

SUMMARY
RORγt is well recognized as the lineage-defining transcription factor for T helper 17 (TH17) cell development. However, the cell-intrinsic mechanisms that negatively regulate TH17 cell development and autoimmunity remain poorly understood. Here, we demonstrate that the transcriptional repressor REV-ERBα is exclusively expressed in TH17 cells, competes with RORγt for their shared DNA consensus sequence, and negatively regulates TH17 cell development via repression of genes traditionally characterized as RORγt dependent, including Il17a. Deletion of REV-ERBα enhanced TH17-mediated proinflammatory cytokine expression, exacerbating experimental autoimmune encephalomyelitis (EAE) and colitis. Treatment with REV-ERB-specific synthetic ligands, which have similar phenotypic properties as RORγ modulators, suppressed TH17 cell development, was effective in colitis intervention studies, and significantly decreased the onset, severity, and relapse rate in several models of EAE without affecting thymic cellularity. Our results establish that REV-ERBα negatively regulates proinflammatory TH17 responses in vivo and identifies the REV-ERBs as potential targets for the treatment of TH17-mediated autoimmune diseases.
Graphical Abstract
In Brief
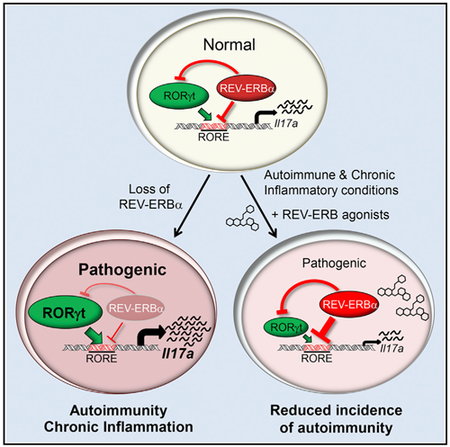
Roles for the circadian protein REV-ERBα have not been extensively explored in the immune system. Amir et al. demonstrate that REV-ERBα acts as a negative regulator of proinflammatory TH17 cell development and function, and REV-ERBα ligands are efficacious in mouse models of autoimmunity.
INTRODUCTION
T helper 17 (TH17) cells are a subset of CD4+ T helper cells that preferentially secrete interleukin 17A (IL-17A), IL-17F, IL-21, and IL-22 and are important during tissue inflammation and anti-microbial and anti-fungal immunity (McGeachy and Cua, 2008). Under homeostatic conditions, TH17 cells have essential roles in protective immunity against extracellular pathogens at mucosal barriers (McGeachy and Cua, 2008). However, TH17 cells have also been associated with the pathogenesis of several autoimmune diseases, including multiple sclerosis and psoriasis (Cho, 2008; Lees et al., 2011; Nair et al., 2009), suggesting that the failure of TH17 cell homeostasis may give rise to disease. A significant amount of work has identified key factors that drive TH17 cell development and pathogenicity. However, cell-intrinsic mechanisms that negatively regulate TH17 cell development and associated inflammatory responses have received less attention. Therefore, a more comprehensive understanding of the factors that both positively and negatively regulate TH17 cell development is necessary to better understand TH17-mediated autoimmunity and would aid in the development of novel therapeutics to treat TH17-mediated diseases.
A number of studies have identified key factors that drive TH17 cell development and pathogenicity, including both the nuclear receptors retinoic-acid-receptor-related orphan receptor α and γt (Ivanov et al., 2006; Yang et al., 2008). RORγt is considered the lineage-defining transcription factor regulating TH17 cell development, and a considerable amount of research has elucidated genomic functions of RORγt. Two other members of the nuclear receptor superfamily, REV-ERBα (NR1D1) and REV-ERBβ (NR1D2), are often co-expressed in the same tissues as the RORs and bind the same DNA response elements, resulting in mutual cross-talk and co-regulation of their shared target genes (Kojetin and Burris, 2014). Outside of the immune system, the RORs and the REV-ERBs modulate a number of physiological processes but are best known for their roles in the regulation of the circadian rhythm, lipid, and glucose metabolic processes. The REV-ERBs are unique within the nuclear receptor superfamily in that they lack the carboxy-terminal tail of their ligand-binding domain (LBD) called the activation function 2 region (AF-2, helix 12), which is required for coactivator recognition. Thus, in contrast to the RORs, which are constitutive activators of transcription, the REV-ERBs are transcriptional repressors (Kojetin and Burris, 2014). Collectively, the balance of expression of the RORs and REV-ERBs is critical for dynamic regulation of their target genes (Kojetin and Burris, 2014). While much is known about RORγt-mediated regulation of TH17 cell development and function, little is known about the role of the REV-ERBs in T cell effector functions, specifically proinflammatory TH17 cell effector functions and autoimmunity.
Most members of the nuclear receptor superfamily are ligand-regulated transcription factors and represent attractive therapeutic targets, including RORγt. After the initial identification of several synthetic RORγ modulators, including SR1001 and digoxin (Huh et al., 2011; Solt et al., 2011), countless other RORγ ligands have been identified, demonstrating the tractability of RORγt-targeted treatment of TH17-mediated auto-immunity (Bronner et al., 2017). The REV-ERBs are also ligand-regulated transcription factors, and the porphyrin heme was identified as the endogenous ligand for both REV-ERBα and REV-ERBβ (Raghuram et al., 2007; Yin et al., 2007). We and others have identified and characterized synthetic ligands that modulate the activity of the REV-ERBs both in vitro and in vivo (Banerjee et al., 2014; Kojetin et al., 2011; Solt et al., 2012). We previously synthesized and characterized SR9009 and SR9011 for their activity and specificity to target the REV-ERBs, demonstrating that in vivo pharmacological modulation of REV-ERB activity affected REV-ERB-mediated processes, including regulation of the circadian rhythm, glucose, and lipid metabolic processes (Solt et al., 2012). Despite the well-documented overlap in genetic programs between the RORs and REV-ERBs in tissues outside of the immune system (Kojetin and Burris, 2014), the role for the REV-ERBs in TH17 cell development is still poorly understood. Furthermore, given the massive pharmaceutical effort focused on developing potent RORγ-modulators, small-molecule modulators of REV-ERB activity could represent a novel therapeutic target for the treatment of TH17-mediated autoimmunity.
While REV-ERBα was previously demonstrated to diurnally regulate TH17 cell frequencies in vivo (Yu et al., 2013), its function in the context of proinflammatory settings and autoimmunity remains poorly defined. Here, we show that REV-ERBα is expressed during TH17 cell development and its presence is required for dampening TH17-mediated proinflammatory cytokine expression. Overexpression of REV-ERBα suppressed TH17 cell development, whereas genetic deletion of REV-ERBα resulted in enhanced TH17 cell development in vitro and exacerbated autoimmune responses in vivo. We found that while REV-ERBα directly repressed Nfil3 (Yu et al., 2013), it also competed with RORγt for binding at the Il17a promoter and CNS-5 enhancer region. We also discovered that REV-ERBα binds within the Rorc promoter region, suggesting potential crosstalk and autoregulation among these receptors for controlling TH17 cytokine expression. Finally, the use of REV-ERB-specific small molecules that we developed suppressed TH17 cell development in vitro and the development of TH17-mediated autoimmunity in vivo and was effective when used in a “treatment mode” in several models of autoimmunity and chronic inflammation. However, unlike RORγ modulators, REV-ERB modulators did not have effects on the thymus. Collectively, our data suggest that REV-ERBα functions outside of its classical role as a core member of the circadian clock under proinflammatory conditions and importantly, is a key cell-intrinsic negative regulator of TH17 cell proinflammatory immune responses.
RESULTS
REV-ERBα Is Upregulated in TH17 Cells
To determine whether the REV-ERBs were expressed during CD4+ T helper cell development, we differentiated naive CD4+ T cells into TH1, TH2, TH17, or inducible T regulatory cells (iTregs). REV-ERBα was upregulated at the mRNA level in TH17 cells, whereas REV-ERBβ was not (Figure 1A). We next performed a kinetic analysis and discovered the expression pattern of REV-ERBα was similar to that of the RORs, whereas REV-ERBβ was downregulated during development (Figures 1B and 1C). The similar expression pattern profiles between the RORs and REV-ERBα are consistent with other published data demonstrating that the RORs drive REV-ERBα expression through conserved ROR response elements (ROREs) within its promoter region (Raspè et al., 2002; Takeda et al., 2012). Furthermore, the disconnect in expression between REV-ERBα and REV-ERBβ is unique given that the REV-ERBs typically have overlapping tissue expression patterns (Kojetin and Burris, 2014), suggesting that REV-ERBα may play a role in the regulation of TH17 cell development.
Figure 1. The REV-ERBs Inhibit TH17 Cell Development.

(A) Quantitative real-time PCR analysis ofThelper cell lineage-specific transcription factorsT-bet (Tbx21), Gata3, Foxp3, RORα (Rora), RORγt (Rorc), REV-ERBα (Nrldl), and REV-ERBβ (Nr1d2) under TH1, TH2, TH17, and inducible T regulatory cell (iTreg) conditions at 48 hr after T cell-activation compared to naive CD4+ T cells (n = 3).
(B) Quantitative real-time PCR of Il17a, Rora, Rorc, Nrldl, and Nr1d2 expression during TH17 cell differentiation. Data represent mean ± SEM (n = 3).
(C) Immunoblot analysis of REV-ERBα and RORγt expression during TH17 cell differentiation (n = 4).
(D) FACS analysis of IL-17A and IL-17F expression in TH17 cells transduced with empty vector (MIGR1), REV-ERBα, or REV-ERBβ. Cells were gated on live, GFP+ cells. Quantitative real-time PCR analysis of REV-ERBα and REV-ERBβ expression from sorted GFP+ TH17 cells transduced with MIGR1, REV-ERBα, or REV-ERBβ (n = 4).
(E) FACS analysis of RORγt expression from T cell cultures shown in (D). Graph (right) indicates median fluorescent intensity (MFI) of RORγt expression in the FACS plots.
(F) Quantitative real-time PCR of sorted GFP+ cells from (D) (n = 3).
(G) Heatmap of differentially expressed genes in TH17 cells (false discovery rate [FDR], <0.05).
(H) KEGG pathway analysis of genes differentially expressed between MIGR1-, REV-ERBα–, and REV-ERBβ-overexpressing TH17 cells. p-actin was used as the internal control for quantitative real-time PCR. *p < 0.05, **p < 0.01, and ***p < 0.001 determined using Student’s t test. ns, not significant (p > 0.05).
The REV-ERBs Repress TH17 Cell Development
To better assess the function of the REV-ERBs in TH17 cells, we retrovirally overexpressed an empty vector (MIGR1), REV-ERBα, or REV-ERBβ in TH17 cells. Although the expression of both REV-ERBs inhibited IL-17A and IL-17F expression, REV-ERBα was more potent (Figure 1D). The inhibitory effect of the REV-ERBs appeared to be TH17-cell specific because it did not affect the expression of Foxp3 in iTreg cells (Figure S1A). Interestingly, overexpression of REV-ERBα but not REV-ERBβ resulted in decreased RORγt expression in transduced TH17 cells (Figure 1E). Quantitative real-time PCR analysis on sorted transduced samples demonstrated a significant decrease in expression of TH17-cell signature genes and Nfil3 (Figure 1F). Although the REV-ERBα-mediated repression on Nfil3 was consistent with previously published work (Yu et al., 2013), the reduction in TH17-cell signature genes was not. Given the striking effects of REV-ERBα on Nfil3 and TH17-mediated gene expression, we wanted to assess how Nfil3 fit into this paradigm. We retrovirally overexpressed or knocked down Nfil3 in TH17 cells and found each to have little effect on IL-17A expression (Figures S1B and S1C). This is consistent with a prior study that demonstrated similar results (Ciofani et al., 2012). Collectively, our data suggest that REV-ERBα may be playing a more direct role in TH17 cell development than previously identified.
To better understand the transcriptional programs dictated by the REV-ERBs during TH17 cell development, we performed RNA sequencing on TH17 cells transduced with an empty vector, REV-ERBα, or REV-ERBβ retrovirus. Consistent with our quantitative real-time PCR data, REV-ERBα transduced cells repressed a number of TH17 cell signature genes, including the “core” RORγt target genes Il17a, Il23r, TGFβ3, Ccl20, and Ltb4r1 (Ciofani et al., 2012), relative to MIGR1 controls (adjusted p value [p. adj.] < 0.05) (Figure 1G and Table S1). KEGG pathway analysis indicated that REV-ERBα differentially regulated genes associated with inflammatory bowel disease (IBD), which is consistent with the involvement of TH17 cells in the pathogenesis of IBD (Harbour et al., 2015; Jostins et al., 2012; Lee et al., 2009), cytokine-cytokine receptor interactions, Jak-STAT signaling pathways, T cell receptor signaling, and the circadian rhythm (Figure 1H). Overexpression of REV-ERBβ had a modest effect on global TH17 cell gene changes relative to REV-ERBα. These data suggest that REV-ERBα is a potent repressor of the TH17 cell genetic program.
Cell Intrinsic Role for REV-ERBα in Restraining TH17 Cell Development and Inflammatory Responses In Vivo
We next used REV-ERBα−/− mice (Chomez et al., 2000) to explore the endogenous role of REV-ERBα in TH17 cell development. Immune phenotyping revealed no overt differences between REV-ERBα+/+ (wild-type [WT]) and REV-ERBα−/− (KO) mice (Figures S2A–S2C). We differentiated WT and KO naive CD4+ T cells under TH1-, TH2-, TH17-, or iTreg-polarizing conditions and observed a significant increase in the frequency of IL-17A+IL-17F+ cells and the expression of RORγt in TH17 cells (Figure 2A). Similar results were observed across various TH17 cell culture conditions, including pathogenic TH17 conditions (Lee et al., 2012; Peters et al., 2011), with little to no effect on the development of TH1 and TH2 cells (Figures S3A–S3C). Although a prior study found that REV-ERBα deficiency resulted in decreased IL-17A production in vitro, differences in the culture conditions, protocols, and mouse strains could account for the different outcomes (Yu et al., 2013). However, REV-ERBα deficiency resulted in reduced expression of Foxp3 and increased expression of RORγt in iTreg cultures (Figure S3D). Because overexpression of REV-ERBα did not affect iTreg development in vitro, these results suggested this was possibly through REV-ERBα-dependent effects on RORγt expression. REV-ERBα deficiency also increased the expression of TH17 signature genes as well as Nfil3 (Figure 2B), consistent with a direct relief of repression expected when a transcriptional repressor is deleted (Cho et al., 2012; Everett and Lazar, 2014). Finally, we performed RNA sequencing to examine the global transcriptional effects of REV-ERBα deficiency on TH17 cell development. Similar to our overexpression studies (Figure 1), REV-ERBα-deficient TH17 cells differentially expressed numerous TH17 cell signature genes, including Il17a, Il17f, Ccl20, and Lta, relative to WT control cells (p. adj. < 0.05) and regulated similar pathways identified by KEGG pathway analysis (Figure 2C and Table S2). Because REV-ERBα and RORγ bind to the same DNA response elements, we compared our data to previously published data that assessed RORγ knockout TH17 cells (Ciofani et al., 2012). Although we observed only a small overlap in genes, most of the overlapping genes were the core RORγt target genes (Ciofani et al., 2012) (Figure 2C). These data indicate that REV-ERBα affects TH17 cell development through several mechanisms, including RORγt-dependent TH17 cell cytokine expression.
Figure 2. Loss of REV-ERBα Leads To Increased TH17-Mediated Autoimmunity.

(A) FACSanalysisfromT|-|17culturesderived from REV-ERBα+/+ (WT) and REV-ERBα−/− (KO) mice. Graphs indicate percent IL-17A+IL-17F+ cells(top) and MFI of RORγt expression in the FACS plots (bottom) (n = 4).
(B) Quantitative real-time PCR of TH17-mediated cytokines in TH17 cell cultures from WT and KO mice. β-actin was used as the internal control (n = 3).
(C) Heatmap of differentially expressed genes between WT and KO TH17 cells. KEGG pathway analysis of genes differentially expressed between WT and KO TH17 cells. Venn diagram depicting the numbers of unique and shared genes differentially regulated in WT/KO and WT/RORγ−/− TH17 cells (FDR, <0.05).
(D) Clinical EAE scores (left) and disease incidence (right) from WT and KO mice subjected to MOG-induced EAE.
(E and F) Graphs summarizing the frequency of TCRβ+CD4+ cells (E) and Foxp3+ cells in the CNS of mice (F).
(G) Graph representing FACS analysis of RORγt expression (left) and total IL-17Aexpression in RORγt+ cells (right) in theTCRp+CD4+ cells in the CNS of WT and KO mice.
(H) FACS analysis and graph depicting the frequency of IL-17A+IFNγ−, IL-17A+IFNγ+, and IL-17A−IFNγ+ cells in the CNS ofWT and KO mice.
Cellsweregated on live, CD45+TCRp+CD4+ cells. Each symbol representsan individual mouse (n = 4/group). Data represent mean ± SEM and are representative of two separate independent experiments generating similar results. Two-way ANOVA (clinical score) and Student’sttests were performed forstatistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine whether REV-ERBα influenced TH17-mediated autoimmunity, we immunized WT and KO mice with a myelin oligodendrocyte glycoprotein peptide (MOG35–55) to induce experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis. Signs of disease were exacerbated, and the onset of disease was slightly earlier in KO mice compared to WT littermate controls (Figure 2D). We observed an increased frequency of CD4+ T cells infiltrating the CNS of the KO mice at the peak of disease (Figure 2E). Of the infiltrating CD4+ T cells in the CNS, the overall frequency of RORγt+ cells was elevated (Figure 2G). This correlated with the increased expression of IL-17A+ T cells in the CNS, whereas the percent of anti-inflammatory Foxp3+ Tregs was significantly decreased (Figures 2F and 2G). Because TH17 cells can give rise to TH1-like cells that produce IFNγ, and acquisition of IFNγ has been linked to their pathogenicity in several models of chronic disease (Ghoreschi et al., 2010; Hirota et al., 2011; Lee et al., 2009), we assessed the frequency of IL-17A+, IL-17A+IFNγ+, and IFNγ+ T cells in the CNS (Figure 2H). There was a significant increase in the IL-17A+IFNγ+ population in KO mice compared to WT controls. These results indicated that the loss of REV-ERBα leads to increased proinflammatory responses in vivo.
REV-ERBα Deficiency Exacerbates the Development of Colitis
To determine the T cell-specific effects of REV-ERBα in vivo, we sorted naive CD4+ T cells from WT and KO mice and adoptively transferred them into Rag1−/− recipients to track the development of colitis, which is another TH17-driven inflammatory disorder (Harbour et al., 2015; Lee et al., 2009) by using PBS-sham injections as a control. The transfer of KO cells resulted in more severe weight loss and intestinal inflammation than WT cells (Figure 3A). Mice receiving KO T cells had an increased frequency of RORγt+ TH17 cells and decreased frequency of Foxp3+ Tregs relative to WT recipient mice in spleens and mesenteric lymph nodes (mLNs) (Figures 3B–3D). Mice receiving KO T cells also had an increased frequency of IL-17A+ and pathogenic IL-17A+IFNγ+ cells (Figure 3E). The expression of TH17-associated cytokines (Il17a, Il17f, and Il22), proinflammatory cytokines (Il1b, Il6, Tnfa, and IFNγ), and genes activated downstream of IL-17A and IL-17F and IL-22 signaling (Cxcl2 and Cxcl10) were increased at the mRNA level in whole-tissue homogenates of the proximal colons of mice receiving KO T cells (Figure 3F). Finally, histological analysis of the proximal colons demonstrated increased cellular infiltration, broadening of the crypts, and epithelial hyperplasia in mice receiving KO T cells compared to WT controls (Figure 3G). Similar results were observed in the distal colon of the recipient mice (data not shown). These results indicate that the loss of REV-ERBα promotes T cell-mediated colitis, leading to increased frequencies of proinflammatory TH17 and IFNγ-producing TH17 cells, reinforcing the role of REV-ERBα as a negative regulator of proinflammatory TH17 responses.
Figure 3. Loss of REV-ERBα in T Cells Exacerbates Colitis.

(A) Percent change in body weight, colon weights, colon lengths, and colon weight versus colon length ratios of Rag1−/− recipient mice over 12 weeks post- adoptive transfer of WT, KO, or control (no cells, PBS) T cells.
(B-D) FACS plots demonstrating frequencies of (B and D) RORγt+ and (C and D) CD25+Foxp3+ cells in the spleens and mesenteric lymph nodes (mLN) of recipient mice at the termination of the experiment.
(E) FACS analysis and frequencies of IL-17A+IFNγ−, IL-17A+IFNγ+, and IL-17A−IFNγ+ cells in the spleens and mLNs of recipient mice. Cells were gated on live, CD45+CD3+CD4+ cells.
(F) Quantitative real-time PCR analysis of cytokines and chemokines expressed in the proximal colon from Rag1−/− mice receiving PBS, WT, or KO CD4+ T cells. 18s was used as the internal control.
(G) Histology scores from colon and representative hematoxylin and eosin stained proximal colon sections from Rag1−/− recipient mice12 weeks after transfer of naive CD4+ T cells. (×10 magnification, scale bars represent 200 mm; ×40 magnification, scale bars represent 50 mm) (n = 13 for WT, n = 15 for KO, and n = 3 for PBS).
Data represent mean ± SEM. Two-way ANOVA (body weight) and Student’s t tests were performed for statistical analysis. *p < 0.05, **p < 0.001, ***p < 0.001, ****p<0.0001.
REV-ERBα Competes with RORγt To Repress TH17 Cell Development
We next sought to delineate the molecular mechanisms underlying REV-ERBα-dependent repression of TH17 cell development. Because the RORs and REV-ERBs bind to the same genomic DNA response element sequence, an RGGTCA half-site preceded by a 5′-AT-rich region (RORE and/or REV-ERB response element [RevRE]), we wanted to determine if REV-ERBα could bind and repress within the Il17a locus, a known RORγt target gene (Figure 4A). Cotransfection assays in HEK293 cells using an Il17a+CNS5 luciferase reporter construct (Zhang et al., 2008) demonstrated that REV-ERBα repressed Il17a+CNS5-luciferase driven activity in a concentration-dependent manner (Figure 4B). This effect was dependent on the ability of REV-ERBα to bind DNA because deletion of its DNA-binding domain (DBD) had no effect on luciferase activity, whereas the REV-ERBα DBD alone also dose-dependently inhibited Il17a+CNS5-luciferase driven activity. We next performed competition experiments in HEK293 cells to determine if REV-ERBα competes with RORγt at their shared RORE and/or RevRE in the Il17a promoter and enhancer region. Consistent with Figure 4B, REV-ERBα competed with RORγt for its shared RORE and/or RevRE in a concentration-dependent manner to regulate Il17a+CNS5-luciferase driven activity (Figure 4C). The reduced repression observed with the DBD construct compared with the full-length REV-ERBα is likely due to its inability to recruit corepressor proteins, such as NCoR, via its LBD for active repression (Yin et al., 2010). Finally, in EL4 mouse T cell thymoma cells that endogenously expresses RORγt, we found that REV-ERBα competes with endogenous RORγt using the Il17a+CNS5-luciferase reporter (Figure 4D).
Figure 4. REV-ERBα Competes with RORγt To Repress TH17 Cell Development.

(A) Schematic demonstrating REV-ERBα and RORγt competition for the Il17a locus.
(B) Schematic of REV-ERBα constructs. Cotransfection assays in HEK293 cells demonstrating full-length (FL) and REV-ERBα DBD dose-dependently sup-presses Il17a + CNS5 luciferase activity. EV refers to empty vector. (n = 5).
(C) Cotransfection assay in HEK293 cellsdemonstratingthat FLand REV-ERBα DBD competeswith RORγt binding attheIl17a + CNS5 RORE. The concentration of RORγt was constant in all conditions labeled with a (+). (n = 5).
(D) Cotransfection assay in EL4 cells demonstrating REV-ERBα competes with endogenous RORγt binding at the Il17a + CNS5 RORE. P+I indicates 18-hr stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin (n = 4).
(E) FACS analysis of IL-17A and IFNγ expression in TH17 cells transduced with MIGR1, FLREV-ERBα, REV-ERBα DBD, or REV-ERBαΔDBD. Cells were gated on live, GFP+ cells (n = 4).
(F) ChIP-qPCR of REV-ERBα binding at various sites in KO and WT TH17 cells collected on day 3. (Il17a-p, Il17a promoter).
Data represent mean ± SEM (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001 determined using Student’s t test. ns, not significant (p > 0.05).
To establish whether REV-ERBα modulates RORγt-mediated IL-17A expression in a DBD-dependent manner in TH17 cells, we retrovirally overexpressed empty vector, full-length REV-ERBα, REV-ERBα DBD, or REV-ERBα ΔDBD in TH17 cells and assessed IL-17A expression by flow cytometry. Both FL and REV-ERBα DBD significantly inhibited IL-17A protein expression, whereas IL-17A expression in the REV-ERBα ΔDBD condition was similar to the empty vector control (Figure 4E). Finally, using an anti-REV-ERBα antibody (Cho et al., 2012), chromatin immunoprecipitation (ChIP) experiments in TH17 cells indicated that endogenous REV-ERBα was bound within the promoter and CNS5 region of the Il17a gene, suggesting that regulation of Il17a transcription is direct (Yang et al., 2008) (Figure 4F). We also observed REV-ERBα bound within the upstream promoter region of Rorc (Mongrain et al., 2008) and Nfil3 (Yu et al., 2013), which is consistent with published results; Cry1 (Cho et al., 2012) and Hprt were used as positive and negative controls, respectively. These data indicate that REV-ERBα negatively regulates TH17 cell development by competing with RORγt at the RORE and/or RevRE sites, which includes the Il17a locus. Furthermore, an additional layer of regulation may occur through REV-ERBα-mediated repression of RORγt.
REV-ERBα-Specific Small Molecules Suppress TH17-Cell Development
We previously developed and characterized SR9009, a proof-of-concept REV-ERB-specific synthetic ligand that demonstrated that the REV-ERBs could be pharmacologically targeted in vivo (Solt et al., 2012). Our efforts to improve SR9009 led to our identification of SR12418, which binds to both receptors in a time-resolved fluorescence resonance energy transfer (TR-FRET) biochemical assay and is more potent at REV-ERBα (half maximal inhibitory concentration [IC50] = 68 nM) and REV-ERBβ (IC50 = 119 nM) in a Bmal1-luciferase reporter assay than SR9009 (Figures S4A–S4C, S5A, and S5B) (Noel et al., 2012). SR12418 demonstrated significantly improved plasma exposure relative to SR9009 (Solt et al., 2012), was more effective at inhibiting IL-17A expression in EL4 cells, demonstrated specificity at the REV-ERBs, and did not exhibit activity at any other nuclear receptors (Figures S5C–S5F) (Solt et al., 2012). SR12418 also showed minimal off-target activity in a CEREP (Eurofins Scientific) panel screen of 84 G-protein coupled receptors, ion channels, and transporters (data not shown). Both SR9009 and SR12418 dose-dependently inhibited Il17a lucif-erase activity at REV-ERBα and REV-ERBβ and inhibited TH17 cell differentiation without affecting viability or other T helper populations (Figures 5A, 5B, S6A, and S6B). Expression of RORγt was also downregulated in drug-treated conditions relative to vehicle control (Figure 5C). Quantitative real-time PCR analysis demonstrated that SR9009 and SR12418 potently repressed TH17-mediated gene expression and Nfil3, which is consistent with REV-ERBα acting as a repressor at RORE and/or RevRE sites (Figure 5D). To establish whether REV-ERB-selective small molecules could override potent RORγt-driven TH17 differentiation, we retrovirally transduced naive CD4+ T cells with RORγt cultured under TH17-polarizing conditions and treated the cells with SR9009, SR12418, or vehicle control. Despite the potent and sustained ectopic RORγt expression, REV-ERB-selective small molecules were able to repress RORγt-mediated IL-17A expression (Figure 5E). These data indicate that REV-ERB-selective small molecules can target the IL-17A pathway in vitro and override a potent RORγt stimulus.
Figure 5. REV-ERBα-Specific Small Molecules Suppress TH17 Cell Development and Function.

(A) Cotransfection assay in HEK293 cells using FL REV-ERBα, REV-ERBβ, and the Il17a + CNS5 luciferase reporter. Graphs demonstrate that SR9009 and SR12418 dose-dependently drive REV-ERB-mediated repression of Il17a + CNS5 luciferase activity. Data represent mean ± SEM (n = 4).
(B) Mouse CD4+ T cells were differentiated under TH17 polarizing conditions and treated with vehicle (DMSO), SR9009, or SR12418. IL-17Aand IFNγ expression were analyzed by flow cytometry. Graphs indicate percent IL-17A+ cells and frequency of live cells in cultures with compound treatment (n = 3).
(C) FACS analysis and graphs depicting MFI of RORγt expression in TH17 cultures treated with SR9009 and SR12418.
(D) Quantitative real-time PCR of TH17-mediated cytokines in cells treated with vehicle (DMSO), SR9009 (5 mm), or SR12418(5 mm). Analysis was performed at 96 hr after T cell activation and compared to naive CD4+ T cells. β-actin was used as the internal control. Data represent mean ± SEM (n = 3).
(E) FACS analysis of IL-17A and IFNγ expression inTH7 cellstransduced with MIGR1 RORγt and treated with vehicle (DMSO), SR9009, or SR12418 for 48 hr at the indicated doses. Cells were gated on live, GFP+ cells (n = 4).
(F) FACS analysis of thymocytes from mice treated with vehicle, SR2211, SR9009, or SR12418 for 72 hr.
Graphs depict quantification of total thymocyte number, double positive percentage, and double positive number in each group (n = 5/group). Data represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 determined using Student’s t test. ns, not significant (p > 0.05).
Currently, RORγ modulators are being developed for the treatment of autoimmunity (Guo et al., 2016). However, RORγt is required for thymocyte survival (Kurebayashi et al., 2000; Sun et al., 2000), and one drawback of treatment with RORγ modulators is thymocyte apoptosis (Chang et al., 2014; Guo et al., 2016). To determine if REV-ERB ligands induced a similar pheno-type, we treated C57BL/6 mice for 3 days with SR2211 (a potent RORγ inverse agonist (Kumar et al., 2012) (20 mg/kg, i.p., b.i.d.), SR9009 (100 mg/kg, intraperitoneal [i.p.], twice a day [b.i.d.]), SR12418 (50 mg/kg, i.p., b.i.d), or vehicle control. Although mice treated with SR2211 demonstrated reduced thymic cellularity, specifically in the double-positive stage, treatment with SR9009 or SR12418 did not display significant defects relative to vehicle control. (Figure 5F). Rorcfl/fl and Rorcfl/fl × CD4 Cre mice were also analyzed as controls, and consistent with published reports, deletion of RORγ in the thymus leads to reduced cellularity and apoptosis in the double-positive stage (Kurebayashi et al., 2000; Sun et al., 2000). (Figure S6C). Thus, RORγ inverse agonists, such as SR2211, largely phenocopy the thymocyte apoptosis observed in RORγ knockout mice, whereas REV-ERB synthetic ligands do not.
REV-ERB-Selective Small Molecules Suppress TH17-Mediated Autoimmunity In Vivo
As proof of concept, we tested whether pharmacologically targeting the REV-ERBs in vivo would affect autoimmune disease course by immunizing C57BL/6 mice to induce EAE. Due to its superior pharmacological profile over SR9009, we focused on SR12418, which was administered daily (50 mg/kg, b.i.d.) versus vehicle control following immunization. Mice treated with SR12418 showed delayed onset and severity of disease compared to the vehicle-control-treated group (Figure 6A). The incidence of disease was greatly diminished in the SR12418 treated group, with approximately 20% of mice developing overt signs of disease compared to 90% of vehicle-control mice. SR12418 treatment did not demonstrate overt signs of toxicity, as the weight of the animals remained relatively constant throughout the experiment. Evaluation of liver demonstrated that SR12418 did not appear to significantly perturb the circadian rhythm in other tissues evaluated (Figure S7A). Evaluation of the draining LNs and CNS indicated a reduction in the frequency and number of CD3+CD4+ T cells at the peak of disease (Figure 6B). Intracellular fluorescence-activated cell sorting (FACS) analysis demonstrated a significantly decreased frequency and number of RORγt+ cells in the LNs and CNS of the SR12418-treated mice relative to vehicle controls (Figure 6D). The absolute number of GM-CSF+ T cells was significantly lower in the SR12418 treated mice as was the frequency and number of IL-17A+ and pathogenic IL-17A+IFNγ+ cells in the LNs and CNS than mice receiving vehicle control (Figures 6C and 6E). Thus, targeted pharmacological modulation of REV-ERB activity in vivo effectively suppresses the development and progression of TH17-driven EAE.
Figure 6. SR12418 Potently Suppresses the Development and Severity of EAE.

(A) Clinical EAE scores (left) from mice subjected to MOG-induced EAE and treated with vehicle (10/10/80 formulation of DMSO/Tween80/H2O) or SR12418 (i.p., 50mg/kg, b.i.d.) for the duration of the experiment. Middle and right graphs demonstrate the percent incidence of disease and percent change in body weight overtime between groups, respectively (n = 8–10/group).
(B) Frequencies and cell counts of CD3+CD4+ cells in the draining LNs and CNS of mice at peak of disease. V, vehicle; SR, SR12418-treated mice (n = 8, V; n = 7, SR).
(C) Graphs depicting the frequencies and cell counts of CD4+GM−CSF+ cells in LN and CNS of mice at peak of disease.
(D) FACS analysis, frequencies, and cell counts of RORγt+ cells in LNs and CNS of mice at peak of disease.
(E) FACS analysis, frequencies, and cell counts of IL-17A+IFNγ−, IL-17A+IFNγ+, and IL-17A−IFNγ+ cells in the LN and CNS of mice at peak of disease.
Cells were gated on live, CD45+CD3+CD4+ cells. Data are mean ± SEM and representative of three separate, independent experiments with similar results. Two-way ANOVA (clinical score) and Student’s t tests were performed for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further evaluate the therapeutic efficacy of SR12418, we first performed intervention studies utilizing the adoptive T cell transfer model of colitis (Kjellev et al., 2006; Lindebo Holm et al., 2012) (Figure 7A). Following procedures described above, at 3-weeks after T cell transfer, SR12418 (50 mg/kg, b.i.d.) or vehicle control was administered twice daily for the duration of the experiment. PBS was used as a control. Neither group displayed any overt difference in body weight, colon weight, colon length, or colon weight/length ratio over time (Figure S7B). However, FACS analysis of colon tissue revealed that SR12418 led to a reduction in the frequency of a4b7+ T cells relative to vehicle controls, suggesting SR12418 hampered the ability of the T cells to home to the intestines (Figure 7B). Intracellular FACS analysis demonstrated a decreased frequency of RORγt+ ells in the colons of the SR12418-treated mice relative to vehicle controls, whereas the frequency of IL-17A+ and pathogenic IL-17A+IFNγ+ cells was also lower than mice receiving vehicle control (Figures 7C and 7D). Interestingly, the frequency of CD25+Foxp3+ Tregs was significantly increased in the colons of SR12418-treated mice relative to the vehicle controls (Figure 7E).
Figure 7. SR12418 Is Effective When Used in Intervention Studies of Colitis and Relapsing-Remitting EAE.

(A) Schematic of the adoptive T cell transfer colitis model treatment design.
(B-E) FACS plots and graphs demonstrating frequencies of (B) α4β7+, (C) RORγt+, (D) IL-17A+IFNγ−, IL-17A+IFNγ+, and IL-17A−IFNγ+, and (E) CD25+Foxp3+ T cells in the colons of recipient mice. Cells were gated on live, CD45+CD3+CD4+ cells. V, vehicle; SR, SR12418. Data are mean ± SEM (n = 10/group; PBS, n = 2).
(F) Clinical EAE scores from mice subjected to PLP-induced EAE and treated with vehicle or SR12418 (i.p., 50 mg/kg, b.i.d.) starting on day 18 and continued for the duration of the experiment (n = 17/group).
(G-K) Graphs demonstrating decreased frequencies and cell numbers of (G) effector CD4 and CD8 T cells and (H) CCR6+ T cells in the CNS of mice treated with SR12418. FACS plots and graphs demonstrating frequencies and/or cell numbers of (I) RORγt+, (J) RORγt+ GM-CSF+, and (K) IL-17A+IFNγ−, IL-17A+IFNγ+, and IL-17A−IFNγ+ T cells in the CNS of SR12418-treated mice relative to vehicle controls.
Cells were gated on live, CD45+CD3+CD4+CD44+cells (n = 7, vehicle; n = 6, SR12418). Data represent mean ± SEM. Two-way ANOVA (clinical score) and Student’s t tests were performed for statistical analysis. *p < 0.05, **p < 0.01, ***p < 0.001.
We next tested the therapeutic capacity of SR12418 for treating ongoing PLP139–151-induced relapsing-remitting EAE (R-EAE) in SJL/J mice. SR12418 (50 mg/kg, b.i.d.) or vehicle control was administered twice daily once mice had recovered from the first wave of disease (day 18 post-immunization). SR12418 resulted in a significant reduction in the relapse severity compared to the animals receiving vehicle control (Figure 7F). Twelve of the 17 mice in the vehicle-treated group relapsed, with the average maximum relapsing clinical score for each mouse recorded between 2.5 and 3.5. In contrast, only 5 of the 17 SR12418-treated animals relapsed, with the maximum clinical score recorded as no greater than 1. FACS analysis of the CNS from the animals revealed a significant decrease in the frequency and number of CD4+ and CD8+ effector cells (CD44hi) in the SR12418-treated animals versus vehicle control (Figure 7G), which may be attributed to a decreased frequency and number of CD4+CCR6+ T cells in the CNS (Figure 7H). Intracellular FACS analysis demonstrated a decreased frequency and number of RORγt+ cells in the CNS of the SR12418-treated mice relative to vehicle controls, whereas the number of RORγt+GM-CSF+ cells was also decreased (Figures 7I and 7J). Finally, SR12418-treated animals demonstrated a decreased frequency of IL-17A+ cells and decreased number of IL-17A+ and pathogenic IL-17A+IFNγ+ cells in CNS compared to mice receiving the vehicle control (Figure 7K). These results offer proof-of-concept that pharmacological modulation of REV-ERB activity post-disease onset can suppress the progression of TH17-driven autoimmunity and chronic inflammatory disorders.
DISCUSSION
In this study, we demonstrated that REV-ERBα is induced exclusively in TH17 cells and acts as a cell-intrinsic negative regulator of proinflammatory TH17-cell function. Our results suggest that the induction of REV-ERBα represents a previously undefined and critical checkpoint during TH17 cell development. Overexpression of REV-ERBα inhibited TH17 cell development whereas its deletion led to increased proinflammatory cytokine production in vitro and in vivo, which is consistent with its role as a repressor of gene transcription (Kojetin and Burris, 2014). Mechanistically, REV-ERBα competes with RORγt for binding to the same RORE and/or RevRE genomic response sequence at the Il17a locus, which was further supported by the overlap in gene signatures between WT/KO and WT/RORγt−/− TH17 cells. REV-ERBα bound upstream of the Rorc transcriptional start site, suggesting it also regulated TH17 cell development through repression of RORγt itself. Finally, REV-ERB-specific small molecules suppressed TH17 cell development in vitro and were extremely effective when used prophylactically in suppressing TH17-mediated autoimmunity in vivo. Importantly, SR12418 was effective when used in a therapeutic mode in two separate models of TH17-mediated inflammatory diseases, colitis, and EAE. Collectively, these findings demonstrate that the nuclear receptor REV-ERBα is a cell-intrinsic negative regulator of TH17 cell development whose activity is critical for dampening proinflammatory cytokine production.
Although cross-talk between the RORs and REV-ERBs is well established in the regulation of the circadian rhythm and metabolic processes, cross-talk between these receptors has not been extensively explored in the immune system. Here, we demonstrate that REV-ERBα competes with RORγt at their shared RORE and/or RevRE sites, including the Il17a locus to regulate IL-17A cytokine expression. Comparison of the KO and ROR0γ−/− TH17 cell RNA sequencing (RNA-seq) datasets suggests there is overlap in the genes that have clear RORE and/or RevREs in their promoter regions, including Il17f, Ccl20, and Ltb4r1. Although many genes did not overlap with the RORγ−/− TH17 cells, many of these were downregulated in KO TH17 cells, likely indicating that REV-ERBα was indirectly regulating those genes. Of the other non-RORγ overlapping genes that were upregulated in KO TH17 cells, these repressive effects could be a function of DNA-independent mechanisms, otherwise known as tethering, in which REV-ERBα binds to cell-type-specific transcription factors to negatively regulate gene transcription to convey a tissue-specific genetic program tailored to the needs of that cell type (Zhang et al., 2015). In metabolic tissues such as the liver, these two different modes of repression enable REV-ERBα the ability to stabilize the circadian oscillations of clock genes at RORE and/or RevREs while coupling metabolism to environmental and metabolic changes (Zhang et al., 2015). Perhaps a comparable phenomenon endows REV-ERBα the ability to function in a similar manner in TH17 cells. More in-depth ChIP and ChIP sequencing studies will need to be performed to establish these different modes of REV-ERB-mediated repression.
REV-ERBα also appears to regulate the expression of RORγt, and although consistent with previously published work (Cho et al., 2012; Mongrain et al., 2008), we do not believe the majority of effects observed in vitro and in vivo on TH17 cell function, including IL-17A expression, are due to REV-ERBα-dependent effects on RORγt expression. Mechanistically, binding of REV-ERBα upstream of Rorc would negatively regulate RORγt expression and, in turn, indirectly regulate IL-17A expression in vitro and in vivo. Therefore, the loss of REV-ERBα would have the opposite effect, and one could argue that this would be the sole mechanism for REV-ERBα-mediated regulation of TH17 cell development. However, decreased IL-17A expression was also observed when RORγt was ectopically overexpressed in TH17 cells and REV-ERB-specific small molecules were added to the culture. Since the expression of RORγt is driven by a retroviral element, REV-ERBα could not regulate RORγt at this level. Thus, our data indicate a level of complexity to the cross-talk between the RORs and REV-ERBs in TH17 cell development that extends beyond simple competition for DNA response elements. At one level, RORγt and REV-ERBα compete for binding at the Il17a locus to contain the inflammatory response. At another level, the RORs and REV-ERBα drive and inhibit each other’s expression (Raspè et al., 2002; Takeda et al., 2012), with the RORs potentially driving REV-ERBα expression upon development in order for REV-ERBα to limit not only cytokine expression but also expression of RORγt itself to help temper TH17 proinflammatory responses.
Our observations that REV-ERBα acts as a potent negative regulator of proinflammatory TH17 cell development and auto-immunity contradicts a previous study by Yu et al. (2013) where they demonstrated that REV-ERBα functionally repressed Nfil3, which, in turn, repressed RORγt to regulate Il17a expression and TH17 cell development, indicating an indirect role for REV-ERBα in the regulation of TH17 cell development. In the study by Yu et al. (2013), KO mice presented with decreased IL-17A expression in vitro and in vivo, and although the different gut flora compositions between the facilities could be a factor affecting the development of TH17 cells (Lee and Kim, 2017; Luo et al., 2017; Yang et al., 2014), or the use of different REV-ERBα KO strains (Chomez et al., 2000; Preitner et al., 2002), it is important to note that the studies performed by Yu et al. (2013) were largely in naive mice with the purpose of elucidating the diurnal regulation of TH17 cell function in vivo. In that setting, REV-ERBα and NFIL3 cooperatively regulated TH17 cells in the gut in a clock-dependent manner (Yu et al., 2013). In contrast, our work was performed under specific autoimmune- or chronic proinflammatory-inducing conditions, which are known to be TH17 driven, to specifically address the role of REV-ERBα outside of its function as a core clock component. Under the types of stressors used in our study here, REV-ERBα is required to restrain proinflammatory responses, and in the absence of REV-ERBα, aberrant TH17 responses and autoimmunity ensue. Supporting this notion, microarray analysis of CNS tissues (Pareek et al., 2011), RNA-sequencing of spinal cords (Sevastou et al., 2016), and mRNA analysis of CNS (Sutton et al., 2017) from mice with active EAE all demonstrate a significant decrease in REV-ERBα expression. These data correlate with the findings from our EAE experiments, wherein the loss of REV-ERBα exacerbates EAE, and enhanced REV-ERB activity, through use small molecules agonists (SR9009 or SR12418), suppresses EAE. Additionally, we showed that T cell-specific loss of REV-ERBα exacerbated the development of colitis, further supporting the notion that REV-ERBα acts as a critical negative regulator of proinflammatory TH17-mediated responses.
Despite the in vivo differences between our study and Yu et al. (2013), we did observe REV-ERB-mediated effects on Nfil3 in TH17 cells, which was likely a function of REV-ERBα binding at the Nfil3 promoter region. However, the effects do not appear to be sufficient to override the direct effects of REV-ERBα at Il17a or RORγt itself, as we observed reduced IL-17A expression in REV-ERBα-overexpressing cells and cells treated with a synthetic REV-ERB ligand. Interestingly, despite the increased expression of Nfil3 in KO TH17 cells, we still observed an increased expression of RORγt and IL-17A (Yu et al., 2013). This could be a function of protein degradation because NFIL3 has been shown to be post-translationally modified and targeted for proteasomal degradation in a CK1ε-dependent manner (Doi et al., 2004). However, overexpression and knock down of Nfil3 also had little effect on IL-17A expression, which is consistent with a previous study (Ciofani et al., 2012) finding that retroviral transduction of Nfil3 did not significantly affect TH17 cell development. This conflicting in vitro data could be due to differences in cell culture conditions, reagents, or transduction protocols. Clearly the network linking REV-ERBα and NFIL3 in TH17 cells is complex and further work is needed to elucidate the interplay between these transcription factors in proinflammatory TH17 cell development. Overall, our data provide a new perspective on REV-ERBα, revealing its negative regulatory role of proinflammatory TH17-mediated immune responses.
Modulation of nuclear receptor activity has proven to be a powerful and effective means to treat a host of diseases (Marciano et al., 2014). We have developed and described several REV-ERB-specific small molecules exhibiting various degrees of in vivo exposure (Solt et al., 2012). Using these small molecules, we demonstrated that REV-ERB agonism is sufficient to inhibit TH17 cell development in vitro by inhibiting TH17-specific genes (e.g., Il17a, Il22, and Il23r) as well as RORγt expression, which is consistent with our genetic experiments. Furthermore, REV-ERB-specific small molecules also inhibit the development of TH17 cells in vivo, ameliorating the signs and incidence of EAE, a TH17-mediated autoimmune disease. Importantly, REV-ERB-specific small molecules are efficacious when used therapeutically, blocking the development of colitis and preventing disease relapse in a relapsing-remitting model of multiple sclerosis. It is possible that some of the in vivo effects observed with SR12418 are a function of systemic exposure, which would target the REV-ERBs in other tissues, including macrophages, which have been demonstrated to express the REV-ERBs and can act as antigen-presenting cells (Eichenfield et al., 2016; Gibbs et al., 2012; Lam et al., 2013). Additionally, SR12418 and SR9009 target both REV-ERBα and REV-ERBβ, and although the focus of this work was on REV-ERBα, our data do not rule out effects on REV-ERBβ. In fact, REV-ERBβ is differentially expressed in TH17 cells, and its overexpression did inhibit TH17-mediated cytokine expression, but it was not as potent as REV-ERBα. Thus, the combined deletion of both REV-ERBs may have a more exacerbated effect in vivo on disease course. By this logic, it would stand to reason that ligands that target both REV-ERBs would also have a greater repressive effect than a ligand that targeted only a single receptor subtype.
In summary, we demonstrate here that the nuclear receptor REV-ERBα is a critical, cell-intrinsic negative regulator of proinflammatory TH17-mediated autoimmunity, competing with RORγt at their shared target gene sequences, of which Il17a is one. Although there is still much work to be done to fully elucidate the function of REV-ERBα in TH17 cells, our work also demonstrates the therapeutic potential for targeting REV-ERBα for the treatment of TH17-mediated autoimmune disorders. Currently, there is a massive pharmaceutical effort to develop RORγ-selective inverse agonists for the treatment of TH17-mediated disorders. However, these RORγ synthetic ligands pheno-copy Rorc−/− mice by rapidly inducing thymic apoptosis, whereas REV-ERB ligands do not. Our data indicate that the use of REV-ERB-specific small molecules may be an effective, alternative approach to treat TH17-mediated diseases.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Laura Solt (lsolt@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
The following mouse strains used were purchased from the Jackson Laboratory and/or were bred at Scripps Research - (Florida). C57BL/6J (B6); SJL/J, B6.Cg-Nr1d1tm1Ven/LazJ (KO) (Chomez et al., 2000); B6.129S7-Rag1tm1Mom/J (Rag1−/−) (Mombaerts et al., 1992); B6(Cg)-Rorctm3Litt/J (Rorcfl/fl) (Ivanov et al., 2006); Tg(Cd4-cre)1Cwi/BfluJ (CD4 Cre). Rorcfl/fl mice were crossed with CD4 Cre mice to generate RORcfl/fl × CD4 Cre mice. All experiments were conducted at controlled temperature (22–23°C), humidity ~60%, and 12h:12h light:dark cycles. Mice had access to regular chow (Harlan 2920X) and water, ad libitum. For all in vitro experiments, both male and female mice (8–10 weeks old) were sacrificed between 8 and 10am. For all in vivo experiments, 7–10 week-old male and female mice were used, and were sacrificed between 7 and 11am. For EAE experiments, mice were immunized between 11am and 1pm. The specific age and sex of the mice for experiments is described under each model’s details below. All mice were maintained under specific pathogen free conditions. All studies conform to and were approved by the Institutional Animal Care and Use Committee (IACUC) at the Scripps Research Institute (Florida).
Cell lines
HEK293 (female), EL4 (unknown), and Plat-E (female) cells were cultured in DMEM supplemented with 10% FBS, 2mm L-glutamine, and 1% penicillin/streptomycin at 37°C, 5% CO2 under standard culture conditions. Lymphocytes were cultured in IMDM medium with 10% FBS, 100IU/mL penicillin, 100 μg/ml streptomycin, 50uM β-mercaptoethanol, and 2mm L-glutamine. The gender of EL4 cells is unavailable at this time.
METHOD DETAILS
Chemical synthesis and reagents
SR9009 has been previously described (Solt et al., 2012). Synthesis of SR12418 is as follows:
(S)-(3-((4-(tert-Butoxy)phenoxy)methyl)-6-fluoro-3,4-dihydroisoquinolin-2(1H)-yl)(naphthalen-1-yl)methanone
Step 1: (S)-3-carboxy-6-fluoro-1,2,3,4-tetrahydroisoquinolin-2-ium chloride
To a suspension of 3-fluoro-L-phenylalanine (10.0 g, 55.4 mmol) in conc. HCl (50 mL) was added aq. formaldehyde solution (37% wt.; 6.2 mL, 83.3 mmol). The reaction mixture was heated to 90°C and stirred for 5 h and the completion of the reaction was monitored by anal. HPLC. The mixture was then cooled to room temperature and filtered to give the title compound as the HCl salt (10.8 g, 84% yield), which was used without further purification. 1H NMR (400 MHz, d-DMSO) δ 14.17 (broad S, 1H), 10.12 (broad S, 2H), 7.37–7.22 (m, 1H), 7.20–7.13 (m, 2H), 4.45–4.41 (m, 1H), 4.33 (m, 2H), 3.37 (dd, J = 16 Hz, 4.8Hz), 3.22–3.15 (m, 1H); 13C NMR (100 MHz, d-DMSO) 169.6, 161.1 (d, J = 242 Hz), 133.5 (d, J = 8.2 Hz), 128.6 (d, J = 8.4 Hz), 124.6 (J = 2.7 Hz), 115.0 (d, J = 21.8 Hz), 114.1(d, J = 21.8 Hz), 52.6, 43.2 and 28.0; 19F NMR (376.5 MHz, DMSO-d6) δ −115.19, −119.13; MS (ESI) 196.1 (M + H).
Step 2: (S)-6-fluoro-3-(hydroxymethyl)-1,2,3,4-tetrahydroisoquinolin-2-ium chloride
BH3.DMS (8.1 mL, 85.6 mmol) was added slowly to ((S)-3-carboxy-6-fluoro-1,2,3,4-tetrahydroisoquinolin-2-ium chloride (6.6 g, 28.5 mmol) and anhydrous THF (60 mL) at RT under argon. The mixture was then heated to 70°C for 3h monitoring the reaction by analytical HPLC. When the starting material was consumed, the reaction was cooled to RT and the reaction was quenched with THF/water (1:1) followed by dilute HCl (2M, 50 mL). The mixture was heated to 80°C for 3h and then cooled and concentrated to obtain the title compound as the HCl salt, which was used without further purification. 1H NMR (400 MHz, d-DMSO) δ 9.88 (broad S, 1H), 9.61 (broad S, 1H), 7.36–7.32 (m, 1H), 7.15–7.10 (m, 2H), 5.61 (broad s, 1H), 4.27 (s, 2H), 3.84–3.68 (m, 2H), 3.51 (broad s, 1H), 3.0 (d, J = 8.0 Hz); 13C NMR (100 MHz, d-DMSO) 161.1 (d, J = 242 Hz), 134.4 (d, J = 8.1 Hz), 128.6 (d, J = 8.4 Hz), 125.0 (J = 2.8 Hz), 115.0 (d, J = 21.3 Hz), 113.7 (d, J = 21.5 Hz), 60.4, 53.8, 43.3 and 27.4; 19F NMR (376.5 MHz, DMSO-d6) d 115.24, 119.22; MS 182 (M + (ESI) H).
Step 3: (S)-(6-Fluoro-3-(hydroxymethyl)-3,4-dihydroisoquinolin-2(1H)-yl)(naphthalen-1-yl)methanone
To a mixture of the above crude (S)-6-fluoro-3-(hydroxymethyl)-1,2,3,4-tetrahydroisoquinolin-2-ium chloride (28.5 mmol) in CH2Cl2 (100 mL) and NaHCO3 (12 g, 142.6 mmol) was slowly added 1-naphthoyl chloride (4.71 mL, 31.4 mmol). The reaction was stirred at RT overnight. Water was added and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2×150 mL). The combined organic layers were washed with brine, dried (Na2SO4), and concentrated. The resulting crude residue was purified by chromatography on silica gel (EtOAc/hexanes) to afford the title compound (6.94 g, 73% yield by 2 steps). 1H NMR (400 MHz, d-DMSO) δ 8.06–6.88 (m, 10H), 5.52–5.10 (m, 1H), 4.44–2.77 (m, 7H); MS (ESI) 336 (M + H).
Step 4: (S)-(3-((4-(tert-Butoxy)phenoxy)methyl)-6-fluoro-3,4-dihydroisoquinolin-2(1H)-yl)(naphthalen-1-yl)methanone
To the solution of (S)-(6-fluoro-3-(hydroxymethyl)-3,4-dihydroisoquinolin-2(1H)-yl)(naphthalen-1-yl)methanone (5.19 g, 15.5 mmol), 4-(tert-butoxy)phenol (2.83 g, 17.0 mmol) and n-Bu3P (7.6 mL, 31.0 mml) in anhydrous THF (100mL) under argon was added dropwise 1,1’-(azodicarbonyl)dipiperidine (ADDP) (7.8 g, 31.0 mmol) in anhydrous THF (70 mL). The reaction was stirred at RT overnight and the completion of the reaction was monitored by analytical HPLC. The precipitate was filtered and washed with hexane. The resulting filtrate was concentrated in vacuo to give the crude product which was purified by chromatography on silica gel (EtOAc/hex) to afford the title compound as a colorless solid (5.2 g, 69% yield); 1H NMR (400 MHz, CDCl3) δ 8.08–6.41 (m, 14H), 5.71–5.51 (m, 1H), 4.46–4.15 (m, 2H), 3.91–3.61 (m, 2H), 3.10–3.02 (m, 2H), 1.33–1.25 (m, 9H); 19F NMR (376.5 MHz, CDCl3) δ −114.88, −114.93, −115.55, −115.57; HRMS (ES+): m/z called for C31H30FNO3 [M+H]: 484.2210; found 484.2294.
Induction and clinical evaluation of EAE
Chronic, MOG-induced EAE was induced in 10-week-old, female WT or KO mice by subcutaneous (s.c.) injections over two sites in the flank with 150mg per mouse of MOG35–55 peptide in an emulsion with CFA supplemented with 2.25 μg/ml Mycobacterium tuberculosis, strain H37Ra (500ug per mouse). Pertussis toxin was dissolved in PBS and injected i.p. at 200 ng per mouse 3 hr post immunization (Day 0) and 24 h later. For chronic, MOG-induced drug treatment experiments, 10-week-old, female B6 mice were immunized using EAE induction kits according to manufacturer’s instructions. For R-EAE, 10-week old, female SJL/J mice were immunized using EAE induction kits according to manufacturer’s instructions. Mice were scored daily on a scale of 0–5 in a double-blinded manner using the following criteria: 0, no clinical disease; 1, limp/flaccid tail; 2, limp tail and hind leg weakness; 3, limp tail and complete paralysis of hind limbs; 4, limp tail, complete hind limb and partial front limb paralysis; 5, quadriplegia or pre-moribund state. Gradations of 0.5 were used when mice exhibited signs that fell between two scores. For drug treatment experiments, SR12418 was dissolved in a 10% DMSO, 10% Tween 80, and 80% H2O solution equaling 5μg/ml and administered intraperitoneally (i.p.) at 50mg/kg as was vehicle control (10% DMSO, 10% Tween 80, and 80% H2O) twice per day (b.i.d). The treatment was started the evening of the immunization (MOG-EAE), or once the animals recovered from the first wave of disease (R-EAE) and continued for the duration of the experiment. For all b.i.d. dosing, animals were dosed at 7am and 7pm (lights on/lights off). At the termination of the experiment T cells were isolated from brain and spinal cord following perfusion of deeply anesthetized mice with cold PBS. CNS was minced before being passed through a 70um mesh cell strainer. Single cell suspensions were centrifuged and resuspended in 37% percoll. After further centrifugation, supernatant was removed and the pelleted mononuclear cells were washed and used for staining.
T cell-transfer colitis model
Spleens were collected from 8- to 10-week-old, female KO or control (WT) mice in order to sort naive CD4+ T cells. 5 × 105 CD4+CD25−CD62LhiCD44lo cells suspended in PBS were adoptively transferred i.p. (100ul/mouse) into 8-week-old, female Rag1−/− recipient mice and monitored bi-weekly for body weight change. Beddings were routinely transferred between cages to distribute microflora and limit bias between cages regarding disease development. For drug treatment experiments, vehicle or SR12418 was administered starting at three-weeks post T cell transfer and continued for the duration of the experiment following procedures and time points described in the EAE experiments above. Mice were sacrificed to assess histological inflammation due to ethical requirements if they reached 80% or less of their original body weight. At the termination of the experiment, the whole colon and ileum (distal 1/3 part of the small intestine) were removed from the mouse. Colon length was measured first before each segment of the intestine was opened longitudinally and the fecal contents gently removed. Next, colon weights were measured prior to dissection longitudinally into two sections. One of the sections was rolled (Swiss roll) from the rectum along with the ileum and fixed in 10% neutral buffered formalin for 24 h. Swiss roll samples were then embedded in paraffin wax and sectioned for histological examination and immunohistochemistry staining. The other section of the colon was dissected in equal halves, designated as ‘proximal’ and ‘distal’ colon and snap frozen in dry ice for RNA analysis. Histological Assessment of colon - following overnight formalin fixation, paraffin-embedding, and standard H&E staining, histological colitis scoring of H&E-stained sections was performed blinded. Assessments of crypt architecture, crypt abscesses, tissue damage, goblet cell depletion, inflammatory cell infiltration, and neutrophil counts were assigned scores with the maximum combined score for each part of the intestine equaling 25(Wang et al., 2015). For cell isolation - whole colons were removed, flushed with PBS to remove fecal contents, and opened longitudinally. Tissues were cut into small segments and incubated for 30 min at room temperature in DMEM without phenol red plus 0.15% DTT. After washing with media, intestines were incubated for 30 min at room temperature in media containing 1 mm EDTA to remove the epithelium. After washing again with media, lamina propria was digested in media containing 0.25 μg/ml liberase TL and 10 U/mL RNase-free DNaseI in a bacterial incubator for 15–25 min at 37°C. Single cell suspensions were passed through 70 mm mesh cell strainer and mononuclear cells were isolated by 70/30% percoll gradient centrifugation. Monocuclear cells were washed two more times, counted, and used for FACS analysis.
Analysis of thymocytes
For the measurement of DP survival kinetics, male, 7–9 week-old, male C57BL/6 mice were treated with vehicle control or compounds for 3 days. All compounds were formulated in 15% cremophor at the following concentrations unless otherwise noted: SR2211 – 2μg/ml; SR9009 – 10μg/ml; SR12418 – 5μg/ml, and administered i.p., b.i.d. At the termination of the experiment, thymii were collected, minced, and passed through a 70um mesh cell strainer. Cell counts were performed prior to staining the cells for flow cytometry analysis.
In vitro CD4+ T cell differentiation
Naive CD4+ T cells from spleen and LNs of 8–10-week-old male and female mice were purified after removing the red blood cells using Lympholyte-M solution. Cells were enriched for naive CD4+ T cells using the mouse naive CD4+ T Cell Isolation Kit according to the manufacturer’s instruction. If sorting was performed (FACS Aria II; BD Bioscience), the CD4+CD25−CD62LhiCD44lo fraction was collected. The conditions for the different TH cell subsets were: For TH0 (neutral conditions): 5 μg/ml anti-IL-4 and 5 μg/ml anti-IFNγ; For TH1 conditions: 5 μg/ml anti-IL-4, 20 ng/ml IL-12 and 10ng/ml IFNγ; For TH2 conditions: 5 μg/ml anti-IFNγ and 10ng/ml IL-4; For TH17 conditions: 5 μg/ml anti-IFNγ, 5 μg/ml anti-IL-4, 1.5ng/ml TGFβ and 30ng/ml IL-6; For iTreg conditions: 5 μg/ml anti-IFNγ, 5 μg/ml anti-IL-4, and 5ng/ml TGFβ. Other cytokines used for various TH17 conditions: IL-1b (10ng/ml), IL-21 (20ng/ml), and IL-23 (20ng/ml). 1 × 106 cells/ml of naive CD4+ T cells were activated with anti-CD3 and anti-CD28 by precoating plates with 100 μg/ml goat anti-hamster IgG. After 48 h, cells were removed from the TCR signal and recultured at a concentration of 1 × 106cells/ml. Four days after activation, all cells were restimulated with 50ng/mL phorbol-12-myristate-13-acetate (PMA)and 1 μg/ml ionomycin for 2 hr with the addition of GolgiStop for an additional 2 hr before intracellular staining. Cells were cultured in IMDM medium with 10% FBS, 100IU/mL penicillin, 100 μg/ml streptomycin, 50uM β-mercaptoethanol, and 2mm L-glutamine. All cultures were performed in a volume of 200ul in 96-well U-bottomed plates.
Flow cytometry
Surface staining: single cell suspensions prepared from spleen, LNs, colons, CNS, etc. were washed and stained with fluorescenceconjugated antibodies for 20 min, washed, then resuspended in FACS buffer (0.5% BSA, 2mm EDTA in PBS). Intracellular cytokine staining: cells were re-stimulated with 50ng/mL PMA and 1 μg/ml ionomycin for 2 hr with the addition of GolgiStop for an additional 2 hr. Cells were then surface stained using procedures outlined above, fixed and permeabilized using the Foxp3 staining kit. Flow cytometric analysis was performed on a BD LSRII (BD Biosciences) instrument and analyzed using FlowJo software.
Retroviral Transduction
To generate murine REV-ERBα or REV-ERBβ retroviral vectors, mouse REV-ERB sequences were inserted into the MIGR1 vector using the XhoI site and further screened for orientation. The DBD truncation construct was generated by PCR amplification of residues 103–225 of REV-ERBα with addition of 5′ XhoI and 3′ HpaI cut sites. The amplified product was cloned into the MIGR1 vector by double digest followed by T4 DNA ligation. The ΔDBD deletion construct was generated by a single PCR reaction of the MIGR1-REV-ERBα construct to delete residues 132–197. To generate the murine NFIL3 retroviral vector, the mouse NFIL3 sequence was inserted into the MIGR1 vector using the XhoI and BamHI sites. In each experiment, MIGR1 empty vector was used as a control. shRNAmirs against mouse CD8 (Cd8a) and Nfil3 were purchased from TransOMIC Technologies. shRNAmirs were PCR amplified and cloned into ametrine-expressing murine retroviral vectors (LMPd) containing the enhanced miR-30 cassette(Fellmann et al., 2013). MIGR1 RORγt retroviral construct was a gift from Dan Littman (Addgene Plasmid #24069). Virus production: Plat-E cells were cultured in DMEM containing 10% fetal bovine serum, 2mm L-glutamine, and 1% penicillin/streptomycin at 37°C under standard culture conditions. Plat-E cells were seeded at 350,000 cells/ml in a 6 well plate the day before transfection. 3 mg total retroviral plasmid DNA (1.5 μg MIGR1 plus 1.5 μg pCL-Eco) was transfected using Fugene6 reagent according to manufacturer’s protocol. Viral supernatant was harvested 48 hr post transfection and used immediately for transduction. For retroviral transduction, naive CD4+ T cells were stimulated as indicated with anti-CD3 and anti-CD28 and cultured under TH17 conditions. At 24 h post TCR priming, the culture medium was replaced with virus supplemented with 8 μg/ml polybrene. Plates were centrifuged at 1,800 rpm for 90 h at 37°C and then incubated at 37°C for 3–4 h. After this time, the medium was replaced with the original media removed before addition of virus.
Luciferase reporter assays
HEK293 cells were plated 24 hr prior to transfection in 96-well plates at a density of 15 × 103 cells/well. Transfections were performed using Lipofectamine 3000 according to manufacturer’s protocol. To generate the DBD REV-ERBα truncation construct, residues 103–224 of the human REV-ERBα were cloned into the pcDNA3.1+ plasmid by HindIII and BamHI double digest. The ΔDBD deletion construct in was generated by PCR deletion of residues 131–197 of pcDNA3.1+ REV-ERBα. For drug treatments - 16 hr post-transfection, cells were treated with vehicle or compound. 24 hr post-treatment luciferase activity was measured using BriteLite and read using an Envision multilabel plate reader (PerkinElmer Life and Analytical Sciences). All values were normalized to DMSO to produce fold induction values. For assays in which drug treatment did not occur, luciferase activity was measured 24h post-transfection by DualGlo firefly and renilla luciferase reagents. Mouse EL4 cells were transfected using the Amaxa cell line nucleofector kit L according to manufacturer’s instructions. 6 hr post-nucleofection, EL4 cells were stimulated with PMA (50ng/ml) and ionomycin (1ug/ml), plated in 12 well plates and incubated overnight for 16 hr. The following day, luciferase activity was measured as indicated for HEK293 cells. The pGL4 mIl17a-2kb promoter + CNS5 was a gift from Warren Strober (Zhang et al., 2008). The pGL3 mBmal1 luciferase reporter has been previously described (Solt et al., 2012; Yu et al., 2002).
ChIP
ChIP assays were performed following manufacturer’s instructions. Briefly, naive CD4+ T cells were differentiated under TH17 conditions for 3 days, fixed in 1% formaldehyde for 10 min at room temperature, quenched in glycine (120mm) on ice for 5 min, and washed with PBS prior to lysis. Chromatin (20 × 106 cells/condition) was sheared with a Misonix S-4000 sonicator for 20 × 10 s cycles at 20% amplitude to yield 100–300 bp DNA fragments. After removing 1% as input DNA, immunoprecipitation was performed by adding anti-REV-ERBα antibody (Cho et al., 2012) (4ug/reaction) along with protein G magnetic beads on an end-to-end rotator at 4°C overnight. KO TH17 cells and rabbit IgG were used as negative controls. Beads were then washed 3X with low salt ChIP buffer, followed by high salt ChIP buffer and eluted by re-suspending the beads in Elution Buffer. Both input and ChIP DNA were then incubated at 65 C for 30 min, followed by addition of Proteinase K and incubated at 65°C to reverse crosslink for 2 hr. DNA was then purified with QIAquick columns per manufacturer’s instructions and resuspended in a 50 mL volume. Real-time PCR detection of immunoprecipitated targets was performed using SYBR Green including passive reference dye (ROX) on a HT7900 Fast Real Time PCR system (Life Technologies, CA). A standard curve was generated for each sample based on amplification of serial dilutions of input DNA. ChIP DNA PCR reactions were performed in duplicates. Melt curves were analyzed to ensure amplification of specific target sequences. The primers used for qRT-PCR can be found in Table S3.
Quantitative real-time PCR
For in vitro cultures, total RNA was extracted using a RNeasy Plus Micro Kit and reverse transcribed using iScript cDNA biosynthesis kit. Tissue samples (snap frozen and homogenized) - following isolation using TRIzol reagent mRNA was purified using RNeasy columns, followed by cDNA synthesis using iScript containing oligo (dT) and random hexamer primers. Real-time PCR was performed using SYBR Green including passive reference dye on a HT7900 Fast Real Time PCR system (Life Technologies, CA). All gene expression data were normalized to the housekeeping gene, b-actin unless otherwise mentioned, using a DD cycle threshold-based algorithm followed by fold change comparison with the average of the control group. Primer efficiencies were determined using complementary DNA and primer dilutions for each gene of interest. Primers sequences for specific genes are provided in Table S3.
RNA-sequencing and data analysis
mRNA was extracted from TH17 cells on Day 2 (WT versus REV-ERBα KO cells) or Day 3 (MIGR1 transduced cells) of in vitro differentiation. Total RNA was extracted using QIAGEN RNeasy kits, quantified using the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA), and run on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) for quality assessment. DNase-treated total RNA (300ng) was depleted of ribosomal RNA (rRNA) using appropriate probes provided by Illumina and further assessed on the bio-analyzer to confirm 18S and 28S rRNA peaks are depleted. rRNA-depleted RNA is processed using the TruSeq Stranded Total RNA sample prep kit. Briefly, RNA samples are chemically fragmented in a buffer containing divalent cations and heating at 94°C for 8 min. The fragmented RNA is random hexamer primed and reverse transcribed to generate first strand cDNA. The second strand is synthesized after removing the RNA template and incorporating dUTP in place of dTTP. cDNA is then end repaired and adenylated at their 3′ ends. A corresponding ‘T’ nucleotide on the adaptors is utilized for ligating the adaptor sequences to the cDNA. The adaptor ligated DNA is purified using magnetic Ampure XP beads and PCR amplified using 12–13 cycles to generate the final libraries. The final libraries are size selected and purified using 1.0 × Ampure XP beads to remove any primer dimers. The final library size is typically 200–600bp with insert sizes ranging from 80–450bp. Final libraries are validated using bioanalyzer DNA chips and qPCR quantified using primers recognizing the Illumina adaptors. Libraries are pooled at equimolar ratios, quantified using qPCR (quantification of only the adaptor-ligated libraries) and loaded onto the NextSeq 500 flow cell at 1.8pM final concentration for pair end 75bp reads. 20–25 million mappable reads per sample were collected. Demultiplexed and quality filtered raw reads (fastq) generated from the NextSeq 500 were trimmed (adaptor sequences) using Flexbar 2.4 and aligned to the reference genome using TopHat version 2.0.9 (Trapnell et al., 2009). HT seq-count version 0.6.1 was used to generate gene counts and differential gene expression analysis was performed using Deseq2 (Anders and Huber, 2010). The normalized gene counts were used to plot the heatmaps using the heatplot package in R. To determine enriched functional groups in the RNA-seq data, KEGG pathway analysis was performed using DAVID (Huang et al., 2009a; Huang et al., 2009b).
TR-FRET corepressor interaction assay
The TR-FRET assay was performed in black low-volume 384-well plates (Greiner). Each well contained 4 nM 6xHistag-REV-ERBα LBD (human; residues 281–614) or 6xHistag-REV-ERBβ LBD (human; residues 212–579) protein expressed in and purified from Escherichia coli using nickel affinity and size exclusion chromatography; 1 nM LanthaScreen Elite Tb-anti-HIS Antibody; and 400 nM FITC-labeled peptide derived from the SMRT corepressor containing a N-terminal FITC label with a six-carbon linker (Ahx) and an amidated C terminus for stability in TR-FRET buffer (20 mm potassium phosphate, pH 7.4, 50 mm potassium chloride, 1 mm dithiothreitol, and 0.005% Tween-20). Ligand stocks were prepared via serial dilution in DMSO, added to wells in triplicate, and plates were incubated at 4°C for 2 h and read using BioTek Synergy Neo multimode plate reader. The Tb donor was excited at 340 nm; its fluorescence emission was monitored at 495 nm, and the acceptor FITC emission was measured at 520 nm; and the TR-FRET ratio was calculated as the signal at 520 nm divided by the signal at 495 nm. Data were plotted using GraphPad Prism as TR-FRET ratio versus ligand concentration and fit to sigmoidal dose response curve equation.
Immunoblot Analysis
T cells were washed once with phosphate-buffered saline and then incubated for 10 min at 4°C in 100 mL of TNT lysis buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, and 1% Triton X-100) containing protease inhibitors. Samples were then vortexed for 30 s and centrifuged (14,000RPM for 10 min). Protein levels in the supernatants were determined using a Coomassie protein assay kit, and 15 mg of protein from each sample was separated by SDSPAGE (10%) and transferred to a PVDF membrane and immunoblotted with primary antibodies: mouse RORγt and Actin. KLH-conjugated (LifeTein, LLC.) peptides were designed to generate paralog-specific antibodies recognizing epitopes spanning amino acid residues 268 to 279 of REV-ERBα and were injected in rabbits, similar to what has been previously described (Cho et al., 2012). Peptides were synthesized by LifeTein, LLC. (Somerset, NJ, USA). Immunization and serum collection were performed by Pocono Rabbit Farm and Laboratories (Canadensis, PA), and antibodies were purified by peptide affinity chromatography. Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson Immunoresearch. Detection of the bound antibody by enhanced chemiluminescence was performed according to the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as the mean ± SEM. All statistical analyses were performed using GraphPad PRISM 6. Student’s t test was used for comparison between two groups. To compare differences between groups in vivo, a 2-way ANOVA with Bonferroni’s multiple comparison test was performed if values were derived from a normal distribution. Where a normal distribution could not be confirmed or sample size was small, nonparametric Mann-Whitney U tests with a post hoc test were performed. A p value of < 0.05 was considered statistically significant. The number of sample replicates and statistical cut-offs used in the analysis of genomics data are indicated in the figure legends or the text.
DATA AND SOFTWARE AVAILABILITY
All next-generation sequencing data generated for this paper were deposited in the Gene Expression Omnibus (GEO) under accession number GEO: GSE122726.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| LanthaScreen Elite Tb-anti-His Antibody | Thermo Fisher | Cat # PV5863 | |
| Mouse RORγt (immunoblot) | eBioscience | Cat # MA5-16227; RRID: AB_2537745 | |
| Actin, clone C4 | EMD Millipore | Cat # MAB1501 | |
| Rabbit polyclonal anti-REV-ERBα | This paper | N/A | |
| Purified anti IL-4, clone 11B11 | Biolegend | Cat# 504115; RRID: AB_2295885 | |
| Purified anti-IFNγ, clone XMG1.2 | Biolegend | Cat# 505834; RRID: AB_11150776 | |
| Soluble anti-CD3, clone 2C11 | eBioscience | Cat# 16-0031-86; RRID: AB_468849 | |
| Soluble anti-CD28, clone 37.51 | eBioscience | Cat# 16-0281-86; RRID: AB_468923 | |
| Goat anti-Hamster IgG | MP Biomedicals | Cat # 856984 | |
| BUV395 Hamster anti-mouse CD3e, clone 145-2C11 | BD Biosciences | Cat# 563565; RRID: AB_2738278 | |
| BV711 anti-mouse CD4, clone RM4-5 | Biolegend | Cat# 100549; RRID:AB_11219396 | |
| FITC anti-mouse CD8a, clone 53-6.7 | eBioscience | Cat# 11-0081-85; RRID: AB_464916 | |
| FITC anti-mouse CD19, clone eBio1D3 | eBioscience | Cat# 11-0193-86; RRID: AB_657665 | |
| BV605 anti-mouse CD25, clone PC61 | Biolegend | Cat# 102035; RRID: AB_11126977 | |
| PerCP-Cy5.5 anti-mouse/human CD44, clone IM7 | Biolegend | Cat# 103031; RRID: AB_2076206 | |
| A700 anti-mouse CD45, clone 30-F11 | Biolegend | Cat# 103128; RRID: AB_493715 | |
| BV421 Rat anti-mouse CD62L, clone MEL-14 | BD Biosciences | Cat# 562910; RRID: AB_2737885 | |
| eFluor 660 anti-mouse CCR6, clone sirx6 | eBioscience | Cat# 50-7196-80; RRID: AB_11218711 | |
| Pe/Cy7 anti-mouse CXCR3, clone CXCR3-173 | Biolegend | Cat# 126515; RRID: AB_2086740 | |
| FITC anti-mouse B220, clone RA3-6B2 | eBioscience | Cat# 11-0452-82; RRID: AB_465054 | |
| eFluor 660 anti-mouse Foxp3, clone FJK-16 s | eBioscience | Cat# 50-5773-82; RRID: AB_11218868 | |
| PE anti-mouse GM-CSF, clone MP1-22E9 | Biolegend | Cat# 505405; RRID: AB_315381 | |
| PE anti-mouse IL-4, clone 11B11 | Biolegend | Cat# 504103; RRID: AB_315317 | |
| PE/Cy7 anti-mouse IL-10 | Biolegend | Cat# 505025; RRID: AB_11149682 | |
| BV421 anti-mouse IL-17A, clone TC11-18H10 | BD Biosciences | Cat# 563354; RRID: AB_2687547 | |
| PE anti-mouse IL-17F, clone eBio18F10 | eBioscience | Cat# 12-7471-82; RRID: AB_1210742 | |
| PE/Cy7 anti-mouse IFNγ, clone AMG1.2 | eBioscience | Cat# 25-7311-82; RRID: AB_469680 | |
| PE-CF594 anti-mouse RORγt, clone Q31-378 | BD Biosciences | Cat# 562684; RRID: AB_2651150 | |
| BV510 anti-mouse TCRβ, clone H57-597 | BD Biosciences | Cat# 563221; RRID: AB_2738078 | |
| PE/Cy7 anti-mouse TNFα, clone MP6-XT22 | eBioscience | Cat# 25-7321-82; RRID: AB_11042728 | |
| Mouse Fc Block (purified anti-CD16/32), clone 24G2 | BD Biosciences | Cat# 553141; RRID: AB_394656 | |
| Bacterial and Virus Strains | |||
| BL21(DE3) E.coli cells | New England Biolabs | Cat # C2527 | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| FITC-SMRT ID2 (FITC-NH-TNMGLEAIIRKALMGKYDQWEE) | LifeTein, LLC. | N/A | |
| MOG 35-55 peptide | LifeTein, LLC. | LT12018, LT051716 | |
| Incomplete Freunds Adjuvant (IFA) | Fisher | Cat#PI-77145 | |
| Pertussis Toxin | List Biological Labs | Cat# 180 | |
| H27Ra TB | Difco | Cat# DF3114338 | |
| SR2211 | Kumar et al., 2012 | Dr. Ted Kamenecka | |
| SR9009 | Solt et al., 2011 | Dr. Ted Kamenecka | |
| Tween 80 | Sigma-Aldrich | P8074 | |
| Kolliphor EL (Crempahor) | Sigma-Aldrich | C5135 | |
| Phorbol 12-myristate 13-acetate (PMA) | Tocris | Cat # 1201 | |
| Lonomycin | Sigma-Aldrich | Cat # I0634 | |
| Lympholyte-M | Accurate Chemicals | Cat# ACL5035 | |
| Percoll | Sigma-Aldrich | Cat #P1644 | |
| TRIzol | Life Technologies | Cat# 10296-028 | |
| FuGene | Promega | Cat #E2311 | |
| Lipofectamine 3000 | Invitrogen | Cat #L-3000008 | |
| Recombinant mouse IL-12 | R & D Systems | Cat# 419-ML | |
| Recombinant mouse IL-4 | R & D Systems | Cat # 404-ML | |
| Recombinant human TGFβ | R & D Systems | Cat# 240-B | |
| Recombinant mouse IL-6 | R & D Systems | Cat# 406-ML | |
| Recombinant mouse IL-23 | R & D Systems | Cat# 1887-ML | |
| Recombinant mouse IFNγ | R & D Systems | Cat# 485-MI-100 | |
| Recombinant mouse IL-1β | R & D Systems | Cat# 410-ML | |
| Recombinant mouse IL-21 | R & D Systems | Cat# 594-ML | |
| Xhol | NEB | Cat# R0146L | |
| Hpal | NEB | Cat# R0105L | |
| BamHI-HF | NEB | Cat# R3136L | |
| Hindlll-HF | NEB | Cat# R3104L | |
| GolgiStop | BD Biosciences | Cat# 554724 | |
| RNase-free DNasel | Sigma-Aldrich | Cat# 4716728001 | |
| Liberase TL | Sigma-Aldrich | Cat# 5401020001 | |
| EDTA | Amresco | Cat# E177 | |
| DTT (Dithiothreitol) | Sigma-Aldrich | Cat# DTTRO | |
| DMEM | Mediatech | Cat# 10-017-CV | |
| IMDM | Life Technologies | Cat# 30980-097 | |
| L-glutamine | Life Technologies | Cat# 25030-081 | |
| Penicillin/streptomycin | Life Technologies | Cat# 15140-122 | |
| FBS | Gemini | Cat# 100-106 | |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M3148 | |
| Fixable viability dye eFluor 780 | eBioscience | Cat# 65-0865-14 | |
| Fixable viability dye eFluor 506 | eBioscience | Cat# 65-0866-14 | |
| 6xHistag REV-ERBα LBD | Homemade | Dr. Doug Kojetin | |
| 6xHistag REV-ERBβ LBD | Matta-Camacho et al., 2014. | Dr. Doug Kojetin | |
| Polybrene | SantaCruz Biotechnology | Cat # sc-134220 | |
| Critical Commercial Assays | |||
| EasySep mouse Naive CD4+ T cell isolation kit | StemCell Tech | Cat# 19765 | |
| RNeasy Plus Micro Kit | QIAGEN | Cat #74034 | |
| RNeasy Mini kit | QIAGEN | Cat # 74106 | |
| qScript cDNA synthesis Kit | QuantaBio/VWR | Cat #101414-100 | |
| PerfeCTa SYBR Green FastMix | QuantaBio/VWR | Cat# 101414-280 | |
| Dual-Glo Luciferase Assay System | Promega | Cat # E2940 | |
| Britelite Plus | Perkin Elmer | Cat #6066769 | |
| Amaxa EL4 Nucleofector kit | Lonza | Cat #VCA-1005 | |
| Zippy Plasmid Maxi Prep Kits | Zymo | Cat # D4028 | |
| QIAquick Gel extraction kit | QIAGEN | Cat# 28704 | |
| Pierce Coomassie (Bradford) Protein Assay kit | Thermo Fisher | Cat# 23200 | |
| TruSeq Stranded Total RNA Ribo-Zero Gold (H/M/R) | Illumina | Cat# RS-122-2301 | |
| TruSeq Stranded Total RNA v2 with rRNA depletion | Illumina | Cat# RS-122-2001 | |
| SuperSignal West Femto Chemiluminescence | Thermo Fisher | Cat# PI34095 | |
| eBioscience Foxp3 Transcription Factor staining kit | Thermo Fisher | Cat # 00-5523-00 | |
| MOG 35-55/CFA Emulsion PTX (EAE induction kit) | Hooke Laboratories | EK-2110 | |
| PLP 139-151/CFA Emulsion PTX (EAE induction kit) | Hooke Laboratories | EK-2120 | |
| SimpleChIP Plus Sonication Chromatin IP kit | Cell Signaling Technology | Cat # 56383 | |
| Deposited Data | |||
| REV-ERB overexpression RNA-seq | This paper | GEO: GSE122726 | |
| REV-ERBα WT versus KO RNA-seq | This paper | GEO: GSE122726 | |
| Experimental Models: Cell Lines | |||
| HEK293 cells | ATCC | Cat# CRL-1573; RRID: CVCL_0045 | |
| PlatE cells | Cell Biolabs, Inc | Cat # RV-101 | |
| EL4 cells | ATCC | ATCC# TIB-39; RRID: CVCL_0255 | |
| Experimental Models: Organisms/Strains | |||
| C57BL/6 | Jackson Laboratories | Stock# 000664 | |
| SJL/J | Jackson Laboratories | Stock # 000686 | |
| KO (B6.Cg-Nr1d1tm1Ven/LazJ) | Jackson Laboratories | Stock # 018447; RRID: IMSR_JAX:018447 | |
| Rag1−/− (B6.129S7-Rag7tm1Mom/J) | Jackson Laboratories | Stock # 003145 | |
| Rorcfi/fi(B6(Cg)-fiorctm3Litt/J) | Jackson Laboratories | Stock # 008771 | |
| CD4 Cre (Tg(Cd4-cre)1Cwi/BfluJ) | Jackson Laboratories | Stock # 017336 | |
| Oligonucleotides | |||
| Primers for qRT-PCR, see Table S3 | This paper | N/A | |
| ChIP primers Il17a promoter, see Table S3 | Zhang et al., 2008 | N/A | |
| ChIP primers Il17a CNS2, see Table S3 | Zhang et al., 2008 | N/A | |
| ChIP primers Cry1, see Table S3 | This paper | N/A | |
| ChIP primers Nfil3, see Table S3 | Yu et al., 2013 | N/A | |
| ChIP primers Rorc, see Table S3 | This paper | N/A | |
| ChIP primers Hprt, see Table S3 | This paper | N/A | |
| Recombinant DNA | |||
| MIGR1 | Addgene | Addgene plasmid # 9044 | |
| MIGR1 REVERBα | Homemade | N/A | |
| MIGR1 REVERBβ | Homemade | N/A | |
| MIGR1 REVERBα DBD | Homemade | N/A | |
| MIGR1 REVERBα ΔDBD | Homemade | N/A | |
| MIGR1 NFIL3 | Homemade | N/A | |
| pLMPd-Ametrine | Chen et al., 2014 | Dr. Matthew Pipkin | |
| ShRNAmir: Cd8a | TransOMIC | Cat# TLMSU1400-12525 | |
| ShRNAmir: Nr1d2 | TransOMIC | Cat# TRMSU2000-353187 | |
| shRNAmir:Nfil3 | TransOMIC | Cat# TRMSU2000-18030 | |
| MIGR1 RORγt | Addgene | Addgene plasmid #24069 | |
| pGL4 mll17a-2kb promoter+CNS5 luciferase | Zhang et al., 2008 | Addgene plasmid # 20128 | |
| pGL3 Bmal1 luciferase | Yu et al., 2002 | Dr. Tom Burris | |
| pGL4.73 Renilla | Promega | Cat# E691A | |
| pcDNA3.1 REV-ERBα | Homemade | N/A | |
| pcDNA3.1 REV-ERBα DBD | Homemade | N/A | |
| pcDNA3.1 REVERBα ΔDBD | Homemade | N/A | |
| pcDNA3.1 RORγt | Homemade | N/A | |
| pCL-Eco packaging vector | Naviaux et al., 1996 | Addgene plasmid #12371 | |
| Software and Algorithms | |||
| GraphPad Prism | GraphPad Software | https://www.graphpad.com | |
| FlowJo (Version 10.4.1) | TreeStar | https://www.flowjo.com | |
| R studio | R studio | N/A | |
| DAVID | LHRI | https://david.noifcrf.gov | |
Highlights.
REV-ERBα is upregulated in TH17 cells
REV-ERBα deficiency exacerbates TH17-mediated diseases, including EAE and colitis
REV-ERBα competes with RORγt to modulate TH17-signature genes, including Il17a
REV-ERBα-specific ligands suppress the development and progression of autoimmunity
ACKNOWLEDGMENTS
We thank the members of the Pipkin and Sundrud labs for their helpful comments and critiques of experimental design and analysis as well as the Scripps Florida Genomics and Scripps Florida and California Bioinformatics Cores for library preparation, RNA-seq, and data analysis. This work was supported by the Margaret Q. Landenberger Foundation and the NIH (1R01AI116885 to L.A.S.; 1R01GM114420 to D.J.K.). L.B.C. was supported by NSF award 1359369. M.A. was supported through the American Association of Immunologists Careers in Immunology Fellowship Program and the Crohn’s and Colitis Foundation of America (546172).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.101.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Wang Y, Solt LA, Griffett K, Kazantzis M, Amador A, El-Gendy BM, Huitron-Resendiz S, Roberts AJ, Shin Y, et al. (2014). Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nat. Commun 5, 5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner SM, Zbieg JR, and Crawford JJ (2017). RORg antagonists and inverse agonists: a patent review. Expert Opin. Ther. Pat 27, 101–112. [DOI] [PubMed] [Google Scholar]
- Chang MR, Lyda B, Kamenecka TM, and Griffin PR (2014). Pharmacologic repression of retinoic acid receptor-related orphan nuclear receptor g is therapeutic in the collagen-induced arthritis experimental model. Arthritis Rheumatol. 66, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Belanger S, Frederick MA, Li B, Johnston RJ, Xiao N, Liu YC, Sharma S, Peters B, Rao A, et al. (2014). In vivo RNA interference screens identify regulators of antiviral CD4(+) and CD8(+) T cell differentiation. Immunity 41, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH (2008). The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol 8, 458–466. [DOI] [PubMed] [Google Scholar]
- Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, et al. (2012). Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 485, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomez P, Neveu I, Mansén A, Kiesler E, Larsson L, Vennström B, and Arenas E (2000). Increased cell death and delayed development in the cerebellum of mice lacking the REV-ERBα(alpha) orphan receptor. Development 127, 1489–1498. [DOI] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M, et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi M, Okano T, Yujnovsky I, Sassone-Corsi P, and Fukada Y (2004). Negative control of circadian clock regulator E4BP4 by casein kinase Iepsilon-mediated phosphorylation. Curr. Biol 14, 975–980. [DOI] [PubMed] [Google Scholar]
- Eichenfield DZ, Troutman TD, Link VM, Lam MT, Cho H, Gosselin D, Spann NJ, Lesch HP, Tao J, Muto J, et al. (2016). Tissue damage drives co-localization of NF-κB, Smad3, and Nrf2 to direct Rev-erb sensitive wound repair in mouse macrophages. eLife 5, e13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LJ, and Lazar MA (2014). Nuclear receptor Rev-erba: up, down, and all around. Trends Endocrinol. Metab 25, 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. (2010). Generation of pathogenic T(H)17 cells in the absence of TGF-b signalling. Nature 467, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs JE, Blaikley J, Beesley S, Matthews L, Simpson KD, Boyce SH, Farrow SN, Else KJ, Singh D, Ray DW, and Loudon AS (2012). The nuclear receptor REV-ERBα mediates circadian regulation of innate immunity through selective regulation of inflammatory cytokines. Proc. Natl. Acad. Sci. USA 109, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, MacIsaac KD, Chen Y, Miller RJ, Jain R, Joyce-Shaikh B, Ferguson H, Wang IM, Cristescu R, Mudgett J, et al. (2016). Inhibition of RORγT skews TCRα gene rearrangement and limits t cell repertoire diversity. Cell Rep 17, 3206–3218. [DOI] [PubMed] [Google Scholar]
- Harbour SN, Maynard CL, Zindl CL, Schoeb TR, and Weaver CT (2015). Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc. Natl. Acad. Sci. USA 112, 7061–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol 12, 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV, et al. (2011). Digoxin and its derivatives suppress Th17 cell differentiation by antagonizing RORgt activity. Nature 472, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, and Littman DR (2006). The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133. [DOI] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. ; International IBD Genetics Consortium (IIBDGC) (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellev S, Lundsgaard D, Poulsen SS, and Markholst H (2006). Reconstitution of Scid mice with CD4+CD25- T cells leads to rapid colitis: an improved model for pharmacologic testing. Int. Immunopharmacol 6, 1341–1354. [DOI] [PubMed] [Google Scholar]
- Kojetin DJ, and Burris TP (2014). REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov 13, 197–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin D, Wang Y, Kamenecka TM, and Burris TP (2011). Identification of SR8278, a synthetic antagonist of the nuclear heme receptor REV-ERB. ACS Chem. Biol 6, 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Lyda B, Chang MR, Lauer JL, Solt LA, Burris TP, Kamenecka TM, and Griffin PR (2012). Identification of SR2211: a potent synthetic RORγ-selective modulator. ACS Chem. Biol 7, 672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurebayashi S, Ueda E, Sakaue M, Patel DD, Medvedev A, Zhang F, and Jetten AM (2000). Retinoid-related orphan receptor gamma (RORgamma) is essential for lymphoid organogenesis and controls apoptosis during thymopoiesis. Proc. Natl. Acad. Sci. USA 97, 10132–10137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, et al. (2013). Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, and Kim WU (2017). Microbiota in T-cell homeostasis and inflammatory diseases. Exp. Mol. Med 49, e340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, and Weaver CT (2009). Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, et al. (2012). Induction and molecular signature of pathogenic Th17 cells. Nat. Immunol 13, 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees CW, Barrett JC, Parkes M, and Satsangi J (2011). New IBD genetics: common pathways with other diseases. Gut 60, 1739–1753. [DOI] [PubMed] [Google Scholar]
- Lindebo Holm T, Poulsen SS, Markholst H, and Reedtz-Runge S (2012). Pharmacological evaluation of the SCID T cell transfer model of colitis: as a model of Crohn’s disease. Int. J. Inflamm 2012, 412178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo A, Leach ST, Barres R, Hesson LB, Grimm MC, and Simar D (2017). The microbiota and epigenetic regulation of T helper 17/regulatory t cells: in search of a balanced immune system. Front. Immunol 8, 417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano DP, Chang MR, Corzo CA, Goswami D, Lam VQ, Pascal BD, and Griffin PR (2014). The therapeutic potential of nuclear receptor modulators for treatment of metabolic disorders: PPARγ, RORs, and Rev-erbs. Cell Metab 19, 193–208. [DOI] [PubMed] [Google Scholar]
- Matta-Camacho E, Banerjee S, Hughes TS, Solt LA, Wang Y, Burris TP, and Kojetin DJ (2014). Structure of REV-ERBβ ligand-binding domain bound to a porphyrin antagonist. J Biol Chem 289, 20054–20066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, and Cua DJ (2008). Th17 cell differentiation: the long and winding road. Immunity 28, 445–453. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Mongrain V, Ruan X, Dardente H, Fortier EE, and Cermakian N (2008). Clock-dependent and independent transcriptional control of the two isoforms from the mouse RORgamma gene. Genes Cells 13, 1197–1210. [DOI] [PubMed] [Google Scholar]
- Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, et al. ; Collaborative Association Study of Psoriasis (2009). Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet 41, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naviaux RK, Costanzi E, Hass M, and Verma IM (1996). ThepCL vector system: rapid production of helper-free, high-titer, recombinant retrovirsus. J. Virol 70, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel R, Song X, Shin Y, Banerjee S, Kojetin D, Lin L, Ruiz CH, Cameron MD, Burris TP, and Kamenecka TM (2012). Synthesis and SAR of tetrahydroisoquinolines as Rev-erbα agonists. Bioorg. Med. Chem. Lett 22, 3739–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareek TK, Belkadi A, Kesavapany S, Zaremba A, Loh SL, Bai L, Cohen ML, Meyer C, Liby KT, Miller RH, et al. (2011). Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep 1, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Lee Y, and Kuchroo VK (2011). The many faces of Th17 cells. Curr. Opin. Immunol 23, 702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, and Schibler U (2002). The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260. [DOI] [PubMed] [Google Scholar]
- Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, McClure DB, Burris LL, Khorasanizadeh S, Burris TP, and Rastinejad F (2007). Identification of heme as the ligand for the orphan nuclear receptors REVERBalpha and REV-ERBbeta. Nat. Struct. Mol. Biol 14, 1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspè E, Mautino G, Duval C, Fontaine C, Duez H, Barbier O, Monte D, Fruchart J, Fruchart JC, and Staels B (2002). Transcriptional regulation of human Rev-erbαlpha gene expression by the orphan nuclear receptor retinoic acid-related orphan receptor alpha. J. Biol. Chem 277, 49275–49281. [DOI] [PubMed] [Google Scholar]
- Sevastou I, Pryce G, Baker D, and Selwood DL (2016). characterisation of transcriptional changes in the spinal cord of the progressive experimental autoimmune encephalomyelitis Biozzi ABH mouse model by RNA sequencing. PLoS One 11, e0157754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt LA, Kumar N, Nuhant P, Wang Y, Lauer JL, Liu J, Istrate MA, Kamenecka TM, Roush WR, Vidović D, et al. (2011). Suppression of Th17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472, 491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, Shin Y, Liu J, Cameron MD, Noel R, et al. (2012). Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 485, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, and Littman DR (2000). Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288, 2369–2373. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Finlay CM, Raverdeau M, Early JO, DeCourcey J, Zaslona Z, O’Neill LAJ, Mills KHG, and Curtis AM (2017). Loss of the molecular clock in myeloid cells exacerbates T cell-mediated CNS autoimmune disease. Nat. Commun 8, 1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Jothi R, Birault V, and Jetten AM (2012). RORg directly regulates the circadian expression of clock genes and downstream targets in vivo. Nucleic Acids Res 40, 8519–8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Hasnain SZ, Tong H, Das I, Che-Hao Chen A, Oancea I, Proctor M, Florin TH, Eri RD, and McGuckin MA (2015). Neutralizing IL-23 is superior to blocking IL-17 in suppressing intestinal inflammation in a spontaneous murine colitis model. Inflamm. Bowel Dis 21, 973–984. [DOI] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. (2008). T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. (2014). Focused specificity of intestinal Th17 cells towards commensal bacterial antigens. Nature 510, 152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, and Lazar MA (2007). Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789. [DOI] [PubMed] [Google Scholar]
- Yin L, Wu N, and Lazar MA (2010). Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl. Recept. Signal 8, e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Nomura M, and Ikeda M (2002). Interactivating feedback loops within the mammalian clock: BMAL1 is negatively autoregulated and upregulated by CRY1, CRY2, and PER2. Biochem. Biophys. Res. Commun 290, 933–941. [DOI] [PubMed] [Google Scholar]
- Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, Rothman PB, Takahashi JS, and Hooper LV (2013). Th17 cell differentiation is regulated by the circadian clock. Science 342, 727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Meng G, and Strober W (2008). Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat. Immunol 9, 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Fang B, Emmett MJ, Damle M, Sun Z, Feng D, Armour SM, Remsberg JR, Jager J, Soccio RE, et al. (2015). Gene regulation. Discrete functions of nuclear receptor Rev-erba couple metabolism to the clock. Science 348, 1488–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All next-generation sequencing data generated for this paper were deposited in the Gene Expression Omnibus (GEO) under accession number GEO: GSE122726.