

Abstract
Fragile X-associated tremor/ataxia syndrome (FXTAS) is an adult-onset neurodegenerative disorder that affects premutation carriers (55–200 CGG repeats) of the fragile X mental retardation 1 (FMR1) gene. Much remains unknown regarding the metabolic alterations associated with FXTAS, especially in the brain, and the most affected region, the cerebellum. Investigating the metabolic changes in FXTAS will aid in the identification of biomarkers as well as in understanding the pathogenesis of disease. To identify the metabolic alterations associated with FXTAS, we took advantage of our FXTAS mouse model that expresses 90 CGG repeats in cerebellar Purkinje neurons and exhibits the key phenotypic features of FXTAS. We performed untargeted global metabolic profiling of age-matched control and FXTAS mice cerebella at 16–20 weeks and 55 weeks. Out of 506 metabolites measured in cerebellum, we identified 186 metabolites that demonstrate significant perturbations due to the (CGG)90 repeat (P<0.05) and found that these differences increase dramatically with age. To identify key metabolic changes in FXTAS pathogenesis, we performed a genetic screen using a Drosophila model of FXTAS. Out of 28 genes that we tested in the fly, 8 genes showed significant enhanced neuronal toxicity associated with CGG repeats, such as Schlank (ceramide synthase), Sk2 (sphingosine kinase) and Ras (IMP dehydrogenase). By combining metabolic profiling with a Drosophila genetic screen to identify genetic modifiers of FXTAS, we demonstrate an effective method for functional validation of high-throughput metabolic data and show that sphingolipid and purine metabolism are significantly perturbed in FXTAS pathogenesis.
Introduction
Fragile X-associated tremor/ataxia syndrome (FXTAS) is a late-onset neurodegenerative disorder characterized by intention tremor, cerebellar ataxia, brain atrophy and parkinsonism (1,2). Neuropathologically, FXTAS is defined by ubiquitin-positive intranuclear inclusions in neurons and astrocytes of the brain, as well as Purkinje neurons of the cerebellum (3,4). The cerebellum is significantly affected in this disease, as demonstrated by Purkinje cell loss and axonal degeneration (5). FXTAS is distinct from other causes of tremor and ataxia in that it is found in carriers of the premutation, individuals with 55–200 expanded CGG repeats in the 5′UTR of fragile X mental retardation 1 (FMR1). Typically, individuals carry 5–54 CGG repeats, and individuals who harbor greater than 200 CGG repeats in FMR1 develop fragile X syndrome due to methylation-mediated silencing of FMR1 transcription and translation (6–9). Premutation carriers are found quite frequently in the population, with an estimated prevalence of 1:259 in females and 1:813 in males (10,11). Interestingly, only a proportion of premutation carriers develop FXTAS; ~40% of males and ~16% of females develop FXTAS in late adulthood (2,12). The incomplete penetrance of FXTAS has puzzled researchers and clinicians for over a decade and identifying the molecular basis of FXTAS pathogenesis will allow us to identify biomarkers to predict who will develop disease and will significantly advance efforts to develop treatment.
Since the identification of FXTAS in 2001 (13), various animal models have been developed to study the molecular and genetic basis of FXTAS. In addition to knock-in mouse models that are available (14,15), we generated a murine model of FXTAS that expresses r(CGG)90 in the context of FMR1 in Purkinje neurons (16). In addition, our group has previously described a Drosophila disease model of FXTAS that expresses the premutation CGG repeat in the context of the human FMR1 5′UTR. For both the mouse and fly model, we have demonstrated that the premutation CGG repeats are sufficient to cause FXTAS pathology (17). Expression of r(CGG)90 under the eye-specific driver in Drosophila, gmr-GAL4, results in a rough-eye phenotype characterized by cell death, loss of pigmentation and ommatidial disruption (17).
Investigations into the molecular basis of the pathogenesis of FXTAS have not previously focused on metabolic changes associated with FXTAS. Metabolism encompasses all the essential aspects of life; chemical reactions catalyzed by enzymes are responsible for providing the power, energy and building blocks to sustain the growth and maintenance of living cells. Through metabolomics, we are able to sensitively measure and analyze the levels of metabolites and obtain a snapshot of the physiological state of the organism (18). Other studies have used metabolomics to gauge the global responses to external factors such as stress, to profile the global metabolic characteristics in a disease state (19–21), to generate hypotheses to uncover mechanisms of disease pathogenesis (22) or to identify biomarkers of disease (23–25).
Studying the metabolomics of FXTAS is of interest because metabolic alterations have been identified in other neurodegenerative Central Nervous System disorders such as Alzheimer’s, Huntington’s and Parkinson’s diseases (20,21,26,27). In addition to using human patient samples, metabolomics studies on animal disease models have proven to be advantageous for the study of neurodegenerative diseases. Model organisms present a more simplified and accessible system to study the metabolic changes in disease, especially in tissues such as the brain, for which human samples are challenging to acquire.
A recent study by Giulivi et al. evaluated the plasma metabolic profiles of human premutation carriers and controls and reported that FXTAS premutation carriers exhibited mitochondrial dysfunction, markers of neurodegeneration and pro-inflammatory damage (28). However, as of yet, there has been no published study investigating the metabolic changes associated with FXTAS in the cerebellum, the most affected tissue in FXTAS. Here, we take advantage of the FXTAS murine model to expand our understanding of metabolic alterations in FXTAS by comparing the metabolites in the cerebellum of FXTAS mice expressing r(CGG)90 in Purkinje cells with wild-type (WT) mice. We demonstrate a distinctive approach to functionally validate the high-throughput metabolomics via a genetic screen using the Drosophila model of FXTAS. We show that significant metabolic changes occur in sphingolipid and purine metabolism in the cerebella of FXTAS mice. More specifically, we demonstrate that Schlank(Cers5), Sk2 (Sphk1) and Ras (Impdh1), which encode enzymes in the sphingolipid and purine metabolism, respectively, are genetic modifiers of CGG toxicity in Drosophila.
Results
Metabolic profiling demonstrates significant metabolic perturbations in the presence of r(CGG)90, especially during aging
In order to identify the metabolic alterations that are associated with CGG toxicity and progression of FXTAS, we performed untargeted global metabolic profiling of WT and FXTAS mice at 16–20 and 55 weeks. The FXTAS mice express a human FMR1 r(CGG)90 repeat in Purkinje neurons and WT littermates were used as controls (16). Out of 506 metabolites detected, 200 showed significant alterations (P < 0.05) in the post hoc pairwise comparisons of an analysis of variance (ANOVA) model when comparing aged WT versus young WT, aged FXTAS versus young FXTAS, young FXTAS versus young WT and aged FXTAS versus aged WT mice (Supplementary Materials, Fig. S1 and Table S1). The 93 most significantly altered metabolites (P < 0.005) are shown in Figure 1A. More than half of the 200 altered metabolites were lipids, followed by amino acids, nucleotides and carbohydrates (Fig. 1B). We also identified the proportion of each superpathway that is significantly altered in the ANOVA contrasts (P < 0.05) out of the total metabolites assessed per pathway (Supplementary Material, Fig. S2). Intriguingly, examining all the proportion of metabolites altered, energy was the most affected superpathway, with 6 out of 9 energy-related metabolites in the cerebellum being altered, followed by carbohydrates (15 out of 34) and lipids (111 out of 253) (Supplementary Material, Fig. S2).
Figure 1.
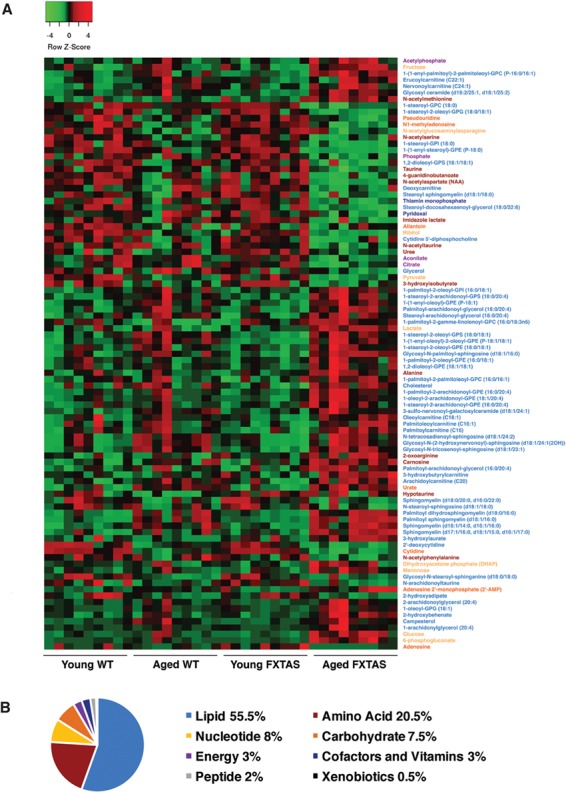
Metabolic profiling demonstrates significant metabolic perturbations in the presence of r(CGG)90 repeats. (A) A heatmap of the top 93 most significantly altered metabolites (P < 0.005) distinguishes the FXTAS and WT mouse cerebellum samples. For the complete heatmap of 200 significantly altered metabolites (P < 0.05), see Supplementary Material, Figure S1. Heatmap was generated using average linkage hierarchical clustering in Heatmapper (52). Red indicates high and green indicates low intensity of the metabolite relative to the median (black). Names of the corresponding metabolites are coded in color to represent the appropriate superpathway: lipid (blue), amino acid (red), nucleotide (yellow), carbohydrate (orange), energy (purple), cofactors and vitamins (navy), peptide (gray) and xenobiotics (black). (B) Representation of the superpathways of metabolism affected in the 200 significantly altered metabolites (P < 0.05).
We used an UpSet plot (Fig. 2) to summarize the significantly altered metabolites that are unique to each comparison as well as the altered metabolites that are seen in multiple comparison groups. Importantly, only one metabolite was altered uniquely in young FXTAS versus young WT mice, demonstrating that early in the disease there are minimal metabolic alterations due to the presence of the r(CGG)90. In contrast, upon aging, metabolic perturbations substantially increase in the progression of FXTAS: the aged FXTAS versus young FXTAS mice comparison demonstrates the highest number of altered metabolites unique to one group, followed by the aged FXTAS versus aged WT mice comparison (Fig. 2). Furthermore, 14 out of the 200 altered metabolites were uniquely perturbed in normal aging (aged WT versus young WT mice comparison) and were excluded from further analyses (Fig. 2). The highest number of metabolites that are shared between comparisons are seen in both the aged FXTAS versus aged WT mice and the aged FXTAS versus young FXTAS mice comparisons, revealing that 51 metabolites are altered due to the presence of the r(CGG)90 toxicity and the progression of FXTAS. Further details about the metabolites altered in each comparison and the superpathways impacted can be found in Supplementary Materials, Figs S3–5.
Figure 2.
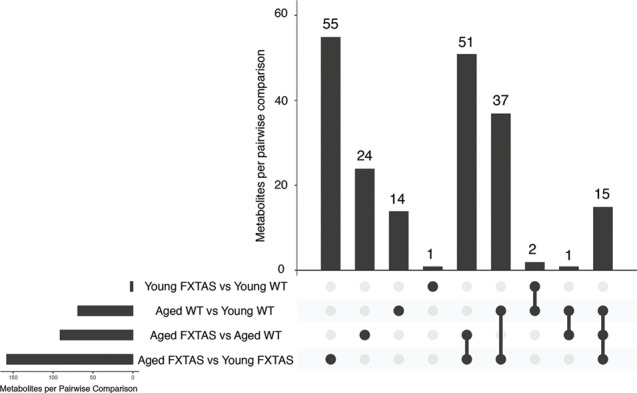
Altered metabolites by pairwise comparison. An UpSet plot was generated in R to visualize the metabolites altered in each comparison (53). The horizontal bars indicate the total number of metabolites per pairwise comparison. The vertical bars indicate the number of metabolites in each combination of pairwise comparisons.
With the results of the high-throughput unbiased global metabolic profiling of FXTAS mice in hand, we were interested in taking advantage of our FXTAS Drosophila model to examine whether these metabolic perturbations in the FXTAS mice cerebella can be attributed to a genetic interaction between the metabolic genes and the r(CGG)90. Hence, we set out to identify metabolic enzyme gene candidates that may be associated with the biochemical alterations observed in our comparisons. After excluding the 14 metabolites that were uniquely altered in normal aging (Fig. 2), 186 of the 506 metabolites detected in the cerebellum showed significant alteration in the FXTAS mice (P < 0.05) (Fig. 2). From these 186 metabolites, we used manual curation to identify enzymes that are responsible for the synthesis/degradation of the metabolite, following selection criteria that are detailed in the Materials and Methods section (Fig. S6), which yielded 28 candidate genes to test for genetic interaction with the CGG repeat (Supplementary Material, Table S2). These 28 candidate genes account for 12.9% of the 186 metabolites (Supplementary Material, Table S2). For these 28 genes, we tested for genetic modulation of the CGG toxicity in our Drosophila FXTAS model by knocking down the candidate gene in the FXTAS fly eye using the UAS-GAL4 system and screening for enhancement or suppression of the eye phenotype.
Drosophila genetic screen identifies eight genes that exhibit significant interaction with FXTAS CGG toxicity
Out of the 28 candidates selected from manual curation, the Drosophila screen identified 8 genes that exhibit enhancement or suppression of CGG associated toxicity in FXTAS Drosophila (Table 1; Supplementary Material, Table S2). Knockdown of seven of the eight genes resulted in enhancement of the rough-eye phenotype: CG8353, ChAT, Pect, Ppat-dpck, Ras, Schlank and Sk2 which are Drosophila orthologs for cytidine deaminase (Cda), choline O-acetyltransferase (Chat), phosphate cytidylyltransferase 2, ethanolamine (Pcyt2), coenzyme A synthase (Coasy), inosine monophosphate dehydrogenase (Impdh1), ceramide synthase (Cers5) and sphingosine kinase (Sphk1), respectively (Table 1; Supplementary Materials, Fig. S7 and Table S2). Interestingly, knockdown of CG4306, the fly ortholog of Gamma-glutamylcyclotransferase (Ggct), demonstrated significant suppression of CGG toxicity (Table 1; Supplementary Material, Fig. S7). Taken together, high-throughput metabolomics followed by the FXTAS Drosophila genetic screen led to the identification of eight genetic modifiers of CGG toxicity in FXTAS. From the eight genetic modifiers of CGG toxicity in FXTAS, we focused on three enzymes based on the correlation between the Drosophila genetic data and the upstream/downstream metabolic alterations observed in the FXTAS mouse model.
Table 1.
Eight candidate genes that exhibit modulation of r(CGG)90 toxicity in Drosophila
| No. | Mouse gene | Drosophila ortholog | BDSC stock | Effect on r(CGG)90 toxicity | |
|---|---|---|---|---|---|
| 1 | Cda | Cytidine deaminase | CG8353 | 51915 | Enhanced |
| 2 | Cers5 | Ceramide synthase 5 | Schlank | 29340 | Enhanced |
| 3 | Chat | Choline O-acetyltransferase | ChAT | 60028 | Enhanced |
| 4 | Coasy | Coenzyme A synthase | Ppat-Dpck | 57231 | Enhanced |
| 5 | Ggct | Gamma-glutamylcyclotransferase | CG4306 | 65890 | Suppressed |
| 6 | Impdh1 | Inosine monophosphate dehydrogenase 1 | Ras | 31653 | Enhanced |
| 7 | Pcyt2 | Phosphate cytidylyltransferase 2, ethanolamine | Pect | 63710 | Enhanced |
| 8 | Sphk1 | Sphingosine kinase 1 | Sk2 | 36741 | Enhanced |
Sphingolipid metabolism is perturbed in FXTAS: knockdown of schlank (Cers5) and Sk2 (Sphk1) result in enhancement of premutation CGG repeat-mediated neurodegeneration
The untargeted global metabolic profiling revealed that a significant number of metabolites were altered in the lipid pathway in FXTAS comparisons (Fig. 1). Among the lipids, the sphingolipid pathway was significantly perturbed in the onset and progression of CGG toxicity in the cerebellum (Fig. 3A and B). Although sphingolipids were not altered early on in the disease (young FXTAS versus young WT mice, Supplementary Material, Fig. S3C), upon progression of FXTAS, aged FXTAS mice cerebella demonstrated significant perturbations in sphingolipids compared with aged WT as well as young FXTAS mice (Fig. 3B).
Figure 3.
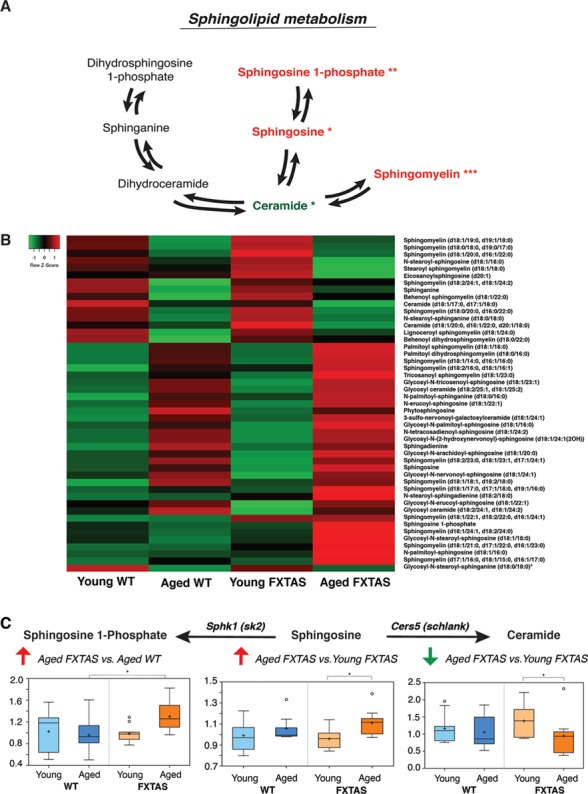
Sphingolipids are significantly altered in FXTAS mice. (A) Schematic depicting key components of the sphingolipid pathway and their perturbations in FXTAS versus WT mice comparisons. Red and green denote increased and decreased fold changes, respectively, in the pairwise comparisons. One asterisk (*) indicates changes in the aged FXTAS versus young FXTAS mice comparison, whereas two asterisks (**) indicate changes in the aged FXTAS versus aged WT mice comparison. Three asterisks (***) indicate changes in both comparisons. (B) A heatmap showing the metabolite alterations in sphingolipid metabolism for young WT, aged WT, young FXTAS and aged FXTAS mice. Heatmap was generated using the average metabolite intensity of each group, using average linkage hierarchical clustering in Heatmapper (52). Red indicates high and green indicates low intensity of the metabolite relative to the median (black). (C) Ceramide synthase [Cers5 (Schlank)] catalyzes the conversion of sphingosine to ceramide. Sphingosine kinase [Sphk1 (Sk2)] catalyzes the conversion of sphingosine into sphingosine-1-phosphate. Box plots display the scaled intensity values (y-axis) for sphingosine-1-phosphate, sphingosine and ceramide (d18:1/20:0, d16:1/22:0 and d20:1/18:0). For sphingosine-1-phosphate, significant alterations were seen in aged FXTAS versus aged WT mice (P = 0.0232, q = 0.119). Significant differences are seen between aged FXTAS versus young FXTAS mice with P = 0.0151 and q = 0.0263 for sphingosine and P = 0.0313 and q = 0.0441 for ceramide. X-axis indicates the genotype.
Ceramide synthase is the enzyme responsible for converting sphingosine to ceramide, known as the chemical backbone of all sphingolipids [Fig. 3A, (29)]. Upon manual curation to take a closer look at the metabolites and enzymes involved, we found that the level of sphingosine, the substrate of ceramide synthase (Cers5), was significantly elevated in the progression of FXTAS (aged FXTAS versus young FXTAS mice, P = 0.0151 and q = 0.0263; Fig. 3C). In contrast, the level of ceramide, the product of the acylation reaction catalyzed by CerS, was significantly decreased in the aged FXTAS mice compared with the younger counterparts (P = 0.0313, q = 0.0441; Fig. 3C). As shown in Figure 3, the elevation in the levels of the substrate sphingosine, coinciding with a reduction in the product ceramide in the aged FXTAS versus young FXTAS mice cerebella, motivated us to test whether CerS function may be perturbed in the progression of FXTAS owing to a genetic interaction between Cers and the expanded CGG repeat in FXTAS (Fig. 3C).
Furthermore, an additional enzyme in the sphingolipid pathway, sphingosine kinase was also selected as a candidate enzyme for the Drosophila screen. Sphingosine kinase catalyzes the phosphorylation of sphingosine into sphingosine 1-phosphate (S1P) (Fig. 3A). In the comparison of aged FXTAS versus aged WT mice, the level of sphingosine was not significantly elevated but instead S1P was significantly elevated (P = 0.0232, q = 0.119; Fig. 3C), which led us to hypothesize that sphingosine kinase may play a role in the toxicity of r(CGG)90 in FXTAS.
To probe for genetic interaction, we employed the Drosophila model of FXTAS which allows for facile genetic screening (17). Knockdown of Schlank (Cers5) and Sk2 (Sphk1) resulted in significant enhancement of the CGG toxicity in the fly progeny, indicating that Schlank and Sk2 interact with the CGG repeat of FXTAS and significantly enhance the CGG-associated neurotoxicity (Fig. 4). Importantly, knockdown of Schlank and Sk2 in the WT fly did not result in change in eye phenotype (Fig. 4).
Figure 4.
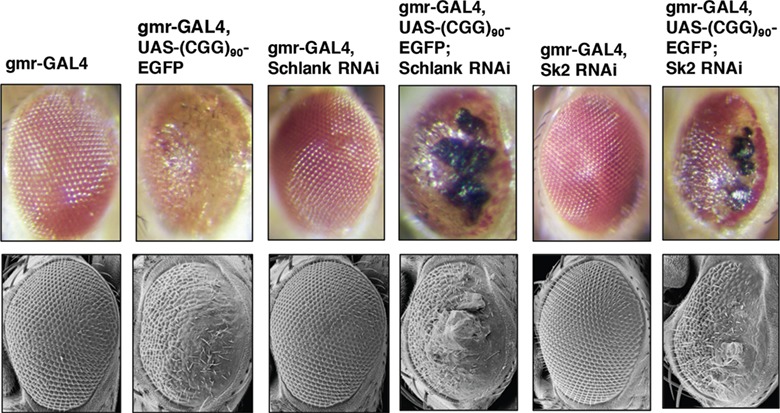
Knockdown of Schlank and Sk2 enhance r(CGG)90-mediated neurodegeneration in FXTAS Drosophila. Knockdown of Schlank and Sk2 (fly ortholog of Cers5 and Sphk1) enhance r(CGG)90 mediated neurodegeneration in FXTAS Drosophila. Top panel displays the light microscopy images from Drosophila of the indicated genotypes crossed to gmr-GAL4 flies and gmr-GAL4, UAS-(CGG)90 EGFP flies. From left to right: WT control (gmr-GAL4) (column 1), (CGG)90 control (gmr-GAL4, UAS-(CGG)90-EGFP) (column 2), WT fly expressing RNAi knockdown of schlank (column 3) does not show any phenotype. The (CGG)90 fly expressing RNAi knockdown of Schlank results in enhancement of the CGG-associated neurotoxicity (column 4). Columns 5 and 6 show RNAi knockdown of Sk2 in WT and (CGG)90 flies, respectively, demonstrating that knockdown of Sk2 in the (CGG)90 fly results in enhancement of CGG-associated toxicity. Bottom panel shows the corresponding scanning electron microscopy images.
Purine metabolism is perturbed in FXTAS: knockdown of Ras (Impdh1) results in enhancement of premutation CGG repeat-mediated neurodegeneration
In addition to sphingolipid metabolism, metabolites in the purine metabolism pathway were also significantly altered in the presence of CGG (aged FXTAS versus aged WT mice, Fig. 5), as well as in the progression of FXTAS (aged FXTAS versus young FXTAS mice, Fig. 5). One of the candidate enzyme genes selected for the Drosophila genetic screen through manual curation was Impdh1, which encodes inosine 5′monophosphate dehydrogenase (Impdh). As the rate-limiting enzyme in guanine nucleotide biosynthesis, Impdh catalyzes the conversion of IMP into xanthosine monophosphate (XMP) [Fig. 5C, (30)]. The metabolomics data indicated a significant increase in the levels of IMP in the aged FXTAS mice compared with that in the aged WT mice (P = 0.0055, q = 0.0591; Fig. 5C), as well as a significant decline in the levels of xanthosine in the same comparison, which is generated from XMP (P = 0.0276, q = 0.1327; Fig. 5C). The increase in IMP coinciding with the decrease in levels of xanthosine in the aged FXTAS versus aged WT mice comparison led us to hypothesize that Impdh function may be significantly altered in FXTAS. When we performed the genetic screen in FXTAS Drosophila, upon knockdown of Ras, the fly ortholog of Imdph1, the fly progeny, demonstrated significant enhancement of the r(CGG)90-associated toxicity, while no toxicity was seen in the WT control (Fig. 6).
Figure 5.
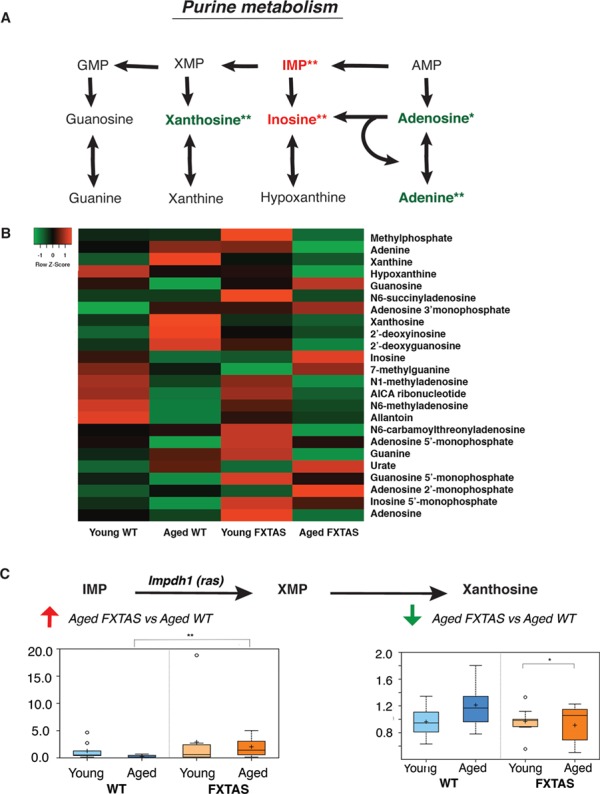
Purine metabolism is significantly perturbed in FXTAS mice. (A) Schematic depicting key components of the purine metabolism pathway and their perturbations in FXTAS versus WT mice comparisons. Red and green denote increased and decreased fold changes, respectively, in the pairwise comparisons. One asterisk (*) indicates changes in the aged FXTAS versus young FXTAS mice comparison, whereas two asterisks (**) indicate changes in the aged FXTAS versus aged WT mice comparison. Three asterisks (***) indicate changes in both comparisons. (B) A heatmap showing the metabolite alterations in purine metabolism for young WT, aged WT, young FXTAS and aged FXTAS mice. Heatmap was generated using the average metabolite intensity of each group, using average linkage hierarchical clustering in Heatmapper (52). Red indicates high and green indicates low intensity of the metabolite relative to the median (black). (C) IMP dehydrogenase [Impdh1 (Ras)] catalyzes the conversion of IMP to XMP in the rate-limiting step of guanine nucleotide biosynthesis. Box plots display significant differences between aged FXTAS versus aged WT mice in IMP (P = 0.0055, q = 0.0591) and xanthosine (P = 0.0276, q = 0.1327). X-axis indicates the genotype.
Figure 6.
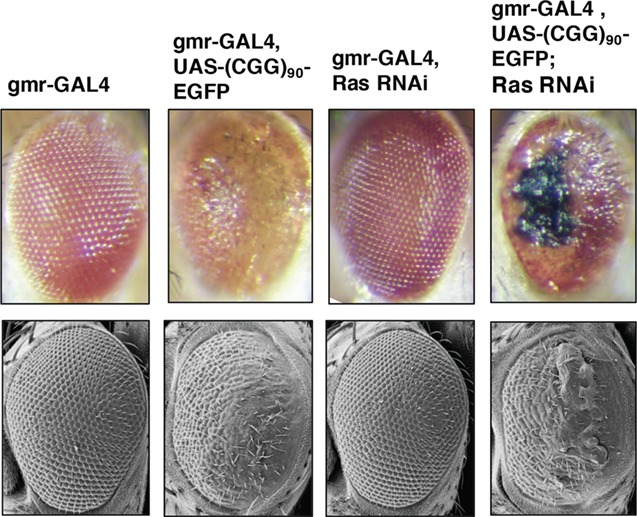
Knockdown of Ras enhances rCGG mediated neurodegeneration in FXTAS Drosophila. Knockdown of Ras (fly ortholog of Impdh) enhances r(CGG)90-mediated neurodegeneration in FXTAS Drosophila. Top panel displays the light microscopy, from left to right: WT control (gmr-GAL4) (column 1) and (CGG)90 control (gmr-GAL4, UAS-(CGG)90-EGFP) (column 2). WT fly expressing RNAi knockdown of Ras demonstrates no toxicity (column 3) while the (CGG)90 fly expressing RNAi knockdown of Ras demonstrates enhancement of the CGG-associated neurotoxicity (column 4). Bottom panel shows the corresponding scanning electron microscopy images.
Discussion
Metabolic profiling of mice cerebella revealed significant metabolic perturbations in the aging of FXTAS mice carrying r(CGG)90 in Purkinje cells, especially in pathways of lipid, amino acid and nucleotide metabolism. This finding correlates with the late-onset phenotype of FXTAS in human premutation carriers, suggesting that metabolic perturbations in the cerebellum may be associated with the neurodegenerative phenotype in human premutation carriers.
Importantly, in this study, we demonstrated successful use of Drosophila genetics for functional validation of the untargeted metabolic profiling data. As a result, we identified eight genetic modifiers of CGG toxicity, which include the enzymes ceramide synthase, sphingosine kinase (sphingolipid metabolism) and IMP dehydrogenase (purine metabolism). To the authors’ knowledge, this paper is the first report of metabolic profiling performed on the cerebella of a FXTAS mouse model and the first identification of Schlank(Cers5), Sk2 (Sphk1), Ras (Impdh1), CG8353 (Cda), CG4306 (Ggct), ChAT (ChAT), Pect (Pcyt2) and Ppat-dpck (Coasy) as genetic modifiers of the expanded CGG premutation repeat-mediated neurodegeneration in a FXTAS Drosophila model.
Sphingolipids have been implicated in multiple neurodegenerative disorders such as Alzheimer’s, Parkinson’s and Huntington’s diseases (24,31–33). Our study has identified the sphingolipid metabolic pathway and, in particular, the levels of ceramide, sphingosine and sphingosine-1-phosphate as being significantly perturbed in the progression of CGG toxicity in the mouse cerebellum (Fig. 3). Ceramide synthase (CerS) plays a central role in sphingolipid metabolism by synthesizing the backbone of all sphingolipids, ceramide, from sphingosine. Six ceramide synthase variants exist in mammals, and among them, Cers1 is expressed in the cerebellum (Purkinje cells), as well as the brain (neurons) and skeletal muscle (34). Schlank is the Drosophila ortholog of Cers1–6. Through Drosophila genetics, we have demonstrated that Schlank modulates CGG neurotoxicity caused by the expanded premutation (CGG)90 repeat. A previous literature has reported evidence linking CerS and cerebellum integrity, suggesting that the genetic interaction between the expanded CGG repeat and Cers may directly lead to disruptions in cerebellum integrity. In fact, Ginkel et al. generated CerS1-deficient mice that exhibited a foliation defect, progressive shrinkage and neuronal apoptosis in the cerebellum (35). In a separate study, Zhao et al. have reported that deficiency of CerS1 in mice results in progressive Purkinje cell loss and that loss of CerS1 significantly affects sphingolipid homeostasis, as well as protein and organelle homeostasis (36). Significantly, their work indicated that defects in sphingolipid metabolism can directly result in Purkinje cell death (36). In this study, we found that while shRNA-mediated knockdown Schlank did not have any significant effect on the WT fly eye, Schlank interacted with the expanded CGG repeat, resulting in a strong enhancement of the FXTAS rough-eye phenotype. Taken together, our data strongly support the hypothesis that in the presence of the expanded CGG premutation in FXTAS, the biological function of CerS is altered, resulting in perturbation of associated metabolites in the sphingolipid pathway and neurotoxicity, especially in Purkinje cells.
Sphingosine kinases are responsible for phosphorylating sphingosine to yield S1P. Sphingosine kinases can be stimulated by diverse stimuli—such as PDGF, VEGF and TNF-α to generate intracellular S1P (37). Once considered to be a mere intermediate, S1P is now known to be a critical signaling molecule for neural and vascular development (38) and plays a prominent role in various cellular processes such as stress response, growth, differentiation, as well as calcium balance (37). Intriguingly, S1P has been found to regulate cell growth (39,40) and to suppress programmed cell death (41), whereas the precursors of S1P—sphingosine and ceramide—have been found to promote growth arrest and cell death (42,43). This observation led Spiegel et al. to propose that the relative levels of ceramide and S1P determine cell fate, functioning as a “sphingolipid rheostat”.
In this study, we observed that S1P is significantly increased in the aged FXTAS versus the aged WT comparison. In our Drosophila genetic screen, knockdown of Sk2, an ortholog of SphK1 (and SphK2 to a lesser extent), resulted in enhanced CGG-associated neurotoxicity while the WT flies were not affected by the knockdown (Fig. 4). A potential explanation is that S1P may play a suppressive role against the neurotoxicity in FXTAS and that, in the presence of the premutation repeat, knockdown of Sk2 prevents the increased production of S1P and results in enhancement of the neurotoxic phenotype. Indeed, in the WT fly, Sk2 knockdown does not have an obvious effect, but in the presence of the CGG repeats, the flies demonstrate significant neurotoxicity. Furthermore, S1P’s role in calcium balance may also be linked to the increased toxicity in FXTAS, as FXTAS has been recently shown to be associated with dysregulation of calcium homeostasis (44). In light of these findings, activating SphK1 and/or increasing the intracellular concentration of S1P may be a potential path toward developing therapeutics for FXTAS.
Moreover, our study has also found that purine metabolism is significantly perturbed in the presence of CGG repeats in aged FXTAS versus young FXTAS mice cerebella. Disruptions in purine metabolism were also reported in the metabolic profiling of plasma from human FXTAS premutation carriers (28). Imbalances in purine synthesis can have significant biological consequences by influencing a myriad of processes such as replication, transcription, as well as DNA repair. Our finding that knockdown of Ras (Impdh1) results in a strong enhanced-eye phenotype in the FXTAS Drosophila suggests that genetic interaction between Impdh and CGG repeat may attribute to the disruptions observed in DNA metabolism in FXTAS.
It has not escaped our attention that Cers and Impdh catalyze the rate-limiting ‘gateway’ reactions for sphingolipid metabolism and guanine nucleotide synthesis. Though warranting further study, the results of our Drosophila genetic screen raise the possibility that the genetic interaction between such critical ‘gateway’ enzymes and the premutation CGG repeat may be the initiating factor in the global metabolic disruptions seen in FXTAS mice as well as human premutation carriers. In the meantime, it would also be beneficial to further explore the enzyme gene candidates validated through Drosophila genetics in this paper as potential treatment targets. This could open up another door to much-needed treatment opportunities for FXTAS patients (45).
Finally, to date, no molecular biomarker of FXTAS has been identified, which has delayed the diagnosis, treatment and prognosis of FXTAS patients. The 186 metabolites that were identified in this study to be significantly altered in the cerebella of FXTAS mice present a novel and useful resource in the search for biomarkers for FXTAS.
In this paper, we present a unique approach of combining high-throughput unbiased metabolic profiling with functional validation in Drosophila. Given the vast breadth and complexity of metabolomics, the interpretation of such data can often be limited to finding global trends or pathway alterations. However, as we demonstrate in this paper, the rapid, inexpensive and high-throughput nature of Drosophila genetic screening affords a facile method to validate the metabolomics findings by testing for genetic interactions. This method can be easily applied to other metabolomics analyses, thanks to the availability of Drosophila models for a variety of human diseases (46–49).
Materials and Methods
Animals
FXTAS mice expressing human FMR1 r(CGG)90 repeat in Purkinje neurons (published previously) and WT control littermates were housed, maintained and euthanized according to Institutional Animal Care and Use Committee guidelines at Emory University and Baylor College of Medicine (16).
Sample preparation for metabolomics
For each of the 4 groups [young WT (16–20 weeks), aged WT (55 weeks), young FXTAS (16–20 weeks), and aged FXTAS mice (55 weeks)], cerebellum tissue was collected from 9 mice and kept at −80°C. A total of 36 mice cerebellum tissues were sent for untargeted biochemical profiling at Metabolon, Inc. (Morrisville, NC). After the samples were homogenized and subjected to methanol extraction, they were split into aliquots for analysis by ultrahigh performance liquid chromatography/mass spectrometry (MS) in the positive (two methods: Pos Early and Pos Late) and negative (two methods: Neg and Polar) mode methods (50).
Compounds identification and quantification
Raw data were extracted, peak-identified and Quality Control-processed using Metabolon’s hardware and software as previously described (51). Compounds were identified by comparison to library entries of purified standards, based on the retention time/index, mass-to-charge ratio (m/z) and chromatographic data (including MS/MS spectral data). Library matches for each compound were checked for each sample and corrected if necessary. Peaks were quantified using area under the curve. The biochemical data were normalized to mass-to-volume extraction ratio, missing values were imputed with minimum observed values for each compound and the results were median scaled.
Statistical analysis of metabolites
Two-way ANOVA models were tested. Post hoc pairwise comparisons were used to identify significantly altered biochemicals differentiating the experimental groups (P < 0.05). The analysis was conducted using log transformed data in ArrayStudio, version 7.2 (OmicsSoft Corporation, Research Triangle Park, NC).
Manual curation selection criteria
Enzyme gene candidates were selected based on the following criteria: (1) the metabolite is present in the KEGG database, (2) there is a known gene encoding the enzyme involved in its synthesis/degradation, (3) there are (<5) enzymes involved in the synthesis/degradation of biochemical, (4) the reaction catalyzed by the enzyme is not denoted to be reversible in KEGG and (5), importantly, in order to test the gene candidates using a Drosophila genetic screen, gene candidates were limited to those that have fly orthologs and RNAi lines available.
Drosophila genetics
Transgenic flies expressing r(CGG)90 were previously described. The gmr-GAL4 and UAS-TRiP lines were obtained from Bloomington Stock Centre (Bloomington, IN, USA). All crosses were grown on standard medium at 25°C. After performing the crosses, progeny was collected and aged to ~7 days. The screen was performed by eye phenotype, which was visualized using light microscopy and confirmed with scanning electron microscopy.
RNA isolation and quantitative RT-PCR
Actin-GAL4 flies were crossed with the corresponding RNAi lines. Larvae were collected and homogenized in Trizol (Invitrogen, Carlsbad, CA) with pellet pestles (Fisher Scientific, Hampton, NH) and total RNA was extracted using the manufacturer’s protocol. cDNA was synthesized with SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA) using random hexamer primers. Real-time PCR was performed with primers in Supplementary Material, Table S3.
Scanning electron microscopy
Whole flies were dehydrated in increasing concentrations of ethanol (25, 50, 75 and 100%), then incubated for 1 h with hexamethyldisilazane (Electron Microscopy Sciences, Hatfield, PA). After removing the hexamethyldisilazane, the flies were dried overnight in a fume hood and subsequently analyzed using the Dual Stage Scanning Electron Microscope DS 130F (Topcon, Tokyo, Japan).
Supplementary Material
Funding
National Institutes of Health (NS051630 to D.L.N. and P.J., U54 HD083082 to D.L.N., 2T32GM007526 to Y.G., NS091859 to E.G.A. and P.J.).
References
- 1. Jacquemont S., Hagerman R.J., Leehey M., Grigsby J., Zhang L., Brunberg J.A., Greco C., Des Portes V., Jardini T., Levine R. et al. (2003) Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet., 72, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hagerman P.J. and Hagerman R.J. (2015) Fragile X-associated tremor/ataxia syndrome. Ann. N. Y. Acad. Sci., 1338, 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greco C.M., Berman R.F., Martin R.M., Tassone F., Schwartz P.H., Chang A., Trapp B.D., Iwahashi C., Brunberg J., Grigsby J. et al. (2006) Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain, 129, 243–255. [DOI] [PubMed] [Google Scholar]
- 4. Ariza J., Rogers H., Monterrubio A., Reyes-Miranda A., Hagerman P.J. and Martínez-Cerdeño V. (2016) A majority of FXTAS cases present with intranuclear inclusions within Purkinje cells. Cerebellum, 15, 546–551. [DOI] [PubMed] [Google Scholar]
- 5. Greco C.M., Hagerman R.J., Tassone F., Chudley A.E., Del Bigio M.R., Jacquemont S., Leehey M. and Hagerman P.J. (2002) Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain, 125, 1760–1771. [DOI] [PubMed] [Google Scholar]
- 6. Kremer E., Pritchard M., Lynch M., Yu S., Holman K., Baker E., Warren S., Schlessinger D., Sutherland G. and Richards R. (1991) Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science, 252, 1711–1714. [DOI] [PubMed] [Google Scholar]
- 7. Verkerk A.J.M.H., Pieretti M., Sutcliffe J.S., Fu Y.-H., Kuhl D.P.A., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F. et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell, 65, 905–914. [DOI] [PubMed] [Google Scholar]
- 8. Hagerman R.J. and Hagerman P.J. (2002) The fragile X premutation: into the phenotypic fold. Curr. Opin. Genet. Dev., 12, 278–283. [DOI] [PubMed] [Google Scholar]
- 9. Colak D., Zaninovic N., Cohen M.S., Rosenwaks Z., Yang W.-Y., Gerhardt J., Disney M.D. and Jaffrey S.R. (2014) Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science, 343, 1002–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dombrowski C., Lévesque S., Morel M.L., Rouillard P., Morgan K. and Rousseau F. (2002) Premutation and intermediate-size FMR1 alleles in 10 572 males from the general population: loss of an AGG interruption is a late event in the generation. Hum. Mol. Genet. 11, 371–378. [DOI] [PubMed] [Google Scholar]
- 11. Rousseau F., Rouillard P., Morel M.L., Khandjian E.W. and Morgan K.. Prevalence of carriers of premutation-size alleles of the FMRI gene—and implications for the population genetics of the fragile X syndrome. Am. J. Hum. Genet., 57, 1006–1018. [PMC free article] [PubMed] [Google Scholar]
- 12. Jacquemont S., Hagerman R.J., Leehey M.A., Hall D.A., Levine R.A., Brunberg J.A., Zhang L., Jardini T., Gane L.W., Harris S.W. et al. (2004) Penetrance of the fragile X–associated tremor/ataxia syndrome in a premutation carrier population. JAMA, 291, 460. [DOI] [PubMed] [Google Scholar]
- 13. Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B. and Hagerman P.J. (2001) Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology, 57, 127–130. [DOI] [PubMed] [Google Scholar]
- 14. Bontekoe C.J.M. (2001) Instability of a (CGG)98 repeat in the Fmr 1 promoter. Hum. Mol. Genet., 10, 1693–1699. [DOI] [PubMed] [Google Scholar]
- 15. Entezam A., Biacsi R., Orrison B., Saha T., Hoffman G.E., Grabczyk E., Nussbaum R.L. and Usdin K. (2007) Regional FMRP deficits and large repeat expansions into the full mutation range in a new fragile X premutation mouse model. Gene, 395, 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hashem V., Galloway J.N., Mori M., Willemsen R., Oostra B.A., Paylor R. and Nelson D.L. (2009) Ectopic expression of CGG containing mRNA is neurotoxic in mammals. Hum. Mol. Genet., 18, 2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin P., Zarnescu D.C., Zhang F., Pearson C.E., Lucchesi J.C., Moses K. and Warren S.T. (2003) RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron, 39, 739–747. [DOI] [PubMed] [Google Scholar]
- 18. Roessner U. and Bowne J. (2009) What is metabolomics all about? Biotechniques, 46, 363–365. [DOI] [PubMed] [Google Scholar]
- 19. Van Assche R., Temmerman L., Dias D.A., Boughton B., Boonen K., Braeckman B.P., Schoofs L. and Roessner U. (2015) Metabolic profiling of a transgenic Caenorhabditis elegans Alzheimer model. Metabolomics, 11, 477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johansen K.K., Wang L., Aasly J.O., White L.R., Matson W.R., Henchcliffe C., Beal M.F. and Bogdanov M. (2009) Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS One, 4, e7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burté F., Houghton D., Lowes H., Pyle A., Nesbitt S., Yarnall A., Yu-Wai-Man P., Burn D.J., Santibanez-Koref M. and Hudson G. (2017) Metabolic profiling of Parkinson’s disease and mild cognitive impairment. Mov. Disord., 32, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patti G.J., Yanes O. and Siuzdak G. (2012) Innovation: metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol., 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peng B., Li H. and Peng X.-X. (2015) Functional metabolomics: from biomarker discovery to metabolome reprogramming. Protein Cell, 6, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mielke M.M. and Lyketsos C.G. (2010) Alterations of the sphingolipid pathway in Alzheimer’s disease: new biomarkers and treatment targets? Neuromolecular Med., 12, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Havelund J.F., Heegaard N.H.H., Færgeman N.J.K. and Gramsbergen J.B. (2017) Biomarker research in Parkinson’s disease using metabolite profiling. Metabolites, 7, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mastrokolias A., Pool R., Mina E., Hettne K.M., Duijn E., Mast R.C., Ommen G., t Hoen P.A.C., Prehn C., Adamski J. et al. (2016) Integration of targeted metabolomics and transcriptomics identifies deregulation of phosphatidylcholine metabolism in Huntington’s disease peripheral blood samples. Metabolomics, 12, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trushina E., Dutta T., Persson X.-M.T., Mielke M.M. and Petersen R.C. (2013) Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS One, 8, e63644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giulivi C., Napoli E., Tassone F., Halmai J. and Hagerman R. (2016) Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem. J., 473, 3871–3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park J.-W., Park W.-J. and Futerman A.H. (1841) Ceramide synthases as potential targets for therapeutic intervention in human diseases. Biochim. Biophys. Acta, 1841, 671–681. [DOI] [PubMed] [Google Scholar]
- 30. Hedstrom L. (2009) IMP dehydrogenase: structure, mechanism, and inhibition. Chem. Rev., 109, 2903–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cutler R.G., Kelly J., Storie K., Pedersen W.A., Tammara A., Hatanpaa K., Troncoso J.C. and Mattson M.P. (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A., 101, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang G., Silva J., Dasgupta S. and Bieberich E. (2008) Long-chain ceramide is elevated in presenilin 1 (PS1M146V) mouse brain and induces apoptosis in PS1 astrocytes. Glia, 56, 449–456. [DOI] [PubMed] [Google Scholar]
- 33. Piccinini M., Scandroglio F., Prioni S., Buccinnà B., Loberto N., Aureli M., Chigorno V., Lupino E., DeMarco G., Lomartire A. et al. (2010) Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol. Neurobiol., 41, 314–340. [DOI] [PubMed] [Google Scholar]
- 34. Tidhar R. and Futerman A.H. (2013) The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim. Biophys. Acta, 1833, 2511–2518. [DOI] [PubMed] [Google Scholar]
- 35. Ginkel C., Hartmann D., vom Dorp K., Zlomuzica A., Farwanah H., Eckhardt M., Sandhoff R., Degen J., Rabionet M., Dere E. et al. (2012) Ablation of neuronal ceramide synthase 1 in mice decreases ganglioside levels and expression of myelin-associated glycoprotein in oligodendrocytes. J. Biol. Chem., 287, 41888–41902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao L., Spassieva S.D., Jucius T.J., Shultz L.D., Shick H.E., Macklin W.B., Hannun Y.A., Obeid L.M. and Ackerman S.L. (2011) A deficiency of ceramide biosynthesis causes cerebellar Purkinje cell neurodegeneration and lipofuscin accumulation. PLoS Genet., 7, e1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spiegel S. and Milstien S. (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol., 4, 397–407. [DOI] [PubMed] [Google Scholar]
- 38. Mizugishi K., Yamashita T., Olivera A., Miller G.F., Spiegel S. and Proia R.L. (2005) Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol., 25, 11113–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang H., Desai N.N., Olivera A., Seki T., Brooker G. and Spiegel S. (1991) Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol., 114, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olivera A. and Spiegel S. (1993) Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature, 365, 557–560. [DOI] [PubMed] [Google Scholar]
- 41. Cuvillier O., Pirianov G., Kleuser B., Vanek P.G., Coso O.A., Gutkind J.S. and Spiegel S. (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature, 381, 800–803. [DOI] [PubMed] [Google Scholar]
- 42. Hannun Y.A. and Obeid L.M. (2002) The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J. Biol. Chem., 277, 25847–25850. [DOI] [PubMed] [Google Scholar]
- 43. Kolesnick R. (2002) The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J. Clin. Invest., 110, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robin G., López J.R., Espinal G.M., Hulsizer S., Hagerman P.J. and Pessah I.N. (2017) Calcium dysregulation and Cdk5-ATM pathway involved in a mouse model of fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet., 26, 2649–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kong H.E., Zhao J., Xu S., Jin P. and Jin Y. (2017) Fragile X-associated tremor/ataxia syndrome: from molecular pathogenesis to development of therapeutics. Front. Cell. Neurosci., 11, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McGurk L., Berson A. and Bonini N.M. (2015) Drosophila as an in vivo model for human neurodegenerative disease. Genetics, 201, 377–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sen A. and Cox R.T. (2017) Fly models of human diseases. Curr. Top. Dev. Biol., 121, 1–27. [DOI] [PubMed] [Google Scholar]
- 48. Prüßing K., Voigt A. and Schulz J.B. (2013) Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener., 8, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wolf M.J., Amrein H., Izatt J.A., Choma M.A., Reedy M.C. and Rockman H.A. (2006) From the cover: drosophila as a model for the identification of genes causing adult human heart disease. Proc. Natl. Acad. Sci., 103, 1394–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Evans A., Bridgewater B.R., Liu, Q., M.W.,R.J, Dai, H., S.J., De Haven C.D. and Miller L.A.D. (2014) High resolution mass spectrometry improves data quantity and quality as compared to unit mass resolution mass spectrometry in high-throughput profiling metabolomics. Metabolomics, 04, 132 [Google Scholar]
- 51. DeHaven C.D., Evans A.M., Dai H. and Lawton K.A. (2010) Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform., 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Babicki S., Arndt D., Marcu A., Liang Y., Grant J.R., Maciejewski A. and Wishart D.S. (2016) Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res., 44, W147–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conway J.R., Lex A., Gehlenborg N. and Hancock J. (2017) UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics, 33, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.