

Abstract
Adaptive ligands, which can adapt their coordination mode to the electronic structure of various catalytic intermediates, offer the potential to develop improved homogeneous catalysts in terms of activity and selectivity. 2,2′-Diphosphinobenzophenones have previously been shown to act as adaptive ligands, the central ketone moiety preferentially coordinating reduced metal centers. Herein, the utility of this scaffold in nickel-catalyzed alkyne cyclotrimerization is investigated. The complex [(p-tolL1)Ni(BPI)] (p-tolL1 = 2,2′-bis(di(para-tolyl)phosphino)-benzophenone; BPI = benzophenone imine) is an active catalyst in the [2 + 2 + 2] cyclotrimerization of terminal alkynes, selectively affording 1,2,4-substituted benzenes from terminal alkynes. In particular, this catalyst outperforms closely related bi- and tridentate phosphine-based Ni catalysts. This suggests a reaction pathway involving a hemilabile interaction of the C=O unit with the nickel center. This is further borne out by a comparative study of the observed resting states and DFT calculations.
Keywords: alkyne cyclotrimerization, nickel complexes, adaptive ligand, π−acceptor ligand, hemilabile ligand
Introduction
The search for improved activity and selectivity in transition-metal based homogeneous catalysts strongly relies on the development of steering ligands to tune the steric and electronic properties of the metal center to the requirements of particular transformations. Recent years have seen the development of more sophisticated adaptive or cooperative ligands that can adjust their binding modes or engage in chemical transformations along a catalytic cycle.1−4 The use of such ligands also plays an important role in the development of catalysts based on inexpensive and nontoxic first-row transition metals that can compete with, or even surpass, their counterparts based on precious metals, contributing to a more sustainable chemistry.5 Perhaps one of the simplest implementations of the concept of adaptive ligands are hemilabile ligands, which are polydentate ligands featuring a weakly binding moiety that can reversibly (de)coordinate the metal center.6 Since their introduction in synthetic chemistry by Jeffrey and Rauchfuss,7 hemilabile ligands have played a significant role in the current transition from noble to base metals in homogeneous catalysis. The hemilabile interaction is important not only for opening up a masked coordination site but also to stabilize catalytic intermediates, by allowing the supporting ligand to adapt its binding mode throughout the reaction coordinate. Most commonly, weak donor groups, such as ether (OR2),7,8 amine (NR3),9 or imine derivatives (R2C=NR),8b,10 are employed as the labile unit, often resulting in enhanced catalytic activity for different kinds of reactions. Examples include olefin oligomerizations and polymerizations,8c,11 (cyclo)trimerization,9c,11 carbonylation of methanol and methyl acetate,12 (transfer) hydrogenation of ketones or dehydrogenation of alcohols,9b,13,14 hydroacylation of alkenes and alkynes,8d,15 and coupling reactions.9c,11−19
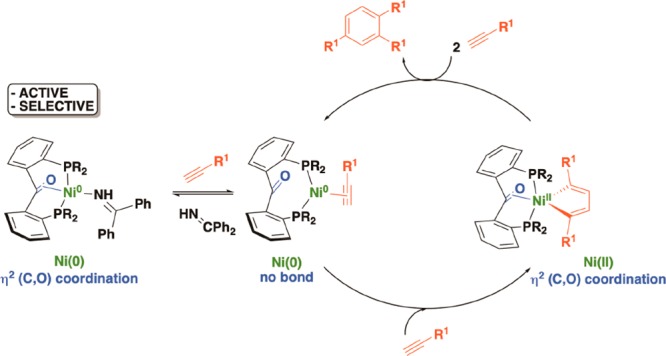
In addition to weak σ-donor moieties, σ-acceptor groups20 (e.g., triarylborane)21 or π-ligands2 (e.g., C=C double bond)22 can also act as hemilabile fragments. Our group recently reported the hemilabile character of the ligand 2,2′-bis(diphenylphosphino)benzophenone23 (PhL1) bound to nickel24 and other first-row transition metals.25PhL1 was shown to function as a hemilabile π-acceptor ligand, with the central C=O bond coordinating to the metal center in its more reduced forms (Figure 1). Ru,23,26 Rh,27−29 Os,30 and Ir31 complexes of PhL1 were also studied, more specifically in the catalytic hydrogenation of ketones,23,26,28,30 in which the formation of a hydroxyalkyl species was accessible via the hydrogenation of the ketone moiety of the ligand.28
Figure 1.
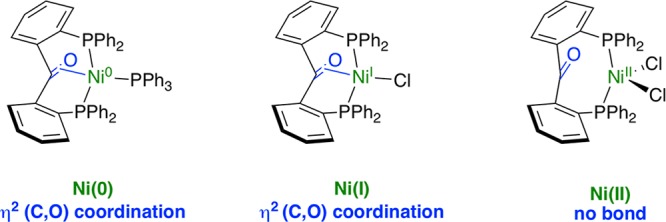
Previous work.24 Coordination of a diphosphine benzophenone ligand, PhL1, to nickel(0), nickel(I), and nickel(II).
In this context, we set out to assess the potential utility of an η2-bound hemilabile π-acceptor moiety incorporated in a pincer type architecture for catalysis, using the cyclotrimerization of alkynes as a benchmark reaction. Since its discovery by Reppe et al.32 using a [Ni(CO)2(PCl3)2] catalyst, intermolecular [2 + 2 + 2] cyclotrimerization of alkynes has been widely studied.33 Metal catalyzed cyclotrimerization is an elegant method for the formation of cyclic frameworks, particularly convenient when the desired product is not accessible via traditional aromatic substitution reactions. Many transition metal systems have been developed (e.g., Ti,34 Fe,35 Co,36 Ni,37 Mo,38 Ru,39 Rh,40 Pd,41 Ir42). Literature precedents for the nickel-catalyzed conversion of alkynes to substituted benzene regio-isomers are known, both from an intramolecular36c,37g−37l,37n,37p,37r,37s and intermolecular approach.9c,32,36a,37a−37f,37m,37o,37q,37t−37z For the [2 + 2 + 2] intermolecular alkyne cyclotrimerization, it starts from the early example by Reppe et al.32 to the combination of divalent nickel halide salts and reductants36a,37f or even activated Ni-particles.37b Phosphine,9c,36a,37j,37m,37t,37v NHC,37v,37x mixed phosphine/imino (P,N),9c or 1,4-diazadiene37c,37e,37u,37w,37y,37z ligands, have been introduced to Ni systems in order to improve selectivity and catalytic activity. Phosphine complexes generally favor the formation of benzene derivatives while 1,4-diazadiene complexes yield cyclooctatetraenes.
Besides their synthetic utility, alkyne condensations are also useful probes for ligand effects in catalysis.43 The possible formation of different products and isomers, such as cyclotrimers, cyclotetramers, linear oligomers, polymers, and other compounds, provides useful information about the reactivity of a particular catalyst. For example, Uyeda37w,44 and Ess44 recently applied this approach to the evaluation of the effect of catalyst nuclearity, and Jones9c applied it to assessing differences in catalytic performance between bidentate PP and PN ligands (Figure 2). In this study, we assess the effect of using hemilabile π-acceptor tridentate ligand (p-tolL1; Figure 3) in alkyne cyclotrimerization through comparison with diverse nickel systems. Ni(0) adopting a strong tridentate architecture (ligand = p-tolL2; Figure 3) and Ni complexes supported by a bidentate ligand (PhL3; Figure 3) were selected for comparison with p-tolL1.
Figure 2.

Examples of Ni(0) complexes used to evaluate the catalytic properties of a ligand–metal system by their reactivity with alkynes.9c,37w Blue bold font = property studied. R = Ph, CF3, tert-butyl.
Figure 3.

Tridentate and bidentate ligands of comparison in this study.
Here we report the synthesis of nickel(0) complexes of p-tolL1, p-tolL2, and PhL3 featuring benzophenone imine (BPI) as a labile protecting ligand. Their stoichiometric reactivity with terminal alkynes is investigated, and the resulting Ni–alkyne complexes are thoroughly characterized. Then, a comparative study of these bi- and tridentate Ni(0)-catalysts in the cyclotrimerization of terminal alkynes is described, aiming to study the effect of a hemilabile π-acceptor system. For all tested alkyne substrates, p-tolL1 outperforms p-tolL2 and PhL3, as well as the structurally more different diphosphine rac-BINAP (PhL4), in terms of selectivity and activity. Reactivity studies combined with DFT calculations provide insight into the increased catalytic activity, suggesting that p-tolL1 can adapt its binding mode throughout the reaction pathway, by the labile (de)coordination of its ketone unit.
Results and Discussion
Ni(0)-Benzophenone Imine Complexes
The diphosphine–benzophenone pincer-type ligand23 2,2′-bis(di(para-tolyl)phosphino)-benzophenone25 (p-tolL1; Figure 3) and the triphosphine pincer-type ligand45 bis(2-(di-(para-tolyl)phosphino)phenyl)phenylphosphine (p-tolL2; Figure 3) were synthesized according to (adapted) literature procedures,24,25,46 by lithiation of o-bromo(diarylphosphino)benzene47 with n-BuLi followed by reaction with the appropriate electrophile (Scheme 1). para-Tolyl substituents on the phosphine were introduced both to improve the solubility of the complexes and to provide a convenient 1H NMR handle for characterization. The 31P NMR spectrum of p-tolL2 indicates the presence of an AB2 system (Δν ≈ JA,B)48 with a coupling constant of 3JA,B = 155 Hz, as previously observed for its phenyl-substituted analogue.46 The diphosphine ether ligand PhL3(49) was obtained from commercial sources and used as received.
Scheme 1. Synthesis of p-tolL1 and p-tolL2 from o-Bromo(di-(para-tolyl)phosphino)benzene.

Coordination of the chelating phosphine ligands to a nickel(0) center was achieved by reaction with Ni(cod)2 (cod = cyclooctatetraene) in the presence of benzophenone imine (BPI), in a stoichiometric ratio, affording respectively complexes (i) [(p-tolL1)Ni(BPI)] (p-tol1; Scheme 2), (ii) [(p-tolL2)Ni(BPI)] (p-tol2; Scheme 2), and (iii) [(PhL3)Ni(BPI)] (Ph3; Scheme 2). BPI was chosen as coligand since it binds strongly enough to Ni(0) to prevent the formation of dimeric species24 but weakly enough to be exchanged with different types of potential substrates such as alkenes, nitriles, and alkynes (see below; Scheme 4).
Scheme 2. Ni0–Benzophenone Imine Complexes.

(A) General synthetic route. (B) Isolated complexes. BPI = benzophenone imine.
Scheme 4. Ligand Exchange Reaction between the Benzophenone Imine from p-tol1 and Different Kinds of Substrates and Coligands.

NMR characterization of p-tol1 indicates that the BPI ligand is bound in an η1 fashion. The 13C NMR signal corresponding to the imine appears as a triplet (169.8 ppm,3JC,P = 6.1 Hz) close to that of the free imine at 177.3 ppm, consistent with retention of C=N double bond character. In the 1H NMR spectra, slight deshielding of the C=N–H proton by 0.24 ppm with respect to free BPI is also in accordance with η1(N) binding. Moreover, the ATR-IR spectrum displays a signal at 3152 cm–1 in the region corresponding the N–H stretch. In contrast, the backbone C=O moiety is bound side-on: the corresponding 13C NMR signal appears at 119.0 ppm (t, 3JC,P = 5.1 Hz), considerably shifted from the value of 197.4 ppm found in the free ligand.25 This large shift is useful as a diagnostic tool to determine whether the CO moiety is coordinated to the Ni center. The 31P NMR spectra of p-tol1 consist of a single singlet signal at 15.5 ppm, indicating that the two phosphorus atoms are equivalent on the NMR time scale. However, from 1H and 13C NMR analysis, it appears that complex p-tol1 contains two chemically different methyl groups from the para-tolyl substituents (2.01 and 1.91 ppm in 1H NMR, 21.3 and 21.1 ppm in 13C NMR). Therefore, the para-tolyl substituents belonging to a single P-donor site are not equivalent on the NMR time scale, which can be used as a secondary indication for ketone coordination.
In a similar fashion, NMR data support η1 binding of the BPI ligand in the triphosphine complex p-tol2. The 13C NMR signal corresponding to the N=C bond is a doublet of triplets at 168.9 ppm (3JC,P = 8.1 Hz, 3JC,P = 7.4 Hz). The imine proton appears at 10.25 ppm in the 1H NMR spectrum (deshielding of 0.48 ppm compared to the free BPI ligand) and couples with the phosphorus nuclei (dt, 3JH,P = 3.2 Hz, 3JH,P = 2.7 Hz). The 31P NMR spectrum is consistent with an AK2 system (Δν ≈ 5JA,K)48 with a coupling constant 2JA,K = 85 Hz. From 1H and 13C NMR analysis, it appears that the complex contains two chemically different para-tolyl substituents (2.14 and 1.95 ppm in 1H NMR, 21.4 and 21.1 ppm in 13C NMR), as also observed with complex p-tol1.
In the 1H NMR of Ph3, the N–H signal from the coordinated imine appears as a broad singlet at δH = 9.71 in the expected 1:38 ratio compared to the aromatic region of the spectrum. The 31P NMR spectrum exhibits a single resonance at 32.4 ppm. No satisfactory 13C NMR data could be obtained due to the low solubility of the complex and its progressive decomposition, making the assignment of the binding mode of BPI in solution uncertain. The side-on binding of BPI observed in the solid state (see below) may be preserved in solution, in which case fast rotation of BPI ligand on the 1H NMR time scale would render the two phosphorus atoms equivalent. The N–H stretch from the bound imine appears as a broad weak band in the ATR-IR spectrum, located at 3200 cm–1.
More insights into the structural and electronic properties of nickel complexes p-tol1 and Ph3 were obtained by X-ray crystal structure determination (Figure 4). Crystals suitable for X-ray diffraction were not accessible with p-tol1 but with the structurally related complex Ph1, in which the para-tolyl ligand p-tolL1 is replaced by the phenyl analogue 2,2′-bis(di(phenyl)phosphino)-benzophenone (PhL1).
Figure 4.

Molecular structures of [(PhL1)Ni(BPI)] (Ph1) and [(PhL3)Ni(BPI)] (Ph3) in the crystal (50% probability level). Hexane solvent molecules (Ph1) and C−H hydrogen atoms are omitted for clarity.
In accord with NMR data, the structure of Ph1 (Figure 4; left) exhibits a side-on bound ketone moiety with Ni1–O11 and Ni1–C71 bond lengths of 2.0217(14) and 1.9760(19) Å, respectively. The C71–O11 bond length (1.320(2) Å) is found between those of unbound PhL1 (1.213(3) Å)23 and a C–O single bond (1.43 Å in ethanol50). The ketone is slightly less activated than in [(PhL1)Ni(PPh3)],24 which displays a C–O bond length of 1.310(2) Å, consistent with less pronounced π-back-donation arising from the weaker donor character of BPI with respect to PPh3. The sum of bond angles (351.3(3)°) around the carbon atom lies between those expected for sp2 (328.5°) and sp3 (360°) hybridization at the carbon, in accordance with the Dewar–Chatt–Duncanson model. The N12–C12 bond length of 1.289(2) Å is close to the value of a C–N double bond (ca. 1.28 Å)50 and comparable to the value found by Zhao et al. (1.294(3) Å)51a for η1-coordination mode in their synthesized NHC-based nickel(0) complex, [(IPr)Ni(η2-BPI)(η1-BPI)] (IPr = 1,3-bis(diisopropylphenyl)-imidazolium), showing that the π-back-donation to the BPI ligand is small. Accordingly, the valence angles around the imine carbon add up to 360.0(3)°. Finally, the torsion angle (∠Ni1–N12–C12–CPh) of 20.0(3)° shows that the nickel atom is slightly out of the plane of BPI, which is probably sterically driven.
In contrast to that of Ph1, the crystal structure of Ph3 (Figure 4, right) reveals a trigonal planar complex in which the imine coligand is coordinated to the nickel in an η2 fashion (Ni1–C37 = 2.018(2) Å, Ni1–N1 = 1.8985(19) Å). The C–N bond axis is parallel to the metal coordination plane, which is thought to maximize π-back-donation from the high-lying in-plane d orbital with significant σ*(P–Ni) character. Accordingly, strong π-back-donation into the π*(N–C) orbital is evidenced by an elongated C37–N1 bond (1.373(3) Å vs 1.28 Å50 in the free imine) and the pyramidalization of the C37 atom, which displays a sum of valence angles of 355.3(3)°, in line with known η2(C,N)-Ni(0) complexes.51 As expected, the central oxygen atom from the ligand is not binding to the metal center (Ni1–O1 = 3.3668(16) Å), as also confirmed by a Wiberg bond index lower than 0.01 calculated by NBO analysis at a B3LYP/def2TZVP level of theory. The distance between the nickel and oxygen atoms is similar to the values reported from Ni(0) complexes bearing this ligand.52 Finally, the measured P1–Ni1–P2 bite angle of 108.59(2)° and the Ni1–O1 distance of 3.3668(16) Å are in the same range as those found for [(PhL1)Ni(Cl2)],24 in which the C=O unit is not bound to the Ni ion (P1–Ni–P2 112.996(13)°; Ni–C = 3.4031(12) Å; Ni–O = 3.1012(10) Å), supporting the use of Ph3 as a model complex for the unbound state of p-tolL1.
In summary, complexes p-tol1 and p-tol2 are geometrically similar and mainly differ by the central coordinating unit the ligand (C=O vs P). Complex Ph3 differs in that the central oxygen atom is not coordinated and can be seen as a structural analogue of the unbound state of ligand RL1 (R = p-tolyl or Ph). Interestingly, moving from tridentate to bidentate mode is coupled to a change in binding mode of the BPI ligand from end-on to side-on, which can be understood from enhanced π-back-donation from a d10 L2Ni fragment (see Supporting Information for more insight by NBO analysis into the different coordination modes of the imine coligand; Table S8).
Ni(0)–Alkyne Complexes
With complexes p-tol1–Ph3 in hand, their reactivity toward terminal alkynes was investigated, leading to the generation of Ni–alkyne complexes [(p-tolL1)Ni(HC≡CPh)] (p-tol4-Ph), [(p-tolL1)Ni(HC≡CCH2OMe)] (p-tol4-CH2OMe), [(p-tolL2)Ni(HC≡CPh)] (p-tol5-Ph), and [(PhL3)Ni(HC≡CPh)] (Ph6-Ph) (Scheme 3). Ni-complexes p-tol5-Ph and Ph6-Ph, bearing phenylacetylene as coligand, are sufficiently stable to be isolated and are discussed first.
Scheme 3. Ni0-Terminal Alkyne Complexes.

(A) General synthetic route. (B) Generated complexes. p-tol4-Ph and p-tol4-CH2OMe were characterized in-situ in solution, while p-tol5-Ph and Ph6-Ph were isolated. BPI = benzophenone imine.
The 31P NMR spectrum of p-tol5-Ph displays two signals at 78.7 ppm (t, 2JP,P = 35.4 Hz) and at 28.1 ppm (d, 2JP,P = 35.4 Hz) in a ratio of 2:1, indicating that the ligand binds in a tridentate fashion. Two 1H NMR singlets of the same intensity at 2.12 and 1.91 ppm correspond to the methyl group from diastereotopic para-tolyl substituents bound to the same phosphorus atom. A single alkyne is bound to Ni as indicated by the terminal proton signal at 6.30 ppm, which appears as a doublet of triplets (3JH,P = 25.6 Hz, 3JH,P = 7.2 Hz) and couples with a carbon nucleus at 92.9 ppm in HMQC (1H–13C). Further confirmation of this assignment is provided by treatment of p-tol2 with d-phenylacetylene, resulting in the disappearance of the signal at 6.30 ppm in an otherwise identical 1H NMR spectrum (Supporting Information; Figure S68 and S76). In agreement with the spectroscopic characterization, the solid-state structure of p-tol5-Ph (Figure 5, left) reveals a pseudo-tetrahedral geometry in which the alkyne moiety is coordinated in η2 fashion.
Figure 5.

Molecular structure of [(p-tolL2)Ni(HC≡CPh)] (p-tol5-Ph) and [(PhL3)Ni(HC≡CPH)] (Ph6-Ph) in the crystal (50% probability level). Toluene solvent molecules (Ph6-Ph) and hydrogen atoms (except alkyne) have been omitted for clarity.
Compound Ph6-Ph, supported by the bidentate diphosphine ether ligand PhL3, displays two signals at 29.2 ppm and at 27.3 ppm coupling to each other (2JP,P = 14.0 Hz) and in a ratio of 1:1 in the 31P NMR spectrum. This is consistent with a trigonal planar geometry in which rotation of the alkyne ligand is slow on the 1H NMR time scale; warming up the sample up to 100 °C does not result in coalescence of the two 31P NMR signals. This interpretation is supported by reaction of Ph3 with 1 equiv of the symmetric alkyne diphenylacetylene, resulting in a symmetrical analogue of Ph6-Ph displaying a single 31P NMR singlet at 28.1 ppm (Supporting Information; Figure S98). The proton from the alkyne gives a 1H NMR signal at 6.9 ppm, as revealed by HMQC (1H–13C) and comparison with deuterated analogue. The solid state structure (Figure 5, right) of Ph6-Ph is in accord with the NMR data and reveals a trigonal planar geometry with a P1–Ni–P2 bite angle of 104.94(2)°, where the oxygen atom is not bound to the nickel center (Ni1–O1 = 3.3317(15) Å). The alkyne is bound in an η2-coordination mode, the C37–C38 axis being in the Ni coordination plane.
While stable tricoordinate Ni(0) alkyne complexes with formally 16 valence electrons analogous to Ph6-Ph are well documented,53,54 compound p-tol5-Ph represents an unusual example of tetracoordinate, 18 VE Ni(0)–alkyne complex, to the best of our knowledge the first to be structurally characterized. Spectroscopic and structural data point toward a considerably weaker activation of the C≡C triple bond in this geometry (Table 1). First, the 13C NMR chemical shifts of the two alkyne carbons (δC = 92.9 and 101.0) in p-tol5-Ph are moderately deshielded in comparison with the free alkyne (δC = 77.5 and 83.3), indicating relatively weak rehybridization from sp to sp2. This contrasts with a stronger activation of the C≡C triple bond of phenylacetylene in Ph6-Ph evidenced by the 13C NMR signals from the coordinated phenylacetylene at 125.7 ppm (C≡C–H, dd, 2JC,P = 40.1 Hz, 2JC,P = 7.1 Hz) and 136.4 ppm (C≡C–Ph, dd, 2JC,P = 38.9 Hz, 2JC,P = 8.4 Hz), which were assigned with the assistance of APT 13C NMR and by comparison with the d-phenylacetylene complex. Second, the acetylic hydrogen signal of p-tol5-Ph (δH = 6.30) is less shifted than that of Ph6-Ph (δH = 6.9), consistent with a stronger rehybridization toward sp2. Third, the C≡C stretch vibration from the alkyne in p-tol5-Ph is found at 1823 cm–1 in the FT-IR spectrum versus 2126 cm–1 for free phenylacetylene and versus 1749 cm–1 for Ph6-Ph. Finally, the slightly elongated C47–C48 distance at 1.231(8) Å (vs 1.182–1.190 Å in free phenylacetylene)55 and the rather large C47–C48–C49 angle at 152.0(6)° are also consistent with a relatively weak activation of the C–C triple bond, contrasting with the longer C37–C38 bond length (1.269(3) Å) and the more acute the C37–C38–C39 angle (144.07(19)°) found for Ph6-Ph. The C47–C48 bond length in p-tol5-Ph is the shortest we are aware of for nickel(0)–alkyne complexes. Together, these data suggest that the π-back-donation from the nickel to phenylacetylene is more pronounced in Ph6-Ph, resulting in a stronger rehybridization toward sp2.
Table 1. Selected Spectroscopic and X-ray Crystal Structure Values of Phenylacetylene, p-tol5-Ph and Ph6-Pha.
| phenylacetylene | p-tol5-Ph | Ph6-Ph | |
|---|---|---|---|
| δC(C≡C–H) [ppm] | 77.5 | 92.9 | 125.7 |
| δC(C≡C–Ph) [ppm] | 83.3 | 101.1 | 136.4 |
| δH(C≡C–H) [ppm] | 2.73 | 6.30 | 6.9 |
| ν(C≡C) [cm–1] | 2126 | 1823 | 1749 |
| C≡C [Å] | 1.182–1.19055 | 1.231(8) | 1.269(3) |
| ∠C≡C–CPh [deg] | 177.39–179.4955 | 152.0(6) | 144.07(19) |
Vibrational frequencies are measured with ATR-IR (neat). NMR chemical shifts are given in C6D6.
The observations made for complex Ph6-Ph parallel those reported for mononuclear, 16 valence electron, tricoordinate Ni–alkyne, in which the C–C bond is significantly more elongated than in p-tol5-Ph, regardless of substituent groups on the alkyne. For example, strong activation of the alkyne is observed for bidentate diimine and mixed P,N supported species (up to 1.296(6) Å),9c,53a,53b,53i,53l,53n,53r while bidentate phosphorus ligands afford C–C bond lengths from 1.260(4) to 1.283(3) Å.52b,53d−53f,53h,53j,53m,53o−53q The substantially weaker rehybridization observed in p-tol5-Ph can be assigned to its unique coordination geometry (i.e., tetracoordinate complex; 3 donor ligands; pseudo-Td geometry) compared to Ph6-Ph and the reported Ni–alkyne complexes (i.e., tricoordinate complexes; 2 donor ligands; trigonal planar geometry). The weak activation of phenylacetylene in p-tol5-Ph is presumably a consequence of the smaller splitting of the d-orbitals in its pseudo-tetrahedral geometry (3 donor ligands), resulting in less pronounced π-back-donation from Ni to the antibonding orbitals of the alkyne. Similar considerations were used by Lee and co-workers to explain the rather weak activation of CO2 in the tetracoordinate pseudo-tetrahedral nickel complex [(PPMeP)Ni(η2-CO2)] (PPMeP = PMe[2-PiPr2–C6H4]2)56 with respect to tricoordinate Ni-η2-CO2 adducts supported by bidentate phosphines such as [(dtbpe)Ni(η2-CO2)] (dtbpe = 1,2-bis(di-tert-butylphophino)-ethane).57
Finally, reaction of the diphosphine-ketone complex p-tol1 with terminal alkynes generally led to a mixture containing the alkyne complex, [(p-tolL1)Ni(HC≡CR1)] (p-tol4-R1; R1 = substituent on the alkyne), the starting material p-tol1, BPI, the substrate, and cyclotrimerization products. This indicates that cyclotrimerization occurs concomitantly with ligand exchange, precluding the isolation of pure alkyne complexes. Nonetheless, in situ characterization of alkyne complexes was possible. Interestingly, 13C NMR of p-tol4-Ph indicates that the ketone moiety is not bound to the metal center: addition of phenylacetylene to p-tol1 causes the disappearance of the triplet at 119.0 ppm, characteristic of the η2(C,O) coordination to nickel and the appearance of a triplet at 202.7 ppm (3JC,P = 4.9 Hz). In addition, all the para-tolyl groups in p-tol4-Ph are equivalent at room temperature, with singlets appearing at 2.00 and at 21.2 ppm in 1H and 13C NMR, respectively. This is consistent with an unbound ketone allowing fast ring inversion of the chelate macrocycle. A single 31P NMR singlet at 33.0 ppm down to −80 °C indicates fast rotation of the alkyne, exchanging the two phosphorus atoms on the NMR time scale even at low temperature (Supporting Information; Figure S59).
The C≡C–H1H NMR signal of the coordinating phenylacetylene could not be located, presumably hidden in the crowded aromatic region of the spectrum. In contrast, the assignment of the acetylic proton of the bound alkyne was possible in the 1H NMR spectrum of p-tol4-CH2OMe where it appears as a triplet of triplets at 6.34 ppm (3JH,P = 15.6, 4JH,H = 1.6 Hz) and integrates in a ratio of 1:12 compared to the four CH3 groups of the ligand, confirming that a single alkyne molecule is bound. The integrals, coupling constants, and multiplicity of CH2 (4.63 ppm, d, 4JH,H = 1.6 Hz) and OCH3 (3.32 ppm, s) of the bound alkyne are in agreement with the proposed structure. Similar to p-tol4-Ph, both 1H and 13C NMR of p-tol4-CH2OMe reveal that the ketone moiety is not bound to the metal center: the ketone resonance appears as a triplet at 203.1 ppm (3JC,P = 4.7 Hz) and the methyl groups from the para-tolyl are all equivalent, with a singlet at 2.03 and a doublet 22.2 ppm (4JC,P = 2.3 Hz) in 1H and 13C NMR, respectively. A singlet 31P NMR signal at 32.7 ppm at room temperature is consistent with fast rotation of the methyl propargyl ether similar to p-tol4-Ph. However, gradually cooling the sample down to −85 °C first shows the appearance of a new unsymmetrical species at ca. −10 °C, as characterized by two doublets (2JP,P = 23.3 Hz) at 32.1 and 34.1 ppm (Supporting Information; Figure S67). The central signal corresponding to p-tol4-CH2OMe does split into two new doublets (2JP,P = 23.3 Hz) with a coalescence temperature between −50 and −60 °C. This indicates that the rotation of methyl propargyl ether can be frozen on the NMR time scale, but also that two distinct species are formed at low temperature. This is tentatively attributed to a secondary interaction of the oxygen atom from H–C≡C–CH2OMe with Ni. In addition, the second species observed at low temperature may be either a diastereomeric conformer or an isomeric structure in which the central ketone fragment binds to the nickel center.
The fast rotation of the alkyne fragment in complex p-tol4-Ph contrasts with the observed behavior of the tricoordinate Ph6-Ph. DFT calculations on the acetylene complex p-tol4-H reveal that the rotation of acetylene is assisted by transient coordination of the ketone moiety to the Ni center, resulting in a low overall rotation energy barrier of 5.5 kcal/mol (Figure 6). The first transition state, p-tol4-H(TS1), from the unbound ketone state p-tol4-H to the bound state p-tol4-H(I1), is characterized by the coordination of the ketone moiety (ν = −113 cm–1; ΔGTS1 = 4.3 kcal/mol) to generate a four-coordinate intermediate p-tol4-H(I1). The second transition state, p-tol4-H(TS2), corresponds to the rotation of acetylene (ν = −95 cm–1; ΔGTS2 = 4.1 kcal/mol) accompanied by partial decoordination of one of the phosphine arms. The low overall energy barrier of ΔGTS,overall = 5.5 kcal/mol is in qualitative agreement with the observed fast rotation of alkynes at room temperature. For comparison, a rotation energy barrier of 25.0 kcal/mol was calculated for the rotation of acetylene in the diphosphine ether complex Ph6-H (Supporting Information; Figure S112), in qualitative agreement with the observation of two 31P NMR signals for Ph6-Ph up to 100 °C.
Figure 6.

(top) Energy diagram for the rotation of acetylene around the Ni-coordination plane (∠C1–C2–Ni–P1) of p-tol4-H through two different transition states, p-tol4-H(TS1) = p-tol4-H(TS1′) and p-tol4-H(TS2), and one intermediate, p-tol4-H(I1) = p-tol4-H(1′). Numbers in parentheses are G° values given in kcal/mol. ΔGTS1 = 4.3 kcal/mol. ΔGTS1′ = 2.9 kcal/mol. ΔGTS2 = 4.1 kcal/mol. ΔGTS,overall = 5.5 kcal/mol. (bottom) Optimized geometry of transition states p-tol4-H(TS1) and p-tol4-H(TS2) as well as intermediate p-tol4-H(I1) at a B3LYP/6-31g(d,p) level of theory under vacuum. The imaginary frequency (−113 cm–1) of p-tol4-H(TS1) shows the coordination of the ketone (C3–O) to Ni, while the imaginary frequency (−95 cm–1) of p-tol4-H(TS2) is the rotation of acetylene around the C1–C2–Ni–P1 torsion angle. Selected bond lengths [Å] for p-tol4-H(TS1): C3–O = 1.25; Ni–C3 = 2.35; Ni–O = 2.55; Ni–P1 = 2.17; Ni–P2 = 2.19; C1–C2 = 1.27. Selected bond lengths [Å] for p-tol4-H(I1): C3–O = 1.30; Ni–C3 = 2.01; Ni–O = 2.06; Ni–P1 = 2.18; Ni–P2 = 2.24; C1–C2 = 1.25. Selected bond lengths [Å] for p-tol4-H(TS2): C3–O = 1.32; Ni–C3 = 1.96; Ni–O = 1.92; Ni–P1 = 2.14; Ni–P2 = 2.45; C1–C2 = 1.24. Hydrogen atoms have been omitted for clarity.
The differences in electronic and structural properties of Ni–alkyne complexes p-tol4-Ph–Ph6-Ph were further investigated by DFT calculations (geometry optimization and NBO analysis), as summarized in Table 2. The optimized geometries of p-tol4-Ph, p-tol5-Ph, and Ph6-Ph (Table 2, entries 1–4) are consistent with the obtained crystal structures in which the C1–C2 bond length, C1–C2–C3 angle and Ni–C1 and Ni–C2 distances calculated for p-tol5-Ph indicate weaker activation of the HC≡CPh substrate. Accordingly, Wiberg bond indexes (WBIs) calculated in the NBO basis at a B3LYP/def2TZVP level of theory show that the binding of phenylacetylene induces a decrease of the C1–C2 WBI (Table 2, entry 5) from 2.82 to 2.30 in p-tol5-Ph versus 2.20 in p-tol4-Ph and 2.17 in Ph6-Ph. The smaller Ni–C1 and Ni–C2 WBIs (Table 2, entries 6 and 7) of 0.31 in p-tol5-Ph corroborate a weaker orbital interaction between the HC≡CPh ligand and the Ni-center in the triphosphine complex p-tol5-Ph compared p-tol4-Ph (WBIs of 0.43 and 0.39) and Ph6-Ph (WBIs of 0.42 and 0.39).
Table 2. Selected DFT Bond Distances and Angles and Wiberg Bond Indexes (WBI)a.

| entry | property | HC≡CPh | p-tol4-Ph | p-tol5-Ph | Ph6-Ph |
|---|---|---|---|---|---|
| DFT Bond Length and Angle [in Å and deg] | |||||
| 1 | C1–C2 | 1.21 | 1.29 | 1.26 | 1.30 |
| 2 | ∠C1–C2–C3 | 180 | 141 | 150 | 141 |
| 3 | Ni–C1 | 1.84 | 1.94 | 1.84 | |
| 4 | Ni–C2 | 1.89 | 1.97 | 1.89 | |
| Wiberg Bond Index (WBI) | |||||
| 5 | C1–C2 | 2.82 | 2.20 | 2.30 | 2.17 |
| 6 | Ni–C1 | 0.43 | 0.31 | 0.42 | |
| 7 | Ni–C2 | 0.39 | 0.31 | 0.39 | |
Geometry optimizations of [(p-tolL1)Ni(HC≡CCPh)] (p-tol4-Ph), [(p-tolL2)Ni(HC≡CPh)] (p-tol5-Ph), and [(PhL3)Ni(HC≡CPh)] (Ph6-Ph) were performed at a B3LYP/6-31g(d,p) level of theory. WBIs were calculated by NBO analysis at a B3LYP/def2TZVP level of theory from the optimized geometries. Hydrogen atoms have been omitted for clarity.
In summary, we showed in this section that ligands p-tolL1–PhL3 all form Ni(0)–alkyne complexes with phenylacetylene. The triphosphine p-tolL2 binds in a tridentate mode in p-tol5-R1, resulting in unusually weak activation of the alkyne, while PhL3 acts as a bidentate ligand in the tricoordinate Ph6-R1, in which a strong Ni–alkyne interaction causes a high rotation barrier around the Ni–alkyne axis. The hemilabile ligand p-tolL1 is able to sample both binding modes: it binds as a bidentate ligand in p-tol4-R1, resulting in a similar geometry as that of Ph6-R1, but the observed low rotation barrier around the Ni–alkyne axis is indicative of the ability of p-tolL1 to transiently adopt a tridentate mode.
Ligand Exchange Reactions on [(p-tolL1)Ni(BPI)]
Having observed that the ketone moiety decoordinates upon binding of an alkyne to the (p-tolL1)Ni fragment, we sought to probe the generality of this hemilabile behavior. NMR-tube experiments showed that the BPI coligand can be reversibly exchanged with several types of ligands, such as benzonitrile (Keq ≈ 2.7 × 10–3; ΔG298.15 ≈ 3.5 kcal/mol), styrene (Keq ≈ 1.5 × 10–2; ΔG298.15 ≈ 2.4 kcal/mol), and diphenylacetylene (Keq ≈ 6.1 × 10–3; ΔG298.15 ≈ 3.1 kcal/mol), as depicted in Scheme 4 (see Supporting Information for more details). The proposed mode of coordination of the benzonitrile ligand in p-tol7-PhCN relies on DFT calculations, which predict the η2-(C,N) binding to be 9.4 kcal/mol energetically less stable than the η1-(N) coordination mode (Supporting Information; Table S11). With triphenylphosphine, the exchange is irreversible. In every case, with the exception of diphenylacetylene, 13C NMR demonstrates that the ketone moiety is still bound to nickel (no peaks between 190 and 210 ppm). The coordinated ketone appears at 120.4 ppm (dt, 2JC,P = 13.8 Hz, 2JC,P = 4.4 Hz) for the PPh3 complex p-tol7-PPh3 and shows two chemically different methyl groups from the para-tolyl substituents at 1.97 ppm (s, 6H) and 1.96 ppm (s, 6H) in 1H NMR and at 21.2 and 21.1 ppm, respectively, in 13C NMR. Unfortunately for the PhCN complex p-tol7-PhCN and for the styrene complex p-tol7-C2H3Ph, the carbon resonance of the bound ketone could not be assigned, as the weak signal is covered by those of the substrate and the starting complex p-tol1. However, for all of these complexes, 1H NMR shows that the two methyl groups from the para-tolyl substituents on the phosphine are not equivalent (δH = 2.04 and 2.03 for p-tol7-PhCN, 2.05 and 2.01 for p-tol7-C2H3Ph), which is also indicative of the pincer-type binding mode of p-tolL1 to nickel, with the central ketone moiety bound, as observed with p-tol1 and p-tol7-PPh3 and in opposition to p-tol4-R1. With the diphenylacetylene complex p-tol7-C2Ph2, the absence of interaction of the ketone is demonstrated by the presence of a triplet at 198.3 of 3JC,P = 4.7 Hz and with all four C–H3 being chemically equivalent (δH = 1.95; δC = 21.2), similar to the closely related terminal alkyne complex p-tol4-CH2OMe. The difference in coordination between olefin and alkyne complexes is consistent with alkynes being stronger π-acceptors than olefins and illustrates the ability of the diphosphine ketone framework to adapt its binding mode to the electronic properties of a substrate.
Catalytic Comparison
Having shown that the hemilabile diphosphine-ketone ligand p-tolL1 is able to adapt its coordination mode to a substrate bound to Ni(0), we turn to its performance as a supporting ligand in the cyclotrimerization of alkynes. We compare hemilabile p-tol1 with the strong tridentate p-tol2 and the bidentate Ph3 to delineate specific effects of the hemilabile behavior. In addition, the smaller bite-angle rac-BINAP (PhL4) system was also tested.58 Terminal alkyne substrate with diverse electronic properties were selected for comparison: first, phenylacetylene (8), as a standard aryl-substituted substrate; second, methyl propiolate (9), as electron poor alkyne and since its additional oligomerization into cyclooctatetraene (COT) regio-isomers is commonly observed;59 and finally, methyl propargyl ether (10) as an electron rich substrate. The catalytic outcome of the reaction of these alkynes with 0.5 mol % of isolated BPI-catalysts p-tol1, p-tol2, and Ph3 and in situ generated PhL4 catalyst at room temperature was analyzed. The organic products were separated from the reaction mixture as a mixture of 1,2,4- (a) and 1,3,5- (b) substituted arenes, in addition to cyclotetramerization products (when applicable), COTs (c), in which their relative ratio was determined by 1H NMR, as described in Table 3. To exclude a particular role of the BPI or cod coligands, the active catalysts were also generated in situ by mixing p-tolL1–PhL3 with 1 equiv of Ni(cod)2 and 200 equiv of the corresponding alkyne. The reactions display similar activity as the values reported in Table 3 (Supporting Information; Table S4) and form the same resting states (respectively, p-tol4-R1, p-tol5-R1, Ph6-R1) under these conditions.
Table 3. Catalytic Comparison for the Cyclotrimerization of Phenylacetylene (8), Methyl Propiolate (9), and Methyl Propargyl Ether (10), Using Catalysts p-tol1 to Ph3, and the PhL4 Systema.

| entry | substrate (-R1) | catalyst | yield 1,2,4- (a) [%] | yield 1,3,5- (b) [%] | yield COTs (c) [%] | ratio a/b/c |
|---|---|---|---|---|---|---|
| 1 | phenyl acetylene, 8 (-Ph) | p-tol1 | 86.9 | 3.2 | 0 | 97:3:0 |
| p-tol2 | 3.1 | 1.9 | 0 | 62:38:0 | ||
| Ph3 | 2.8 | 0.2 | 0 | 94:6:0 | ||
| PhL4 + Ni(cod)2 | 4.6 | 1.9 | 0 | 70:30:0 | ||
| 2 | methyl propiolate, 9 (−CO2Me) | p-tol1 | 90.2 | 6.3 | 2.5 | 91:7:2 |
| p-tol2 | 24.5 | 2.1 | 7.5 | 72:6:22 | ||
| Ph3 | 65.0 | 12.3 | 6.5 | 77:15:8 | ||
| PhL4 + Ni(cod)2 | 14.0 | 3.0 | 32 | 29:6:65 | ||
| 3 | methyl propargyl ether, 10 (−CH2OMe) | p-tol1 | 71.6 | 6.9 | 0 | 90:10:0 |
| p-tol2 | <1 | <1 | 0 | b | ||
| Ph3 | <1 | <1 | 0 | b | ||
| PhL4 + Ni(cod)2 | <1 | <1 | 0 | b |
Similar results were obtained when the active catalysts were generated in situ using p-tolL1 to PhL3, 1 equiv of Ni(cod)2, and the corresponding terminal alkyne (Supporting Information; Table S4). Yields and ratios were averaged over two runs (Supporting Information; Tables S2 and S3). Yields are isolated yields. Ratios were determined by 1H NMR.
For systems p-tol2, Ph3, and PhL4, only trace amounts of products 10a and 10b were detected in which an accurate determination of the ratio between the two regio-isomers was not possible.
Mixing phenylacetylene (8) with 0.5 mol % of p-tol1 in toluene at room temperature for 16 h yields 86.9% of 1,2,4-triphenylbenzene (8a) and 3.2% of 1,3,5-triphenylbenzene (8b), in an isomeric ratio of 97:3. Under the same conditions, the complexes p-tol2 and Ph3 and the PhL4-Ni system afforded only low yields of 8a: 3.1%, 2.8%, and 4.6%, respectively. In addition, a lower regioselectivity toward the 1,2,4 isomers of 62:38 and 70:30 compared to the 1,3,5 isomer was observed with p-tol2 and PhL4 + Ni(cod)2, respectively. With the more electron poor alkyne methyl propiolate (9), the reaction catalyzed by p-tol1 led to nearly exclusive cyclotrimerization with the formation of the 1,2,4-isomer 9a in a ratio of 91:7 compared to its 1,3,5 analogue 9b and with a yield of 90.2%. Only small amounts of the cyclotetramers, tetramethyl-cyclooctatetraene-tetracarboxylates (9c) are observed, in a ratio of 2:98 compared to the trimerization products. In contrast, the yields obtained for 9a with the other catalysts in the series range from 14.0% to 65.0%. More importantly, catalysts p-tol2 and Ph3 and the PhL4 system display lower selectivity. The tridentate triphosphine catalyst p-tol2 exhibits a regioselectivity of 92:8 (1,2,4-/1,3,5-isomer) and produces cyclotetramers in a 22:78 ratio compared to the cyclotrimerization products. The bidentate systems Ph3 and PhL4 are also less selective and lead to the formation of 9a in 84:16 and 82:18, respectively, compared to 1,3,5-benzene product 9b. The bidentate catalyst Ph3 displays good chemoselectivity (92:8) toward cyclotrimerization, whereas the other diphosphine system PhL4 appears to even favor cyclotetramerization products 9c (35:65). Finally, methyl propargyl ether (10), bearing an electron rich substituent was also tested and afforded lower yields than 8 and 9. With the ketone-based catalyst p-tol1, 10 is converted into 1,2,4-tris(methoxymethyl)benzene (10a) in a yield of 71.6%, and the regioselectivity toward 10a remains high (90:10). Under the same conditions, the other complexes showed poor activity (less than 1% total yield). Overall, for the three examined substrates, catalyst p-tol1, bearing the hemilabile diphosphine benzophenone ligand p-tolL1, was shown to be more active and more selective toward the formation of the 1,2,4-trisubstituted benzene product.
Additional Substrates
In addition to the substrates described in Table 3, catalyst p-tol1 was applied to the cyclotrimerization of terminal alkynes 11a to 13a with high selectivity for 1,2,4-substituted benzenes and in good isolated yields (>65%, Table 4). Ethyl propiolate (11) (entry 1) affords triethylbenzene-1,2,4-tricarboxylate (11a) at room temperature. At 50 °C, 4-ethynylanisole (13) produces the desired 1,2,4-cyctrotrimerization product in a lower yield than the weakly electron-withdrawing analogue 1-ethynyl-4-fluorobenzene (12), supporting a preference for electron-withdrawing substrate (entries 2 and 3). The preference for electron poor alkynes is commonly observed in Ni-catalyzed cyclotrimerization of terminal alkynes.37m,37v−37y In comparison with the Ni-catalysts reported in literature,37 complex p-tol1 is a competitive catalyst in terms of activity and regioselectivity (from 90:10 to 97:3). A catalyst loading of p-tol1 of 0.05 mol % mediated the cyclotrimerization of ethyl propiolate (11) into triethylbenzene-1,2,4-tricarboxylate (11a) in 87% isolated yield, reaching a TON of 1740 after 16 h.
Table 4. Additional Substrates for Alkyne Cyclotrimerization Catalyzed by p-tol1c.


Reaction temperature = room temperature.
Reaction temperature = 50 °C.
Reported yields are isolated yield for the 1,2,4-trisubstituted benzene isomer. Isomeric ratios between the 1,2,4 and 1,3,5 isomer and between the trimers and tetramers were determined by 1H NMR. Yields and ratios were averaged over two runs (Supporting Information; Table S5). Catalytic method A was applied (see experimental part).
Mechanistic Considerations
The comparison study between p-tol1 and the other Ni-systems (p-tol2, Ph3, and PhL4) strongly suggests that the hemilabile π-acceptor moiety contributes to the high catalytic performance. In this section, the role of the ketone fragment in the improved catalytic performance of p-tol1 is studied and supported by computational modeling of some targeted compounds. To address the question, an overview of the general mechanism in metal-catalyzed [2 + 2 + 2] alkyne cyclotrimerization is discussed first (Scheme 5), followed by differences in reactivity among the three catalysts tested in the comparison study.
Scheme 5. Proposed Intermediates for the Transition-Metal-Catalyzed [2 + 2 + 2] Cyclotrimerization of Acetylene, as Commonly Reported in Literature33.

Scheme 5 shows a commonly proposed mechanism for the cyclotrimerization of acetylene catalyzed by transition metals.33 For base metals, most mechanistic studies have been carried out using cobalt complexes as catalysts.60 From the acetylene metal complex 14-C2H2, the association of a second molecule of acetylene generates complex 14-(C2H2)2, which requires an open coordination site in 14-C2H2. Next, the oxidative coupling of the two coordinated alkyne fragments from 14-(C2H2)2 gives rise to the key metallacyclopentadiene intermediate. This step is generally thought to be rate determining. The metallacyclopentadiene intermediate can be best described as one of two resonance structures, 14-MCP or 14-MCP′, depending on the nature of metal complex used. For example Saá et al. calculated that CpRuCl (Cp = cyclopentadiene)-catalyzed alkyne cyclotrimerization proceeds via 14-MCP′, while the cobalt system CpCo proceeds via 14-MCP.60d Late transitions metals tend to favor the concerted-oxidative cyclization between two alkynes and the low-valent metal center.60a However, a stepwise zwitterionic diradical pathway, involving the formation of a σ(C–C) bond and one Ni–C bond cannot be excluded. When acetylene is substituted, the formation of the substituted metallacyclopentadiene is of importance as different 14-MCP regio-isomers can be generated. The substituted 14-MCP will thus dictate the overall selectivity toward the product formation of the 1,2,4- or 1,3,5-trisubstituted benzene regio-isomer (more precisely the transition state TS1 between the bisalkyne and metallacyclopentadiene intermediate).61 In the last step (insertion of the third alkyne), different mechanisms have been proposed.62 The insertion of a third alkyne could either proceed via a Diels–Alder type [4 + 2] cycloaddition, in which the metal does not participate in the bond formation (Scheme 5, pathway a),60e or with the assistance of the metal, prior to the new C–C bond formation (Scheme 5; pathway b). 7-Metallanorbornadiene (14-[4 + 2])63 is frequently presented as an unstable species and immediately collapses to arene, forming a metal–arene adduct. As regards pathway b, the coordination of the third alkyne is followed by either migratory insertion (metallacycloheptatriene 14-I) or [2 + 2] cycloaddition (cycloadduct 14-[2 + 2]). These complexes can also be formed in a concerted way, without involving the direct formation of an acetylene metallacyclopentadiene complex 14-AMCP, but still with participation of the metal center. It has also been suggested that an additional pathway could occur via an intermolecular [4 + 2] cycloaddition forming the η4-bound intermediate, 14-[4 + 2]′.60d Pathway a is likely to happen for strong donor ligands or solvents.60d The insertion of a fourth alkyne (especially if the intermediate formed is 14-I) leads to formation of COT side products.61a Eventually, the formation of the benzene product happens by either reductive elimination or ligand exchange of benzene with a molecule of acetylene (cycloadduct 14-[2 + 2] rearranges itself prior to the reductive elimination).
In this overall picture, several effects of the hemilabile π-acceptor ketone moiety ion complex p-tol1 can be envisioned. First, the hemilabile interaction of the ketone unit can be thought to facilitate the alkyne uptake, especially in comparison with the tridentate phosphine system p-tol2. In situ NMR spectroscopy experiments with the three complexes p-tol1, p-tol2, and Ph3 showed that the corresponding monoalkyne complexes p-tol4-R1–Ph6-R1 are the resting states of the catalyst in all cases. For the ketone catalyst p-tol1 and the bidentate catalysts Ph3, these resting states (p-tol4-R1 and Ph6-R1) are 16 VE species that can readily accept an incoming alkyne molecule, while the corresponding triphosphine complex p-tol5-R1 is saturated (18 VE). Most probably, coordination of a second equivalent of alkyne to p-tol5-R1 requires decoordination of one of the phosphorus atoms, which is likely to raise the overall reaction barrier and result in lower activity under the same conditions (Supporting Information; Scheme S4). The unique feature of the ketone unit is that the tricoordinate alkyne complex can be accessed directly from the tetracoordinate precursor p-tol1. A similar process can be thought to happen at the end of the catalytic cycle: the ketone may accelerate product release by transient coordination to Ni(0), which would explain the lower propensity of p-tol1 to form cyclooctatetraenes.
Second, the interaction of the ketone with the nickel center could help to stabilize transient intermediates, such as the key metallacycle 14-MCP species. In order to assess structural differences between the metallacyclopentadiene supported by the different ligands, p-tolL1, p-tolL2, and PhL3, we made use of geometry optimization by DFT calculation at a B3LYP/6-31g(d,p) level of theory using acetylene as a model substrate.
Unsurprisingly, the ketone moiety is not bound in the bisalkyne diphosphine benzophenone complex p-tol15-(C2H2)2. However, geometry optimization of the metallacyclopentadiene intermediate formed by oxidative coupling reveals a pentacoordinate geometry in which the ketone is bound to the metal (p-tol16-MCP, Scheme 6), suggesting hemilabile behavior of p-tolL1 in the key oxidative coupling step. Respective Ni–O and Ni–C distances of 1.93 and 1.99 Å, as well as an elongated C–O bond (1.35 Å), indicate a strong interaction with the metal center. In addition, a P1–Ni–P2 angle of 171° results in an approximate trigonal-bipyramidal geometry with apical P atoms. The fact that the C=O unit binds side-on to a formal Ni(II) center can be surprising at first sight in view of its low propensity to bind to divalent metal halides.24,25 This can be understood by a synergistic interaction between a strongly σ-donating bidentate hydrocarbyl ligand and the strongly π-accepting ketone ligand in the equatorial plane of the trigonal bipyramid.
Scheme 6. C–C Coupling Reaction Step, as Modeled by DFT Calculations.
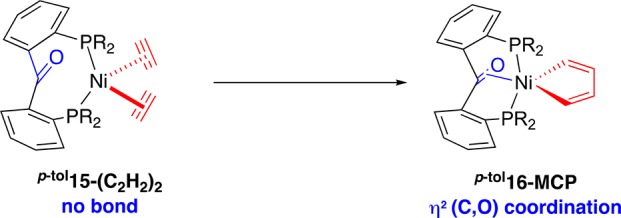
R = para-tolyl.
For comparison, the geometry of the metallacycles supported by the triphosphine p-tolL2 and the diphosphine ether PhL3 ligands were also optimized (Figure 7). The triphosphine-supported [(p-tolL2)Ni(C4H4)] (p-tol17-MCP) adopts a similar trigonal bipyramid geometry to p-tol16-MCP. This binding mode may contribute to explain the differences in reactivity with the rac-BINAP system, for which this mode is inaccessible. In contrast, [(PhL3)Ni(C4H4)] (Ph18-MCP) exhibits a distorted square planar geometry, in which the PhL3 is bound in bidentate manner (N–O = 3.20 Å; WBI(Ni–O) < 0.01). Hence, the metallacycle in Ph18-MCP is not stabilized by its central ether donor group,64 which may partly explain the lower regioselectivity of Ph3 is toward the 1,2,4-trisubstituted arene cyclotrimerization product. Furthermore, no significant modifications of the P1–O–P2 bite angle from phenylacetylene analogue Ph6-Ph are visible.
Figure 7.

C–C coupling reaction step. Comparison of metallacyclopentadiene molecular structures bearing the p-tolL1, p-tolL2, and PhL3 ligands, optimized at a B3LYP/6-31g(d,p) level of theory. Hydrogen atoms have been omitted for clarity.
At this point, we showed that the hemilabile character of the ketone ligand p-tolL1 contributes to higher activity and selectivity in the cyclotrimerization of terminal alkynes. The combination of the two effects presented in this section, that is, decoordination and coordination of the C=O unit to assist the substrate uptake and stabilize the key metallacyclopentadiene intermediate, can explain its superior performance. Exchanging the C=O moiety in complex p-tol1 with a stronger donor atom, like a phosphine group (P–Ph) or with a bidentate ligand (O as central atom) decreases either the activity or the selectivity of the overall process.
Therefore, based on the computational and experimental observations obtained from this study, a simplified catalytic cycle is proposed for the cyclotrimerization of terminal alkynes catalyzed by the nickel diphosphine benzophenone system (Scheme 7). The resting state, p-tol4-R1, can be generated by ligand exchange from p-tol1 or in situ from the ligand and Ni(cod)2 (1). The in situ system and p-tol1 operate at similar rates of product formation under the same tested conditions (similar percentage of isolated yields), showing that the dissociation of the coligand (cod vs BPI) and competitive binding with the metal center does not affect the final yields, as long as p-tol4-R1 can be formed. At this step, the ketone is not bound, which facilitates coordination of the second alkyne molecule to generate Ni-bisalkyne p-tol15-(C2HR1)2 (2). The next step (3) involves the C–C oxidative coupling, which is coupled to coordination of the ketone to nickel in η2-fashion. This interaction may favor the formation of the 2,5-disubstituted metallacyclopentadiene p-tol16-MCP-R1. The R1 substituents have been arbitrarily positioned in 2,5 positions as they promote the selective formation of the 1,2,4-trisubstituted benzene. From this intermediate, the insertion of the third alkyne to form the final benzene product could go via either [4 + 2] cycloaddition, migratory insertion, or [2 + 2] cycloaddition, followed by reductive elimination of the trisubstituted benzene and ligand exchange to regenerate p-tol1 for a new turnover (4). Throughout the reaction coordinate, the ligand can adapt its geometry by the labile interaction of C=O to Ni and stabilize intermediates.
Scheme 7. Proposed Simplified Catalytic Cycle for Cyclotrimerization of Terminal Alkynes Catalyzed by [(p-tolL1)Ni(BPI)] or by in Situ Generation of the Active Intermediate with p-tolL1 + Ni(cod)2.

R = para-tolyl.
Conclusions
In conclusion, we have reported here the synthesis and characterizations of Ni(0) complexes, incorporating a diphosphine ketone (p-tolL1), trisphosphine (p-tolL2), and diphosphine ether (PhL3) ligand, in which the binding mode of the stabilizing imine or alkyne coligand can change according to the structural and electronic characteristics of the supporting ligand. The characterization of [(p-tolL2)Ni(HC≡CPh)] (p-tol5-Ph) provides a rare example of weak activation of alkynes by nickel complexes, attributed to its unique coordination geometry. We show in this study that [(p-tolL1)Ni(BPI)] (p-tol1) is an effective catalyst in the [2 + 2 + 2] cyclotrimerization of terminal alkynes. In contrast, related Ni(0) complexes [(p-tolL2)Ni(BPI)] (p-tol2) and [(PhL3)Ni(BPI)] (Ph3) are less active or less selective toward the 1,2,4-trisubstitued benzene cyclotrimerization product under similar conditions. We attribute the enhanced reactivity of p-tol1 to the hemilabile character of its diphosphine benzophenone ligand. In situ NMR spectroscopy and DFT calculations suggest that C=O hemilability may facilitate substrate uptake and assist the key oxidative coupling step. A more detailed mechanistic study of the reaction and applications of diphosphine-ketone ligands to other catalytic processes are actively investigated in our laboratories.
Experimental Section
Chemicals and Reagents
Unless otherwise noted, all reactions were carried out under an inert N2(g) atmosphere, using standard Schlenk line or glovebox techniques, and stirred magnetically. Silica gel P60 (SiliCycle) was used for column chromatography. Analytical thin layer chromatography was performed using SiliCycle 60 F254 silica gel (precoated sheets, 0.20 mm thick) from Merck KGaA (Darmstadt, Germany). Deuterated solvents were purchased from Cambridge Isotope Laboratory Incorporation (Cambridge, USA) and were degassed by standard freeze–thaw–pump procedure65 and subsequently stored over molecular sieves. Common solvents were purified using a MBRAUN MB SPS-80 purification system or by standard distillation techniques or both.65 They were degassed by bubbling N2(g) through the liquid for at least 30 min and then stored over molecular sieves. Non-halogenated solvents were tested with a standard purple solution of sodium benzophenone ketyl in tetrahydrofuran to confirm effective oxygen and moisture removal. Other solvents were checked for water content by the Karl Fischer titration or by 1H NMR. Liquid chemicals were first degassed by standard freeze–pump–thaw procedures or purged with N2(g) and then stored over molecular sieves prior to use. Phosphorus-containing compounds were checked for oxidation by 31P NMR before use. O-(Bromophenyl)-diphenylphosphine,24,47o-(bromophenyl)-di-p-tolylphosphine,25 2,2′-bis(diphenylphosphino)benzophenone (PhL1),24 and 2,2′-bis(di(p-tolyl)phosphino)benzophenone (p-tolL1)25 were synthesized according to reported procedures. All other reagents and starting materials were purchased from commercial sources and used without further purification, except when specified.
Physical Methods
The 1H, 13C, 31P, and 19F NMR (400, 100, 161, and 400 MHz, respectively) spectra were recorded at 297 K on an Agilent MRF 400 spectrometer. All chemical shifts are reported in the standard δ notation of parts per million, referenced to residual peak of the solvent, as determined relative to Me4Si (δ = 0 ppm).66 Variable-temperature (VT) NMR were recorded in d-toluene from −85 °C to room temperature. Infrared spectra were recorded using a PerkinElmer Spectrum Two FT-IR spectrometer. For air-sensitive compounds, a N2 flow was used. Absorption spectra were recorded using a Lambda 35 UV–vis spectrometer. The UV–vis solutions were prepared in the glovebox, using degassed and dried solvent, and then stored in a cuvette sealed with a Teflon cap. The acquisition and analysis of the UV–vis data were performed with PerkinElmer UW WinLab and UV WinLab Data Processor and Viewer software. GC-MS measurements were conducted on a PerkinElmer Clarus 680 GC (column PE, Elite 5MS, 15 m × 0.25 mm ID × 0.25 μm) equipped with Clarus SQ8T MS and analyzed with TurboMass software. ESI-MS analysis was recorded with a Water LCT Premier XE spectrometer. Elemental analysis was provided by Mikroanalytisches Laboratorium Kolbe, Mülheim an der Ruhr, Germany, and Medac Ltd., Surrey, UK.
Computational Methods
DFT (density functional theory) results were obtained using the Gaussian 09 software package.67 Restricted (R) geometry optimizations use the B3LYP (Becke, three-parameter, Lee–Yang–Parr) functional and the 6-31g(d,p) basis set on all atoms. The structures were optimized without any symmetry restraints and are either minima or transition states. Frequency analyses were performed on all calculations. The transition states search was performed using the QST3 (synchronous transit-guided quasi Newton number 3) method. For NBO (natural bond orbital) calculation, the NBO6 program,68 up to the NLMO (natural localized molecular orbital) basis set, was used at B3LYP/def2TZVP level of theory from the optimized geometry. Pictures derived from DFT calculations have been generated using the GaussView software. The B3LYP functional was chosen as it has been shown to be accurate for geometry optimization of related metal compounds,69 including closely related PhL1–metal systems.24,25
Synthetic Methods
Ligand: Bis(2-di-p-tolyl)phosphinophenyl) Phenylphosphine (p-tolL2)
Adapted from the procedure by Koshevoy et al.46 To a suspension of (o-bromophenyl)di-p-tolylphosphine (5.01 g, 16.6 mmol) in dried and degassed THF (76 mL), a hexane solution of n-BuLi (1.6 M, 8.50 mL, 13.5 mmol) was added dropwise within 10 min at −78 °C under a N2 atmosphere. The reaction mixture was stirred at −78 °C for 1 h, and PPhCl2 (0.92 mL, 6.77 mmol) was added dropwise. The mixture was stirred at this temperature for one additional hour and then allowed to slowly warm up to room temperature. The solution was stirred at room temperature for three more hours and then quenched with MeOH (30 mL). The volatiles were removed in vacuo, and the yellow amorphous residue was washed with MeOH (5 × 15 mL) to afford p-tolL2 as a white solid, which was dried overnight under vacuum (3.52 g, 5.10 mmol, 76%). 1H NMR (400 MHz, C6D6, 25 °C): δH 7.40–7.33 (ArH, m, 6H), 7.33–7.25 (ArH, m, 6H), 7.19–7.16 (ArH, m, 2H), 7.01–6.94 (ArH, m, 5H), 6.93–6.87 (ArH, m, 6H), 6.84 (ArH, d, 3JH,H = 7.9 Hz, 4H), 2.03 (CH3, s, 6H), 2.01 (CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP – 14.8 (p-tolP, 2P), – 16.9 (PhP, 1P) [AB2 system, 2JA,B = 155 Hz]. 13C NMR (100 MHz, C6D6, 25 °C): δC 145.6–144.8 (m), 138.4–138.1 (m), 138.0 (d, JC,P = 12.7 Hz), 135.2–134.9 (m), 134.7 (t, JC,P = 4.4 Hz), 134.6–134.3 (m), 129.6, 129.4 (t, JC,P = 3.3 Hz), 129.1, 128.9, 128.2, 129.9, 21.2 (CH3), 21.2 (CH3). ATR-IR: ν [cm–1] = 3043, 1494, 1439, 1184, 1090, 806, 752, 504. HRMS (ESI, CH3CN, AgNO3): m/z calcd for [M + Ag]+ 793.1472; found 793.1605.
Nickel Complexes
[(PhL1)Ni(BPI)] (Ph1)
Ni(cod)2 (249 mg, 0.91 mmol), PhL1 (502 mg, 0.91 mmol), and benzophenone imine (181 mg, 0.92 mmol) were dissolved in dried degassed toluene (10 mL) under an inert atmosphere. The reaction mixture was stirred at room temperature for 20 min. Dried and degassed hexane (5 mL) was added to the resulting black solution, causing the precipitation of a black solid. The precipitate was collected by filtration, washed with hexane (3 × 4 mL), and dried under vacuum to afford Ph1 as a black powder (597 mg, 0.76 mmol, 83%). Single crystals suitable for X-ray diffraction and elemental analysis were obtained by slow exchange of hexane into a concentrated THF solution of Ph1. 1H NMR (400 MHz, C6D6, 25 °C): δH 9.88 (NH, br s, 1H), 7.90 (ArH, d, 3JH,H = 7.2 Hz, 2H), 7.83 (ArH, d, 3JH,H = 7.6 Hz, 2H), 7.75–7.70 (ArH, m, 4H), 7.23–7.19 (ArH, m, 2H) 7.09–6.79 (ArH, m, 26H), 6.73 (ArH, t, 3JH,H = 7.6 Hz, 2H). 31P NMR (161 MHz, C6D6, 25 °C): δP 16.4 (s, 2P). 13C NMR (100 MHz, d8-THF, 25 °C): δC 170.7 (C=N, t, 3JC,P = 5.4 Hz), 156.0 (t, JC,P = 18.0 Hz), 141.1, 140.2 (t, JC,P = 17.0 Hz), 138.7, 136.7 (t, JC,P = 11.2 Hz), 136.4 (t, JC,P = 13.7 Hz), 132.9 (t, JC,P = 7.7 Hz), 132.6, 131.8 (t, JC,P = 6.6 Hz), 129.62 (t. JC,P = 4.0 Hz), 128.9–126.7 (m) 125.8, 125.5 (t, JC,P = 8.1 Hz), 117.2 (C=O, t, 3JC,P = 7.6 Hz). ATR-IR: ν [cm–1]: 3163, 3050, 1583, 1404, 1432, 1478, 1432, 1303, 1249, 1091, 912, 778, 739, 692, 515. UV–vis (toluene): λmax [nm] 365, 574. Elemental analysis, Anal. Calcd for C50H39NNiOP2·1/2 hexane: C, 76.37; H, 5.56; N, 1.68. Found: C, 76.33; H, 5.58; N, 1.52. The crystal structure contains channels along the c-axis, which are filled with disordered hexane molecules (Supporting Information; Figure S110).
[(p-tolL1)Ni(BPI)] (p-tol1)
Ni(cod)2 (108 mg, 0.39 mmol), p-tolL1 (238 mg, 0.39 mmol), and benzophenone imine (71 mg, 0.39 mmol) were dissolved in dried degassed toluene (10 mL) under inert atmosphere. The reaction mixture was stirred at room temperature for 20 min. The solvent was evaporated, and the crude mixture was subsequently dissolved in dried and degassed THF (5 mL). Dried and degassed hexane (5 mL) was added to the resulting black solution, causing the precipitation of a black solid. The precipitate was filtered, washed with hexane (3 × 4 mL), and dried under vacuum to afford p-tol1 as a black powder (221 mg, 0.31 mmol, 79%). Single crystals for elemental analysis were obtained by slow exchange of hexane into a concentrated toluene solution of p-tol1. 1H NMR (400 MHz, C6D6, 25 °C): δH 10.01 (NH, s, 1H), 7.98 (ArH, d, 3JH,H = 7.4 Hz, 2H), 7.88 (ArH, dd, 3JH,H = 7.7, 4JH,H = 1.3, 2H), 7.66 (ArH, dt, 3JH,H = 7.9, 4JH,P = 4.6, 4H), 7.30–7.25 (ArH, m, 2H), 7.05 (ArH, dt, 3JH,H = 7.9 Hz, 4JH,P = 4.4 Hz), 6.97–6.88 (ArH, m, 6H), 6.86–6.73 (ArH, m, 14H), 2.11 (CH3, s, 6H), 2.09 (CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 15.5 (s, 2P). 13C NMR (100 MHz, C6D6, 25 °C): δC 169.8 (C=N, t, 3JC,P = 6.1 Hz), 156.7 (t, JC,P = 18.1 Hz), 141.6 (t, JC,P = 2.3 Hz), 141.2 (t, JC,P = 16.9 Hz), 138.6 (Ar, t, JC,P = 2.4 Hz,), 138.1, 137.2, 134.7 (t, JC,P = 12.1 Hz), 133.9 (t, JC,P = 14.7 Hz), 133.6, 133.5, 133.5, 133.4, 132.7 (t, JC,P = 6.8 Hz), 130.5 (t, JC,P = 4.3 Hz), 129.1 (t, JC,P = 4.4 Hz), 129.0 (t, JC,P = 4.0 Hz), 128.8, 127.5, 126.8, 126.5 (t, JC,P = 8.1 Hz), 119.0 (C=O, t, JC,P = 5.1 Hz), 21.3 (CH3), 21.1(CH3). ATR-IR: ν [cm–1]: 3152, 3053, 2917, 2860, 1598, 1496, 1448, 1394, 1250, 1185, 1092, 1018, 912, 803, 693, 627, 514. Elemental analysis, Anal. Calcd for C54H47NNiOP2: C, 76.61; H, 5.66. Found: C, 76.55; H, 5.93.
[(p-tolL2)Ni(BPI)] (p-tol2)
Ni(cod)2 (163 mg, 0.59 mmol), p-tolL2 (405 mg, 0.59 mmol), and benzophenone imine (108 mg, 0.59 mmol) were combined together and dissolved in dried degassed toluene (7 mL) under inert atmosphere. The reaction mixture was stirred at room temperature for 1 h, and dried degassed hexane (7 mL) was added. The resulting solution was left in the freezer at −35 °C for 16 h, during which the precipitation of a black solid was observed. The solid was filtered, washed with cold hexane (5 × 4 mL), and dried under vacuum to afford p-tol2 as a black powder (315 mg, 0.34 mmol, 58%). 1H NMR (400 MHz, C6D6, 25 °C): δH 10.25 (NH, dt, 3JH,P = 3.2 Hz, 3JH,P = 2.7 Hz, 1H), 8.49 (ArH, d, 3JH,H = 7.6 Hz, 2H), 8.05 (ArH, dd, 3JH,H = 7.2 Hz, 3JH,P = 4.4 Hz, 2H), 7.66–7.60 (ArH, m, 5H), 7.55 (ArH, t, 3JH,H = 8.0 Hz, 2H), 7.12–6.86 (ArH, m, 18H), 6.83 (ArH, d, 3JH,H = 8.4 Hz, 6H), 6.72 (ArH, d, 3JH,H = 7.6 Hz, 4H), 2.14 (CH3, s, 6H), 1.95 (CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 37.1 (p-tolP, 2P) 28.7 (PhP, 1P) [AK2 system, 2JA,K = 85 Hz]. 13C NMR (100 MHz, C6D6, 25 °C): δC 168.5 (C=N, dt, 3JC,P = 8.1 Hz, 3JC,P = 7.4 Hz), 149.6–147.8 (m), 143.2, 141.3 (dt, JC,P = 11.1 Hz, JC,P = 2.7 Hz), 137.8 (dt, JC,P = 13.9 Hz, JC,P = 4.0 Hz), 137.2, 137.1–136.4 (m), 136.3, 132.8–132.6 (m), 132.4 (d, JC,P = 14.1 Hz), 131.8 (t, JC,P = 6.7 Hz), 131.3 (d, JC,P = 13.8 Hz), 130.6, 129.9–129.5 (m), 129.1 (t, JC,P = 4.2 Hz), 128.8–128.6 (m) 127.6, 127.1, 126.3, 21.4 (CH3), 21.1 (CH3). ATR-IR: ν [cm–1] 3048, 2972, 2917, 2864, 1664, 1598, 1496, 1445, 1187, 1114, 804, 695, 513. Elemental analysis, Anal. Calcd for C59H52NNiP3: C, 76.47; H, 5.66; N, 1.51. Found: C 75.97 H 5.49 N 1.56.
[(PhL3)Ni(BPI)] (Ph3)
Under inert atmosphere, a dried and degassed toluene solution (7 mL) of benzophenone imine (169 mg, 0.93 mmol) was added to a vial containing Ni(cod)2 (255 mg, 0.93 mmol), and PhL3 (500 mg, 0.93 mmol). The reaction mixture was stirred at room temperature for 16 h in which a solid spontaneously precipitated. The resulting solid was filtered, washed with cold hexane (5 × 4 mL), and dried under vacuum to afford Ph3 as an orange powder (582 mg, 0.75 mmol, 81%). Single crystals suitable for X-ray diffraction and elemental analysis were obtained by slow diffusion of hexane into a C6D6 solution of Ph3. 1H NMR (400 MHz, C6D6, 25 °C): δH 9.71 (NH, s, 1H), 8.00–7.85 (ArH, br s, 2H), 7.70–7.64, (ArH, br s, 2H), 7.61–7.53 (ArH, br s, 6H), 7.13–6.90 (ArH, br s, 22H), 6.78–6.72 (ArH, br m, 4H), 6.56–6.52 (ArH, br m, 2H). 31P NMR (161 MHz, C6D6, 25 °C): δP 32.4 (s, 2P). ATR-IR: ν [cm–1] 3200, 3052, 1589, 1565, 1462, 1434, 1363, 1259, 1216, 1095, 880, 747, 693, 622, 505. Elemental analysis, Anal. Calcd for C49H29NNiOP2: C, 75.60; H, 5.05; N, 1.80. Found: C, 75.22; H, 5.69; N, 2.05. Due to poor solubility and progressive decomposition of Ph3 in common solvents, no 13C NMR was recorded.
In Situ Generation of [(p-tolL1)Ni(HC≡CR1)] (p-tol4-R1)
Under an inert atmosphere, 1 equiv of [(p-tolL1)Ni(BPI)] (p-tol1) was mixed with 1 equiv of a terminal alkyne and dissolved in dried degassed C6D6 (0.6 mL). The solution was transferred into a Young-type NMR tube, and the mixture was analyzed. 31P and 1H NMR show the appearance of one new single species, [(p-tolL1)Ni(HC≡CR1)] (p-tol4-R1), in addition to the partial release of BPI. High in situ yield was obtained with methyl propargyl ether (R1 = CH2OMe) as alkyne reactant, leading to the formation of [(p-tolL1)Ni(HC≡CH2OMe)] (p-tol4-CH2OMe). The solution contains a mixture of p-tol4-CH2OMe, p-tol1, BPI, and methyl propargyl ether. 1H NMR (400 MHz, C6D6, 25 °C): δH 10.01 (p-tol1, NH, s, 1H), 9.82 (BPI, NH, br. s, 1H) 8.03 (p-tol1, ArH, d, 3JH,H = 7.4 Hz, 2H), 7.92 (p-tol1, ArH, dd, 3JH,H = 7.7, 4JH,H = 1.3, 2H), 7.70 (p-tol1, ArH, dt, 3JH,H = 7.9, 4JH,P = 4.6, 4H), 7.54 (p-tol4-CH2OMe, p-tol-ArH, td, 3JH,P = 8.0 Hz, 3JH,P = 7.2 Hz, 8H), 7.35–7.29 (p-tol1, ArH, m, 2H), 7.10 (p-tol1, ArH, dt, 3JH,H = 7.9 Hz, 4JH,P = 4.4 Hz), 7.02–6.92 (p-tol1, ArH, m, 6H), 6.91–6.76 (p-tol1, ArH, m, 14H), 6.74 (p-tol4-CH2OMe, ArH, t, 3JH,H = 7.6 Hz, 2H), 6.66 (p-tol4-CH2OMe, ArH, td, 3JH,H = 7.6 Hz, 4JH,H = 1.6 Hz, 2H), 6.34 ppm (p-tol4-CH2OMe, ≡CH,3JH,P 15.6, 4JH,H = 1.6 Hz, 1H) 4.63 (p-tol4-CH2OMe, CH2, d, 4JH,H = 1.6 Hz, 2H), 3.70 (HC≡CCH2OMe, CH2,4JH,H = 1.6 Hz, 2H) 3.32 (p-tol4-CH2OMe, OCH3, s, 3H), 3.03 (HC≡CCH2OMe, OCH3, s, 1H); 2.10 (p-tol1, CH3, s, 6H), 2.03 (p-tol4-CH2OMe, CH3, s, 12H), 1.96 (HC≡CCH2OMe, ≡CH,4JH,H = 1.6 Hz, 1H), 1.91 (p-tol1, CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 33.3 (p-tol4-CH2OMe, s, 2P), 15.5 (p-tol1, s, 2P).
13C NMR was acquired in order to locate the resonances from the carbonyl and methyl groups of the ligand of p-tol4-CH2OMe. 13C NMR (100 MHz, C6D6, 25 °C): δC 203.1 (C=O, t, 4JC,P = 4.7 Hz), 22.2 (H3C, d, 4JC,P = 2.3 Hz).
[(p-tolL2)Ni(HC≡CPh)] (p-tol5-Ph)
Under inert atmosphere, [(p-tolL2)Ni(BPI)] (p-tol2) (100 mg, 0.11 mmol), and phenylacetylene (11 mg, 0.11 mmol) were mixed together and dissolved in dried degassed toluene (5 mL). The reaction mixture was subsequently stirred at room temperature for 1 h. The solvent was removed under vacuum, and the crude residue was dissolved in dried degassed THF (3 mL). Dried degassed hexane (3 mL) was added, and the mixture was put in the freezer at −35 °C for 16 h, after which a red precipitate was observed. The solid was isolated by filtration and washed with cold hexane (5 × 2 mL) to afford p-tol5-Ph as a red powder (59 mg, 0.07 mmol, 66%). Single crystals suitable for X-ray diffraction were obtained by slow evaporation of a concentrated THF solution of p-tol5-Ph. 1H NMR (400 MHz, C6D6, 25 °C): δH 7.92 (ArH, dd, 3JH,H = 8.0 Hz, 3JH,P = 6.8 Hz, 2H), 7.63–7.53 (ArH, m, 9H), 7.45–7.39 (ArH, m, 2H), 7.10–6.95 (ArH, m, 1H), 6.88 (ArH, tt, 3JH,H = 7.2 Hz, 4JH,P 1.2 Hz, 1H), 6.83 (ArH, d, 3JH,H = 7.6 Hz, 4H), 6.72 (ArH, d, 3JH,H = 7.6 Hz, 4H), 6.29 (≡CH, dt, 3JH,P = 25.6 Hz, 3JH,P = 7.2 Hz, 1H), 2.12 (CH3, s, 6H), 1.91 (CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 78.7 (PhP, t, 2JP,P = 35.4 Hz, 1P), 28.1 (p-tolP, d, 2JP,P = 35.4 Hz, 2P). 13C NMR (100 MHz, C6D6, 25 °C): δC 150.0 (dt, JC,P = 47.1 Hz, JC,P = 16.4 Hz), 146.9 (d, JC,P = 36.0 Hz) 146.4 (dd, JC,P = 61.6 Hz, JC,P = 34.3 Hz),140.1 (dt, JC,P = 41.6 Hz, JC,P = 26.0 Hz), 137.9, 137.0–136.6 (m), 136.8, 133.7, 133.1 (t, JC,P = 28.0) Hz, 132.5 (t, JC,P = 28.0 Hz), 131.8–131.5 (m), 129.8, 129.7–128.9 (m) 101.1 (≡CPh, d, 2JC,P = 20.0 Hz), 92.9 (CH, br s), 21.3 (CH3, d, JC,P = 4.0 Hz), 21.1 (CH3, d, JC,P = 4.0 Hz). ATR-IR: ν [cm–1] 3042, 2918, 2861, 1823, 1589, 1479, 1440,1425, 1394, 1185, 1100, 1085, 1118, 805, 758, 690, 666, 625, 612, 539, 519. Due to the high sensitivity of p-tol6-Ph, no elemental analysis data was obtained. The purity and identity of the compound was established by NMR and by its X-ray diffraction structure.
Alternative Synthesis of p-tol5-Ph
Under inert atmosphere, Ni(cod)2 (80 mg, 0.29 mmol), p-tolL2 (200 mg, 0.29 mmol), and phenylacetylene (32 mg, 0.31 mmol) were mixed together and dissolved in dried degassed toluene (5 mL). The reaction mixture was subsequently stirred at room temperature for 1 h. Precipitation of a red solid was observed after adding dried degassed hexane (10 mL) and leaving the solution to stand overnight in the freezer at −35 °C. The resulting solid was filtered, washed with cold hexane (5 × 5 mL), and dried under vacuum to afford p-tol5-Ph as a red powder (169 mg, 0.20 mmol, 70%).
Synthesis of Deuterated Analogue of p-tol5-Ph, [(p-tolL2)Ni(DC≡CPh)]
The same procedure as for the synthesis of p-tol5-Ph was applied from Ni(cod)2 (30 mg, 0.10 mmol), p-tolL2 (70 mg, 0.10 mmol), and d-phenylacetylene (11 mg, 0.10 mmol). [(p-tolL2)Ni(DC≡CPh)] was isolated as a red solid in a 74% yield (63 mg, 0.07 mmol). [(p-tolL2)Ni(DC≡CPh)] can also be generated from reaction of p-tol2 with d-acetylene in a 1:1 stoichiometry. 1H NMR (400 MHz, C6D6, 25 °C): δH 7.92 (ArH, dd, 3JH,H = 8.0 Hz, 3JH,P = 6.8 Hz, 2H), 7.63–7.53 (ArH, m, 9H), 7.45–7.39 (ArH, m, 2H), 7.10–6.95 (ArH, m, 14H), 6.88 (ArH, tt, 3JH,H = 7.2 Hz, 4JH,P 1.2 Hz, 1H), 6.83 (ArH, d, 3JH,H = 7.6 Hz, 4H), 6.72 (ArH, d, 3JH,H = 7.6 Hz, 4H), 2.12 (CH3, s, 6H), 1.91 (CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 78.7 (PhP, t, 2JP,P = 35.4 Hz, 1P), 28.1 (p-tolP, d, 2JP,P = 35.4 Hz, 2P). 13C NMR (100 MHz, C6D6, 25 °C): δC 150.0 (dt, JC,P = 47.1 Hz, JC,P = 16.4 Hz), 146.9 (d, JC,P = 36.0 Hz) 146.4 (dd, JC,P = 61.6 Hz, JC,P = 34.3 Hz), 140.1 (dt, JC,P = 41.6 Hz, JC,P = 26.0 Hz), 137.9, 137.0–136.6 (m), 136.8, 133.7, 133.1 (t, JC,P = 28.0) Hz, 132.5 (t, JC,P = 28.0 Hz), 131.8–131.5 (m), 129.8, 129.7–128.9 (m) 101.1 (≡CPh, br m, 21.3 (≡CH3, d, JC,P = 4.0 Hz), 21.1 (CH3, d, JC,P = 4.0 Hz). ATR-IR: ν [cm–1] 3333, 3042, 2918, 2861, 1823, 1761, 1589, 1479, 1440,1425, 1394, 1185, 1100, 1085, 1118, 805, 758, 690, 666, 625, 612, 539, 519.
[(PhL3)Ni(HC≡CPh)] (Ph6-Ph)
Under inert atmosphere and at room temperature, a suspension of [(PhL3)Ni(BPI)] (Ph3) (200 mg, 0.26 mmol) in dried degassed toluene (3 mL) was combined with a toluene solution (3 mL) of phenylacetylene (26 mg, 0.26 mmol), resulting in a light yellow solution. The reaction mixture was subsequently stirred at room temperature for 10 min. Precipitation of a yellow solid was observed after adding dried degassed hexane (6 mL) and leaving the solution to stand overnight. The resulting solid was filtered, washed with hexane (5 × 5 mL), and dried under vacuum to afford Ph6-Ph as a yellow powder (112 mg, 0.16 mmol, 74%). Single crystals suitable for X-ray diffraction were obtained by slow diffusion of hexane into a concentrated toluene solution of Ph6-Ph. 1H NMR (400 MHz, C6D6, 25 °C): δH 7.94–7.87 (ArH, m, 4H), 7.46–7.40 (ArH, m, 4H), 7.30 (ArH, dd, 3JH,H = 6.8 Hz, 4JH,H = 1.4 Hz, 2H), 7.05–6.80 (ArH and ≡CH, m, 18H), 6.75 (ArH, ddd, 3JH,H = 7.6 Hz, 4JH,P = 4.4 Hz, 4JH,H = 1.2 Hz, 1H), 6.66–6.56 (ArH, m, 3H), 6.39 (ArH, ddd, 3JH,H = 7.2 Hz, 4JH,P = 4.4 Hz, 4JH,H = 1.2 Hz, 1H), 6.32 (ArH, tt, 3JH,H = 7.6 Hz, 4JH,H = 1.0 Hz, 1H). 31P NMR (161 MHz, C6D6, 25 °C): δP 29.2 (d, 2JP,P = 22.5 Hz, 1P), 27.3 (d, 2JP,P = 22.5 Hz, 1P). 13C NMR (100 MHz, C6D6, 25 °C) δC 160.1 (d, 2JC,P = 9.1 Hz), 159.8 (d, 2JC,P = 11.4 Hz), 136.6 (≡CPh, dd, 2JC,P = 34.9 Hz, 2JC,P = 4.8 Hz), 136.1, 135.5 (dd, JC,P = 34.8 Hz, JC,P = 4.1 Hz), 134.9 (d, JC,P = 14.5 Hz), 133.8 (d, JC,P = 12.7 Hz), 132.9 (d, JC,P = 1.8 Hz), 130.7, 130.0, 129.8 (d, JC,P = 3.8 Hz), 129.6, 129.4 (d, JC,P = 1.7 Hz), 129.3, 129.0 (d, JC,P = 3.1 Hz), 128.8 (d, JC,P = 1.5 Hz), 125.3, 125.7(≡CH, dd, 2JC,P = 34.9 Hz, 2JC,P = 4.8 Hz) 124.9 (d, JC,P = 4.0 Hz), 124.1 (d, JC,P = 4.0 Hz), 122.9 (d, JC,P = 4.9 Hz), 118.3 (d, JC,P = 3.4 Hz). ATR-IR: ν [cm–1] 3286, 3171, 3052, 1749, 1588, 1564, 1480, 1461, 1434, 1259, 1213, 1095, 882, 834, 745, 692, 554, 503. Due to the high sensitivity of Ph6-Ph, no elemental analysis data was obtained. The purity and identity of the compound was established by NMR and by its X-ray diffraction structure.
Alternative Synthesis of Ph6-Ph
Under inert atmosphere, Ni(cod)2 (292 mg, 1.06 mmol), PhL3 (567 mg, 1.05 mmol), and phenylacetylene (108 mg, 1.06 mmol) were mixed together and dissolved in dried degassed toluene (7 mL). The reaction mixture was subsequently stirred at room temperature for 20 min. Precipitation of a yellow solid was observed after adding dried degassed hexane (10 mL) and leaving the solution to stand overnight. The resulting solid was filtered, washed with hexanes (5 × 5 mL), and dried under vacuum to afford Ph6-Ph as a yellow powder (387 mg, 0.55 mmol, 53%).
Synthesis of Deuterated Analogue of Ph6-Ph, [(PhL3)Ni(DC≡CPh)]
The same experimental procedure as for the synthesis of Ph6-Ph was followed, using Ni(cod)2 (50 mg, 0.18 mmol), PhL3 (99 mg, 0.18 mmol), and d-phenylacetylene (19 mg, 0.18 mmol) as starting materials. [(PhL3)Ni(DC≡CPh)] was isolated as a yellow powder (60 mg, 0.08 mmol, 48%). [(PhL3)Ni(DC≡CPh)] can also be generated from reaction of Ph3 with d-acetylene in a 1:1 stoichiometry. 1H NMR (400 MHz, C6D6, 25 °C): δH 7.94–7.87 (ArH, m, 4H), 7.46–7.40 (ArH, m, 4H), 7.30 (ArH, dd, 3JH,H = 6.8 Hz, 4JH,H = 1.4 Hz, 2H), 7.05–6.80 (ArH, m, 17H), 6.75 (ArH, ddd, 3JH,H = 7.6 Hz, 4JH,P = 4.4 Hz, 4JH,H = 1.2 Hz, 1H), 6.66–6.56 (ArH, m, 3H), 6.39 (ArH, ddd, 3JH,H = 7.2 Hz, JH,P = 4.4 Hz, 4JH,H = 1.2 Hz, 1H), 6.32 (ArH, tt, 3JH,H = 7.6 Hz, 4JH,H = 1.0 Hz, 1H). 31P NMR (161 MHz, C6D6, 25 °C): δP 29.2 (d, 2JP,P = 22.5 Hz, 1P), 27.3 (d, 2JP,P = 22.5 Hz, 1P). 13C NMR (100 MHz, C6D6, 25 °C): δC 160.1 (d, 2JC,P = 9.1 Hz), 159.8 (d, 2JC,P = 11.4 Hz), 136.4 (≡CPh, dd, JC,P = 34.9 Hz, JC,P = 4.8 Hz), 136.1, 135.5 (dd, JC,P = 34.8 Hz, JC,P = 4.1 Hz), 134.9 (d, JC,P = 14.5 Hz), 133.8 (d, JC,P = 12.7 Hz), 132.9 (d, JC,P = 1.8 Hz), 130.7, 130.0, 129.8 (d, JC,P = 3.8 Hz), 129.6, 129.4 (d, JC,P = 1.7 Hz), 129.3, 129.0 (d, JC,P = 3.1 Hz), 128.8 (d, JC,P = 1.5 Hz), 125.3, 124.9 (d, JC,P = 4.0 Hz), 124.1 (d, JC,P = 4.0 Hz), 122.9 (d, JC,P = 4.9 Hz), 118.3 (d, JC,P = 3.4 Hz). ATR-IR: ν [cm–1] 3052, 1588, 1564, 1461, 1434, 1259, 1213, 1095, 882, 745, 692, 503.
In Situ Generation of [(PhL3)Ni(PhC≡CPh)] for Analytical Comparison with [(PhL3)Ni(HC≡CPh)] (Ph6-Ph)
Under inert atmosphere, Ni(cod)2 (7.6 mg, 0.028 mmol), PhL3 (15.6 mg, 0.029 mmol), and diphenylacetylene (5.0 mg, 0.028 mmol) were combined together and diluted in C6D6 (0.6 mL) at room temperature, turning the solution red. The mixture was transferred into a Young-type NMR tube and measured after 30 min of reaction. NMR analysis showed full conversion of the starting reagents and the release of cod, in addition to the single generation of a new species, [(PhL3)Ni(PhC≡CPh)]. 1H NMR (400 MHz, C6D6, 25 °C): δH 7.71–7.61 (m, 8H), 7.18–7.13 (m, 4H), 6.97–7.88 (m, 12H), 6.86–6.82 (m, 6H), 6.73–6.66 (m, 4H), 6.55 (dd, 3JH,H = 8.0 Hz, 4JH,P = 2.8 Hz, 2H), 6.44 (t, 3JH,H = 7.6 Hz, 2H), 5.58 (cod, s, 4H), 2.21 (cod, s, 8H). 31P NMR (161 MHz, C6D6, 25 °C): δP 28.1 (s, 2P). 13C NMR (100 MHz, C6D6, 25 °C): δC 159.8 (t, 2JC,P = 5.0 Hz), 136.3 (≡CPh, t, 2JC,P = 6.8 Hz), 136.3, 136.0, 135.8 135.3, 135.2, 135.0 (d, JC,P = 3.2 Hz), 134.8, 134.7, 134.5 (t, JC,P = 7.0 Hz), 140.0, 130.4, 129.6, 129.5 (d, JC,P= 3 Hz), 129.3, 129.1, 128.8, 128.7, 124.4 (cod), 123.9, 121.1, 28.4 (cod).
Ligand Exchange Reactions (Scheme 4)
In Situ Generation of [(p-tolL1)Ni(PPh3)] (p-tol7-PPh3)
In the glovebox, p-tol1 (5 mg, 5.9 μmol) and 1 equiv of PPh3 (1.6 mg, 5.9 μmol) were dissolved in 0.6 mL of C6D6. The solution was transferred into a Young-type NMR tube, and an NMR spectrum was recorded approximately 15 min after the two reactants have been mixed together, showing full conversion of p-tol1. The analyzed solution contains a mixture of p-tol7-PPh3 and BPI in a 1:1 ratio. [(p-tolL1)Ni(PPh3)] (p-tol7-PPh3). [(PhL1)Ni(PPh3)], with phenyl substituents on the phosphine ligand, has previously been reported.241H NMR (400 MHz, C6D6, 25 °C): δH 7.86 (ArH, d, 3JH,H = 8.0 Hz, 2H), 7.62 (ArH, t, 3JH,P = 8.8 Hz, 6H), 7.44–7.39 (ArH, m, 2H), 7.38–7.33 (ArH, m, 4H), 7.25–7.19 (ArH, m, 4H), 6.93–7.80 (ArH, m, 9H), 6.67 (p-tolArH, d, 3JH,H = 7.6 Hz, 2H), 6.61 (p-tolArH, d, 3JH,H = 7.6 Hz, 2H), 1.97 (CH3, s, 6H), 1.96 (CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 38.5 (Ph3P, t, 2JP,P = 25.7 Hz 1P), 17.1 (p-tol2P, d, 2JP,P = 25.7 Hz, 2P). 13C NMR (100 MHz, C6D6, 25 °C): δC 155.1 (dd, JC,P = 19.1 Hz, JC,P = 16.0 Hz), 142.8 (dt, JC,P = 8.4 Hz, JC,P = 16.8 Hz), 138.4, 137.6 (dt, JC,P = 29.8 Hz, JC,P = 4.6 Hz), 136.9, 135.2 (t, JC,P = 11.4 Hz), 134.3–136.0 (m), 132.8 (t, JC,P = 7.0 Hz), 132.2, 130.6, 128.7–128.5 (m), 126.8, 120.4 (C=O, dt, 2JC,P = 13.8 Hz, 2JC,P = 4.4 Hz) 21.2 (CH3) 21.1 (CH3).
In Situ Generation of [(p-tolL1)Ni(PhCN)] (p-tol7-PhCN) and Keq Determination for the Reaction p-tol1 + PhCN ⇔ p-tol7-PhCN + BPI
In the glovebox, p-tol1 (4.9 mg, 5.8 μmol) was dissolved in 0.6 mL of C6D6, and 1 equiv of PhCN was added via a microsyringe (0.6 μL, 5.8 μmol). The solution was transferred to a Young-type NMR tube, and an NMR spectrum was recorded approximately 15 min after the two reactants have been mixed together. 31P NMR was recorded with a relaxation time of 21 s. For 1H NMR, the singlets at 10.01 ppm (1H), 2.10 ppm (6H), and 1.91 ppm (6H) for p-tol1 and the triplet at 6.63 (3H) for PhCN were selected to determine the relative concentration of the reactants. As regards the determination of the concentration of products, the two singlets at 2.04 ppm (6H) and 2.03 ppm (6H) for p-tol7-PhCN and the singlet at 9.82 (1H) for BPI were selected. The same procedure was repeated at different stoichiometry of PhCN (i.e., 50 equiv and 200 equiv compared to p-tol1). 1H NMR (400 MHz, C6D6, 25 °C): δH 10.01 (p-tol1, NH, s, 1H), 9.82 (BPI, NH, br. s, 1H), 8.03 (p-tol1, ArH, d, 3JH,H = 7.4 Hz, 2H), 7.92 (p-tol1, ArH, dd, 3JH,H = 7.7, 4JH,H = 1.3, 2H), 7.70 (p-tol1, ArH, dt, 3JH,H = 7.9, 4JH,P = 4.6, 4H), 7.35–7.29 (p-tol1, ArH, m, 2H), 7.10 (p-tol1, ArH, dt, 3JH,H = 7.9 Hz, 4JHP = 4.4 Hz), 7.02–6.92 (p-tol1, ArH, m, 6H), 6.91–6.76 (p-tol1, ArH, m, 14H), 6.63 (PhCN, ArH, t, 3JH,H = 7.6, 2H) 2.10 (p-tol1, CH3, s, 6H), 2.04 (p-tol7-PhCN, CH3, s, 6H), 2.03 (p-tol7-PhCN, CH3, s, 6H), 1.91 (p-tol1, CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 15.5 (p-tol1, s, 2P), 12.7 (p-tol7-PhCN, s, 2P).
13C NMR was acquired in order to locate the resonances from the carbonyl and methyl groups of the ligand of p-tol7-PhCN. 13C NMR (100 MHz, C6D6, 25 °C): δc 22.8 (CH3). No carbonyl peak around 200 ppm was detected.
In Situ Generation of [(p-tolL1)Ni(C2H3Ph)] (p-tol7-C2H3Ph) and Keq Determination for the Reaction p-tol1 + C2H3Ph ⇔ p-tol7-C2H3Ph + BPI
In the glovebox, p-tol1 (4.4 mg, 5.2 μmol) was dissolved in 0.6 mL of C6D6 and 1 equiv of styrene was added via a microsyringe (0.6 μL, 5.2 μmol). The solution was transferred to a Young-type NMR tube, and an NMR spectrum was recorded approximately 15 min after the two reactants have reacted. 31P NMR was recorded with a relaxation time of 21 s. For 1H NMR, the singlet at 10.01 ppm (1H) for p-tol1 and the four sets of peaks at 7.23 ppm (2H), 6.58 ppm (1H), 5.60 ppm (1H), and 5,07 ppm (1H) for C2H3Ph were selected to determine the relative concentration of the reactants. As regards the determination of the concentration of products, the four sets of peaks at 6.34 ppm (2H), 5.02–4.92 ppm (1H), 4.53 ppm (1H), 3.78–3.73 (1H) for p-tol7-C2H3Ph and the singlet at 9.82 (1H) for BPI were selected. The same procedure was repeated at different stoichiometry of styrene (i.e., 50 equiv and 200 equiv compared to p-tol1). 1H NMR (400 MHz, C6D6, 25 °C): δH 10.01 (p-tol1, NH, s, 1H), 9.82 (BPI, NH, br. s, 1H), 8.03 (p-tol1, ArH, d, 3JH,H = 7.4 Hz, 2H), 7.92 (p-tol1, ArH, dd, 3JH,H = 7.7, 4JH,H = 1.3, 2H), 7.70 (p-tol1, ArH, dt, 3JH,H = 7.9, 4JH,P = 4.6, 4H), 7.35–7.29 (p-tol1, ArH, m, 2H), 7.23 (C2H3Ph, PhH, d, 3JH,H = 7.2 Hz, 2H); 7.10 (p-tol1, ArH, dt, 3JH,H = 7.9 Hz, 4JHP = 4.4 Hz), 7.02–6.92 (p-tol1, ArH, m, 6H), 6.91–6.76 (p-tol1, ArH, m, 14H), 6.58 (C2H3Ph, =CH, dd, 3JH,H = 17.6 Hz, 3JH,H = 10.8 Hz, 1H), 6.34 (p-tol7-C2H3Ph, PhH, d, 3JH,H = 7.2 Hz, 2H), 5.60 (C2H3Ph, =CH2, d, 3JH,H = 17.6 Hz, 1H), 5.07 (C2H3Ph, =CH2, d, 3JH,H = 10.8 Hz, 1H), 5.02–4.92 (p-tol7-C2H3Ph, =CH, m, 1H), 4.53 (p-tol7-C2H3Ph, =CH2, J = 9.6 Hz, d, 1H); 3.78–3.73 (p-tol7-C2H3Ph, =CH2, m, 1H), 2.10 (p-tol1, CH3, s, 6H); 2.05 (p-tol7-C2H3Ph, CH3, s, 6H), 2.01 (p-tol7-C2H3Ph, CH3, s, 6H), 1.91 (p-tol1, CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 39.2 (p-tol7-C2H3Ph, d, 2JP,P = 33.8 Hz, 1P), 15.9 (p-tol7-C2H3Ph, d, 2JP,P = 33.8 Hz, 1P), 15.4 (p-tol1, s, 2P).
13C NMR was acquired in order to locate the resonances from the carbonyl and methyl groups of the ligand of p-tol7-C2H3Ph. 13C NMR (100 MHz, C6D6, 25 °C): δC 21.9 (CH3), 21.8 (CH3). No carbonyl peak around 200 ppm was detected.
In Situ Generation of [(p-tolL1)Ni(C2Ph2)] (p-tol7-C2Ph2) and Keq Determination for the Reaction p-tol1 + C2Ph2 ⇔ p-tol7-C2Ph2 + BPI
In the glovebox, p-tol1 (4.7 mg, 5.7 μmol) and 1 equiv of diphenylacetylene (1.0 mg, 5.7 μmol) was dissolved in 0.6 mL of C6D6. The solution was transferred to a Young-type NMR tube, and an NMR spectrum was recorded approximately 15 min after the two reactants have reacted. 31P NMR was recorded with a relaxation time of 21 s. For 1H NMR, the singlets at 10.01 ppm (1H), 2.10 ppm (6H), and 1.91 ppm (6H) for p-tol1 and the doublet of doublets at 7.52 (2H) for C2Ph2 were selected to determine the relative concentration of the reactants. As regards the determination of the concentration of products, the singlets at 1.95 ppm (12H) for p-tol7-C2Ph2 and the singlet at 9.82 (1H) for BPI were selected. The same procedure was repeated at different stoichiometry of diphenylacetylene (i.e., 50 equiv and 200 equiv compared to p-tol1). 1H NMR (400 MHz, C6D6, 25 °C): δH 10.01 (p-tol1, NH, s, 1H), 9.82 (BPI, NH, br. s, 1H), 8.03 (p-tol1, ArH, d, 3JH,H = 7.4 Hz, 2H), 7.92 (p-tol1, ArH, dd, 3JH,H = 7.7, 4JH,H = 1.3, 2H), 7.70 (p-tol1, ArH, dt, 3JH,H = 7.9, 4JH,P = 4.6, 4H), 7.52 (C2Ph2, PhH, dd, 3JH,H = 8.0 Hz, 4JH,H = 2.4 Hz, 2H), 7.35–7.29 (p-tol1, ArH, m, 2H), 7.23 (C2H3Ph, PhH, d, 3JH,H = 7.2 Hz, 2H), 7.10 (p-tol1, ArH, dt, 3JH,H = 7.9 Hz, 4JHP = 4.4 Hz), 7.02–6.92 (p-tol1, ArH, m, 6H), 6.91–6.76 (p-tol1, ArH, m, 14H), 6.70 (p-tol7-C2Ph2, ArH, d, 3JH,H = 8.0 Hz 8H), 2.10 (p-tol1, CH3, s, 6H), 1.95 (p-tol7-C2Ph2, CH3, s, 12H), 1.91 (p-tol1, CH3, s, 6H). 31P NMR (161 MHz, C6D6, 25 °C): δP 34.0 (p-tol7-C2Ph2, s, 2P), 15.4 (p-tol1, s, 2P).
13C NMR was acquired in order to locate the resonances from the carbonyl and methyl groups of the ligand of p-tol7-C2Ph2. 13C NMR (100 MHz, C6D6, 25 °C): δC 198.3 (C=O, t, 4JC,P = 4.7 Hz); 21.2 (CH3).
Catalysis: General procedure for alkyne cyclotrimerization (Table 3 and Table 4)
Method A
In the glovebox, a toluene solution (3 mL) of alkyne was added slowly, within 1 min, into a toluene solution (3 mL) containing 0.5 mol % of the nickel catalyst (Ni-cat. p-tol1 to Ni-cat. Ph3). The solution was stirred at room temperature (substrates 8–11) or 50 °C (substrates 12 and 13) for 16 h. The solution was then opened to air, and the organic layer was extracted with HCl 1 M (1 × 10 mL) and water (2 × 10 mL). The aqueous layer was washed with Et2O (3 × 10 mL), and the organic portions were combined together, dried over MgSO4, filtered, and concentrated under vacuum. The product was extracted with Et2O and filtered through a silica plug. The final product was dried in vacuo and analyzed by GC-MS and NMR. The NMR characterization values of the organic catalytic products were compared with literature. The isomeric ratios were calculated and are reported according to 1H NMR. The reported values (yields and isomeric ratios) presented in Table 3 and Table 4 are the average values over two runs.
Method B
In the glovebox, a toluene solution (3 mL) of alkyne was added slowly, within 1 min, into a toluene solution (3 mL) containing 0.5 mol % of the ligand (p-tolL1 to PhL4) and 0.5 mol % of Ni(cod)2. The solution was stirred at room temperature for 16 h. The solution was then opened to air, and the organic layer was extracted with HCl 1 M (1 × 10 mL) and water (2 × 10 mL). The aqueous layer was washed with Et2O (3 × 10 mL), and the organic portions were combined together, dried over MgSO4, filtered, and concentrated under vacuum. The product was extracted with Et2O and filtered through a silica plug. The final product was dried under vacuum and analyzed by GC-MS and NMR. The NMR characterization values of the organic catalytic products were compared with literature. The ratios were calculated and are reported according to 1H NMR.
TON Experiment for the Cyclotrimerization of Ethyl Propiolate Catalyzed by p-tol1
In the glovebox, a toluene solution (3 mL) of ethyl propiolate (11, 232 mg, 2.37 mmol) was added slowly, within 1 min, into a toluene solution (3 mL) containing 0.05 mol % of the nickel catalyst p-tol1 (1 mg, 1.2 μmol). The solution was stirred at room temperature for 16 h. The solution was then opened to air, and the organic layer was extracted with HCl 1 M (1 × 10 mL) and water (2 × 10 mL). The aqueous layer was washed with Et2O (3 × 10 mL), and the organic portions were combined together, dried over MgSO4, filtered, and concentrated under vacuum. The product was extracted with Et2O and filtered through a silica plug. The ratios between 1,2,4- (11a) and 1,3,5- (11b) regioisomers in addition to a minor amount of tetraethyl-cyclooctatetraene-tetracarboxylates (11c) were determined by 1H NMR in a ratio of 91:7:2. Triethylbenzene-1,2,4-tricarboxylate (11a) was obtained in a yield of 87% (202 mg, 0.69 mmol).
Acknowledgments
The X-ray diffractometer has been financed by The Netherlands Organization for Scientific Research (NWO). We acknowledge funding from NWO council under grant agreement ECHO-STIP, project no. 717.014.009. This work was sponsored by NWO Exacte en Natuurwetenschappen (Physical Sciences) for the use of supercomputer facilities, with financial support from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research, NWO). The DFT work was carried out on the Dutch national e-infrastructure with the support of the SURF Foundation. The authors thank Dr. J. T. B. H. Jastrzebski for assistance with NMR spectroscopic analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b05025.
The authors declare no competing financial interest.
Notes
CCDC 1882146–1882149 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
References
- van der Vlugt J. I. Cooperative Catalysis with First-Row Late Transition Metals. Eur. J. Inorg. Chem. 2012, 2012, 363–375. 10.1002/ejic.201100752. [DOI] [Google Scholar]
- Verhoeven D. G. A.; Moret M.-E. Metal–Ligand Cooperation at Tethered π-Ligands. Dalt. Trans. 2016, 45, 15762–15778. 10.1039/C6DT02184K. [DOI] [PubMed] [Google Scholar]
- Grützmacher H. Cooperating Ligands in Catalysis. Angew. Chem., Int. Ed. 2008, 47, 1814–1818. 10.1002/anie.200704654. [DOI] [PubMed] [Google Scholar]
- The Organometallic Chemistry of Transition Metals, 6th ed.; Crabtree R. H., Ed.; John Wiley and Sons: Hoboken, USA, 2014. [Google Scholar]
- Catalysis Without Precious Metals; Bullock R. M., Ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Slone C. S.; Weinberger D. A.; Mirkin C. A. The Transition Metal Coordination Chemistry of Hemilabile Ligands. Prog. Inorg. Chem. 2007, 48, 233–350. 10.1002/9780470166499.ch3. [DOI] [Google Scholar]
- Jeffrey J. C.; Rauchfuss T. B. Metal Complexes of Hemilabile Ligands. Reactivity and Structure of Dichlorobis(o-(diphenylphosphino)anisole)ruthenium(II). Inorg. Chem. 1979, 18, 2658–2666. 10.1021/ic50200a004. [DOI] [Google Scholar]
- a Adams G. M.; Weller A. S. POP-type ligands: Variable Coordination and Hemilabile Behaviour. Coord. Chem. Rev. 2018, 355, 150–172. 10.1016/j.ccr.2017.08.004. [DOI] [Google Scholar]; b Oliveri C. G.; Ulmann P. A.; Wiester M. J.; Mirkin C. A. Heteroligated Supramolecular Coordination Complexes Formed via the Halide-Induced Ligand Rearrangement Reaction. Acc. Chem. Res. 2008, 41, 1618–1629. 10.1021/ar800025w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bader A.; Lindner E. Coordination Chemistry and Catalysis with Hemilabile Oxygen-Phosphorus Ligands. Coord. Chem. Rev. 1991, 108, 27–110. 10.1016/0010-8545(91)80013-4. [DOI] [Google Scholar]; d Moxham G. L.; Randell-Sly H. E.; Brayshaw S. K.; Woodward R. L.; Weller A. S.; Willis M. C. A Second-Generation Catalyst for Intermolecular Hydroacylation of Alkenes and Alkynes Using β-S-Substituted Aldehydes: The Role of a Hemilabile P-O-P Ligand. Angew. Chem., Int. Ed. 2006, 45, 7618–7622. 10.1002/anie.200603133. [DOI] [PubMed] [Google Scholar]
- a Lindner R.; van den Bosch B.; Lutz M.; Reek J. N. H.; van der Vlugt J. I. Tunable Hemilabile Ligands for Adaptive Transition Metal Complexes. Organometallics 2011, 30, 499–510. 10.1021/om100804k. [DOI] [Google Scholar]; b Choualeb A.; Lough A. J.; Gusev D. G. Hemilabile Pincer-Type Hydride Complexes of Iridium. Organometallics 2007, 26, 5224–5229. 10.1021/om700550x. [DOI] [Google Scholar]; c Müller C.; Lachicotte R. J.; Jones W. D. Catalytic C–C Bond Activation in Biphenylene and Cyclotrimerization of Alkynes: Increased Reactivity of P,N- versus P,P-Substituted Nickel Complexes. Organometallics 2002, 21, 1975–1981. 10.1021/om0110989. [DOI] [Google Scholar]; d Fryzuk M. D.; MacNeil P. A.; Rettig S. J.; Secco A. S.; Trotter J. Tridentate Amido Phosphine Derivatives of the Nickel Triad: Synthesis, Characterization, and Reactivity of Nickel(II), Palladium(II), and Platinum(II) Amide Complexes. Organometallics 1982, 1, 918–930. 10.1021/om00067a006. [DOI] [Google Scholar]
- a Braunstein P.; Naud F. Hemilability of Hybrid Ligands and the Coordination Chemistry of Oxazoline-Based Systems. Angew. Chem., Int. Ed. 2001, 40, 680–699. 10.1002/1521-3773(20010216)40:4<680::AID-ANIE6800>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; b van der Vlugt J. I.; Pidko E. A.; Vogt D.; Lutz M.; Spek A. L.; Meetsma A. T-Shaped Cationic CuI Complexes with Hemilabile PNP-Type Ligands. Inorg. Chem. 2008, 47, 4442–4444. 10.1021/ic800298a. [DOI] [PubMed] [Google Scholar]; c van der Vlugt J. I.; Pidko E. A.; Vogt D.; Lutz M.; Spek A. L. CuI Complexes with a Noninnocent PNP Ligand: Selective Dearomatization and Electrophilic Addition Reactivity. Inorg. Chem. 2009, 48, 7513–7515. 10.1021/ic901032c. [DOI] [PubMed] [Google Scholar]
- Deckers P. J. W.; Hessen B.; Teuben J. H. Switching a Catalyst System from Ethene Polymerization to Ethene Trimerization with a Hemilabile Ancillary Ligand. Angew. Chem., Int. Ed. 2001, 40, 2516–2519. 10.1002/1521-3773(20010702)40:13<2516::AID-ANIE2516>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Lindner E.; Sickinger A.; Wegner P. Neuartige Basische Liganden für die Homogenkatalytische Methanolcarbonylierung: XVII. Dioxanylmethyldiorganylphosphane als Steuerliganden bei der Methanol(hydro) Carbonylierung zu Acetaldehyd und Essigsäure. J. Organomet. Chem. 1988, 349, 75–94. 10.1016/0022-328X(88)80439-X. [DOI] [Google Scholar]
- Zhang J.; Leitus G.; Ben-david Y.; Milstein D. Efficient Homogeneous Catalytic Hydrogenation of Esters to Alcohols. Angew. Chem. 2006, 118, 1131–1133. 10.1002/ange.200503771. [DOI] [PubMed] [Google Scholar]
- Spasyuk D.; Vicent C.; Gusev D. G. Chemoselective Hydrogenation of Carbonyl Compounds and Acceptorless Dehydrogenative Coupling of Alcohols. J. Am. Chem. Soc. 2015, 137, 3743–3746. 10.1021/ja512389y. [DOI] [PubMed] [Google Scholar]
- Pawley R. J.; Huertos M. A.; Lloyd-Jones G. C.; Weller A. S.; Willis M. C. Intermolecular Alkyne Hydroacylation. Mechanistic Insight from the Isolation of the Vinyl Intermediate that Precedes Reductive Elimination. Organometallics 2012, 31, 5650–5659. 10.1021/om300647n. [DOI] [Google Scholar]
- Weng Z.; Teo S.; Hor T. S. A. Metal Unsaturation and Ligand Hemilability in Suzuki Coupling. Acc. Chem. Res. 2007, 40, 676–684. 10.1021/ar600003h. [DOI] [PubMed] [Google Scholar]
- van der Vlugt J. I.; Siegler M. A.; Janssen M.; Vogt D.; Spek A. L. A Cationic AgI(PNPtBu) Species Acting as PNP Transfer Agent: Facile Synthesis of Pd(PNPtBu)(alkyl) Complexes and Their Reactivity Compared to PCPtBu Analogues. Organometallics 2009, 28, 7025–7032. 10.1021/om900862w. [DOI] [Google Scholar]
- Gunanathan C.; Ben-David Y.; Milstein D. Direct Synthesis of Amides from Alcohols and Amines with Liberation of H2. Science 2007, 317, 790–792. 10.1126/science.1145295. [DOI] [PubMed] [Google Scholar]
- Gunanathan C.; Milstein D. Selective Synthesis of Primary Amines Directly from Alcohols and Ammonia. Angew. Chem. 2008, 120, 8789–8792. 10.1002/ange.200803229. [DOI] [PubMed] [Google Scholar]
- For reviews on σ-acceptor ligands, see; a Braunschweig H.; Dewhurst R. D.; Schneider A. Electron-Precise Coordination Modes of Boron-Centered Ligands. Chem. Rev. 2010, 110, 3924–3957. 10.1021/cr900333n. [DOI] [PubMed] [Google Scholar]; b Amgoune A.; Bourissou D. σ-Acceptor, Z-Type Ligands for Transition Metals. Chem. Commun. 2011, 47, 859–871. 10.1039/C0CC04109B. [DOI] [PubMed] [Google Scholar]
- For examples of triarylborane-supported metal complexes, see ref (20a) and; a Fontaine F.-G.; Boudreau J.; Thibault M. H. Coordination Chemistry of Neutral (Ln)-Z Amphoteric and Ambiphilic Ligands. Eur. J. Inorg. Chem. 2008, 2008, 5439–5454. 10.1002/ejic.200800784. [DOI] [Google Scholar]; b Moret M.-E.; Peters J. C. Terminal Iron Dinitrogen and Iron Imide Complexes Supported by a Tris(phosphino)borane Ligand. Angew. Chem., Int. Ed. 2011, 50, 2063–2067. 10.1002/anie.201006918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of C=C π-ligands, see; a Bennett M. A.; Clark P. W. Tridentate Chelate π-Bonded Complexes of Rhodium(I), Iridium(I), and Iridium(III) Chelate σ-Bonded Complexes of Nickel(II), Palladium(II), and Platinum (II) Formed by Intramolecular Hydrogen Abstarction Reactions. J. Organomet. Chem. 1976, 110, 367–381. 10.1016/S0022-328X(00)82497-3. [DOI] [Google Scholar]; b Bennett M. A.; Johnson R. N.; Tomkins I. B. Additions to Metal Atom and to Coordinated Ligand in Complexes of Rhodium(I) and Iridium(I) Formed by a Tridentate Olefinic Ditertiary Phosphine: Chelate Olefin Complexes and σ-Alkyls of Rhodium(III) and Iridium(III). J. Organomet. Chem. 1976, 118, 205–232. 10.1016/S0022-328X(00)92155-7. [DOI] [Google Scholar]; c Linden A.; Llovera L.; Herrera J.; Dorta R.; Agrifoglio G.; Dorta R. Chiral-at-Metal” Hemilabile Nickel Complexes with a Latent d10-ML2 Configuration: Receiving Substrates with Open Arms. Organometallics 2012, 31, 6162–6171. 10.1021/om300484z. [DOI] [Google Scholar]; d Barrett B. J.; Iluc V. M. Coordination of a Hemilabile Pincer Ligand with an Olefinic Backbone to Mid-to-Late Transition Metals. Inorg. Chem. 2014, 53, 7248–7259. 10.1021/ic500549z. [DOI] [PubMed] [Google Scholar]; e Barrett B. J.; Iluc V. M. Group 10 Metal Complexes Supported by Pincer Ligands with an Olefinic Backbone. Organometallics 2014, 33, 2565–2574. 10.1021/om500256r. [DOI] [Google Scholar]; f Comanescu C. C.; Vyushkova M.; Iluc V. M. Palladium Carbene Complexes as Persistent Radicals. Chem. Sci. 2015, 6, 4570–4579. 10.1039/C5SC01441G. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Campos J.; Ortega-Moreno L.; Conejero S.; Peloso R.; Lõpez-Serrano J.; Maya C.; Carmona E. Reactivity of Cationic Agostic and Carbene Structures Derived from Platinum(II) Metallacycles. Chem. - Eur. J. 2015, 21, 8883–8896. 10.1002/chem.201500756. [DOI] [PubMed] [Google Scholar]
- For the first synthetic report of PhL1, see; Jing Q.; Sandoval C. A.; Wang Z.; Ding K. Complete Chiral Induction from Enantiopure 1,2-Diamines to Benzophenone-Based Achiral Bisphosphane Ligands in Noyori-Type RuII Catalysts. Eur. J. Org. Chem. 2006, 2006, 3606–3616. 10.1002/ejoc.200600299. [DOI] [Google Scholar]
- Saes B. W. H; Verhoeven D. G. A.; Lutz M.; Klein Gebbink R. J. M.; Moret M.-E. Coordination of a Diphosphine–Ketone Ligand to Ni(0), Ni(I), and Ni(II): Reduction-Induced Coordination. Organometallics 2015, 34, 2710–2713. 10.1021/acs.organomet.5b00264. [DOI] [Google Scholar]
- Verhoeven D. G. A.; van Wiggen M. A. C.; Kwakernaak J.; Lutz M.; Klein Gebbink R. J. M.; Moret Periodic Trends in the Binding of a Phosphine-Tethered Ketone Ligand to Fe, Co, Ni, and Cu. Chem. - Eur. J. 2018, 24, 5163–5172. 10.1002/chem.201703254. [DOI] [PubMed] [Google Scholar]
- Mikami K.; Wakabayashi K.; Aikawa K. Achiral” Benzophenone Ligand for Highly Enantioselective Ru Catalysts in Ketone Hydrogenation. Org. Lett. 2006, 8, 1517–1519. 10.1021/ol052936o. [DOI] [PubMed] [Google Scholar]
- Mikami K.; Wakabayashi K.; Yusa Y.; Aikawa K. Achiral Benzophenone Ligand-Rhodium Complex with Chiral Diamine Activator for High Enantiocontrol in Asymmetric Transfer Hydrogenation. Chem. Commun. 2006, 22, 2365–2367. 10.1039/B517585B. [DOI] [PubMed] [Google Scholar]
- Sung S.; Boon J. K.; Lee J. J. C.; Rajabi N. A.; Macgregor S. A.; Krämer T.; Young R. D. Convergent (De)hydrogenative Pathways via a Rhodium α-Hydroxylalkyl Complex. Organometallics 2017, 36, 1609–1617. 10.1021/acs.organomet.7b00158. [DOI] [Google Scholar]
- Sung S.; Joachim T.; Krämer T.; Young R. D. Protonolysis of an α-Hydroxyl Ligand for the Generation of a PCcarbeneP Pincer Complex and Subsequent Reactivity Studies. Organometallics 2017, 36, 3117–3124. 10.1021/acs.organomet.7b00461. [DOI] [Google Scholar]
- Barbato C.; Baldino S.; Ballico M.; Figliolia R.; Magnolia S.; Siega K.; Herdtweck E.; Strazzolini P.; Chelucci G.; Baratta W. OsXCl(phosphine)2 (diamine) and OsXCl(diphosphine)(diamine) (X = Cl, H) Complexes for Ketone Hydrogenation. Organometallics 2018, 37, 65–77. 10.1021/acs.organomet.7b00737. [DOI] [Google Scholar]
- Sung S.; Young R. D. Facile Generation of Iridium PCcarbeneP Pincer Complexes: Via Water Elimination from an Alcohol Proligand. Dalton Trans. 2017, 46, 15407–15414. 10.1039/C7DT03690F. [DOI] [PubMed] [Google Scholar]
- Reppe W.; Schlichting O.; Klager K.; Toepel T. Cyclisierende Polymerisation von Acetylen. I. Über Cyclooctatetraen. Justus Liebigs Ann. Chem. 1948, 560, 1–92. 10.1002/jlac.19485600102. [DOI] [Google Scholar]
- For reviews on metal-catalyzed alkyne cyclotrimerization reactions, see; a Broere D.; Ruijter E. Recent Advances in Transition-Metal-Catalyzed [2 + 2 + 2]-Cyclo(co)trimerization Reactions. Synthesis 2012, 44, 2639–2672. 10.1055/s-0032-1316757. [DOI] [Google Scholar]; b Domínguez G.; Pérez-Castells J. Recent Advances in [2 + 2+2] Cycloaddition Reactions. Chem. Soc. Rev. 2011, 40, 3430–44. 10.1039/c1cs15029d. [DOI] [PubMed] [Google Scholar]; c Saito S.; Yamamoto Y. Recent Advances in the Transition-Metal-Catalyzed Regioselective Approaches to Polysubstituted Benzene Derivatives. Chem. Rev. 2000, 100, 2901–2915. 10.1021/cr990281x. [DOI] [PubMed] [Google Scholar]; d Chopade P. R.; Louie J. [2 + 2+2] Cycloaddition Reactions Catalyzed by Transition Metal Complexes. Adv. Synth. Catal. 2006, 348, 2307–2327. 10.1002/adsc.200600325. [DOI] [Google Scholar]; e Shibata T.; Tsuchikama K. Recent Advances in Enantioselective [2 + 2 + 2] Cycloaddition. Org. Biomol. Chem. 2008, 6, 1317–1323. 10.1039/b720031e. [DOI] [PubMed] [Google Scholar]; f Galan B. R.; Rovis T. Beyond Reppe: Building Substituted Arenes by [2 + 2+2] Cycloadditions of Alkynes. Angew. Chem., Int. Ed. 2009, 48, 2830–2834. 10.1002/anie.200804651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of titanium-catalyzed cyclotrimerization of alkynes, see; a Johnson E. S.; Balaich G. J.; Fanwick P. E.; Rothwell I. P. Trimerization of tert-Butylacetylene to 1,3,6-Tri(tert-butyl)fulvene Catalyzed by Titanium Aryloxide Compounds. J. Am. Chem. Soc. 1997, 119, 11086–11087. 10.1021/ja972306k. [DOI] [Google Scholar]; b Ozerov O. V.; Patrick B. O.; Ladipo F. T. Highly Regioselective [2 + 2 + 2] Cycloaddition of Terminal Alkynes Catalyzed by η6-Arene Complexes of Titanium Supported by Dimethylsilyl-Bridged p-tert-Butyl Calix[4]arene Ligand. J. Am. Chem. Soc. 2000, 122, 6423–6431. 10.1021/ja994543o. [DOI] [Google Scholar]; c Ladipo F. T.; Sarveswaran V.; Kingston J. V.; Huyck R. A.; Bylikin S. Y. S. Y.; Carr S. D.; Watts R.; Parkin S. Synthesis, Characterization, and Alkyne Cyclotrimerization Chemistry of Titanium Complexes Supported by Calixarene-Derived Bis(aryloxide) Ligation. J. Organomet. Chem. 2004, 689, 502–514. 10.1016/j.jorganchem.2003.10.039. [DOI] [Google Scholar]; d Morohashi N.; Yokomakura K.; Hattori T.; Miyano S. Highly Regioselective [2 + 2+2] Cycloaddition of Terminal Alkynes Catalyzed by Titanium Complexes of p-tert-Butylthiacalix[4]arene. Tetrahedron Lett. 2006, 47, 1157–1161. 10.1016/j.tetlet.2005.12.034. [DOI] [Google Scholar]; e See X. Y.; Beaumier E. P.; Davis-Gilbert Z. W.; Dunn P. L.; Larsen J. A.; Pearce A. J.; Wheeler T. A.; Tonks I. A. Generation of TiII Alkyne Trimerization Catalysts in the Absence of Strong Metal Reductants. Organometallics 2017, 36, 1383–1390. 10.1021/acs.organomet.7b00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of iron-catalyzed cyclotrimerization of alkynes, see; a Breschi C.; Piparo L.; Pertici P.; Caporusso A. M.; Vitulli G. (η6-Cyclohepta-1,3,5-triene)(η4-cycloocta-1,5-diene)iron(0) Complex as Attractive Precursor in Catalysis. J. Organomet. Chem. 2000, 607, 57–63. 10.1016/S0022-328X(00)00211-4. [DOI] [Google Scholar]; b Liu Y.; Yan X.; Yang N.; Xi C. Highly Regioselective Cyclotrimerization of Terminal Alkynes Catalyzed by Fe(II) Complexes Bearing 2-(Benzimidazolyl)-6-(1-(arylimino)ethyl)pyridines. Catal. Commun. 2011, 12, 489–492. 10.1016/j.catcom.2010.11.013. [DOI] [Google Scholar]; c Minakawa M.; Ishikawa T.; Namioka J.; Hirooka S.; Zhou B.; Kawatsura M. Iron-Catalyzed [2 + 2 + 2] Cycloaddition of Trifluoromethyl Group Substituted Unsymmetrical Internal Alkynes. RSC Adv. 2014, 4, 41353–41356. 10.1039/C4RA06973K. [DOI] [Google Scholar]; d Lipschutz M. I.; Chantarojsiri T.; Dong Y.; Tilley T. D. Synthesis, Characterization, and Alkyne Trimerization Catalysis of a Heteroleptic Two-Coordinate FeI Complex. J. Am. Chem. Soc. 2015, 137, 6366–6372. 10.1021/jacs.5b02504. [DOI] [PubMed] [Google Scholar]; e Bhatt D.; Chowdhury H.; Goswami A. Atom-Economic Route to Cyanoarenes and 2,2′-Dicyanobiarenes via Iron-Catalyzed Chemoselective [2 + 2 + 2] Cycloaddition Reactions of Diynes and Tetraynes with Alkynylnitriles. Org. Lett. 2017, 19, 3350–3353. 10.1021/acs.orglett.7b01217. [DOI] [PubMed] [Google Scholar]; f Brenna D.; Villa M.; Gieshoff T. N.; Fischer F.; Hapke M.; Jacobi von Wangelin A. Iron-Catalyzed Cyclotrimerization of Terminal Alkynes by Dual Catalyst Activation in the Absence of Reductants. Angew. Chem., Int. Ed. 2017, 56, 8451–8454. 10.1002/anie.201705087. [DOI] [PubMed] [Google Scholar]
- For examples of cobalt-catalyzed cyclotrimerization of alkynes, see; a Chini P.; Santambrogio A.; Palladino N. Cyclisation of 2-Methylbut-3-yn-2-ol. Part I. Cyclisation to Aromatic Compounds. J. Chem. Soc. C 1967, 830–835. 10.1039/j39670000830. [DOI] [Google Scholar]; b Sigman M. S.; Fatland A. W.; Eaton B. E. Cobalt-Catalyzed Cyclotrimerization of Alkynes in Aqueous Solution. J. Am. Chem. Soc. 1998, 120, 5130–5131. 10.1021/ja9807284. [DOI] [Google Scholar]; c Teplý F.; Stará I. G.; Starý I.; Kollárovič A.; Šaman D.; Rulíšek L.; Fiedler P. Synthesis of [5]-, [6]-, and [7]Helicene via Ni(0)- or Co(I)-Catalyzed Isomerization of Aromatic cis,cis-Dienetriynes. J. Am. Chem. Soc. 2002, 124, 9175–9180. 10.1021/ja0259584. [DOI] [PubMed] [Google Scholar]; d Dosa P. I.; Whitener G. D.; Vollhardt K. P. C.; Bond A. D.; Teat S. J. Cobalt-Mediated Synthesis of Angular [4] Phenylene: Structural Characterization of a Metallacyclopentadiene(Alkyne) Intermediate and Its Thermal and Photochemical Conversion. Org. Lett. 2002, 4, 2075–2078. 10.1021/ol025956o. [DOI] [PubMed] [Google Scholar]; e Yong L.; Butenschön H. The First Cobalt Catalyzed [2 + 2 + 2] Alkyne Cyclotrimerization in Aqueous Medium at Room Temperature. Chem. Commun. 2002, 2, 2852–2853. 10.1039/B207978J. [DOI] [PubMed] [Google Scholar]; f Hilt G.; Vogler T.; Hess W.; Galbiati F. A Simple Cobalt Catalyst System for the Efficient and Regioselective Cyclotrimerisation of Alkynes. Chem. Commun. 2005, 100, 1474–1475. 10.1039/b417832g. [DOI] [PubMed] [Google Scholar]; g Hilt G.; Hess W.; Vogler T.; Hengst C. Ligand and Solvent Effects on Cobalt(I)-Catalysed Reactions: Alkyne Dimerisation versus [2 + 2+2]-Cyclotrimerisation versus Diels-Alder Reaction versus [4 + 2+2]-Cycloaddition. J. Organomet. Chem. 2005, 690, 5170–5181. 10.1016/j.jorganchem.2005.03.067. [DOI] [Google Scholar]; h Chang H. T.; Jeganmohan M.; Cheng C. H. One-Pot Synthesis of Benzolactones and Lactams Via a Cobalt-Catalyzed Regioselective [2 + 2 + 2] Cocyclotrimerization of Alkynyl Alcohols and Amines with Propiolates. Chem. Commun. 2005, 39, 4955–4957. 10.1039/b509731b. [DOI] [PubMed] [Google Scholar]; i Lombardo M.; Pasi F.; Trombini C.; Seddon K. R.; Pitner W. R. Task-Specific Ionic Liquids as Reaction Media for the Cobalt-Catalysed Cyclotrimerisation Reaction of Arylethynes. Green Chem. 2007, 9, 321–322. 10.1039/b616427g. [DOI] [Google Scholar]; j Hilt G.; Hengst C.; Hess W. Solvent-Dependent Regiochemical Cyclotrimerisation of Phenylacetylene with Cobalt Catalysts Containing Disulfide Ligands: A Case Study. Eur. J. Org. Chem. 2008, 2008, 2293–2297. 10.1002/ejoc.200800106. [DOI] [Google Scholar]; k Geny A.; Agenet N.; Iannazzo L.; Malacria M.; Aubert C.; Gandon V. Air-Stable {(C5H5)Co} Catalysts for [2 + 2+2] Cycloadditions. Angew. Chem., Int. Ed. 2009, 48, 1810–1813. 10.1002/anie.200806001. [DOI] [PubMed] [Google Scholar]; l Eichman C. C.; Bragdon J. P.; Stambuli J. P. Exploiting Guanidine as a Ligand in Cobalt-Catalyzed Alkyne Cyclotrimerizations. Synlett 2011, 2011, 1109–1112. 10.1055/s-0030-1259931. [DOI] [Google Scholar]; m Xu L.; Yu R.; Wang Y.; Chen J.; Yang Z. Highly Regioselective Syntheses of Substituted Triphenylenes from 1,2,4-trisubstituted Arenes via a Co-Catalyzed Intermolecular Alkyne Cyclotrimerization. J. Org. Chem. 2013, 78, 5744–5750. 10.1021/jo400570b. [DOI] [PubMed] [Google Scholar]; n Jungk P.; Fischer F.; Hapke M. In Situ-Generated Chiral Co(I)-Catalyst for Asymmetric [2 + 2+2] Cycloadditions of Triynes. ACS Catal. 2016, 6, 3025–3029. 10.1021/acscatal.6b00560. [DOI] [Google Scholar]; o Wang Y. I.; Hsu W. L.; Ho F. C.; Li C. P.; Wang C. F.; Chen H. H. Efficient Regioselective Approach for Polysubstituted Benzene Derivatives by Intermolecular [2 + 2+2] Cyclotrimerization of Alkynes. Tetrahedron 2017, 73, 7210–7216. 10.1016/j.tet.2017.11.012. [DOI] [Google Scholar]; p Kumon T.; Yamada S.; Agou T.; Kubota T.; Konno T. Highly Regioselective Cobalt-Catalyzed [2 + 2+2] Cycloaddition of Fluorine-Containing Internal Alkynes to Construct Various Fluoroalkylated Benzene Derivatives. J. Fluorine Chem. 2018, 213, 11–17. 10.1016/j.jfluchem.2018.06.004. [DOI] [Google Scholar]; q Xu F.; Si X. J.; Wang X. N.; Kou H. D.; Chen D. M.; Liu C.-S.; Du M. A High-Activity Cobalt-Based MOF Catalyst for [2 + 2 + 2] Cycloaddition of Diynes and Alkynes: Insights Into Alkyne Affinity and Selectivity Control. RSC Adv. 2018, 8, 4895–4899. 10.1039/C7RA12136A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of nickel-catalyzed cyclotrimerization of alkynes, see refs (9c, 36a, and 36c) and; a Meriwether L. S.; Leto M. F.; Colthup E. C.; Kennerly G. W. The Polymerization of Acetylenes by Nickel-Carbonyl-Phosphine Complexes. IV. Kinetics and Polymerization Mechanisms. J. Org. Chem. 1962, 27, 3930–3941. 10.1021/jo01058a043. [DOI] [Google Scholar]; b Simons L. H.; Lagowski J. J. Novel Oligomers of Propyne: Tetramethylcyclooctatetraenes and (Z)-2,4-Dimethyl-1,3-heptadien-5-yne. J. Org. Chem. 1978, 43, 3247–3248. 10.1021/jo00410a035. [DOI] [Google Scholar]; c Diercks R.; Stamp L.; Kopf J.; tom Dieck H. Elementary Steps in Catalytic 1-alkyne Coupling to Substituted Cyclooctatetraenes. Angew. Chem., Int. Ed. Engl. 1984, 23, 893–894. 10.1002/anie.198408931. [DOI] [Google Scholar]; d Dieck H. T.; Dietrich J. Diazadienes as Controlling Ligands in Catalysis. Synthesis of Chiral Diazadienes R* – N = CR’ – CR’ = N – R*. Chem. Ber. 1984, 117, 694–701. 10.1002/cber.19841170222. [DOI] [Google Scholar]; e Diercks R.; Dieck tom H. Diazadiene als Steuerliganden in der Homogenen Katalyse. Katalytische Cyclotetramerisierung von Propiolsäureestern. Chem. Ber. 1985, 118, 428–435. 10.1002/cber.19851180203. [DOI] [Google Scholar]; f Lawrie C. J.; Gable K. P.; Carpenter B. K. Catalytic and Stoichiometric Formation of Cyclooctatetraenes from Soluble Nickel Complexes. Organometallics 1989, 8, 2274–2276. 10.1021/om00111a030. [DOI] [Google Scholar]; g Bhatarah P.; Smith E. H. Oxygen Ligation in the Nickel-Mediated [2 + 2 + 2] Co-cyclisation of a Hepta-1,6-diyne and Alkynol Derivatives. J. Chem. Soc., Perkin Trans. 1 1990, 26, 2603–2606. 10.1039/P19900002603. [DOI] [Google Scholar]; h Bhatarah P.; Smith E. H. A Singular Substituent Effect in the Nickel(0) Mediated Cocyclisation of Octa-1,7-diynes with Alkynols. One-Step Synthesis of Tetralin Lactones from Acyclic Precursors. J. Chem. Soc., Chem. Commun. 1991, 5, 277–278. 10.1039/c39910000277. [DOI] [Google Scholar]; i Bhatarah P.; Smith E. H. Nickel(0)-Promoted Synthesis of Tetralin Lactones from the Co-cyclisation of Monoynes and Octa-1,7-diynes Terminally Substituted with Ester or Amide Groups. J. Chem. Soc., Perkin Trans. 1 1992, 14, 2163–2168. 10.1039/p19920002163. [DOI] [Google Scholar]; j Sato Y.; Nishimata T.; Mori M. Asymmetric Synthesis of Isoindoline and Isoquinoline Derivatives Using Nickel(0)-Catalyzed [2 + 2 + 2] Cocyclization. J. Org. Chem. 1994, 59, 6133–6135. 10.1021/jo00100a003. [DOI] [Google Scholar]; k Duckworth D. M.; Lee-Wong S.; Slawin A. M. Z.; Smith E. H.; Williams D. J. Co-cyclizations of Nitrogen-Containing Acetylenes Induced by a Nickel Triphenylphosphine Complex to Give Aminoindane, Isoindoline and Isoindolinone Derivatives. J. Chem. Soc., Perkin Trans. 1 1996, 1, 815–821. 10.1039/p19960000815. [DOI] [Google Scholar]; l Sato Y.; Ohashi K.; Mori M. Synthesis of Biaryls using Nickel-Catalyzed [2 + 2+2] Cocyclization. Tetrahedron Lett. 1999, 40, 5231–5234. 10.1016/S0040-4039(99)00945-4. [DOI] [Google Scholar]; m Mori N.; Ikeda S. I.; Odashima K. Chemo- and Regioselective Cyclotrimerization of Monoynes Catalyzed by a Nickel(0) and Zinc(II) Phenoxide System. Chem. Commun. 2001, 54, 181–182. 10.1039/b008882j. [DOI] [Google Scholar]; n Hocek M.; Stará I. G.; Starý I.; Dvořáková H. Cyclotrimerization of 6-Ethynylpurines. Synthesis of 1,2,4- and 1,3,5-Tris(purin-6-yl)benzenes as Novel Hoogsteen-Triplet Analogues. Tetrahedron Lett. 2001, 42, 519–521. 10.1016/S0040-4039(00)01989-4. [DOI] [Google Scholar]; o Saito S.; Kawasaki T.; Tsuboya N.; Yamamoto Y. Highly Regioselective Cyclotrimerization of 1-Perfluoroalkylenynes Catalyzed by Nickel. J. Org. Chem. 2001, 66, 796–802. 10.1021/jo0056242. [DOI] [PubMed] [Google Scholar]; p Jeevanandam A.; Korivi R. P.; Huang I.; Cheng C.-H. Ni-Catalyzed Highly Regio- and Chemoselective Cocycloaddition of Nonconjugated Diynes with 1,3-Diynes: A Novel Method for Polysubstituted Arylalkynes. Org. Lett. 2002, 4, 807–810. 10.1021/ol017300l. [DOI] [PubMed] [Google Scholar]; q Rosenthal U.; Schulz W. Zusammenhånge Zwischen Alkinkomplexierung und Aktivierung in Komplexen des Types (ligand)2Ni(alkin). J. Organomet. Chem. 1987, 321, 103–117. 10.1016/0022-328X(87)80329-7. [DOI] [Google Scholar]; r Deaton K. R.; Gin M. S. Regioselective [2 + 2 + 2] Cycloaddition of a Nickel-Benzyne Complex with 1,3-Diynes. Org. Lett. 2003, 5, 2477–2480. 10.1021/ol034720x. [DOI] [PubMed] [Google Scholar]; s Turek P.; Kotora M.; Tislerová I.; Hocek M.; Votruba I.; Císarová I. Cocyclotrimerization of 6-Alkynylpurines with α,ω-Diynes as a Novel Approach to Biologically Active 6-Arylpurines. J. Org. Chem. 2004, 69, 9224–33. 10.1021/jo0486342. [DOI] [PubMed] [Google Scholar]; t Teske J. A.; Deiters A. Microwave-Mediated Nickel-Catalyzed Cyclotrimerization Reactions: Total Synthesis of Illudinine. J. Org. Chem. 2008, 73, 342–345. 10.1021/jo7020955. [DOI] [PubMed] [Google Scholar]; u Xi C.; Sun Z.; Liu Y. Cyclotrimerization of Terminal Alkynes Catalyzed by the System of NiCl2/Zn and (Benzimidazolyl)-6-(1-(arylimino)ethyl)pyridines. Dalton Trans. 2013, 42, 13327–13330. 10.1039/c3dt51674a. [DOI] [PubMed] [Google Scholar]; v Rodrigo S. K.; Powell I. V.; Coleman M. G.; Krause J. A.; Guan H. Efficient and Regioselective Nickel-Catalyzed [2 + 2 + 2] Cyclotrimerization of Ynoates and Related Alkynes. Org. Biomol. Chem. 2013, 11, 7653–7657. 10.1039/c3ob41872c. [DOI] [PubMed] [Google Scholar]; w Pal S.; Uyeda C. Evaluating the Effect of Catalyst Nuclearity in Ni-Catalyzed Alkyne Cyclotrimerizations. J. Am. Chem. Soc. 2015, 137, 8042–8045. 10.1021/jacs.5b04990. [DOI] [PubMed] [Google Scholar]; x Xue F.; Loh Y. K.; Song X.; Teo W. J.; Chua J. Y. D.; Zhao J.; Hor T. S. A. Nickel-Catalyzed Facile [2 + 2+2] Cyclotrimerization of Unactivated Internal Alkynes to Polysubstituted Benzenes. Chem. - Asian J. 2017, 12, 168–173. 10.1002/asia.201601469. [DOI] [PubMed] [Google Scholar]; y Hollingsworth R. L.; Bheemaraju A.; Lenca N.; Lord R. L.; Groysman S. Divergent Reactivity of a New dinuclear Xanthene-Bridged Bis(iminopyridine) Di-Nickel Complex with Alkynes. Dalton Trans. 2017, 46, 5605. 10.1039/C6DT04532D. [DOI] [PubMed] [Google Scholar]; z Beesam R.; Nareddula D. R. Pd and Ni Complexes of a Novel Vinylidene β-diketimine Ligand: Their Application as Catalysts in Heck Coupling and Alkyne Trimerization. Appl. Organomet. Chem. 2017, 31, e3696. 10.1002/aoc.3696. [DOI] [Google Scholar]
- For examples of molybdenum-catalyzed cyclotrimerization of alkynes, see; a Kaneta N.; Hirai T.; Mori M. Reaction of Alkyne Having Hydroxyphenyl Group with Mo(CO)6. Chem. Lett. 1995, 24, 627–628. 10.1246/cl.1995.627. [DOI] [Google Scholar]; b Matusiak R.; Keller A. Cyclotrimerization of Phenylacetylene and Living Polymerization of tert-Butylacetylene by Mo2(O2CCH3)4-Lewis Acid Systems. J. Mol. Catal. A: Chem. 2003, 195, 29–35. 10.1016/S1381-1169(02)00540-X. [DOI] [Google Scholar]; c Kotha S.; Sreevani G. Molybdenum Hexacarbonyl: Air Stable Catalyst for Microwave Assisted Intermolecular [2 + 2+2] Co-Trimerization Involving Propargyl Halides. Tetrahedron Lett. 2015, 56, 5903–5908. 10.1016/j.tetlet.2015.09.029. [DOI] [Google Scholar]
- For examples of ruthenium-catalyzed cyclotrimerization of alkynes, see; a Rüba E.; Schmid R.; Kirchner K.; Calhorda M. J. Ruthenium-Mediated Cyclotrimerization of Alkynes Utilizing the Cationic Complex [RuCp(CH3CN)3]PF6. J. Organomet. Chem. 2003, 682, 204–211. 10.1016/S0022-328X(03)00783-6. [DOI] [Google Scholar]; b Ura Y.; Sato Y.; Shiotsuki M.; Kondo T.; Mitsudo T. A. Ruthenium-Catalysed Synthesis of o-Phthalates by Highly Chemoselective Intermolecular [2 + 2 + 2] Cycloaddition of Terminal Alkynes and Dimethyl Acetylenedicarboxylate. J. Mol. Catal. A: Chem. 2004, 209, 35–39. 10.1016/j.molcata.2003.08.007. [DOI] [Google Scholar]; c Yamamoto Y.; Ishii J. I.; Nishiyama H.; Itoh K. Cp*RuCl-Catalyzed Formal Intermolecular Cyclotrimerization of Three Unsymmetrical Alkynes through a Boron Temporary Tether: Regioselective Four-Component Coupling Synthesis of Phthalides. J. Am. Chem. Soc. 2005, 127, 9625–9631. 10.1021/ja051377d. [DOI] [PubMed] [Google Scholar]; d Cadierno V.; García-Garrido S. E.; Gimeno J. Efficient Intermolecular [2 + 2 + 2] Alkyne Cyclotrimerization in Aqueous Medium Using a Puthenium(IV) Precatalyst. J. Am. Chem. Soc. 2006, 128, 15094–15095. 10.1021/ja066552k. [DOI] [PubMed] [Google Scholar]; e Dutta B.; Curchod B. F. E.; Campomanes P.; Solari E.; Scopelliti R.; Rothlisberger U.; Severin K. Reactions of Alkynes with [RuCl(cyclopentadienyl)] Complexes: The important First Steps. Chem. - Eur. J. 2010, 16, 8400–8409. 10.1002/chem.201000855. [DOI] [PubMed] [Google Scholar]; f Öztürk B. Ö.; Karabulut S.; Imamoğlu Y. Activity of Homobimetallic Ruthenium Alkylidene Complexes on Intermolecular [2 + 2+2] Cyclotrimerisation Reactions of Terminal Alkynes. Inorg. Chim. Acta 2011, 378, 257–263. 10.1016/j.ica.2011.09.023. [DOI] [Google Scholar]; g Kawatsura M.; Yamamoto M.; Namioka J.; Kajita K.; Hirakawa T.; Itoh T. Ruthenium-Catalyzed Regioselective [2 + 2 + 2] Cyclotrimerization of Trifluoromethyl Group Substituted Internal Alkynes. Org. Lett. 2011, 13, 1001–1003. 10.1021/ol1030734. [DOI] [PubMed] [Google Scholar]; h Geetharani K.; Tussupbayev S.; Borowka J.; Holthausen M. C.; Ghosh S. A Mechanistic Study of the Utilization of Arachno-Diruthenaborane [(Cp*RuCO)2B2H6] as an Active Alkyne-Cyclotrimerization Catalyst. Chem. - Eur. J. 2012, 18, 8482–9. 10.1002/chem.201200291. [DOI] [PubMed] [Google Scholar]; i Chen C. R.; Lin Y. C. Syntheses of Arylphosphonium Salts from Cyclotrimerization of Terminal Aryl Alknyes by a Ruthenium Pentadienyl Complex and Revisiting the Catalytic Dimerization. Organometallics 2014, 33, 6408–6412. 10.1021/om500745y. [DOI] [Google Scholar]
- For examples of rhodium-catalyzed cyclotrimerization of alkynes, see; a Amer I.; Bernstein T.; Eisen M.; Blum J.; Vollhardt K. P. C. Oligomerization of Alkynes by the RhCl3-Aliquat 336 Catalyst System. Part 1. Formation of Benzene Derivatives. J. Mol. Catal. 1990, 60, 313–321. 10.1016/0304-5102(90)85254-F. [DOI] [Google Scholar]; b Tanaka K.; Shirasaka K. Highly Chemo- and Regioselective Intermolecular Cyclotrimerization of Alkynes Catalyzed by Cationic Rhodium(I)/Modified BINAP Complexes. Org. Lett. 2003, 5, 4697–4699. 10.1021/ol035963s. [DOI] [PubMed] [Google Scholar]; c Tanaka K.; Toyoda K.; Wada A.; Shirasaka K.; Hirano M. Chemo- and Regioselective Intermolecular Cyclotrimerization of Terminal Alkynes Catalyzed by Cationic Rhodium(I)/Modified BINAP Complexes: Application to One-Step Synthesis of Paracyclophanes. Chem. - Eur. J. 2005, 11, 1145–1156. 10.1002/chem.200401017. [DOI] [PubMed] [Google Scholar]; d Yoshida K.; Morimoto I.; Mitsudo K.; Tanaka H. RhCl3/Amine-Catalyzed [2 + 2+2] Cyclization of Alkynes. Tetrahedron 2008, 64, 5800–5807. 10.1016/j.tet.2008.03.079. [DOI] [Google Scholar]; e Dachs A.; Torrent A.; Pla-Quintana A.; Roglans A.; Jutand A. Rates and Mechanism of Rhodium-Catalyzed [2 + 2+2] Cycloaddition of Bisalkynes and a Monoalkyne. Organometallics 2009, 28, 6036–6043. 10.1021/om900544u. [DOI] [Google Scholar]; f Shibata Y.; Tanaka K. Rhodium-Catalyzed [2 + 2+2] Cycloaddition of Alkynes for the Synthesis of Substituted Benzenes: Catalysts, Reaction Scope, and Synthetic Applications. Synthesis 2012, 44, 323–350. 10.1055/s-0031-1289665. [DOI] [Google Scholar]; g Heya A.; Namba T.; Hara J.; Shibata Y.; Tanaka K. Cationic Rhodium(I)/BIPHEP Complex-Catalyzed Cross-Cyclotrimerization of Silylacetylenes and Unsymmetrical Electron-Deficient Internal Alkynes. Tetrahedron Lett. 2015, 56, 4938–4942. 10.1016/j.tetlet.2015.06.096. [DOI] [Google Scholar]; h Hayase N.; Miyauchi Y.; Aida Y.; Sugiyama H.; Uekusa H.; Shibata Y.; Tanaka K. Synthesis of [8]Cycloparaphenylene-Octacarboxylates via Rh-Catalyzed Stepwise Cross-Alkyne Cyclotrimerization. Org. Lett. 2017, 19, 2993–2996. 10.1021/acs.orglett.7b01231. [DOI] [PubMed] [Google Scholar]; i Nishigaki S.; Shibata Y.; Tanaka K. Rhodium-Catalyzed Chemo- and Regioselective Intermolecular Cross-Cyclotrimerization of Nonactivated Terminal and Internal Alkynes. J. Org. Chem. 2017, 82, 11117–11125. 10.1021/acs.joc.7b02121. [DOI] [PubMed] [Google Scholar]
- For examples of palladium-catalyzed cyclotrimerization of alkynes, see; a Jhingan A. K.; Maier W. F. Homogeneous Catalysis with a Heterogeneous Palladium Catalyst. An Effective Method for the Cyclotrimerization of Alkynes. J. Org. Chem. 1987, 52, 1161–1165. 10.1021/jo00382a039. [DOI] [Google Scholar]; b Gevorgyan V.; Radhakrishnan U.; Takeda A.; Rubina M.; Rubin M.; Yamamoto Y. Palladium-Catalyzed Highly Chemo- and Regioselective Formal [2 + 2 + 2] Sequential Cycloaddition of Alkynes: A Renaissance of the Well Known Trimerization Reaction?. J. Org. Chem. 2001, 66, 2835–2841. 10.1021/jo0100392. [DOI] [PubMed] [Google Scholar]; c Li J.; Jiang H.; Chen M. CuCl2-Induced Regiospecifical Synthesis of Benzene Derivatives in the Palladium-Catalyzed Cyclotrimerization of Alkynes. J. Org. Chem. 2001, 66, 3627–3629. 10.1021/jo0017382. [DOI] [PubMed] [Google Scholar]; d Carvalho M. F. N. N.; Almeida F. M. T.; Galvão A. M.; Pombeiro A. J. L. Benzene Ring Assembly Promoted by a Camphor Derived Palladium Complex. J. Organomet. Chem. 2003, 679, 143–147. 10.1016/S0022-328X(03)00557-6. [DOI] [Google Scholar]; e Cheng J. S.; Jiang H. F. Palladium-Catalyzed Regioselective Cyclotrimerization of Acetylenes in Supercritical Carbon Dioxide. Eur. J. Org. Chem. 2004, 2004, 643–646. 10.1002/ejoc.200300299. [DOI] [Google Scholar]; f Li J. H.; Xie Y. X. Carbon Dioxide Promoted Palladium-Catalyzed Cyclotrimerization of Alkynes in Water. Synth. Commun. 2004, 34, 1737–1743. 10.1081/SCC-120030761. [DOI] [Google Scholar]; g Cheng J.-S.; Wei G.-F.; Huang S.-W.; Jin-Zhao; Jiang H.-F. Solvent-Controlled Chemoselective Palladium-Catalyzed Oligomerization of tert-Butyl Acetylene. J. Mol. Catal. A: Chem. 2007, 263, 169–174. 10.1016/j.molcata.2006.08.060. [DOI] [Google Scholar]; h Chinchilla R.; Nájera C. Chemicals from Alkynes with Palladium Catalysts. Chem. Rev. 2014, 114, 1783–1826. 10.1021/cr400133p. [DOI] [PubMed] [Google Scholar]
- For examples of iridium-catalyzed cyclotrimerization of alkynes, see; a Takeuchi R.; Tanaka S.; Nakaya Y. Iridium Complex-Catalyzed [2 + 2+2] Cycloaddition of α,ω-Diynes with Monoalkynes: A New and Efficient Catalyst for Cyclotrimerization of Alkynes. Tetrahedron Lett. 2001, 42, 2991–2994. 10.1016/S0040-4039(01)00338-0. [DOI] [Google Scholar]; b Takeuchi R.; Nakaya Y. Iridium Complex-Catalyzed Highly Selective Cross [2 + 2 + 2] Cycloaddition of Two Different Monoynes: 2:1 Coupling versus 1:2 Coupling. Org. Lett. 2003, 5, 3659–3662. 10.1021/ol035330d. [DOI] [PubMed] [Google Scholar]; c Farnetti E.; Marsich N. Regioselective Cyclotrimerization of Phenylacetylenes to 1,2,4-Triarylbenzenes Catalyzed by Iridium-Diphosphine Complexes. J. Organomet. Chem. 2004, 689, 14–17. 10.1016/j.jorganchem.2003.10.004. [DOI] [Google Scholar]; d Hashimoto T.; Okabe A.; Mizuno T.; Izawa M.; Takeuchi R. Iridium-Catalyzed [2 + 2+2] Cycloaddition of α,ω-Diynes with Alkynyl Ketones and Alkynyl Esters. Tetrahedron 2014, 70, 8681–8689. 10.1016/j.tet.2014.09.038. [DOI] [Google Scholar]
- Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations; Trost B. M., Li C. J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Kwon D.-H.; Proctor M.; Mendoza S.; Uyeda C.; Ess D. H. Catalytic Dinuclear Nickel Spin Crossover Mechanism and Selectivity for Alkyne Cyclotrimerization. ACS Catal. 2017, 7, 4796–4804. 10.1021/acscatal.7b00978. [DOI] [Google Scholar]
- For the first synthetic report of the phenyl-substituted analogue of p-tolL2, see; Hartley J. G.; Venanzi L. M.; Goodall D. C. The Preparation and Complex-Forming Properties of One Tritertiary and One Tetratertiary Phosphine. J. Chem. Soc. 1963, 3930–3936. 10.1039/jr9630003930. [DOI] [Google Scholar]
- Chakkaradhari G.; Chen Y.-T.; Karttunen A. J.; Dau M. T.; Jänis J.; Tunik S. P.; Chou P.-T.; Ho M.-L.; Koshevoy I. O. Luminescent Triphosphine Cyanide d10 Metal Complexes. Inorg. Chem. 2016, 55, 2174–2184. 10.1021/acs.inorgchem.5b02581. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Wang L.; Chang S.-H.; Huang K.-L.; Chi Y.; Hung W.-Y.; Chen C.-M.; Lee G.-H.; Chou P.-T. Phosphorescent Ir(III) Complexes with Both Cyclometalate Chromophores and Phosphine-Silanolate Ancillary: Concurrent Conversion of Organosilane to Silanolate. Dalt. Trans. 2013, 42, 7111–7119. 10.1039/c3dt32408g. [DOI] [PubMed] [Google Scholar]
- Basic One- and Two-Dimensional NMR Spectroscopy, 5th ed.; Friebolin H., Ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- For the first synthetic report of PhL3, see:; Kranenburg M.; van der Burgt Y. E. M.; Kamer P. C. J.; van Leeuwen P. W. N. M.; Goubitz K.; Fraanje J. New Diphosphine Ligands Based on Heterocyclic Aromatics Inducing Very High Regioselectivity in Rhodium-Catalyzed Hydroformylation: Effect of the Bite Angle. Organometallics 1995, 14, 3081–3089. 10.1021/om00006a057. [DOI] [Google Scholar]
- Chemical Bonds and Bond Energy; Sanderson R. T., Ed.; Academic Press Inc.: New York, USA, 1976. [Google Scholar]
- For examples of η2(C,N)-bound imine to Ni(0), see; a Manan R. S.; Kilaru P.; Zhao P. Nickel-Catalyzed Hydroimination of Alkynes. J. Am. Chem. Soc. 2015, 137, 6136–6139. 10.1021/jacs.5b02272. [DOI] [PubMed] [Google Scholar]; b Hoberg H.; Krüger C.; Tsay Y.-H. Darstellung, Eigenschaften und Bintiungsverh- & ltnisse von Benzophenoneimino-Nickel(0)-Komplexen. J. Organomet. Chem. 1979, 169, 209–217. 10.1016/S0022-328X(00)81160-2. [DOI] [Google Scholar]; c Weng Z.; Teo S.; Koh L. L.; Hor T. S. A. Ethylene Oligomerization at Coordinatively and Electronically Unsaturated Low-Valent Nickel. Angew. Chem., Int. Ed. 2005, 44, 7560–7564. 10.1002/anie.200502532. [DOI] [PubMed] [Google Scholar]; d Cámpora J.; Matas I.; Palma P.; Álvarez E.; Graiff C.; Tiripicchio A. Monomeric Alkoxo and Amido Methylnickel(II) Complexes. Synthesis and Heterocumulene Insertion Chemistry. Organometallics 2007, 26, 3840–3849. 10.1021/om7002909. [DOI] [Google Scholar]; e Iglesias A. L.; Muñoz-Hernández M.; García J. J. Fluoro Aromatic Imine Nickel(0) Complexes: Synthesis and Structural Studies. J. Organomet. Chem. 2007, 692, 3498–3507. 10.1016/j.jorganchem.2007.04.026. [DOI] [Google Scholar]; f Hoshimoto Y.; Ohata T.; Ohashi M.; Ogoshi S. Nickel-Catalyzed Synthesis of N-Aryl-1,2-dihydropyridines by [2 + 2+2] Cycloaddition of Imines with Alkynes Through T-Shaped 14-Electron Aza-Nickelacycle Key Intermediates. Chem. - Eur. J. 2014, 20, 4105–4110. 10.1002/chem.201304830. [DOI] [PubMed] [Google Scholar]; g Reed B. R.; Yousif M.; Lord R. L.; McKinnon M.; Rochford J.; Groysman S. Coordination Chemistry and Reactivity of Bis(aldimino)pyridine Nickel Complexes in Four Different Oxidation States. Organometallics 2017, 36, 582–593. 10.1021/acs.organomet.6b00793. [DOI] [Google Scholar]; h Verhoeven D. G. A.; Negenman H. A.; Orsino A. F.; Lutz M.; Moret M.-E. Versatile Coordination and C-C Coupling of Diphosphine-Tethered Imine Ligands with Ni(II) and Ni(0). Inorg. Chem. 2018, 57, 10846–10856. 10.1021/acs.inorgchem.8b01478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of PhL3-substituted Ni(0) complexes, see; a Wilting J.; Müller C.; Hewat A. C.; Ellis D. D.; Tooke D. M.; Spek A. L.; Vogt D. Nickel-Catalyzed Isomerization of 2-Methyl-3-butenenitrile. Organometallics 2005, 24, 13–15. 10.1021/om049206p. [DOI] [Google Scholar]; b Minami Y.; Yoshiyasu H.; Nakao Y.; Hiyama T. Highly Chemoselective Carbon-Carbon σ-Bond Activation: Nickel/Lewis Acid Catalyzed Polyfluoroarylcyanation of Alkynes. Angew. Chem., Int. Ed. 2013, 52, 883–887. 10.1002/anie.201207880. [DOI] [PubMed] [Google Scholar]
- For examples of structurally characterized monomeric Ni(0) complexes of internal alkynes, see refs (9c and 52b) and; a Diercks R.; Kopf J.; tom Dieck H. (2–3-η-2-Butyne-1,4-diol)[N,N’-ethanediylidenebis(2,6-diisopropylaniline)]nickel(0), [Ni(C26H36N2)(C4H6O2)]. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1984, 40, 363–365. 10.1107/S0108270184004145. [DOI] [Google Scholar]; b Diercks R.; Dieck tom H. Diazadiene als Steuerliganden in der Homogenen Katalyse. IX. Katalytische Cyclotetramerisierung von Propiols. Chem. Ber. 1985, 118, 428–435. 10.1002/cber.19851180203. [DOI] [Google Scholar]; c Dickson R. S.; Ibers J. A. The Structure of Bis(tert-butyl isocyanide)(diphenylacetylene)nickel(0), Ni(C6H5C≡CC6H5)[(CH3)3C–N≡C]2. J. Organomet. Chem. 1972, 36, 191–207. 10.1016/S0022-328X(00)85135-9. [DOI] [Google Scholar]; d Rosenthal U.; Oehme G.; et al. Crystal and Molecular Structure of 2–3-η2- (iPr3P)2Ni(HOCH2C≡CCH2OH) - Some Peculiarities of the Complexation of 2-Butyne-1,4-diol with Nickel(0) Complexes. J. Organomet. Chem. 1991, 409, 299–306. 10.1016/0022-328X(91)86155-J. [DOI] [Google Scholar]; e Bartik T.; Happ B.; Iglewsky M.; Bandmann H.; Boese R.; Heimbach P.; Hoffmann T.; Wenschuh E. Synthesis and Characterization of Bis(phosphine)nickel(0) Complexes Containing Nonsymmetrically Substituted Acetylenes. Organometallics 1992, 11, 1235–1241. 10.1021/om00039a033. [DOI] [Google Scholar]; f Walther D.; Schmidt A.; Klettke T.; Imhof W.; Gorls H. Bis(tetramethylbutynediol)nickel(0), the First Pure Monoalkyne Complex of Nickel and Its Chemistry. Angew. Chem., Int. Ed. Engl. 1994, 33, 1373–1376. 10.1002/anie.199413731. [DOI] [Google Scholar]; g Walther D.; Klettke T.; Görls H.; Imhof W. Homoleptische Alkin-Nickel(0) Komplexe: Kombination von Nickel(0) Zentren mit Alkinylsilanen R1nSi(-C≡C-R2)4-n zu Nickelorganschen Analoga von Siloxanen. J. Organomet. Chem. 1997, 534, 129–137. 10.1016/S0022-328X(96)06922-7. [DOI] [Google Scholar]; h Edelbach B. L.; Lachicotte R. J.; Jones W. D. Catalytic Carbon-Carbon and Carbon-Silicon Bond Activation and Functionalization by Nickel Complexes. Organometallics 1999, 18, 4660–4668. 10.1021/om990457l. [DOI] [Google Scholar]; i Eisch J. J.; Ma X.; Han K. I.; Gitua J. N.; Krüger C. Mechanistic Comparison of the Nickel(0)-Catalyzed Homo-Oligomerization and Co-Oligomerization of Alkynes and Nitriles. Eur. J. Inorg. Chem. 2001, 2001, 77–88. 10.1002/1099-0682(20011)2001:1<77::AID-EJIC77>3.0.CO;2-G. [DOI] [Google Scholar]; j Waterman R.; Hillhouse G. L. Formation of Phosphirenes by Phosphinidene Group-Transfer Reactions from (dtbpe)Ni = (dmp) to Alkynes. Organometallics 2003, 22, 5182–5184. 10.1021/om0341754. [DOI] [Google Scholar]; k Schaub T.; Backes M.; Radius U. Nickel(0) Complexes of N-Alkyl-Substituted N-Heterocyclic Carbenes and Their Use in the Catalytic Carbon-Carbon Bond Activation of Biphenylene. Organometallics 2006, 25, 4196–4206. 10.1021/om0604223. [DOI] [Google Scholar]; l Weng Z.; Teo S.; Hor T. S. A. Stabilization of Nickel(0) by Hemilabile P,N-Ferrocene Ligands and Their Ethylene Oligomerization Activities. Organometallics 2006, 25, 4878–4882. 10.1021/om060607+. [DOI] [Google Scholar]; m Ruhland K.; Obenhuber A.; Hoffmann S. D. Cleavage of Unstrained C(sp2)–C(sp2) Single Bonds with Ni(0) Complexes using Chelating Assistance. Organometallics 2008, 27, 3482–3495. 10.1021/om800054m. [DOI] [Google Scholar]; n Sgro M. J.; Stephan D. W. Synthesis and Exchange Reactions of Ni-Dimine-COD, Acetylene and Olefin Complexes. Dalton Trans. 2010, 39, 5786–5794. 10.1039/c001312a. [DOI] [PubMed] [Google Scholar]; o Hatnean J. A.; Beck R.; Borrelli J. D.; Johnson S. A. Carbon-Hydrogen Bond Oxidative Addition of Partially Fluorinated Aromatics to a Ni(PiPr3)2 Synthon: The Influence of Steric Bulk on the Thermodynamics and Kinetics of C-H Bond Activation. Organometallics 2010, 29, 6077–6091. 10.1021/om1008499. [DOI] [Google Scholar]; p Barrios-Francisco R.; Benítez-Páez T.; Flores-Alamo M.; Arévalo A.; García J. J. Nickel(0) Complexes with Fluorinated Alkyne Ligands and Their Reactivity Towards Semihydrogenation and Hydrodefluorination with Water. Chem. - Asian J. 2011, 6, 842–849. 10.1002/asia.201000601. [DOI] [PubMed] [Google Scholar]; q Staudaher N. D.; Stolley R. M.; Louie J. Synthesis, Mechanism of Formation, and Catalytic Activity of Xantphos Nickel π-Complexes. Chem. Commun. 2014, 50, 15577–15580. 10.1039/C4CC07590K. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Reed B. R.; Yousif M.; Lord R. L.; McKinnon M.; Rochford J.; Groysman S. Coordination Chemistry and Reactivity of Bis(aldimino)pyridine Nickel Complexes in Four Different Oxidation States. Organometallics 2017, 36, 582–593. 10.1021/acs.organomet.6b00793. [DOI] [Google Scholar]
- For examples of structurally characterized monomeric Ni(0) complexes of terminal alkynes, see ref (22c) and; a Pörschke K.-R. Coupling of Two Ethyne Molecules at a Nickel Center to Form a Nickelacyclopentadiene Complex. Angew. Chem., Int. Ed. Engl. 1987, 26, 1288–1290. 10.1002/anie.198712881. [DOI] [Google Scholar]; b Bonrath B. W.; Porschke K. R.; Wilke C.; Angermund K.; Kruger C. Mono- and Dinuclear Nickel(0) Complexes of Butadiyne. Angew. Chem., Int. Ed. Engl. 1988, 27, 833–835. 10.1002/anie.198808331. [DOI] [Google Scholar]
- For examples of crystal structure determinations of a range of disordered molecules of phenylacteylene, see; Thakur T. S.; Sathishkumar R.; Dikundwar A. G.; Guru Row T. N.; Desiraju G. R. Third Polymorph of Phenylacetylene. Cryst. Growth Des. 2010, 10, 4246–4249. 10.1021/cg101076k. [DOI] [Google Scholar]
- Kim Y. E.; Kim J.; Lee Y. Formation of a Nickel Carbon Dioxide Adduct and Its Transformation Mediated by a Lewis Acid. Chem. Commun. 2014, 50, 11458–11461. 10.1039/C4CC04800H. [DOI] [PubMed] [Google Scholar]
- Anderson J. S.; Iluc V. M.; Hillhouse G. L. Reactions of CO2 and CS2 with 1,2-Bis(di-tert-butylphosphino)ethane Complexes of Nickel(0) and Nickel(I). Inorg. Chem. 2010, 49, 10203–10207. 10.1021/ic101652e. [DOI] [PubMed] [Google Scholar]
- Ge S.; Hartwig J. F. Nickel-Catalyzed Asymmetric α-Arylation and Heteroarylation of Ketones with Chloroarenes: Effect of Halide on Selectivity, Oxidation State, and Room-Temperature Reactions. J. Am. Chem. Soc. 2011, 133, 16330–16333. 10.1021/ja2082087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Xi Z. Metal Mediated Synthesis of Substituted Cyclooctatetraenes. Chem. Commun. 2007, 5119–5133. 10.1039/b709839a. [DOI] [PubMed] [Google Scholar]
- For mechanistic studies on cobalt-catalyzed cyclotrimerzation reactions, see; a Hardesty J. H.; Koerner J. B.; Albright T. A.; Lee G. Y. Theoretical Study of the Acetylene Trimerization with CpCo. J. Am. Chem. Soc. 1999, 121, 6055–6067. 10.1021/ja983098e. [DOI] [Google Scholar]; b Dahy A. R. A.; Koga N. Theoretical Study on the Transformation of Bis(acetylene)cobalt to Cobaltacyclopentadiene and the Regioselectivity in this Transformation. Bull. Chem. Soc. Jpn. 2005, 78, 781–791. 10.1246/bcsj.78.781. [DOI] [Google Scholar]; c McAlister D. R.; Bercaw J. E.; Bergman R. G. Parallel Reaction Pathways in the Cobalt-Catalyzed Cyclotrimerization of Acetylenes. J. Am. Chem. Soc. 1977, 99, 1666–1668. 10.1021/ja00447a075. [DOI] [Google Scholar]; d Varela J. A.; Saá C. CpRuCl- and CpCo-Catalyzed or Mediated Cyclotrimerizations of Alkynes and [2 + 2+2] Cycloadditions of Alkynes to Alkenes: A Comparative DFT Study. J. Organomet. Chem. 2009, 694, 143–149. 10.1016/j.jorganchem.2008.10.032. [DOI] [Google Scholar]; e Agenet N.; Gandon V.; Vollhardt K. P. C.; Malacria M.; Aubert C. Cobalt-Catalyzed Cyclotrimerization of Alkynes: The Answer to the Puzzle of Parallel Reaction Pathways. J. Am. Chem. Soc. 2007, 129, 8860–8871. 10.1021/ja072208r. [DOI] [PubMed] [Google Scholar]
- For studies on metallacyclopentadiene-forming transition states (TS1 from Scheme 5) that concertedly form a C–C σ bond and two metal–vinyl carbon bonds, see refs (44, 60d, and 60e) and; a Kirchner K.; Calhorda M. J.; Schmid R.; Veiros L. F. Mechanism for the Cyclotrimerization of Alkynes and Related Reactions Catalyzed by CpRuCl. J. Am. Chem. Soc. 2003, 125, 11721–11729. 10.1021/ja035137e. [DOI] [PubMed] [Google Scholar]; b Orian L.; Swart M.; Bickelhaupt F. M. Indenyl Effect due to Metal Slippage? Computational Exploration of Rhodium-Catalyzed Acetylene [2 + 2+2] Cyclotrimerization. ChemPhysChem 2014, 15, 219–228. 10.1002/cphc.201300934. [DOI] [PubMed] [Google Scholar]; c Liu Z.; Cheng R.; He X.; Liu B. Reactivity and Regioselectivity of Methylacetylene Cyclotrimerization Over the Phillips Cr/Silica Catalyst: A DFT Study. ACS Catal. 2013, 3, 1172–1183. 10.1021/cs400129g. [DOI] [Google Scholar]; d Guo C. H.; Wu H. S.; Hapke M.; Jiao H. Theoretical Studies on Acetylene Cyclotrimerization into Benzene Catalyzed by CpIr Fragment. J. Organomet. Chem. 2013, 748, 29–35. 10.1016/j.jorganchem.2013.03.036. [DOI] [Google Scholar]; e Calhorda M. J.; Costa P. J.; Kirchner K. A. Benzene and Heterocyclic Rings Formation in Cycloaddition Reactions Catalyzed by RuCp Derivatives: DFT studies. Inorg. Chim. Acta 2011, 374, 24–35. 10.1016/j.ica.2011.03.062. [DOI] [Google Scholar]; f Dachs A.; Osuna S.; Roglans A.; Solà M. Density Functional Study of the [2 + 2+2] Cyclotrimerization of Acetylene Catalyzed by Wilkinson’s Catalyst, RhCl(PPh3)3. Organometallics 2010, 29, 562–569. 10.1021/om900836b. [DOI] [Google Scholar]; g Orian L.; Van Stralen J. N. P.; Bickelhaupt F. M. Cyclotrimerization Reactions Catalyzed by Rhodium(I) Half-Sandwich Complexes: A Mechanistic Density Functional Study. Organometallics 2007, 26, 3816–3830. 10.1021/om7004222. [DOI] [Google Scholar]; h Rüba E.; Schmid R.; Kirchner K.; Calhorda M. J. Ruthenium-Mediated Cyclotrimerization of Alkynes Utilizing the Cationic Complex [RuCp(CH3CN)3]PF6. J. Organomet. Chem. 2003, 682, 204–211. 10.1016/S0022-328X(03)00783-6. [DOI] [Google Scholar]; i Becker E.; Mereiter K.; Puchberger M.; Schmid R.; Kirchner K.; Doppiu A.; Salzer A. Novel [2 + 2+1] Cyclotrimerization of Alkynes Mediated by Bidentate Cyclopentadienyl-Phosphine Ruthenium Complexes. Organometallics 2003, 22, 3164–3170. 10.1021/om030297k. [DOI] [Google Scholar]; j Ma W.; Yu C.; Chen T.; Xu L.; Zhang W.-X.; Xi Z. Metallacyclopentadienes: Synthesis, Structure and Reactivity. Chem. Soc. Rev. 2017, 46, 1160–1192. 10.1039/C6CS00525J. [DOI] [PubMed] [Google Scholar]; k Fang B.; Hou G.; Zi G.; Fang D.-C.; Walter M. D. A Thorium Metallacyclopentadiene Complex: a Combined Experimental and Computational Study. Dalton Trans. 2015, 44, 7927–7934. 10.1039/C5DT00838G. [DOI] [PubMed] [Google Scholar]; l Fang B.; Hou G.; Zi G.; Ding W.; Walter M. D. Steric and Electronic Influences of Internal Alkynes on the Formation of Thorium Metallacycles: A Combined Experimental and Computational Study. Organometallics 2016, 35, 1384–1391. 10.1021/acs.organomet.5b00945. [DOI] [Google Scholar]; m Baldridge K. K.; Siegel J. S.; O’Connor J. M. Structure and Dynamics in Unsymmetrically Substituted Five-Coordinate Iridacyclopentadiene Complexes. J. Phys. Org. Chem. 2015, 28, 199–202. 10.1002/poc.3356. [DOI] [Google Scholar]
- For studies on the general mechanisms proposed for the insertion of the third alkyne involving mononuclear metallacyclopentadiene intermediates, see refs (60c−60e and 61a−61i) and; a Calderazzo F.; Pampaloni G.; Pallavicini P.; Strähle J.; Wurst K. Reactions of Zirconium(η6-benzene)(AlCl4)2 with Alkynes: Cyclooligomerization Reactions and Crystal and Molecular Structure of the Seven-Membered Metallacycle [ZrCPh(CPh)4CPh][(μ-Cl)2AlCl2]2. Organometallics 1991, 10, 896–901. 10.1021/om00050a019. [DOI] [Google Scholar]; b Takahashi T.; Xi Z.; Yamazaki A.; Liu Y.; Nakajima K.; Kotora M. Cycloaddition Reaction of Zirconacyclopentadienes to Alkynes: Highly Selective Formation of Benzene Derivatives from Three Different Alkynes. J. Am. Chem. Soc. 1998, 120, 1672–1680. 10.1021/ja970869q. [DOI] [Google Scholar]; c Álvarez E.; Gómez M.; Paneque M.; Posadas C. M.; Poveda M. L.; Rendón N.; Santos L. L.; Rojas-Lima S.; Salazar V.; Mereiter K.; Ruiz C. Coupling of Internal Alkynes in TpMe2Ir Derivatives: Selective Oxidation of a Noncoordinated Double Bond of the Resulting Iridacycloheptatrienes. J. Am. Chem. Soc. 2003, 125, 1478–1479. 10.1021/ja0290375. [DOI] [PubMed] [Google Scholar]; d Paneque M.; Poveda M. L.; Rendón N.; Mereiter K. Isolation of a Stable 1-Iridabicyclo [3.2.0] Hepta-1,3,6-triene and Its Reversible Transformation into an Iridacycloheptatriene. J. Am. Chem. Soc. 2004, 126, 1610–1611. 10.1021/ja0393527. [DOI] [PubMed] [Google Scholar]; e Hill J. E.; Balaich G.; Fanwick P. E.; Rothwell I. P. The Chemistry of Titanacyclopentadiene Rings Supported by 2,6-Diphenylphenoxide Ligation: Stoichiometric and Catalytic Reactivity. Organometallics 1993, 12, 2911–2924. 10.1021/om00032a012. [DOI] [Google Scholar]; f Collman J. P.; Kang J. W.; Little W. F.; Sullivan M. F. Metallocyclopentadiene Complexes of Iridium and Rhodium and Their Role in the Catalytic Cyclotrimerization of Disubstituted Acetylenes. Inorg. Chem. 1968, 7, 1298–1303. 10.1021/ic50065a007. [DOI] [Google Scholar]; g Yamamoto K.; Nagae H.; Tsurugi H.; Mashima K. Mechanistic Understanding of Alkyne Cyclotrimerization on Mononuclear and Dinuclear Scaffolds: [4 + 2] Cycloaddition of the Third Alkyne Onto Metallacyclopentadienes and Dimetallacyclopentadienes. Dalton Trans. 2016, 45, 17072–17081. 10.1039/C6DT03389J. [DOI] [PubMed] [Google Scholar]
- Schore N. E. Transition Metal-Mediated Cycloaddition Reactions of Alkynes in Organic Synthesis. Chem. Rev. 1988, 88, 1081–1119. 10.1021/cr00089a006. [DOI] [Google Scholar]
- For examples of PhL3-substituted complexes in which the cental oxygen atom binds to the metal center, see ref (8d) and; a Stevens T. E.; Smoll K. A.; Goldberg K. I. Direct Formation of Carbon(sp3)–Heteroatom Bonds from Rh(III) to Produce Methyl Iodide, Thioethers, and Alkylamines. J. Am. Chem. Soc. 2017, 139, 7725–7728. 10.1021/jacs.7b04169. [DOI] [PubMed] [Google Scholar]; b Hooper J. F.; Chaplin A. B.; González-Rodríguez C.; Thompson A. L.; Weller A. S.; Willis M. C. Aryl Methyl Sulfides as Substrates for Rhodium-Catalyzed Alkyne Carbothiolation: Arene Functionalization with Activating Group Recycling. J. Am. Chem. Soc. 2012, 134, 2906–2909. 10.1021/ja2108992. [DOI] [PubMed] [Google Scholar]; c Dallanegra R.; Chaplin A. B.; Weller A. S. Rhodium Cyclopentyl Phosphine Complexes of Wide-Bite-Angle Ligands DPEphos and Xantphos. Organometallics 2012, 31, 2720–2728. 10.1021/om201034m. [DOI] [Google Scholar]; d Kuang S. M.; Fanwick P. E.; Walton R. A. Unsymmetrical Dirhenium Complexes that Contain [Re2]6+ and [Re2]5+ Cores Complexed by Tridentate Ligands with P2O and P2N Donor Sets. Inorg. Chem. 2002, 41, 405–412. 10.1021/ic010983g. [DOI] [PubMed] [Google Scholar]; e Van Leeuwen P. W. N. M.; Zuideveld M. A.; Swennenhuis B. H. G.; Freixa Z.; Kamer P. C. J.; Goubitz K.; Fraanje J.; Lutz M.; Spek A. L. Alcoholysis of Acylpalladium(II) Complexes Relevant to the Alternating Copolymerization of Ethene and Carbon Monoxide and the Alkoxycarbonylation of Alkenes: The Importance of Cis-Coordinating Phosphines. J. Am. Chem. Soc. 2003, 125, 5523–5539. 10.1021/ja029341y. [DOI] [PubMed] [Google Scholar]
- Purification of Laboratory Chemicals; Armarego W. L. F., Chai C. L. L., Eds.; Elsevier Science: Oxford, UK, 2003. [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
- All calculations were performed with Frish M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Amado C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, 2013.
- All NBO analyses are performed with Glendening E. D.; Badenhoop J. K.; Reed A. E.; Carpenter J. E.; Bohmann J. A.; Morales C. M.; Landis C. R.; Weinhold F.. NBO 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2013.
- Sperger T.; Sanhueza I. A.; Kalvet I.; Schoenebeck F. Computational Studies of Synthetically Relevant Homogeneous Organometallic Catalysis Involving Ni, Pd, Ir, and Rh: An Overview of Commonly Employed DFT Methods and Mechanistic Insights. Chem. Rev. 2015, 115, 9532–9586. 10.1021/acs.chemrev.5b00163. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.