

Abstract
Charcot–Marie–Tooth (CMT) neuropathies are a group of genetic disorders that affect the peripheral nervous system with heterogeneous pathogenesis and no available treatment. Axonal neuregulin 1 type III (Nrg1TIII) drives peripheral nerve myelination by activating downstream signaling pathways such as PI3K/Akt and MAPK/Erk that converge on master transcriptional regulators of myelin genes, such as Krox20. We reasoned that modulating Nrg1TIII activity may constitute a general therapeutic strategy to treat CMTs that are characterized by reduced levels of myelination. Here we show that genetic overexpression of Nrg1TIII ameliorates neurophysiological and morphological parameters in a mouse model of demyelinating CMT1B, without exacerbating the toxic gain-of-function that underlies the neuropathy. Intriguingly, the mechanism appears not to be related to Krox20 or myelin gene upregulation, but rather to a beneficial rebalancing in the stoichiometry of myelin lipids and proteins. Finally, we provide proof of principle that stimulating Nrg1TIII signaling, by pharmacological suppression of the Nrg1TIII inhibitor tumor necrosis factor-alpha-converting enzyme (TACE/ADAM17), also ameliorates the neuropathy. Thus, modulation of Nrg1TIII by TACE/ADAM17 inhibition may represent a general treatment for hypomyelinating neuropathies.
Introduction
Myelin is an electrically insulating sheath formed in the peripheral nervous system (PNS) by a protrusion of the Schwann cell plasma membrane, which enwraps axons facilitating nerve impulse transmission and supporting long-term integrity of axons (1,2). Charcot–Marie–Tooth (CMT) diseases are the most common demyelinating and dysmyelinating hereditary neuropathies, with onset in childhood or early adulthood. CMTs are characterized by muscular weakness, tremor, hind-limbs atrophy and foot deformities, at present there is no treatment (3–5). CMTs are due to mutations in at least 70 different genes, and although clinical and pathological features partially overlap among CMT subtypes, the analysis of the molecular mechanism has revealed a highly heterogeneous pathogenesis (6). As such, it would be very useful to identify unifying therapeutic strategies. Most CMT neuropathies display altered levels of myelination and, therefore, the development of strategies aimed at restoring normal levels of myelin could result in an effective approach for their collective treatment.
Neuregulin 1 type III (Nrg1TIII), a member of the NRG1 family of proteins, is an essential instructive signal in peripheral myelination. Axonal Nrg1TIII drives a Schwann cell binary choice between a myelinating or non-myelinating phenotype (7), and the levels of Nrg1TIII determine the amount of myelin and its thickness (7,8). Nrg1TIII mostly acts through signaling pathways that control master regulators such as the transcription factor Krox20 (Egr2 gene), which activates genes for the synthesis of myelin proteins and lipids (9,10). Nrg1TIII activity is regulated by proteolytic shedding, the β-secretase BACE1 cleaves Nrg1 and positively regulates myelination (11,12), whereas the α-secretase TACE cleaves Nrg1TIII to inhibit myelination (13). Importantly, recent work has indicated that genetic overexpression or soluble administration of Nrg1 overcomes impaired nerve development in rodent models of CMT1A (14).
Among CMTs, those due to myelin protein zero (MPZ) mutations (CMT1B) are relatively common (4,15). MPZ encodes for P0, the most abundant glycoprotein of peripheral myelin (16,17). At least 200 mutations in MPZ have been identified and the phenotype varies widely from mild to more severe forms (15,18,19). We have generated a series of transgenic mouse models of CMT1B that present various degrees of hypomyelination, demyelination and inefficient remyelination of peripheral nerves and shown that the molecular mechanisms include both loss and gain of toxic function (20). While it is reasonable to expect that augmenting Nrg1 signaling rescues neuropathies due to P0 loss of function, it is important to understand if Nrg1 signaling can also overcome the intoxicated pathways and ameliorate neuropathy when the mutations determine a complex gain-of-function scenario. This is very relevant since in humans pure P0 loss-of-function mutations are very rare, and for most mutations the mechanism includes toxic gain of function. For example, deletion of serine 63 in the extracellular domain of P0 (P0S63del) in transgenic mice causes a CMT1B-like neuropathy that mimics the corresponding human disease (20). In a normal condition, P0 is targeted from the synthetic pathway to the myelin sheath (21,22). In contrast, P0S63del is misfolded and almost completely retained in the endoplasmic reticulum (ER), where it elicits an unfolded protein response (UPR) (20,23,24). Importantly, intracellular retention of mutant proteins has been described for CMT1B (25) and for several other CMT disease genes (for example PMP22 and Cx32) and some of these also cause an UPR (26–29).
Here we show that genetic or pharmacological modulation of Nrg1TIII activity improves hypomyelination and neurophysiology in the P0S63del model, without exacerbating the ER stress due to the toxic gain of mutant P0 function. Surprisingly, this improvement is independent of Krox20 regulation. Our data open new prospects for the identification of a therapy for hypomyelinating CMTs.
Results
Overexpression of Nrg1TIII ameliorates electrophysiological and morphological parameters in a CMT1B mouse model
In many CMT neuropathies, the disease mechanism includes toxic gain of function (4). For example in S63del mice, a model of CMT1B that manifests developmental hypomyelination followed by progressive demyelination, the misfolded P0S63del protein accumulates in the ER and activates an UPR (20,23). To test whether augmenting the key pro-myelinating axonal signal Nrg1TIII (7) could overcome the intoxicated process and ameliorate hypomyelination, demyelination and remyelination, we crossed S63del mice with transgenic mice that overexpress an HA-Nrg1TIII (HANI) fusion protein (30,31). Western blot (WB) analysis on spinal cord lysates, probed with anti-HA antibody, confirmed the expression of the HANI transgene in HANI/+ and HANI/+//S63del mice (Supplementary Material, Fig. S1A).
S63del mice manifest impaired motor capacity, reduced nerve conduction velocity (NCV) and an increased F-wave length (FWL), similar to the alterations found in patients (20,32). Neurophysiological analysis on 6-month-old mice showed that overexpressing Nrg1TIII (HANI/+//S63del) in S63del mice resulted in a significant increase of NCV, that returned closer to wild-type (WT) values as compared with neuropathic mice (Fig. 1A and D). Similarly, the FWL was decreased to almost WT level in HANI/+//S63del (Fig. 1B and C). No significant modifications were observed in either compound motor action potential (CMAP) amplitude or distal latency (Fig. 1D). These results indicate that genetic overexpression of Nrg1TIII leads to a significant amelioration of neurophysiological parameters in a mouse model of CMT1B.
Figure 1.
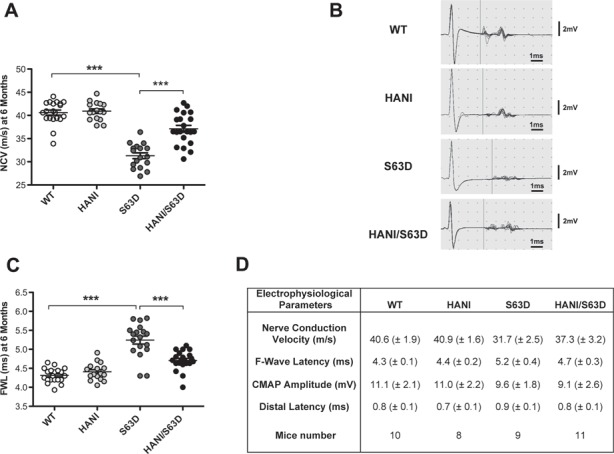
Overexpression of Nrg1TIII ameliorates neurophysiological parameters in P0S63del-CMT1B mice. Analyses were performed on 6-month-old mice. (A) NCV is reduced in S63del mice as compared with WT and is rescued by Nrg1TIII overexpression. (B) Representative original recording indicating the onset of the F-wave (vertical bar). (C) F-wave latency is increased in S63del mice as compared with WT and shows a significant amelioration in HANI/+//S63del as compared with S63del mice. (D) CMAP amplitude and distal latency do not show changes among the genotypes. Data represent the mean ± SEM. Number of animals per genotype is reported in (D). ***P-value < 0.001 by one-way ANOVA with Bonferroni’s multiple comparison test.
To corroborate the electrophysiological findings, we performed morphological examinations of sciatic nerves starting at postnatal day 28 (P28). As expected, electron microscopy (EM) showed hypomyelination in S63del nerves as compared with wild-type (20), whereas HANI/+ nerves were clearly hypermyelinated (Fig. 2A) (8,31). Remarkably, myelin thickness was increased in HANI/+//S63del nerves as compared with S63del (Fig. 2A). Morphometric analysis confirmed these following observations: g-ratios (axon diameter/fiber diameter) were decreased in HANI/+ and HANI/+//S63del as compared with WT and S63del, respectively (WT 0.65 ± 0.006, HANI/+ 0.54 ± 0.012, S63del 0.69 ± 0.005, HANI/+//S63del 0.62 ± 0.002) (Fig. 2B and C). The increase in myelin thickness in HANI/+//S63del was predominant in the small to medium caliber axons (from 1 to 3.9 μm), whereas those with larger diameter (>4 μm) remained hypomyelinated with a g-ratio similar to S63del nerves (Supplementary Material, Fig. S1B). We also observed that Nrg1TIII overexpression led to a small increase in the number of myelinated small caliber fibers (1–1.9 μm) in both WT and S63del backgrounds (Supplementary Material, Fig. S1C), suggesting that some of the axons in the proximity of 1 μm of diameter, normally not myelinated, may become myelinated when they express higher levels of Nrg1TIII.
Figure 2.
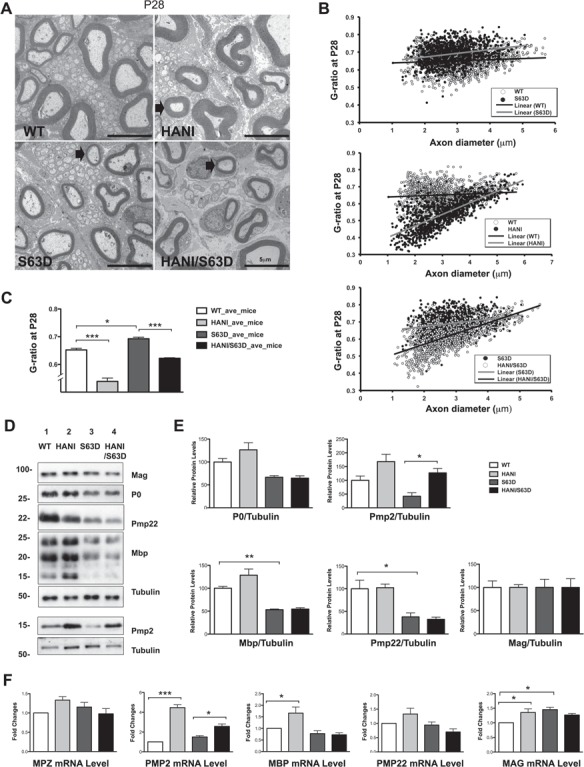
Overexpression of Nrg1TIII leads to thicker myelin without increase in the expression of most myelin proteins. (A) EM of P28 sciatic nerves. HANI mice show thicker myelin (arrow) as compared with WT. Hypomyelination in S63del is rescued in small and medium caliber axons (arrow) in HANI/+//S63del nerves (size bar, 5 μm). (B) G-ratio analysis performed on sciatic nerve cross sections at P28 and (C) relative quantification (average g-ratio values WT 0.64 ± 0.006, HANI/+ 0.53 ± 0.012, S63del 0.69 ± 0.005, S63del//HANI/+ 0.62 ± 0.002). Eight to ten microscopic fields per mouse were analyzed. Three to five mice per genotype were used. (D) Western analysis on sciatic nerve lysates at P28 shows no significant increase in myelin proteins in mice overexpressing Nrg1TIII, except for PMP2. Tubulin was used as loading control. One representative blot of three is shown. (E) Protein levels as measured by densitometric analysis. (F) mRNA expression levels of myelin proteins measured via qRT-PCR. PMP2 mRNA is increased in mice overexpressing Nrg1TIII, MBP shows a small increase in HANI versus WT. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA with Bonferroni’s multiple comparison test; data represent the mean ± SEM. Each experiment was repeated three times on different pools of three nerves per genotype.
Nrg1TIII overexpression was also accompanied by whole nerve hypertrophy (Supplementary Material, Fig. S1D). Previous works have shown that Thy-1.2 promoter-driven overexpression of Nrg1TIII does not alter Schwann cell numbers (8,31), and the overall number of myelinated axons per nerve section was not significantly changed in HANI as compared with WT (WT 3951 ± 210, HANI 4298 ± 35, P = 0.15) or in HANI/+//S63del as compared with S63del (S63del 3210 ± 58, HANI/+//S63del 3372 ± 105, P = 0.25) (Supplementary Material, Fig. S1F). This suggests that the increase in nerve size is mostly due to hypermyelination as well as reduced fibers density (Supplementary Material, Fig. S1G), indicative of increased extracellular matrix deposition. EM analysis confirmed the increase in spacing in between myelinated fibers in HANI as compared with WT and in HANI/+//S63del as compared with S63del, with evident collagen deposition (Supplementary Material, Fig. S2A). Intriguingly, among the extracellular matrix components, we detected a significant increase of fibronectin, known to be abundantly expressed in regenerating peripheral nerves (33), in HANI/+//S63del (Supplementary Material, Fig. S2B), suggesting that NRG1TIII overexpression may provide a more regenerative-permissive environment in neuropathy.
To evaluate whether Nrg1TIII overexpression was affecting myelin maintenance, we performed both morphological and morphometric analysis on sciatic nerves at 6 months, nerves overexpressing the HANI transgene were still slightly hypertrophic, although to a lesser extent as compared with P28 (Supplementary Material, Fig. S1D and E). Thicker myelin was still a feature of HANI/+ mice (Supplementary Material, Fig. S3A) and rescue of hypomyelination was again found mostly in small and medium caliber axons in HANI/+//S63del mice (Supplementary Material, Fig. S3B and C). Since CMT1B is characterized by progressive demyelination (20), we also evaluated the number of demyelinated fibers and onion bulbs at 6 months of age, but we did not find significant differences in HANI/+//S63del as compared with S63del nerves (Supplementary Material, Fig. S4C). This suggests that increased Nrg1TIII expression partially corrects hypomyelination without rescuing demyelination. All the aforementioned observations were confirmed at 12 months of age, indicating that, even at later stages, Nrg1TIII overexpression does not negatively affect myelin maintenance (Supplementary Material, Fig. S4D).
Overexpression of Nrg1TIII may act through a Krox20-independent pathway
During peripheral nerve development the binding of Nrg1TIII to ErbB tyrosine kinase receptors (ErbB2/3 heterodimers) on the Schwann cell surface, triggers the activation of intracellular signaling pathways, such as PI3K/p-Akt and Erk1/2-MAPK, that lead to myelin protein and lipid gene expression, mostly acting through the transcription factor Krox20 (9,10). It would be expected that the increase in myelin thickness due to Nrg1TIII overexpression should be accompanied by a concomitant increase in myelin proteins or lipids. We therefore performed both gene expression and biochemical analysis on P28 sciatic nerves extracts for the major proteins of compact and non-compact myelin. Surprisingly, WB as well as mRNA analysis showed only a small increase of P0, Mbp, Pmp22 and Mag expression in HANI/+ as compared with WT, whereas no differences were found in HANI/+//S63del as compared with S63del mice (Fig. 2D–F). Interestingly, only peripheral myelin protein 2 (Pmp2, P2 or Fabp8), a protein with lipid binding activity in peripheral nerve myelin (34–36), showed a strong increase of gene and protein expression in HANI/+ and HANI/+//S63del mice as compared with controls (Fig. 2D–F). Similar results were found in sciatic nerves lysates at 6 months of age, where we could detect only a slight increase of P0 and Pmp22 protein expression in S63del mice overexpressing the HANI transgene (Supplementary Material, Fig. S5A and B). Overall, our data indicated that despite Nrg1TIII overexpression there is only a minor effect on myelin mRNA and proteins. Therefore, we checked for the activation of the pathways downstream Nrg1TIII. The HANI transgene is highly expressed in spinal cord extracts (Supplementary Material Fig. S1A), but the HA-tag is barely detectable in nerves (31) (data not shown). However, by probing P28 sciatic nerve extracts with an antibody against the C-terminus of Nrg1 (all isoforms), we could detect, especially in the S63del background, an increase of the cleaved active isoform of Nrg1 in mice expressing the transgene (Fig. 3B). As expected, the levels of phosphorylation of both Akt (Ser473) and Erk1/2 were significantly increased in HANI/+ and HANI/+//S63del nerves as compared with controls, suggesting that HA-tagged Nrg1TIII was correctly activating the PI3K and MAPK signaling pathways (Fig. 3B). Paradoxically, however, we did not find any increase of Krox20 mRNA and protein expression (Fig. 3B), which could explain why myelin genes and proteins are not significantly affected in sciatic nerves overexpressing Nrg1TIII. This may suggest that Nrg1TIII rescues hypomyelination in S63del nerves through a Krox20-independent mechanism. Finally, no significant differences in phospho-Akt (Ser473), phospho-Erk1/2 and Krox20 levels were detected at 6 months of age (Supplementary Material Fig. S4A and B and data not shown), suggesting that Nrg1TIII signaling gets attenuated with time.
Figure 3.
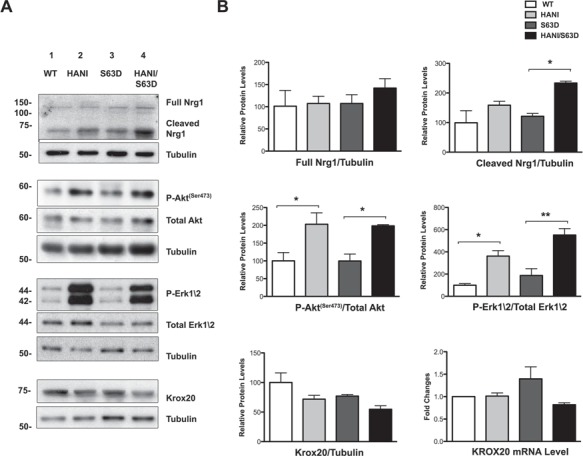
The Akt and Erk1/2 signaling pathways are upregulated by Nrg1TIII overexpression. (A) WB analysis of P28 sciatic nerves lysates. No differences in the endogenous full-length form of Ngr1 (140 kDa) were detected among the four genotypes, whereas the cleaved (65–70 kDa) active form was increased in particular in HANI/+//S63del as compared with S63del. In HANI/+ and HANI/+//S63del mice, both Akt (Ser473) and Erk1/2 phosphorylation are increased as compared with S63del and WT mice, respectively. Krox20 protein expression is slightly, but not significantly decreased in HANI/+//S63del as compared with S63del mice. No differences were detected in Krox20 mRNA expression by qRT-PCR (B, lower right graph). Tubulin was used as loading control. One representative experiment of three is shown. (B) Densitometric quantification of full-length and cleaved Nrg1 forms, p-Akt (ser473), p-Erk1/2 and Krox20 protein levels. *P < 0.05 and **P < 0.01 by one-way ANOVA with Bonferroni’s multiple comparison test.
Nrg1TIII overexpression increases myelin lipid content
One possible explanation for the increase in myelin thickness without corresponding increase in myelin proteins is an alteration in myelin packing; we thus checked myelin periodicity in sciatic nerves at P30. No gross alteration of the myelin sheath periodicity was found among all four genotypes (Supplementary Material, Fig. S6), suggesting that myelin is normally compacted. These observations could therefore be predictive of an altered protein density in the myelin sheath. Indeed, immuno-EM analysis (IEM) revealed a trend towards a reduction of P0 density in the myelin of mice overexpressing Nrg1TIII, particularly in the S63del background (Fig. 4A and B).
Figure 4.
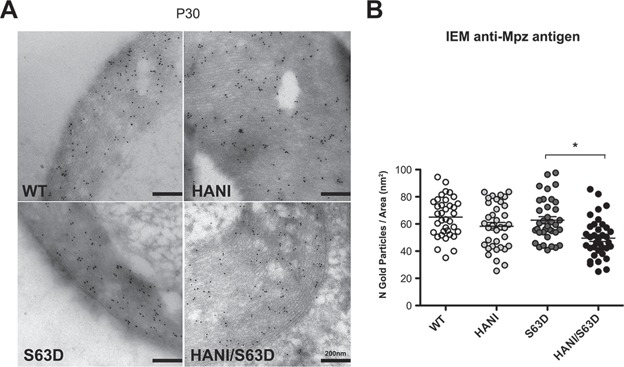
P0 protein density is reduced in mice expressing the HANI transgene. (A) IEM for P0 in transverse sections of P30 sciatic nerves. (B) Quantification of the number of gold granules per myelin area shows a trend towards the reduction of P0 density in HANI/+ and HANI/+//S63del myelin as compared with WT and S63del, respectively. Twelve sciatic nerve images per genotype were counted from three mice per genotype. *P-value = 0.03 by Student’s t-test; error bars represent SEM.
In contrast to most biological membranes, myelin is characterized by a higher lipids to proteins ratio (~75:25) (37,38). Thicker myelin, with reduced protein density could imply a relative increase of the total lipid content. To test this hypothesis, we performed a lipidomic analysis on P28 sciatic nerves. We detected increased levels of free cholesterol (typical of the myelin sheath) in mice overexpressing the HANI transgene as compared with WT, whereas no significant differences were found between S63del and HANI/+//S63del mice (Fig. 5A). Moreover, most saturated (palmitic acid, C16:0; stearic acid, C18:0; behenic acid, C22:0 and lignoceric acid, C24:0) and unsaturated (oleic acid, C18:1; linoleic acid, C18:2; erucic acid, C22:1 and nervonic acid, C24:1) fatty acids showed increased levels in mice harboring the HANI transgene (Fig. 5B). Lipids are important determinants of myelin structure and function, and proper lipid stoichiometry can be indicative of myelin integrity. We calculated the desaturation index (ratio 18:0/18:1) and the membrane fluidity index (ratio 18:1/18:2) (35) that showed no alteration, in accordance with a thicker, but still properly compact myelin. Taken together these data suggest that overexpression of Nrg1TIII determines a thickening of the myelin sheath that appears to be more dependent on an increase in lipids rather than proteins content.
Figure 5.
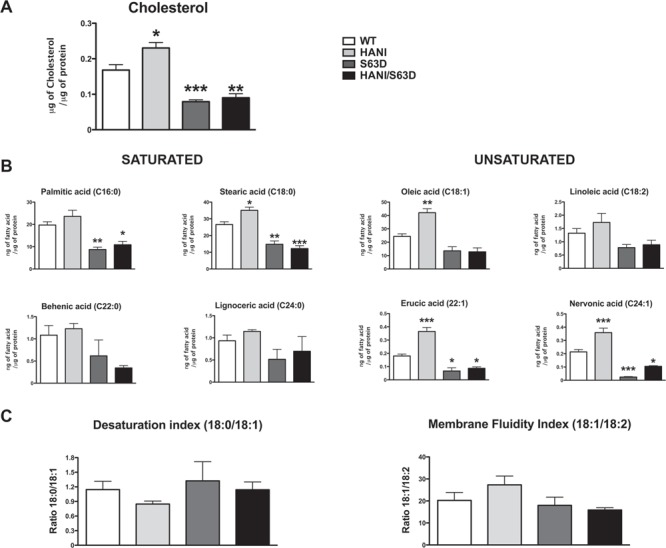
Nrg1TIII overexpression increases cholesterol and fatty acids levels. Lipidomic analysis was performed on P28 sciatic nerves. (A) The level of free cholesterol increased in HANI as compared with WT, whereas no differences were detected between S63del and HANI/+//S63del nerves. (B) Levels of total saturated (palmitic acid, C16:0; stearic acid, C18:0; behenic acid, C22:0 and lignoceric acid, C24:0) and total unsaturated (oleic acid, C18:1; linoleic acid, C18:2; erucic acid, C22:1 and nervonic acid, C24:1) fatty acids and (C) the relative desaturation index (ratio 18:0/18:1) and membrane fluidity index (ratio 18:1/18:2). Data are expressed as μg of cholesterol or ng of fatty acid normalized to μg of total proteins. *P < 0.05, **P < 0.01, ***P < 0.001 versus WT by one-way ANOVA with Bonferroni’s multiple comparison test. Five sciatic nerves from different animals per genotype were analyzed.
Nrg1TIII overexpression does not alter the stress response in S63del Schwann cells
P0S63del is a misfolded protein that fails to be incorporated into myelin and is retained in the ER where it activates a dose-dependent UPR (20,23). In our model, P0S63del is expressed by an MPZ-based transgene that faithfully parallels WT P0 expression (39). It was therefore conceivable that overexpressing Nrg1TIII in S63del mice could increase P0 mutant protein expression, further intensifying the toxic gain of function and potentially worsening the phenotype. However, we observed an amelioration of the neuropathic phenotype coupled to a lack of increase in myelin genes and proteins expression. Importantly, an allelic discrimination assay showed that the ratio between WT and mutant P0 mRNA remained unaltered in S63del and HANI/+//S63del nerves (Supplementary Material, Fig. S7D) strongly suggesting that, just like the other myelin proteins, P0S63del expression was not significantly affected by Nrg1TIII overexpression. To further assess this, we analyzed the expression of UPR markers in HANI/+//S63del and littermate control mice at P28 and 6 months. As previously shown, immunoglobulin heavy chain-binding protein (BiP), Grp94 and P-eIF2alpha proteins, as well as BiP, CAATT enhancer-binding protein homologous protein (Chop) and Xbp1s mRNAs were strongly increased in S63del nerves (20,23,24). However, we did not detect any change in the expression of these UPR markers in HANI/+//S63del indicating that after Nrg1TIII overexpression, the UPR was still active at levels comparable to those of S63del nerves (Fig. 6A–C and Supplementary Material, Fig. S7A–C).
Figure 6.
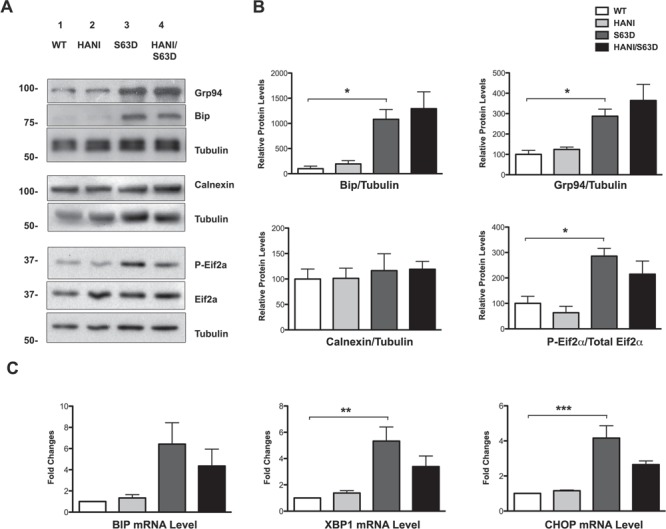
ER stress levels do not increase in S63del/HANI/+ mice. (A) WB analysis of P28 sciatic nerves lysates shows activation of the ER stress markers Bip/Grp78, Grp94 and P-eIF2-alpha in S63del mice as compared with WT. No further increase was detected in the expression levels of these markers between S63del and HANI/+//S63del mice. Tubulin was used as loading control. One representative experiment of three is shown. (B) Densitometric quantification of Bip, Grp94, calnexin and p-eIF2-alpha relative protein levels. (C) qRT-PCR for BiP, CHOP and spliced XBP-1 mRNA in P28 sciatic nerves. All the stress makers are increased in S63del as compared with WT, but we detected no differences in their levels of expression between S63del and HANI/+//S63del nerves. Each experiment was repeated three times on different pools of three nerves per genotype. *P < 0.05, **P-value < 0.01 and ***P < 0.001 by one-way ANOVA with Bonferroni’s multiple comparison test.
Recently, we have also shown that in S63del Schwann cells the expression of negative regulators of myelination such as Sox2 and Id2, which are normally downregulated after birth, remains sustained into adulthood (40). Importantly, this overexpression is an adaptive mechanism by which the Schwann cell reduces mutant P0 toxicity and limits demyelination (40). We thus wondered whether this response was altered by Nrg1TIII overexpression. qRT-PCR and WB analysis, performed on P28 sciatic nerve extracts, showed that as expected both Sox2 and Id2 were increased in S63del nerves, but that they remained similarly elevated in HANI/+/S63del (Fig. 7A and B).
Figure 7.
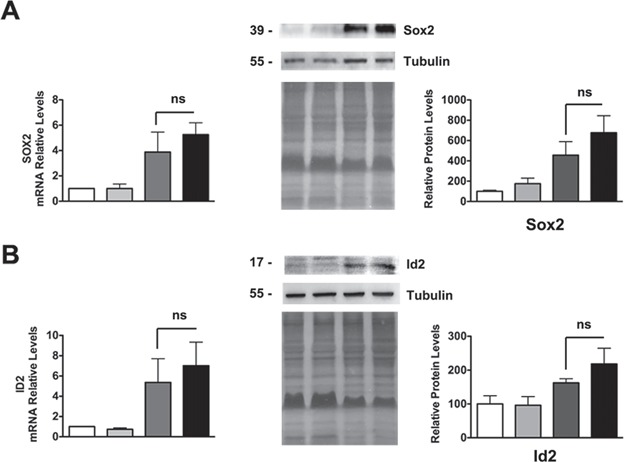
The expression of negative regulators of myelination is not altered by Nrg1TIII overexpression. qRT-PCR and WB analysis (with relative densitometric quantification) on P28 nerves for Sox2 (A) and Id2 (B). Both factors are increased in S63del nerves as compared with WT, but remained similarly increased in HANI/+//S63del mice. Each qRT-PCR experiment was repeated three times on different pools of three nerves per genotype. For WB, one representative experiment of three is shown. Tubulin was used as loading control, equal loading is also shown by coomassie staining; ns, not significant, by one-way ANOVA with Bonferroni’s multiple comparison test.
Overall, our data suggest that Nrg1TIII overexpression ameliorates the phenotype of a CMT1B mouse model without further intensifying the toxic gain-of-function mechanism.
Inhibition of TACE ameliorates myelination in neuropathic DRG explant cultures
Taken together our data suggest that the modulation of Nrg1TIII activity may represent a suitable approach to treat MPZ-related neuropathies. La Marca and colleagues have shown that the α-secretase TACE cleaves Nrg1TIII thus limiting the amount of functional Nrg1TIII expressed on the axonal surface and inhibiting myelination in early development (13). TACE null mice are hypermyelinated, partially mimicking the phenotype observed in transgenic mice overexpressing Nrg1TIII. This, together with our data, predicts that inhibitors of TACE would activate Nrg1TIII and promote myelination. BMS-561392 is a highly selective TACE inhibitor, already employed in phases I and II clinical trials, as a treatment for rheumatoid arthritis (41,42). In a preliminary set of experiments, we treated P15 WT and S63del mice with daily intraperitoneal (i.p.) injections of BMS-561392 for 10 days. Unfortunately, bioavailability analysis revealed almost undetectable levels of BMS-561392 in both WT and S63del sciatic nerves suggesting that this drug does not efficiently cross the blood nerve barrier (BNB) (Supplementary Material, Fig. S8B). Thus, to test the potential of BMS-561392 in promoting myelination, we used organotypic explant cultures of myelinating dorsal root ganglia (DRGs), a reliable model of ex vivo myelination. We treated WT and S63del DRGs with 1 μm BMS-561392 for 2 weeks along with controls (untreated and DMSO-treated DRGs) (Fig. 8A). Staining for myelin basic protein (MBP) clearly showed an increase in the number of myelinated segments in both S63del (Fig. 8C) and WT-treated samples (Fig. 8B and Supplementary Material, Fig. S8A), indicating that TACE inhibition ameliorates myelination, and suggesting that novel inhibitors with improved BNB permeability could represent promising therapeutic candidates for hypomyelinating CMTs.
Figure 8.
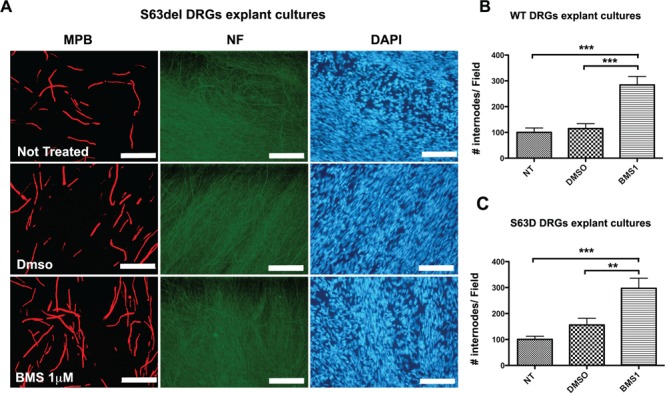
Pharmacological treatment with the TACE inhibitor BMS-561392 ameliorates myelination in DRGs explant cultures. Myelinating DRGs were treated with 1 μm BMS (BMS1) for 2 weeks. As controls, not treated and DMSO-treated DRGs were analyzed. (A) Immunostaining for MBP showing the increase in the number of MBP-positive segments in S63del-treated samples (size bar, 100 μm). Quantification of internode number in (B) WT (see also Supplementary Material, Fig. 8A) and (C) S63del DRGs, normalized to the untreated control. Note how treatment with BMS increases the number of internodes in both the genotypes with similar magnitude. Ten to fifteen DRGs per condition from three independent dissections were analyzed. **P < 0.01 and ***P < 0.001 by one-way ANOVA with Bonferroni’s multiple comparison test; error bars represent SEM.
Discussion
The analysis of the molecular mechanism underlying CMTs has revealed an extensive variety in pathogenesis (20,43,44) so that the development of a specific therapy for each subtype would be difficult to achieve. The identification of common approaches for the treatment of hereditary neuropathies, independently of their pathogenetic mechanism, is therefore highly desirable.
As an essential regulatory signal-driving myelination in the PNS (7,8), Nrg1TIII is an excellent candidate for the development of general therapeutic approaches for CMTs. Indeed, recent work has shown that the genetic or pharmacological reduction of Nrg1TIII signaling rescues the phenotype in mouse models characterized by focal hypermyelination (45).
Relying on the same concept, we hypothesized that increasing Nrg1TIII could ameliorate demyelinating neuropathies characterized by reduced levels of myelination. However, whereas the increase of Nrg1 signaling is expected to rescue neuropathies where loss of function is the prevalent mechanism (see accompanying paper from Belin et al.), in the presence of a toxic myelin protein it could potentially have detrimental effects. Here we showed that the genetic overexpression of Nrg1TIII ameliorates the neuropathic phenotype of a preclinical mouse model of CMT1B characterized by P0 misfolding and UPR activation (20,23) without aggravating the toxic gain of function. In this context, Nrg1TIII appears to act through a Krox20-independent mechanism, which determines a readjustment in myelin protein–lipid stoichiometry. Finally, we provide proof of principle that pharmacological enhancement of Nrg1 signaling may represent an appealing therapeutic approach.
Nrg1TIII overexpression activates Krox20-independent signaling pathways
PI3K/Akt and MAPK/Erk1/2 pathways are known downstream effectors of Nrg1 and thought to trigger Krox20-dependent myelin and lipids genes activation (9,10). For instance, the upregulation of Akt phosphorylation has been directly linked to an increased activation of Nrg1TIII, both in vivo and in vitro (13,46,47). However, following HANI transgene expression, the activation of both Akt(Ser473) and Erk1/2 was uncoupled from Krox20 and myelin genes levels. Intriguingly, recent work has revealed a Krox20-independent signaling pathway in a mouse model overexpressing a constitutively active Akt1 isoform (MyrAkt) in Schwann cells. The regulatory mechanism appears to involve mammalian target of rapamycin (mTORc1) in regulating myelin thickness in the PNS, without affecting Egr2/Krox20 transcription levels (48). Interestingly, a mild but sustained constitutive expression of the protein kinase Mek1 in Schwann cells (Mek1DD mouse), leads to a progressive and redundant hypermyelination in peripheral nerves. In these mice, the enhancement of Erk1/2 phosphorylation, but not Akt, induces robust protein synthesis without significantly affecting myelin mRNA levels. This effect is partially mediated by an mTORc1-dependent regulatory mechanism (49). These observations corroborate the idea that the exogenous overexpression of Nrg1TIII could enhance myelin growth in sciatic nerves through either the PI3K/Akt(Ser473) or the MAPK/Erk1/2 pathway, independently from Krox20 and myelin genes levels. However, in our context we did not observe any significant alteration downstream of the mTORc1 signaling pathway (data not shown), thus ruling out that the HANI transgene overexpression may act through mTOR to enhance protein translation and suggesting a different mechanism, possibly acting on lipids (see below).
The uncoupling of Krox20 from upstream Nrg1TIII signaling may paradoxically underlie the rescue in the S63del/HANI nerves. In fact, in S63del mice, the UPR and the resulting neuropathic phenotype are remarkably dose-dependent. Mice with higher levels of expression of P0S63del (S63del-H mice, 200% overexpression) have higher stress levels and a very severe phenotype as compared with the S63del mice used in this study (S63del-L, 60% overexpression) (20). The fact that, like most myelin genes the expression levels of P0S63del were not directly affected, as confirmed by lack of increase in UPR markers, probably allowed Schwann cells to increase myelination without further intoxicating the system.
The overexpression of Nrg1TIII might determine a shift in myelin proteins–lipids stoichiometry
The thicker myelin of HANI overexpressing mice had no alterations in periodicity and compaction, despite the reduction in major myelin proteins, as epitomized by the decrease of P0 protein density. This suggests a general change in myelin composition. Normally, myelin is composed by 20% of proteins and by 80% of lipids such as free-cholesterol and fatty acids (35,37,50,51). In the mice overexpressing Nrg1TIII, cholesterol along with saturated and unsaturated fatty acids was increased. Interestingly, the increase in lipids appeared more evident in the WT than in S63del background. This could depend on the fact that P0S63del expression leads to a general downregulation in cholesterol and lipid synthesis genes (24), which may partially counteract the effects of Nrg1TIII. In agreement with our observations, recent work has identified the transcription factor Maf as a crucial player in regulating myelination and cholesterol biosynthesis downstream from Nrg1 (52).
Cholesterol is one of the most important regulators of lipid organization in the membrane, and mice that lack cholesterol biosynthesis in glia cells show abnormal myelin structures (53,54). It is intriguing to notice that Pmp2, a member of the fatty acid binding protein family with a role in the binding and transport of lipids to the membrane (35), was the only myelin protein strongly increased by Nrg1TIII overexpression. While Pmp2 does not appear to have a role in myelin structure, it may be important for myelin lipids homeostasis (36). Thus, it is possible that high Pmp2 protein levels could enhance lipids transport and myelin compaction (35,36,55). Interestingly, similar observations were made in the Mek1DD mice (49), which suggests that PMP2 is mostly regulated through the MAPK/ERK1/2 axis.
The increase in myelin thickness and the change in myelin composition, however, did not appear to affect demyelination. We have previously shown that demyelination in the S63del mice can be corrected by ablation of the UPR effectors Chop and Gadd34 (23,24). The fact that the UPR is virtually unchanged by Nrg1TIII overexpression could explain why demyelination is not ameliorated. Along the same line, we have recently shown that in S63del Schwann cells the sustained expression of negative regulators of myelination such as Sox2 and Id2 protects from the deleterious effects of excessive levels of toxic P0S63del protein, limiting dysmyelination and demyelination (40). In HANI/+/S63del nerves, the levels of Sox2 and Id2 remain comparable to those in S63del, again suggesting that overexpression of Nrg1TIII does not significantly alter the molecular signature of S63del Schwann cells.
The Nrg1TIII-driven reduction in g-ratio was more pronounced for axon with small/medium diameter. The reasons for this are not entirely clear, but it has been previously hypothesized that it may depend on the fact that an exponential increase in myelin synthesis would be required to match the linear increase in axon diameter (8). It is therefore possible that the Thy-1.2 promoter driving Nrg1TIII expression in the HANI mice does not allow for enough Nrg1 to be expressed and transported into larger caliber axons to become visibly hypermyelinated (Velanac 2012). Still, the rescue of hypomyelination in this subset of axons was sufficient to significantly ameliorate important neurophysiological parameters, such as NCV and FWL. Unfortunately, we were not able to measure whether there was also an improvement in motor function. In fact, the mice expressing the HANI transgene, even in a WT background, were incapable to perform well on Rotarod (Supplementary Material, Fig. S9). This may depend on the expression of the HANI transgene in areas of the central nervous system such as the cerebellum and hippocampus (30), which may affect the equilibrium and suggests that expression of Nrg1TIII should be locally confined to the PNS. However, it should also be noted that recent works suggest that Nrg1 ameliorates cognitive function impairments and neuropathology in different Alzheimer’s disease (AD) mouse models, indicating that it may serve as potential candidate for the prevention and treatment of AD (56–58).
Neuregulin modulation as therapeutic target
Overall, our data indicate that Nrg1TIII increase may partially overcome the myelin defects due to the expression of a toxic myelin protein. Notably, recent work has indicated that the genetic overexpression of the Nrg1TI, but not of the Nrg1TIII isoform, was effective in ameliorating the impaired nerve development defect in the EC61-CMT1A-CH mouse model (14). The reason of this discrepancy could rely on the fact that Fledrich and colleagues showed an imbalanced activity of the PI3k/Akt and the Mek/Erk1/2 signaling pathways in CMT1A models that leads Schwann cells to acquire a persistent differentiation defect during early postnatal development, leading to dysmyelination (14). In contrast, in the S63del-CMT1B mouse, despite an increase of dedifferentiation markers (24,40), we never observed any imbalance between PI3K/Akt and Mek/Erk1/2 activation.
Nrg1TIII shedding by the α-secretase TACE/ADAM17 negatively regulates PNS myelination (13). Whereas directly increasing Nrg1 may have detrimental side effects, the selective modulation of its secretase is an interesting therapeutic approach for the treatment of human neuropathies. Indeed, a recent study showed promising results in treating CMTs mouse models characterized by excessive redundant myelin thickness with Niaspan, an FDA-approved drug known to enhance TACE/ADAM17 activity (45). Similarly, treatment of P0S63del DRG explant cultures with the TACE inhibitor BMS-561392 showed promising results in ameliorating myelination. Future goals will include the synthesis of specific chemical compounds able to cross the barrier and target the nerves.
Conclusions
We provided proof of principle that modulating Nrg1 is a potential general therapeutic approach for hereditary neuropathies. The exogenous overexpression of Nrg1TIII ameliorated the neuropathic phenotype of a CMT1B mouse model characterized by accumulation of misfolded P0S63del and activation of the UPR, without exacerbating the toxic mechanism. We also showed that Nrg1TIII overexpression may act through a Krox20-independent pathway that affects myelin lipids synthesis rather than protein content. The identification of new small molecules that directly modulate Nrg1TIII activity will have relevant clinical implications to treat hypomyelinating CMTs.
Materials and Methods
Transgenic mice
All experiments involving animals were performed in accordance with experimental protocols approved by the San Raffaele Scientific Institute Animal Care and Use Committee. P0S63del mice and their genotype analysis were previously described (Wrabetz et al., 2006). HANI transgenic mice (31) were generated by insertion of a transgene containing Nrg1TIII fused to an N-terminal HA-tag under a constitutive neuronal Thy-1.2 promoter (30). P0S63del mice were maintained on the FVB/N background, whereas HANI mice were on the C57B6/N genetic background. The analysis of WT, HANI/+, S63del or HANI/+//S63del mice was performed in F1 hybrid background FVB//C57B/6. In all the experiments, littermates were used as controls.
Myelinating DRG explant cultures and immunohistochemistry analysis
Mouse DRG explants were isolated from E13.5 WT and S63del embryos, seeded on collagen-coated plates and maintained in culture as described (7). Myelination was induced with 50 μg/ml ascorbic acid (Sigma-Aldrich, Milan, Italy). Pharmacological treatment with BMS-561392, reconstituted in DMSO, was performed for 10 days parallel to the induction of myelination. Immunofluorescent staining on myelinated DRG explant cultures was performed as previously described (24). The following antibodies were used: chicken anti-Neurofilament (1:1000, BioLegend, San Diego, CA, USA) and rat anti-MBP (smi99/94, 1:5, BioLegend, San Diego, CA, USA), FITC- and Rhodamine-conjugated (1:200, Jackson ImmunoResearch, Baltimore, MD, USA). The number of MBP-positive internodes was counted from 10 fields per DRG, from three DRG cultures per genotype. Immunofluorescence was imaged with a Leica DM5000 microscope, and images were acquired with a Leica DFC480 digital camera and processed with Adobe Photoshop CS4 (Adobe Systems, San Jose, CA).
WB analysis
Sciatic nerves from transgenic mice were dissected and frozen in liquid nitrogen. Protein extraction and protein content analysis were previously described (59). Rabbit polyclonal antibodies recognized MAG (1:1000, Invitrogen_Thermo Fisher Scientific, Waltham, MA, USA), PMP22 (1:2000, Abcam, Cambridge, UK), PMP2 (1:1000, Abcam, Cambridge, UK), Krox20 (1:1000, Covance), Akt and P-Akt (Ser473) (1:1000, Cell Signaling, Boston, MA, USA), Erk1/2 and P-Erk1/2 (1:1000, Cell Signaling), Grp78/Bip (1:1000, Novus Biological, Centennial, CO, USA), Calnexin (1:2000, Sigma Aldrich) and Nrg1 C-terminal domain (1:500, Santa Cruz Biotechnology, Dallas, Texas, USA), Id2 (1:500, Abcam) and Sox2 (1:500, Millipore, Billerica, MA, USA). Rabbit monoclonal antibodies recognized eIF2α and P-eIF2α (1:2000, Cell Signaling XP-Technology). Rat monoclonal antibodies recognized Grp94 (1:2000, Abcam) and HA-tag (1:2000, Roche Diagnostic GmbH, Germany). Chicken monoclonal recognized P0 (1:2000, Aves). Mouse monoclonal recognized MBP (1:5000, Covance) and ß-tubulin (1:5000, Sigma). Peroxidase-conjugated secondary antibodies (1:3000, Sigma Aldrich) were visualized using the ECL method with autoradiography film (GE Healthcare, Amersham, UK). Densitometric quantification was performed with ImageJ.
Electrophysiological analyses
The electrophysiological evaluation was performed on 8–10 mice/genotype at 6 months of age with a specific electromyography (EMG) system (NeuroMep Micro, Neurosoft, Russia), as previously described (60). Mice were anesthetized with trichloroethanol, 0.02 ml/g of body weight (25–30 gr) and placed under a heating lamp to maintain constant body temperature. Sciatic NCV was obtained by stimulating the nerve with steel monopolar needle electrodes. A pair of stimulating electrodes was inserted subcutaneously near the nerve at the ankle. A second pair of electrodes was placed at the sciatic notch to obtain two distinct sites of stimulation, proximal and distal along the nerve. CMAP was recorded with a pair of needle electrodes, the active electrode was inserted in muscles in the middle of the paw, whereas the reference was placed in the skin between the first and second digit. Sciatic nerve F-wave latency measurement was obtained by stimulating the nerve at the ankle and recording the responses in the paw muscle, with the same electrodes employed for the NCV study.
Morphological and morphometric analyses
Transgenic and control littermates were sacrificed at the ages indicated and sciatic nerves were dissected. Semi-thin section and electron microscope analyses of sciatic nerves, at P28 and 6 months, were performed as previously described (61,62). The number of demyelinated axons and onion bulbs were counted blind to genotype from 6-month-old sciatic nerve semi-thin sections (0.5–1 μm thick) stained with toluidine blue, images were taken with a 100× objective. 800–1600 fibers in 10–20 fields for each animal in three nerves per genotype were analyzed. G-ratio analysis (axonal diameter/fiber diameter) and distribution of fiber diameters for myelinated axons were performed with a semi-automated system (Leica QWin V3) as described (23,24). 10–15 microscopic fields from nerves of three to five animals per genotype at each time point were analyzed.
Immunoelectron microscopy
IEM was performed on transgenic and control nerves at P30. After left ventricular perfusion with 4% paraformaldehyde/2.5% glutaraldehyde in 0.08 M sodium phosphate buffer (pH 7.3), sciatic nerves were dissected and fixed in the same solution overnight at 4°C. Samples were infiltrated with increasing gradients (1 M, 1.5 M, 2 M, 2.3 M) of sucrose, 30% polyvinylpyrrolidone (PVP) solution for 24 h for each incubation with constant agitation and then stored at 4°C in 2.3 M sucrose/30% PVP for 3 days before cutting. Tissues were mounted on specimen pins and frozen in liquid nitrogen. Ultrathin cryosections (60 nm thick) were sectioned using an ultracut ultramicrotome and processed as described previously (63,64) with the following modifications: the cryosections were collected over nickel formvar-coated grids and treated with gelatin 2% and PGB (0.1 M glycine, 1% bovine serum albumine (BSA), in PBS 1X) and stained with primary antibody (chicken P0, Aves, 1:600 in PGB) for 1 h at 37°C. Sections were then rinsed in PGB and incubated with secondary antibody (1:200 rabbit anti-chicken, 10 nm gold particles conjugated, British BioCell International) for 1 h at room temperature. After rinsing in cacodylate buffer 0.12 M, samples were incubated in osmium 1% in cacodylate buffer 0.12 M and then stained in saturated uranyl acetate. Tissues were then rinsed in water, dehydrated in increasing gradients of ethanol and finally incubated in ethanol 100%, LR White 1:1 and then in LR white quickly, and the grids were left overnight at 60°C. Cryosections were then stained with saturated uranyl acetate and lead citrate and examined by EM.
TaqMan® quantitative polymerase chain reaction analysis
Sciatic nerves from WT and transgenic mice were frozen in liquid nitrogen after dissection. Total RNA from at least three sciatic nerves (P28 or 6 months) for each genotype was extracted using TRIzol (Roche Diagnostic GmbH, Germany) and reverse transcription was performed as described previously (20,24). Quantitative PCR was performed according to manufacturer’s instructions (TaqMan, PE Applied Biosystems Instruments) on an ABI PRISM 7700 sequence detection system (Applied Biosystems Instruments). The relative standard curve method was applied using WT mice as reference. Normalization was performed using 18S rRNA as reference gene. Target and reference gene PCR amplification were performed in separate tubes with Assay on Demand™ (Applied Biosystems Instruments): 18S assay, Hs99999901_s1; MPZ assay, Mm00485139_m1; MAG assay, Mm00487541_m1; PMP22 assay, Mm01333393_m1; MBP assay, Rn00690431_m1; Ddit3/Chop assay, Mm00492097_m1; Xbp-1u assay, Mm00457357_m1; Xbp-1s assay, Mm03464496_m1; Hspa5/BiP assay, Mm00517691_m1; Egr2/Krox20 assay, Mm00456650_m1; Id2 assay, Mm007011781_m1; Sox2 assay, mm00488369_m1. To quantify the relative abundance of WT and mutant P0, we designed the following allelic discrimination assays: P0wt assay forward TGCTCCTTCTGGTCCAGTGAAT, reverse GGAAGATCGAAATGGCATCTCT and probe Mgb ATGACATCTCTTTTACCTGG; P0mut assay forward TGCTCCTTCTGGTCCAGTGAAT, reverse GGAAGATCGAAATGGCATCTCT and probe Mgb ATGACATCTTTACCTGGC.
Lipidomic analysis
For quantitative analysis of fatty acids and cholesterol, sciatic nerves were lysed and extracted in methanol/acetonitrile (MeOH:ACN) 1:1 volume to volume (v/v) after addition of internal standards (IS; heneicosanoic acid, C21:0 for saturated fatty acids, 13C18-labeled linoleic acid C18:2 for unsaturated fatty acid and 5α-cholestane for cholesterol, Sigma-Aldrich). Then, the pellets were removed and used for total protein quantification by Bradford method. Fractions for the quantitative analysis of free cholesterol were first derivatized with a mixture of bis-trimethylsilyltrifluoroacetamide:pyridine (4:1 v/v) for 30 min at 60°C and then injected into a gas chromatograph–mass spectrometer (GC-MS, Varian Saturn 2100). The MS was operated in the electron impact ionization mode. GC-MS analyses were performed as follows: 1 μl sample was injected in splitless mode (inlet was kept at 270°C with the helium flow at 1.0 ml/min) at the initial 180°C. The oven was first kept at 180°C for 1 min, ramped at 50°C/min to 240°C, then at 5°C/min to 300°C for 6 min. The ions used for the quantification of cholesterol were at mass/charge ratio (m/z) 368 for cholesterol and m/z 357 for 5α-cholestane, the IS. The selection of ions for selective ion monitoring (SIM) analysis was based on mass spectra of pure standards and the quantification was based on calibration curves freshly prepared using a fixed concentration of the IS and different concentrations of cholesterol, in a range from 0 to 10 μg/μl.
Total fatty acids were obtained from samples by acid hydrolysis. Briefly, chloroform/MeOH 1:1 v/v and 1 M HCl:MeOH (1:1, v/v) was added to the total lipid extracts and shook for 2 h. After, chloroform:water (1:1 v/v) was added, and the lower organic phase was collected, transferred into tubes and dried under nitrogen flow. The residue was resuspended in 1 ml of MeOH (65). Fatty acid quantification was performed on an API-4000 triple quadrupole mass spectrometer (AB SCIEXAB SCIEX, Milan, Italy) coupled with a HPLC system (Agilent, Milan, Italy) and CTC PAL HTS autosampler (PAL System). The liquid chromatography mobile phases were (A) 10 mm isopropyl-ethyl-amine, 15 mm acetic acid in water:MeOH 97:3, (B) MeOH. The gradient (flow rate 0.5 ml/min) was as follows: T0: 20% A, T20: 1% A, T25: 1% A, T25.1: 20% A, T30: 20% A. The Hypersil GOLD C8 column (100mm × 3 mm, 3 μm) was maintained at 40°C. The injection volume was 10 μl and the injector needle was washed with MeOH/ACN 1:1 (v/v). Peaks off the liquid chromatography–tandem mass spectrometry (LC-MS/MS) were evaluated using a Dell workstation by means of the software Analyst 1.6.2. The mass spectrometer was operated in negative ion mode with the ESI source using nitrogen as sheath, auxiliary and sweeps gas, respectively. The mass spectrometer was operated in SIM/SIM based on mass spectra of pure standards and the quantification was achieved using calibration curves freshly prepared containing fixed concentration of the IS and different concentrations of the analyzed fatty acids (66).
Bioavailability assay
Three WT and three S63del mice were treated with daily i.p. injection of BMS (48 mg/kg) for 10 days, starting at P15. Blood was collected in tubes with lithium heparin from three animals, centrifuged at 2500 rpm at 4°C to isolate plasma. Sciatic nerves were collected, weighed and frozen with liquid nitrogen. To test BMS bioavailability, samples were analyzed by LC-MS with calibration standards (1–2000 ng/ml) prepared from blank tissue homogenate (Eurofins ADME Bioanalyses, Vergèze, France).
Statistical analysis
No statistical methods were used to predetermine sample sized, but our sample size are similar to those generally used in the field. Graphs and data were analyzed using GraphPad Prism Software. Data show the mean ± standard error of mean (SEM). One-way ANOVA with post hoc analysis (when specified in the text) was used. Significance levels were marked on figures as follows: P-values (P), *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Supplementary Material
Acknowledgements
We thank Maria Carla Panzeri and the Alembic facility at the San Raffaele Scientific Institute for excellent technical support.
Conflict of Interest statement. None declared.
Funding
National Institute of Health (R01 NS55256 and R56NS096104 to L.W.); the Charcot–Marie–Tooth Association (L.W.); Fondazione Telethon (GPP10007 to L.W., M.L.F. and C.T., GGP14147 to M.D.A. and GGP15012 to M.D.A. and C.T.); the Italian Ministry of Health (GR-2011-02346791 to M.D.A.) and the Umberto Veronesi Foundation (Fellowship Award 2014 to C.S.). MHS holds a Heisenberg Fellowship from the Deutsche Forschungsgemeinschaft (DFG) and acknowledges funding by a DFG research grant (SCHW741/4-1).
References
- 1. Jessen K.R. and Mirsky R. (2005) The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci., 6, 671–682. [DOI] [PubMed] [Google Scholar]
- 2. Nave K.A. (2010) Myelination and support of axonal integrity by glia. Nature, 468, 244–252. [DOI] [PubMed] [Google Scholar]
- 3. Barisic N., Claeys K.G., Sirotkovic-Skerlev M., Lofgren A., Nelis E., De Jonghe P. and Timmerman V. (2008) Charcot–Marie–Tooth disease: a clinico-genetic confrontation. Ann. Hum. Genet., 72, 416–441. [DOI] [PubMed] [Google Scholar]
- 4. Scherer S.S. and Wrabetz L. (2008) Molecular mechanisms of inherited demyelinating neuropathies. Glia, 56, 1578–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reilly M.M. and Shy M.E. (2009) Diagnosis and new treatments in genetic neuropathies. J. Neurol. Neurosurg. Psychiatry, 80, 1304–1314. [DOI] [PubMed] [Google Scholar]
- 6. Rossor A.M., Polke J.M., Houlden H. and Reilly M.M. (2013) Clinical implications of genetic advances in Charcot–Marie–Tooth disease. Nat. Rev. Neurol., 9, 562–571. [DOI] [PubMed] [Google Scholar]
- 7. Taveggia C., Zanazzi G., Petrylak A., Yano H., Rosenbluth J., Einheber S., Xu X., Esper R.M., Loeb J.A., Shrager P. et al. (2005) Neuregulin-1 type III determines the ensheathment fate of axons. Neuron, 47, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Michailov G.V., Sereda M.W., Brinkmann B.G., Fischer T.M., Haug B., Birchmeier C., Role L., Lai C., Schwab M.H. and Nave K.A. (2004) Axonal neuregulin-1 regulates myelin sheath thickness. Science, 304, 700–703. [DOI] [PubMed] [Google Scholar]
- 9. Parkinson D.B., Bhaskaran A., Droggiti A., Dickinson S., D'Antonio M., Mirsky R. and Jessen K.R. (2004) Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J. Cell Biol., 164, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leblanc S.E., Srinivasan R., Ferri C., Mager G.M., Gillian-Daniel A.L., Wrabetz L. and Svaren J. (2005) Regulation of cholesterol/lipid biosynthetic genes by Egr2/Krox20 during peripheral nerve myelination. J. Neurochem., 93, 737–748. [DOI] [PubMed] [Google Scholar]
- 11. Willem M., Garratt A.N., Novak B., Citron M., Kaufmann S., Rittger A., DeStrooper B., Saftig P., Birchmeier C. and Haass C. (2006) Control of peripheral nerve myelination by the beta-secretase BACE1. Science, 314, 664–666. [DOI] [PubMed] [Google Scholar]
- 12. Hu X., Hicks C.W., He W., Wong P., Macklin W.B., Trapp B.D. and Yan R. (2006) Bace1 modulates myelination in the central and peripheral nervous system. Nat. Neurosci., 9, 1520–1525. [DOI] [PubMed] [Google Scholar]
- 13. La Marca R., Cerri F., Horiuchi K., Bachi A., Feltri M.L., Wrabetz L., Blobel C.P., Quattrini A., Salzer J.L. and Taveggia C. (2011) TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci., 14, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fledrich R., Stassart R.M., Klink A., Rasch L.M., Prukop T., Haag L., Czesnik D., Kungl T., Abdelaal T.A., Keric N. et al. (2014) Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot–Marie–Tooth disease 1A. Nat. Med., 20, 1055–1061. [DOI] [PubMed] [Google Scholar]
- 15. Shy M.E. (2006) Peripheral neuropathies caused by mutations in the myelin protein zero. J. Neurol. Sci., 242, 55–66. [DOI] [PubMed] [Google Scholar]
- 16. D'Urso D., Brophy P.J., Staugaitis S.M., Gillespie C.S., Frey A.B., Stempak J.G. and Colman D.R. (1990) Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron, 4, 449–460. [DOI] [PubMed] [Google Scholar]
- 17. Shapiro L., Doyle J.P., Hensley P., Colman D.R. and Hendrickson W.A. (1996) Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron, 17, 435–449. [DOI] [PubMed] [Google Scholar]
- 18. Timmerman V., Strickland A.V. and Zuchner S. (2014) Genetics of Charcot–Marie–Tooth (CMT) disease within the frame of the Human Genome Project success. Genes (Basel), 5, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanmaneechai O., Feely S., Scherer S.S., Herrmann D.N., Burns J., Muntoni F., Li J., Siskind C.E., Day J.W., Laura M. et al. (2015) Genotype–phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain, 138, 3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wrabetz L., D'Antonio M., Pennuto M., Dati G., Tinelli E., Fratta P., Previtali S., Imperiale D., Zielasek J., Toyka K. et al. (2006) Different intracellular pathomechanisms produce diverse myelin protein zero neuropathies in transgenic mice. J. Neurosci., 26, 2358–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapp B.D., Itoyama Y., Sternberger N.H., Quarles R.H. and Webster H. (1981) Immunocytochemical localization of P0 protein in Golgi complex membranes and myelin of developing rat Schwann cells. J. Cell Biol., 90, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Trapp B.D., Kidd G.J., Hauer P., Mulrenin E., Haney C.A. and Andrews S.B. (1995) Polarization of myelinating Schwann cell surface membranes: role of microtubules and the trans-Golgi network. J. Neurosci., 15, 1797–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pennuto M., Tinelli E., Malaguti M., Del Carro U., D'Antonio M., Ron D., Quattrini A., Feltri M.L. and Wrabetz L. (2008) Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot–Marie–Tooth 1B mice. Neuron, 57, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. D'Antonio M., Musner N., Scapin C., Ungaro D., Del Carro U., Ron D., Feltri M.L. and Wrabetz L. (2013) Resetting translational homeostasis restores myelination in Charcot–Marie–Tooth disease type 1B mice. J. Exp. Med., 210, 821–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bai Y., Wu X., Brennan K.M., Wang D.S., D'Antonio M., Moran J., Svaren J. and Shy M.E. (2018) Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann. Clin. Transl. Neurol., 5, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Okamoto Y., Pehlivan D., Wiszniewski W., Beck C.R., Snipes G.J., Lupski J.R. and Khajavi M. (2013) Curcumin facilitates a transitory cellular stress response in trembler-j mice. Hum. Mol. Genet., 22, 4698–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giambonini-Brugnoli G., Buchstaller J., Sommer L., Suter U. and Mantei N. (2005) Distinct disease mechanisms in peripheral neuropathies due to altered peripheral myelin protein 22 gene dosage or a Pmp22 point mutation. Neurobiol. Dis., 18, 656–668. [DOI] [PubMed] [Google Scholar]
- 28. Yum S.W., Kleopa K.A., Shumas S. and Scherer S.S. (2002) Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol. Dis., 11, 43–52. [DOI] [PubMed] [Google Scholar]
- 29. Deschenes S.M., Walcott J.L., Wexler T.L., Scherer S.S. and Fischbeck K.H. (1997) Altered trafficking of mutant connexin32. J. Neurosci., 17, 9077–9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caroni P. (1997) Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J. Neurosci. Methods, 71, 3–9. [DOI] [PubMed] [Google Scholar]
- 31. Velanac V., Unterbarnscheidt T., Hinrichs W., Gummert M.N., Fischer T.M., Rossner M.J., Trimarco A., Brivio V., Taveggia C., Willem M. et al. (2012) Bace1 processing of NRG1 type III produces a myelin-inducing signal but is not essential for the stimulation of myelination. Glia, 60, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller L.J., Patzko A., Lewis R.A. and Shy M.E. (2012) Phenotypic presentation of the Ser63Del MPZ mutation. J. Peripher. Nerv. Syst., 17, 197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Previtali S.C., Malaguti M.C., Riva N., Scarlato M., Dacci P., Dina G., Triolo D., Porrello E., Lorenzetti I., Fazio R. et al. (2008) The extracellular matrix affects axonal regeneration in peripheral neuropathies. Neurology, 71, 322–331. [DOI] [PubMed] [Google Scholar]
- 34. Uyemura K., Yoshimura K., Suzuki M. and Kitamura K. (1984) Lipid binding activities of the P2 protein in peripheral nerve myelin. Neurochem. Res., 9, 1509–1514. [DOI] [PubMed] [Google Scholar]
- 35. Chrast R., Saher G., Nave K.A. and Verheijen M.H. (2011) Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J. Lipid Res., 52, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zenker J., Stettner M., Ruskamo S., Domenech-Estevez E., Baloui H., Medard J.J., Verheijen M.H., Brouwers J.F., Kursula P., Kieseier B.C. et al. (2014) A role of peripheral myelin protein 2 in lipid homeostasis of myelinating Schwann cells. Glia, 62, 1502–1512. [DOI] [PubMed] [Google Scholar]
- 37. Greenfield S., Brostoff S., Eylar E.H. and Morell P. (1973) Protein composition of myelin of the peripheral nervous system. J. Neurochem., 20, 1207–1216. [DOI] [PubMed] [Google Scholar]
- 38. Siegel D.P. (1999) The modified stalk mechanism of lamellar/inverted phase transitions and its implications for membrane fusion. Biophys. J., 76, 291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feltri M.L., D'Antonio M., Quattrini A., Numerato R., Arona M., Previtali S., Chiu S.Y., Messing A. and Wrabetz L. (1999) A novel P0 glycoprotein transgene activates expression of lacZ in myelin-forming Schwann cells. Eur. J. Neurosci., 11, 1577–1586. [DOI] [PubMed] [Google Scholar]
- 40. Florio F., Ferri C., Scapin C., Feltri M.L., Wrabetz L. and D'Antonio M. (2018) Sustained expression of negative regulators of myelination protects Schwann cells from dysmyelination in a Charcot–Marie–Tooth 1B mouse model. J. Neurosci., 38, 4275–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grootveld M. and McDermott M.F. (2003) BMS-561392. Bristol-Myers Squibb. Curr. Opin. Investig. Drugs, 4, 598–602. [PubMed] [Google Scholar]
- 42. Moss M.L., Sklair-Tavron L. and Nudelman R. (2008) Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol., 4, 300–309. [DOI] [PubMed] [Google Scholar]
- 43. Jerath N.U. and Shy M.E. (2015) Hereditary motor and sensory neuropathies: understanding molecular pathogenesis could lead to future treatment strategies. Biochim. Biophys. Acta, 1852, 667–678. [DOI] [PubMed] [Google Scholar]
- 44. Baets J., De Jonghe P. and Timmerman V. (2014) Recent advances in Charcot–Marie–Tooth disease. Curr. Opin. Neurol., 27, 532–540. [DOI] [PubMed] [Google Scholar]
- 45. Bolino A., Piguet F., Alberizzi V., Pellegatta M., Rivellini C., Guerrero-Valero M., Noseda R., Brombin C., Nonis A., D'Adamo P. et al. (2016) Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol. Med., 8, 1438–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maurel P. and Salzer J.L. (2000) Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J. Neurosci., 20, 4635–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ogata T., Iijima S., Hoshikawa S., Miura T., Yamamoto S., Oda H., Nakamura K. and Tanaka S. (2004) Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J. Neurosci., 24, 6724–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Domenech-Estevez E., Baloui H., Meng X., Zhang Y., Deinhardt K., Dupree J.L., Einheber S., Chrast R. and Salzer J.L. (2016) Akt regulates axon wrapping and myelin sheath thickness in the PNS. J. Neurosci., 36, 4506–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sheean M.E., McShane E., Cheret C., Walcher J., Muller T., Wulf-Goldenberg A., Hoelper S., Garratt A.N., Kruger M., Rajewsky K. et al. (2014) Activation of MAPK overrides the termination of myelin growth and replaces Nrg1/ErbB3 signals during Schwann cell development and myelination. Genes Dev., 28, 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garbay B., Heape A.M., Sargueil F. and Cassagne C. (2000) Myelin synthesis in the peripheral nervous system. Prog. Neurobiol., 61, 267–304. [DOI] [PubMed] [Google Scholar]
- 51. Schmitt S., Castelvetri L.C. and Simons M. (2015) Metabolism and functions of lipids in myelin. Biochim. Biophys. Acta, 1851, 999–1005. [DOI] [PubMed] [Google Scholar]
- 52. Kim M., Wende H., Walcher J., Kuehnemund J., Cheret C., Kempa S., McShane E., Selbach M., Lewin G.R. and Birchmeier C. (2018) Maf links neuregulin1 signaling to cholesterol synthesis in myelinating Schwann cells. Genes. Dev., 32, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Saher G., Quintes S. and Nave K.A. (2011) Cholesterol: a novel regulatory role in myelin formation. Neuroscientist, 17, 79–93. [DOI] [PubMed] [Google Scholar]
- 54. Saher G., Quintes S., Mobius W., Wehr M.C., Kramer-Albers E.M., Brugger B. and Nave K.A. (2009) Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J. Neurosci., 29, 6094–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Majava V., Polverini E., Mazzini A., Nanekar R., Knoll W., Peters J., Natali F., Baumgartel P., Kursula I. and Kursula P. (2010) Structural and functional characterization of human peripheral nervous system myelin protein P2. PLoS One, 5, e10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xu J., Winter F., Farrokhi C., Rockenstein E., Mante M., Adame A., Cook J., Jin X., Masliah E. and Lee K.F. (2016) Neuregulin 1 improves cognitive deficits and neuropathology in an Alzheimer's disease model. Sci. Rep., 6, 31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ryu J., Hong B.H., Kim Y.J., Yang E.J., Choi M., Kim H., Ahn S., Baik T.K., Woo R.S. and Kim H.S. (2016) Neuregulin-1 attenuates cognitive function impairments in a transgenic mouse model of Alzheimer's disease. Cell Death Dis., 7, e2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang Q., Chen S., Hu C., Huang P., Shen H. and Zhao W. (2016) Neuregulin-1 (Nrg1) signaling has a preventive role and is altered in the frontal cortex under the pathological conditions of Alzheimer's disease. Mol. Med. Rep., 14, 2614–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wrabetz L., Feltri M.L., Quattrini A., Imperiale D., Previtali S., D'Antonio M., Martini R., Yin X., Trapp B.D., Zhou L. et al. (2000) P(0) glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J. Cell Biol., 148, 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Biffi A., De Palma M., Quattrini A., Del Carro U., Amadio S., Visigalli I., Sessa M., Fasano S., Brambilla R., Marchesini S. et al. (2004) Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest., 113, 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quattrini A., Previtali S., Feltri M.L., Canal N., Nemni R. and Wrabetz L. (1996) Beta 4 integrin and other Schwann cell markers in axonal neuropathy. Glia, 17, 294–306. [DOI] [PubMed] [Google Scholar]
- 62. Ferri C., Quattrini A. and D'Antonio M. (2018) Electron microscopy for the analysis of peripheral nerve myelin. Methods Mol. Biol., 1791, 3–13. [DOI] [PubMed] [Google Scholar]
- 63. Trapp B.D., Andrews S.B., Cootauco C. and Quarles R. (1989) The myelin-associated glycoprotein is enriched in multivesicular bodies and periaxonal membranes of actively myelinating oligodendrocytes. J. Cell Biol., 109, 2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yin X., Kidd G.J., Wrabetz L., Feltri M.L., Messing A. and Trapp B.D. (2000) Schwann cell myelination requires timely and precise targeting of P(0) protein. J. Cell Biol., 148, 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cermenati G., Abbiati F., Cermenati S., Brioschi E., Volonterio A., Cavaletti G., Saez E., De Fabiani E., Crestani M., Garcia-Segura L.M. et al. (2012) Diabetes-induced myelin abnormalities are associated with an altered lipid pattern: protective effects of LXR activation. J. Lipid Res., 53, 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cermenati G., Audano M., Giatti S., Carozzi V., Porretta-Serapiglia C., Pettinato E., Ferri C., D'Antonio M., De Fabiani E., Crestani M. et al. (2015) Lack of sterol regulatory element binding factor-1c imposes glial fatty acid utilization leading to peripheral neuropathy. Cell Metab., 21, 571–583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.