DeMarzo AM, Tibshirani R, Botstein D, Brown PO, Brooks JD, Pollack JR. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci. 2014;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu P, Ramachandran S, Seyed MA, Scharer CD, Laycock N, Dalton WB, Williams H, Karanam S, Datta MW, Jaye DL, Moreno CS. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;46:4011–4019. doi: 10.1158/0008-5472.CAN-05-3055. [DOI] [PubMed] [Google Scholar]
- 90.Jansz Natasha, Chen Kelan, Murphy James M, Blewitt Marnie E. The epigenetic regulator SMCHD1 in development and disease. Trends Genet. 2017;33(4):233–243. doi: 10.1016/j.tig.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 91.Jin LY, Dong YM, Wu XM, Cao GX, Wang GL. Versatile and amplified biosensing through enzymatic cascade reaction by coupling alkaline phosphatase in situ generation of photoresponsive nanozyme. Anal Chem. 2015;87:10429–10436. doi: 10.1021/acs.analchem.5b02728. [DOI] [PubMed] [Google Scholar]
- 92.Ramaswamy G, Rao VR, Krishnamoorthy L, Ramesh G, Gomathy R, Renukadevi D. Serum levels of bone alkaline phosphatase in breast and prostate cancers with bone metastasis. Indian J Clin Biochem. 2000;15:110. doi: 10.1007/BF02883737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lin XY, Choi MS, Porter AG. Expression analysis of the human caspase-1 subfamily reveals specific regulation of the CASP5 gene by lipopolysaccharide and interferon-gamma. J Biol Chem. 2000;275:39920–39926. doi: 10.1074/jbc.M007255200. [DOI] [PubMed] [Google Scholar]
- 94.Connolly DJ, Bowie AG. The emerging role of human PYHIN proteins in innate immunity: Implications for health and disease. Biochem Pharmacol. 2014;92(3):405–414. doi: 10.1016/j.bcp.2014.08.031. [DOI] [PubMed] [Google Scholar]
- 95.Wang X, Pei L, Yan H, Wang Z, Wei N, Wang S, et al. Intervention of death-associated protein kinase 1-p53 interaction exerts the therapeutic effects against stroke. Stroke. 2014;45(10):3089–3091. doi: 10.1161/STROKEAHA.114.006348. [DOI] [PubMed] [Google Scholar]
- 96.Pei L, Shang Y, Jin H, Wang S, Wei N, Yan H, et al. DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J Neurosci. 2014;34(19):6546–6556. doi: 10.1523/JNEUROSCI.5119-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pei L, Wang S, Jin H, Bi L, Wei N, Yan H, et al. A novel mechanism of spine damages in stroke via DAPK1 and Tau Cereb. Cortex. 2015;25(11):4559–4571. doi: 10.1093/cercor/bhv096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kang BN, Ahmad AS, Saleem S, et al. Death-associated protein kinase-mediated cell death modulated by interaction with DANGER. J Neurosci. 2010;30(1):93–98. doi: 10.1523/JNEUROSCI.3974-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Keller Nadine, Ozmadenci Duygu, Ichim Gabriel, Stupack Dwayne. Caspase-8 function, and phosphorylation, in cell migration. Semin Cell Dev Biol. 2018;82:105–117. doi: 10.1016/j.semcdb.2018.01.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary S1-PAS protein sequence.
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article.