

Abstract
Background
Diabetic nephropathy is a predominant cause of renal failure, which is an important chronic complication of diabetes. Pyridoxamine (PM) has been reported to protect renal tubular epithelial cells against oxidative damage and delay or inhibit the development and generation of glucose-induced renal insufficiency at the early stage of disease. In this study, we attempted to explore the protection mechanism of PM on human proximal tubular epithelial cells (HK-2 cells) induced by high glucose.
Material/Methods
HK-2 cells were cultivated by high glucose medium in the absence or presence of PM. Cell Counting Kit-8 was used to investigate the most appropriate drug concentration of PM by detecting the cell viability of HK-2 cells. The expression of autophagy-related protein Beclin-1, LC-3II, and p62 was measured by western blot analysis, reverse transcription-quantitative polymerase chain reaction (RT-qPCR), and immunofluorescence. The expression and localization of Beclin-1 and p62 were also detected via immunofluorescence. The intracellular reactive oxygen species generation was detected using the reactive oxygen species assay kit. The effects of PM on antioxidant defenses were evaluated with glutathione peroxidase (GPx), manganese superoxide dismutase (MnSOD) activity, and glutathione/glutathione disulfide (GSH/GSSG) ratio.
Results
High glucose levels were able to upregulate the expression of oxidative stress associated protein and inhibit autophagy-associated changes verified by western blotting, RT-qPCR and immunofluorescence. Administration of PM reversed the high glucose-induced low-expressed Beclin-1 and LC-3II, and overexpressed p62 and intracellular reactive oxygen species levels. Furthermore, non-enzymatic antioxidant defenses and enzymatic antioxidant defenses were turned on by the application of PM.
Conclusions
Treatment with PM could reverse high glucose-induced inhibition of autophagy and oxidative stress.
MeSH Keywords: Autophagy, Glucose, Oxidative Stress, Pyridoxamine
Background
Diabetic nephropathy is a frequently microangiopathy complication of diabetes, which affects the diabetic population. Autophagic dysfunction, as well as the disorders of oxidative stress and antioxidant defense system, have been revealed as important pathogenic mechanisms [1,2]. Under physiological conditions, glomerular podocytes show a high basal level of autophagy, while tubular cells have detectable but low levels of autophagy [3]. Autophagy is a fundamental physiological process that maintains structural and functional integrity of the cell. It can be induced in stress reactions and serves as an adaptive response for cell survival. High glucose (HG) increases intracellular reactive oxygen species (ROS) levels to promote oxidative stress in renal cells [4]. A previous study demonstrated that the increased level of oxidative stress observed in renal tubular epithelial cells was associated with HG environments, which might result in renal dysfunction [5]. Under stress conditions, tubular epithelial cells utilize autophagic machinery as a survival mechanism, which predispose tubular cells to eliminate protein aggregates and damaged organelles [6]. The process of autophagy activation contributes to delay the progression of diabetic renal injury. Several studies have suggested that autophagy is an evolutionarily conserved mechanism in which lysosomes mediate bulk degradation of superfluous or aberrant cytoplasmic components as an adaptive response, and that it might be of importance to the pathogenesis of diabetic nephropathy [7]. Therefore, the HG-induced oxidative stress and autophagic dysfunction in renal tubular epithelial cells are significant pathogenic processes that might contribute to the genesis and progression of diabetic renal injury.
PM is one of the vitamin B6 analogues that has been identified as an anti-glycating agent [8]. It has been suggested that there are potential mechanisms of benefit for in reducing hyperglycemias and alleviating the symptoms of diabetic complications [9,10]. In models of diabetic atherosclerosis, delayed intervention with pyridoxamine (PM) decreased plaque areas in diabetic ApoE (−/−) mice compared with untreated mice. Through inhibiting of advanced glycation end (AGE) product accumulation, PM significantly attenuated the progression of established diabetes-associated atherosclerosis [11]. Moreover, AGEs immunoreactivity was significantly diminished by PM treatment in diabetic rats. The administration of PM had a significant beneficial effect on the upregulation of the antioxidative marker heme oxygenase-1 and the induction of glial fibrillary acidic protein production in Müller glia [12]. Previous data have shown that PM retards the development of diabetic nephropathy in animal models of diabetes [13]. In a clinical trial, change from baseline in serum creatinine and urinary transforming growth factor (TGF)-β1 excretion were reduced by PM after a 6-month treatment in 212 patients with diabetes as well as nephropathy [14]. In animal studies, in addition to levels of serum glucose, creatinine, interleukin (IL)-1α, IL-6, C-reactive protein (CRP), and urine microalbumin, PM induced significant decrease in kidney malondialdehyde content and the gene expression of tumor necrosis factor (TNF)-α and TGF-β1. Certain improvements of histopathology of kidney tissues were also made by PM [15]. However, whether the protective effect of PM was mediated via autophagy or other possible mechanisms still warrant further exploration. In the present study, the HG-induced human proximal tubular epithelial cells (HK-2) cells were chosen to examine and assess the autophagic function and oxidative stress level. The novel investigation of PM might provide updated evidence to indicate that PM performs therapeutic effects on type-2 diabetes mellitus (T2DM)-induced nephropathy.
Material and Methods
Cell culture and treatment
The HK-2 cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The HK-2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, CA, USA) with 10% fetal bovine serum (FBS) and maintained in a humidified incubator filled with 5% CO2 and 95% air at 37°C. PM was obtained from Sigma-Aldrich (St. Louis, MO, USA). After confirming that the HK-2 cells were seeded at 80% confluence, they were cultured in 2% FBS DMEM. After fasting for 24 hours, the HK-2 cells were exposed to DMEM-containing 5.5 mM glucose (the normal glucose group or NG group) or 30 mM glucose (the high glucose group or HG group) and in the presence or absence of PM for 48 hours. After all treatments, the cells were washed 3 times with ice-cold 1×phosphate-buffered saline (PBS) and harvested.
Cell viability assay
Cell Counting Kit-8 (CCK-8) method was used to investigate the most appropriate drug concentration of PM by detecting the cell viability of HK-2 cells. Most appropriate drug concentration of PM was adopted for the following experiments. Cell viability was detected by a CCK-8 (MedChem Express, USA). Briefly, after digestion with 0.25% trypsin, cells were plated at a density of 104 to 105 cells per well in a 96-well plate, and then cells were treated with normal (NG) or high glucose (HG) and/or PM (0.01 mM, 0.1 mM, 1 mM, 10 mM, or 100 mM) for 24 hours. According to the manufacturer’s instructions, the cells were further incubated in fresh medium for 48 hours, and then 10 mL CCK-8 solution was added to the culture medium. The plates were incubated at 37°C in 5% CO2 for 1 hour. Cell viability was analyzed according to the absorbance at 450 nm, measured by using a Microplate Reader.
Western blot analysis
Following washing 3 times with ice-cold 1×PBS, the HK-2 cells were lysed in RIPA lysis buffer and total protein extracts was collected in centrifuge tubes. After centrifugation (13 000 rpm for 15 minutes at 4°C) protein concentration was determined using the bicinchoninic acid (BCA) reagent method. Then 40 μg of total protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes. After that, the membranes were blocked in 5% non-fat milk for 1 hour at 37°C and then incubated overnight at 4°C with primary antibodies. Primary antibodies used in this study were: anti-Beclin-1 (1: 1000; Abcam); anti-LC-3II (1: 1000; Abcam); anti-p62 (1: 1000; Abcam); and anti-β-actin (1: 1000; Abcam). After washing 3 times for 15 minutes with TBST, the membranes were incubated with HRP-conjugated secondary antibodies (1: 1000; Cell Signaling Technology, Inc.) for 2 hours at room temperature. Following development with Odyssey Fc System (LICOR, USA), Western blots were densitometrically analyzed using ImageJ software.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Total cellular RNA isolation (Invitrogen; Thermo Fisher Scientific, Inc.) and reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed to detect the expression of Beclin-1, LC-3II, and P62 mRNA according to the manufacturer’s instructions. Three independent experiments were performed for each group. Specific primer sequences were shown as follows: Beclin-1 forward 5′-AATGACTTTTTTCCTTAGGGGG-3′ and reverse 5′-GTGGCTTTTGTGGATTTTTTCT-3′; LC-3II forward 5′-AGTGCC TGTGTTGTTACGGA-3′ and reverse 5′-GCAGAAGGGAGTGTGTC TGA-3′; P62 forward 5′-AGTCGGAGCGGGTTCTCTAT-3′ and reverse 5′-GTGACACACATTCCAGCGAT-3′; β-actin forward 5′-TGACGTGGACATCCGCAAAG-3′ and reverse 5′-CTGGAA GGTGGACAGCGAGG-3′.
Using cDNA which was synthesized using 2 μg of total RNA in a 25 μL total reaction volume as the template, PCR amplification was performed under the following conditions: 95°C for 5 minutes, followed by 40 cycles each at 95°C for 10 seconds, 60°C for 30 seconds, and 72°C for 45 seconds. Relative gene expression was normalized to β-actin for reaction efficiency. The 2−ΔΔCt method [16] was used to determine the relative quantities of products.
Cell immunofluorescence
The HK-2 cells were seeded on coverslips in 6-well plates. The cells were fixed in 4% paraformaldehyde and then blocked with goat serum for 30 minutes at room temperature. Immunofluorescence analysis was carried out using anti-Beclin-1 polyclonal antibody (ab62557, Abcam) and anti-p62 polyclonal antibody (ab91526, Abcam). After incubated with primary antibody at 4°C overnight, the coverslips were incubated with DyLight 549 goat anti-rabbit IgG (ZSGB-bio, Beijing, China) for 1 hour and DAPI (4,6-diamino-2-phenyl indole) for 10 minutes at room temperature on the following day. Images were visualized and captured under a fluorescence microscope.
Intracellular ROS detection
The ROS generation was evaluated by the Reactive Oxygen Species Assay kit, according to the product instructions. Following exposed to NG or HG and in the presence or absence of PM for 48 hours, the HK-2 cells were cultured in their respective medium contained a fluorescent probe dichlorodihydrofluorescein diacetate (DCFH-DA) with a final concentration of 5 μmol/L. After incubated at 37°C, 5% CO2 incubator for 30 minutes, the cells were fixed with 1% paraformaldehyde for 10 minutes in the fridge with 4°C. After deacetylated by intracellular non-specific esterase, DCFH-DA was then oxidized by ROS to generate the fluorescent compound, 2,7-dichlorofluorescein (DCF). The DCF fluorescence intensity was detected using flow cytometry and analyzed by FlowJo software version 7.6.5.
Measurement of glutathione peroxidase (GPx) and manganese superoxide dismutase (MnSOD) activity
The effects of PM on enzymatic antioxidant defenses were evaluated with glutathione peroxidase (GPx) and manganese superoxide dismutase (MnSOD) activity. GPx activity was determined spectrophotometrically in HK-2 cells according to the method of Flohé and Gunzler [17]. Briefly, GPx was determined at 340 nm, by the reduction of tert-Butyl hydroperoxide (tBOOH) by oxidation of reduced glutathione (GSH). MnSOD activity was determined spectrophotometrically at 340 nm. Following the procedure described by Flohe and Otting [18], 100 μL of cells was added into a test tube. The assay mixture containing sodium carbonate buffer (50 mM, pH 10.0), 0.1 mM xanthine, 0.025 mM nitroblue tetrazolium, 0.1 mM EDTA, xanthine oxidase was added in the tube. MnSOD activity was determined spectrophotometrically at 550 nm. The activity of MnSOD was calculated with standard curve method. And GPx and MnSOD activity was both expressed as nmol/min/mg protein.
Measurement of glutathione (GSH)/glutathione disulfide (GSSG)
The effects of PM on non-enzymatic antioxidant defenses were evaluated with glutathione (GSH) and glutathione disulfide (GSSG). The intracellular expression of GSH and GSSG was detected using GSH/GSSG Ratio Detection Assay Kit (no. ab138881, Abcam). The plates were set up in duplicate for GSH standard (50 μL), GSSG standard (50 μL), and samples (50 μL). To prepare GSH Assay Mixture (GAM), Thiol Green Indicator in Assay Buffer was diluted at 1/100 dilution. Then 50 μL of GAM solution was added to the GSH standard and the sample wells. To prepare the total glutathione assay mixture (TGAM), GSSG probe was diluted in GAM solution at 1/25 dilution. Then 50 μL of GAM solution was added to the GSSG standard and the sample wells. Plates were incubated at room temperature for 10 to 60 minutes. Fluorescence at Ex/Em=490/520 nm was monitored with a fluorescence microplate reader. Then the GSH/GSSG ratio was calculated.
Statistical analyses
All experiments were done in triplicate. Data are expressed as the mean ± standard deviation. Data were analyzed using SPSS software version 23.0 (SPSS, Inc., Chicago, IL, USA). The differences among groups were analyzed for statistical significance using one-way analysis of variance (ANOVA), followed by the Student-Newman-Keuls (SNK) post-hoc test for multiple comparisons. Differences with P<0.05 were considered significant.
Results
PM alleviated HG-reduced decrease of HK-2 cells viability
HK-2 cells were manipulated with increasing doses of PM (Figure 1) in HG or NG environment, and then cell viability was measured to evaluate the functional effects of PM on cell viability. Compared with the control group, at doses of 0.01 mM (P<0.05), 0.1 mM (P<0.01) and 1 mM (P<0.01), PM caused a significant increase in the cell viability in both HG and NG environments. However, PM at 10 mM and 100 mM dramatically reduced cell viability compared with cells treated with PM at 1 mM. Thus, PM at 1 mM was used for further study.
Figure 1.
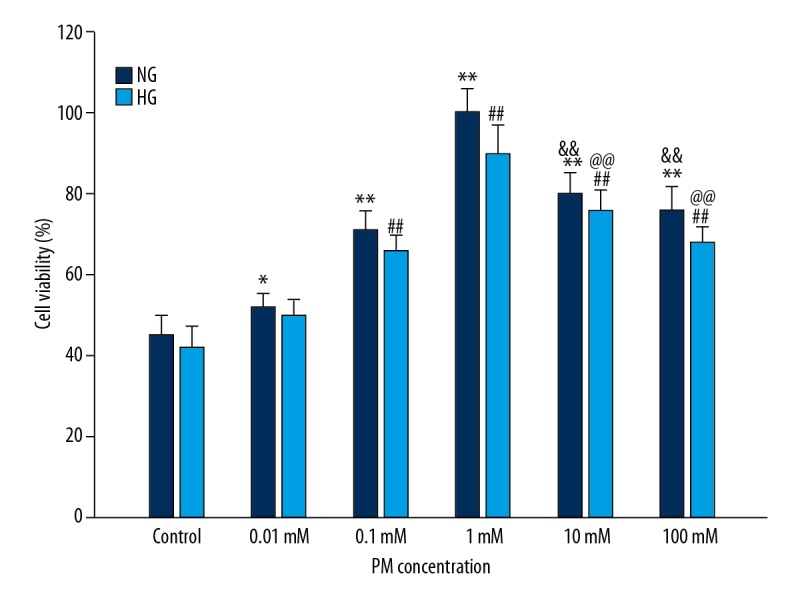
PM affected increases of HK-2 cells viability. Values are presented as means ± standard deviation. NG – normal glucose, 5.5 mM glucose; HG – high glucose, 30 mM glucose. (* P<0.05, ** P<0.01 compared with NG of control group. # P<0.05, ## P<0.01 compared with HG of control group. & P<0.05, && P<0.01 compared with NG of PM at 1 mM. @ P<0.05, @@ P<0.01 compared with HG of PM at 1 mM). PM – pyridoxamine; HK-2 – human proximal tubular epithelial cells.
PM activated HG-induced HK-2 cells autophagy
To determine the effect of PM on autophagy activation in HK-2 cells, we measured expression of Beclin-1, LC-3II, and p62 protein and mRNA via western blot analysis and RT-qPCR. HG dramatically decreased expressions of Beclin-1 and LC-3II protein and mRNA, and increased the protein and mRNA expression of P62 in HK-2 cells (Figure 2; P<0.01). The expression of autophagy-associated protein increased in HK-2 cells after 48 hours of HG treatment compared to the NG group, and then were reversed by PM administration (1 mM). Similar results were found in the immunofluorescent tests (Figure 3). Initially, compared with the NG group, the HG-cultured HK-2 cells decreased the fluorescence intensity of Beclin-1 and increased the fluorescence intensity of P62. Nevertheless, HG-induced autophagic inhibition was ameliorated when treated with 1 mM PM. which was indicated by the reduced fluorescence intensity of P62, and the increased fluorescence intensity of Beclin-1.
Figure 2.

Impacts of PM on the expression of autophagy-associated proteins and mRNA were determined by western blotting and RT-qPCR in HK-2 cells. β-actin was used as an internal control. Expression of (A, B) Beclin-1, (C, D) LC-3II, and (E, F) P62 was determined by western blotting and RT-qPCR. NG – normal glucose, 5.5 mM glucose; HG – high glucose, 30 mM glucose; NG+PM – PM (1 mM) plus 5.5 mM glucose; HG+PM – PM (1 mM) plus 30 mM glucose. (# P<0.05, ## P<0.01 compared with NG. * P<0.05, ** P<0.01 compared with the HG group). PM – pyridoxamine; RT-qPCR – reverse transcription-quantitative polymerase chain reaction; HK-2 – human proximal tubular epithelial cells.
Figure 3.

Effects of PM on the expression of Beclin-1 (A) and P62 (B) were determined by immunofluorescence test. HK-2 cells were cultured in NG and then stimulated by HG for 48 hours. Beclin-1 and P62 (green) was defined by staining cells with anti-Beclin-1 and anti-p62 antibody. DAPI staining (blue) was used to determine the nucleus position. Values are expressed as means ± standard deviation. NG – normal glucose, 5.5 mM glucose; HG – high glucose, 30 mM glucose; NG+PM – PM (1 mM) plus 5.5 mM glucose; HG+PM – PM (1 mM) plus 30 mM glucose. (# P<0.05, ## P<0.01 compared with NG. * P<0.05, ** P<0.01 compared with the HG group). PM – pyridoxamine; HK-2 – human proximal tubular epithelial cells; DAPI – 4,6-diamino-2-phenyl indole.
PM inhibit ROS generation in HG-induced HK-2 cells
The effects of HG and PM on intracellular ROS levels are shown in Figure 4. Compared with the NG group, ROS production was higher in the HG group (P<0.01). However, ROS production was significantly lower in the presence of PM (P<0.01).
Figure 4.
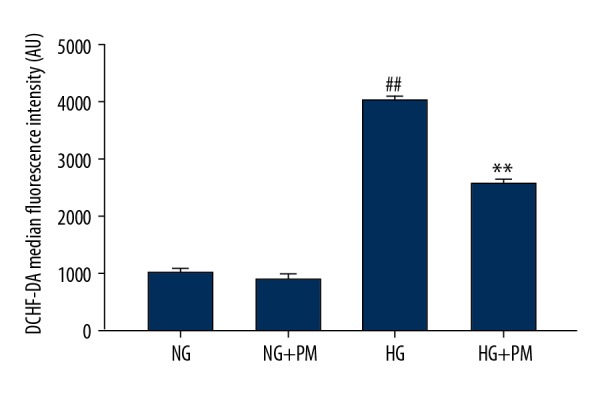
PM inhibits ROS generation in HG-induced HK-2 cells. DCHF-DA fluorescence intensity was analyzed quantificationally by flow cytometry. Data were expressed as means ± standard deviation. NG – normal glucose, 5.5 mM glucose; HG – high glucose, 30 mM glucose; NG+PM – PM (1 mM) plus 5.5 mM glucose; HG+PM – PM (1 mM) plus 30 mM glucose; (# P<0.05, ## P<0.01 compared with NG. * P<0.05, ** P<0.01 compared with the HG group). PM – pyridoxamine; ROS – reactive oxygen species; HK-2 – human proximal tubular epithelial cells; DCHF-DA – dichlorodihydrofluorescein diacetate.
Effects of PM in the antioxidant defense system of HK-2 cells
To explore the effects of PM administration in ROS-defense network of HK-2 cells, we analyzed antioxidant enzymes, namely GPx and MnSOD, and also nonenzymatic antioxidants, namely GSH and GSSG. As shown in the Figure 5, GPx and MnSOD activities were significantly affected by PM treatment. Regarding the nonenzymatic antioxidants, we observed that GSH/GSSG ratio was decreased in HG-cultured HK-2 cells compared to NG-treated cells. Nevertheless, there were no overall effects on GSH/GSSG ratio in presence of PM (Figure 6).
Figure 5.

PM improved enzymatic antioxidant defenses in HG-induced HK-2 cells. GPx (A) and MnSOD (B) activities were evaluated in cultured HK-2 cells. Data were reported as means ± standard deviation. NG – normal glucose, 5.5 mM glucose; HG – high glucose, 30 mM glucose; NG+PM – PM (1 mM) plus 5.5 mM glucose; HG+PM – PM (1 mM) plus 30 mM glucose; (# P<0.05, ## P<0.01 compared with NG. * P<0.05, ** P<0.01 compared with the HG group). PM – pyridoxamine; HK-2 – human proximal tubular epithelial cells; GPx – glutathione peroxidase; MnSOD – manganese superoxide dismutase.
Figure 6.
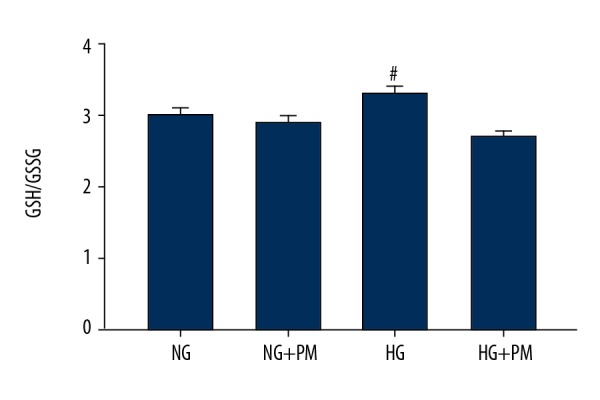
PM did not improve non-enzymatic antioxidant defenses in HG-induced HK-2 cells. GSH/GSSG were evaluated in HK-2 cells. Data were expressed as means ± standard deviation. NG – normal glucose, 5.5 mM glucose; HG – high glucose, 30 mM glucose; NG+PM – PM (1 mM) plus 5.5 mM glucose; HG+PM – PM (1 mM) plus 30 mM glucose; (# P<0.05, ## P<0.01 compared with NG group). PM – pyridoxamine; HK-2 – human proximal tubular epithelial cells; GSH – glutathione; GSSG – oxidized glutathione.
Discussion
The pathogenesis of diabetic nephropathy is the joint result of many factors, and formation of AGEs products, abnormal polyol pathway, oxidative stress, change of glomerular hemodynamics, dyslipidemia and inhibition of autophagy as results of long-term hyperglycemia are considered the causes of diabetic nephropathy [19,20]. Despite several clinical trials and fundamental research studies, the underlying etiology and pathogenesis remains unclear, and associated mechanisms of interventions and therapy have yet to be identified.
In recent years, studies have focused on the HG-induced autophagic response and its biological function and molecular regulation in renal tubular epithelial cells [1,21]. Autophagy is a fundamental cellular process that functions to recycle nutrients and remove damaged organelles and protein aggregates. It has recently aroused widespread attention due to its association with damage of hyperglycemic renal tubular epithelial cells [22]. Autophagy predispose nephrocytes to accumulate damaged organelles and protein aggregates, especially under HG conditions. Many suppositions have been proposed to explicate the underlying molecular mechanism of autophagy exerting its defensive function in nephrocytes. For example, AGEs activated autophagy resulting in LC-3 cleavage increased Atg5 expression, and decreased P62 expression, which are essential for the formation of autophagosomes [23,24]. Another study provided evidence to suggest that increased autophagic activity in tubular cells after kidney injury [25]. Deficiency of Atg5 dramatically sensitizes tubules to ischemic injury, thus caused increasing of damaged mitochondria and increased tubular cell apoptosis. These changes suggest that autophagy might play an important part in maintaining tubular cell integrity during stress conditions. In kidney biopsy of patients with diabetic nephropathy, transmission electron microscopy confirmed the presence of diffuse vacuolization and autophagosomal structures in proximal tubular cells in diabetic nephropathy. Compared with other nephritis, immunohistochemistry of Beclin-1 and LC-3 in reno-puncture tissues of diabetic nephropathy highlighted increased protein expression of all these autophagic factors at the tubular level in diabetic nephropathy [26]. In the present study, HG treatment inhibited autophagy and activated oxidative stress, evidenced by low-expression of Beclin-1 and LC-3II, and overexpression of P62, as well as the highly expressed oxidative stress-related proteins in the HK-2 cells. The present study and multiple researches could be explicated by the fact that tubular epithelial cells utilize autophagic machinery as a survival mechanism under stress conditions.
Some novel agents for preventing diabetic nephropathy development and progression, such as PM, ruboxistaurin, and sulodexide, Janus kinase inhibitors and other new types of glucose-lowering agents are developed and investigated in recent years [20]. As the AGE products being studied extensively in the etiology of diabetic complications, PM have gradually become a promising treatment. It has previously been reported that PM has a preventive effect on diabetic nephropathy [27]. PM had been shown to reduce glomerular hypertrophy, mesangial matrix, and albuminuria in female diabetic db/db mice [28]. In addition, the combined administration of PM and an ACE inhibitor (enalapril) provided added protection against mortality and renal injury compared with the use of enalapril alone. Furthermore, PM also elicit a beneficial influence on activation of autophagy. A previous study showed the administration of PM activated the formation and turnover of autophagosomes in podocytes by inhibiting mTOR and activation of the nuclear translocation of transcription factor EB (TFEB) [29].In addition, PM increased expression of the markers of autophagosome formation (LC-3II and Beclin-1) in vitro, which is in accordance with our study suggesting upregulation of LC-3II and Beclin-1 expression and downregulation of P62 expression in PM-treated HK-2 cells.
Oxidative stress caused by overproduction of mitochondrial ROS is often deemed to be of importance to diabetic complications [30], which closely related to mitochondrial energy generation. Research increasingly has demonstrated that PM can effectively regulate oxidative stress in a variety tissues or cells affected by hyperglycemia. Tuttle et al. observed that PM induced significant decrease in ROS production and the expression of TGF-β1 in rat mesangial cells of renal cortex exposure to HG [31]. Other data have demonstrated that PM significantly enhanced the oxidative stress by directly increased GPx and MnSOD activities [32], which can be regarded as biomarkers of enzymatic antioxidant defenses. The present study demonstrated that HK-2 cells incubated in HG medium exhibited overproduction of ROS. Treatment for 48 hours with 1 mM PM ameliorated ROS production. In addition, the administration of PM increased GPx and MnSOD activities compared with the HG group. GSH/GSSG ratio is responsible for GSH regeneration. However, in our study, there was no obvious change in the GSH/GSSG ratio in the HG+PM group compared with the HG group. These results suggest that to minimize ROS-induced damage, PM needs helped to strengthen complex antioxidant defenses of enzymatic nature but not non-enzymatic nature, which is similar to a previous study that reported impacts of PM in a rat model of type 2 diabetes [33]. In diabetic lysed erythrocyte preparations, administration with PM in vivo did not increase levels of GSH, but decrease levels slightly compared with the diabetes mellitus group. Nevertheless, the role of PM and its potential molecular mechanisms in activating autophagy and improving anti-oxidation has not been fully understood. Moreover, the anti-inflammatory effect and regulation of autophagy of PM in vivo model needs to be explored in depth. Furthermore, further investigation to identify and characterize the effect of PM in multiple proximal tubular epithelial cell lines is warranted.
Conclusions
In conclusion, PM induced the increase in autophagy regulators and GPx and MnSOD activities, in addition to overexpression of ROS production. The present study verified partially that PM might becoming a promising option for future diabetic nephropathy treatment.
Acknowledgements
The authors would like to thank the Department of Pathology of Hebei Medical University (Shijiazhuang, China) for their technical support for the experiments.
Footnotes
Source of support: The present study was supported by National Natural Science Foundation of China (grant no. 81770717)
Conflict of interest
None.
References
- 1.Lin F. Autophagy in renal tubular injury and repair. Acta Physiol (Oxf) 2017;220(2):229–37. doi: 10.1111/apha.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, et al. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018;9(2):119. doi: 10.1038/s41419-017-0135-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L, Wang ZV, Hill JA, Lin FM. New autophagy reporter mice reveal dynamics of proximal tubular autophagy. Autophagy. 2014;25(4):305–15. doi: 10.1681/ASN.2013040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yanjuan H, Ming W, Jinying W, et al. CD36 is involved in high glucose-induced epithelial to mesenchymal transition in renal tubular epithelial cells. Biochem Biophys Res Commun. 2015;468(1–2):281–86. doi: 10.1016/j.bbrc.2015.10.112. [DOI] [PubMed] [Google Scholar]
- 5.Yaribeygi H, Butler AE, Barreto GE, Sahebkar A. Antioxidative potential of antidiabetic agents: A possible protective mechanism against vascular complications in diabetic patients. J Cell Physiol. 2018;234(3):2436–46. doi: 10.1002/jcp.27278. [DOI] [PubMed] [Google Scholar]
- 6.Small DM, Bennett NC, Coombes J, et al. [Mitochondrial homeostasis is impeded by degradation and autophagy in oxidative stress-induced renal cell injury]. Rev Esp Reum Enferm Osteoartic. 1966;11(8):67–73. [in Spanish] [Google Scholar]
- 7.Kitada M, Ogura Y, Monno I, et al. Regulating autophagy as a therapeutic target for diabetic nephropathy. Curr Diab Rep. 2017;17(7):53. doi: 10.1007/s11892-017-0879-y. [DOI] [PubMed] [Google Scholar]
- 8.Booth AA, Khalifah RG, Hudson BG. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: comparison with aminoguanidine. Biochem Biophys Res Commun. 1996;220(1):113–19. doi: 10.1006/bbrc.1996.0366. [DOI] [PubMed] [Google Scholar]
- 9.Kim HH, Kang YR, Lee JY, et al. The postprandial anti-hyperglycemic effect of pyridoxine and its derivatives using in vitro and in vivo animal models. Nutrients. 2018;10(3) doi: 10.3390/nu10030285. pii: E285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed N, Thornalley PJ. Advanced glycation end products: What is their relevance to diabetic complications? Diabetes Obesity & Metabolism. 2010;9(3):233–45. doi: 10.1111/j.1463-1326.2006.00595.x. [DOI] [PubMed] [Google Scholar]
- 11.Watson AMD, Soro-Paavonen A, Sheehy K, et al. Delayed intervention with AGE inhibitors attenuates the progression of diabetes-accelerated atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia. 2011;54(3):681–89. doi: 10.1007/s00125-010-2000-9. [DOI] [PubMed] [Google Scholar]
- 12.Curtis TM, Hamilton R, Yong PH, et al. Müller glial dysfunction during diabetic retinopathy in rats is linked to accumulation of advanced glycation end-products and advanced lipoxidation end-products. Diabetologia. 2011;54(3):690–98. doi: 10.1007/s00125-010-1971-x. [DOI] [PubMed] [Google Scholar]
- 13.Chiazza F, Cento AS, Collotta D, et al. Protective effects of pyridoxamine supplementation in the early stages of diet-induced kidney dysfunction. Biomed Res Int. 2017;2017 doi: 10.1155/2017/2682861. 2682861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams ME, Bolton WK, Khalifah RG, et al. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol. 2007;27(6):605–14. doi: 10.1159/000108104. [DOI] [PubMed] [Google Scholar]
- 15.Youns NNM. Pyridoxamine, an inhibitor of protein glycation, in relation to microalbuminuria and proinflammatory cytokines in experimental diabetic nephropathy. Exp Biol Med. 2013;238(8):881–88. doi: 10.1177/1535370213494644. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25(4):402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Flohé L, Günzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105(1):114–21. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 18.Flohé L, Otting F. Superoxide dismutase assays. Methods Enzymol. 1984;105:93–104. doi: 10.1016/s0076-6879(84)05013-8. [DOI] [PubMed] [Google Scholar]
- 19.Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol. 2015;224(1):R15–30. doi: 10.1530/JOE-14-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin YC, Chang YH, Yang SY, et al. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc. 2018;117(8):662–75. doi: 10.1016/j.jfma.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 21.Khan S, Bhat ZR, Jena G. Role of autophagy and histone deacetylases in diabetic nephropathy: Current status and future perspectives. Genes Dis. 2016;3(3):211–19. doi: 10.1016/j.gendis.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu WJ, Huang WF, Ye L, et al. The activity and role of autophagy in the pathogenesis of diabetic nephropathy. Eur Rev Med Pharmacol Sci. 2018;22(10):3182–89. doi: 10.26355/eurrev_201805_15079. [DOI] [PubMed] [Google Scholar]
- 23.Chiang CK, Wang CC, Lu F, et al. Involvement of endoplasmic reticulum stress, autophagy, and apoptosis in advanced glycation end products-induced glomerular mesangial cell injury. Sci Rep. 2016;6:34167. doi: 10.1038/srep34167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaaf MB, Keulers TG, Vooijs MA, Rouschop KMA. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30(12):3961–78. doi: 10.1096/fj.201600698R. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Hartleben B, Kretz O, et al. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 2012;8(5):826–37. doi: 10.4161/auto.19419. [DOI] [PubMed] [Google Scholar]
- 26.Pontrelli P, Oranger A, Barozzino M, et al. Deregulation of autophagy under hyperglycemic conditions is dependent on increased lysin63 ubiquitination: A candidate mechanism in the progression of diabetic nephropathy. J Mol Med (Berl) 2018;96(7):645–59. doi: 10.1007/s00109-018-1656-3. [DOI] [PubMed] [Google Scholar]
- 27.Abouzed TK, Munesue S, Harashima A, et al. Preventive effect of salicylate and pyridoxamine on diabetic nephropathy. J Diabetes Res. 2016;2016 doi: 10.1155/2016/1786789. 1786789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng F, Zeng YJ, Plati AR, et al. Combined AGE inhibition and ACEi decreases the progression of established diabetic nephropathy in B6 db/db mice. Kidney Int. 2006;70:116–19. doi: 10.1038/sj.ki.5001578. [DOI] [PubMed] [Google Scholar]
- 29.Zhao X, Chen Y, Tan X, et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J Pathol. 2018;245(2):235–48. doi: 10.1002/path.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panigrahy SK, Bhatt R, Kumar A. Reactive oxygen species: Sources, consequences and targeted therapy in type 2 diabetes. J Drug Target. 2017;25(2):93–101. doi: 10.1080/1061186X.2016.1207650. [DOI] [PubMed] [Google Scholar]
- 31.Tuttle KR, Anderberg RJ, Cooney SK, Meek RL. Oxidative stress mediates protein kinase C activation and advanced glycation end product formation in a mesangial cell model of diabetes and high protein diet. A J Nephrol. 2009;29(3):171–80. doi: 10.1159/000154470. [DOI] [PubMed] [Google Scholar]
- 32.Cardoso S, Carvalho C, Marinho R, et al. Effects of methylglyoxal and pyridoxamine in rat brain mitochondria bioenergetics and oxidative status. J Bioenerg Biomembr. 2014;46(5):347–55. doi: 10.1007/s10863-014-9551-2. [DOI] [PubMed] [Google Scholar]
- 33.Nagaraj RH, Sarkar P, Mally A, et al. Effect of pyridoxamine on chemical modification of proteins by carbonyls in diabetic rats: Characterization of a major product from the reaction of pyridoxamine and methylglyoxal. Arch Biochem Biophys. 2002;402(1):110–19. doi: 10.1016/S0003-9861(02)00067-X. [DOI] [PubMed] [Google Scholar]